

Abstract
How long does it take to equilibrate the unfolded state of a protein? The answer to this question has important implications for our understanding of why many small proteins fold with two state kinetics. When the equilibration within the unfolded state U is much faster than the folding, the folding kinetics will be two state even if there are many folding pathways with different barriers. Yet the mean first passage times (MFPTs) between different regions of the unfolded state can be much longer than the folding time. This seems to imply that the equilibration within U is much slower than the folding. In this communication we resolve this paradox. We present a formula for estimating the time to equilibrate the unfolded state of a protein. We also present a formula for the MFPT to any state within U, which is proportional to the average lifetime of that state divided by the state population. This relation is valid when the equilibration within U is very fast as compared with folding as it often is for small proteins. To illustrate the concepts, we apply the formulas to estimate the time to equilibrate the unfolded state of Trp-cage and MFPTs within the unfolded state based on a Markov State Model using an ultra-long 208 microsecond trajectory of the miniprotein to parameterize the model. The time to equilibrate the unfolded state of Trp-cage is ∼100 ns while the typical MFPTs within U are tens of microseconds or longer.
Keywords: protein unfolded state, protein folding, mean first passage time, Markov state model
Introduction
How long does it take to equilibrate the unfolded state of a protein? The answer to this question has important implications for our understanding of why many small proteins fold with two state kinetics.1–28 The protein folding funnel picture provides key insights.3,5,6 When a protein folds along multiple pathways as suggested by the funnel picture, the folding kinetics will still be two-state regardless of differences in the intrinsic barriers along each pathway if the equilibration within the unfolded state ensemble is much faster than the time it takes to fold. Yet the mean first passage times (MFPTs) between different regions of the unfolded state ensemble are typically much longer than the folding time; this suggests that the time to equilibrate the unfolded state ensemble is much longer than the time to fold.29,30 So there is a paradox: the single exponential kinetics can be explained by very fast equilibration within the unfolded state U relative to folding, but the long MFPTs within U seem to imply that the equilibration of the unfolded state is slow relative to folding. In this communication we resolve this paradox. It arises when the average time for a single molecule trajectory to hit a specific location (the MFPT to state i) within U, is compared with the time for population fluctuations within the unfolded state to relax. This relaxation time provides a quantitative measure of the time to equilibrate the unfolded state. We will show that the MFPT to any state within the unfolded ensemble is approximately equal to the time to equilibrate the unfolded state divided by the population of the target state. The smaller the size of the target state, the longer the MFPT to that state, even though the equilibration of the unfolded state ensemble is very fast. For the Trp-cage example we use for discussion, MFPTs between different regions of the unfolded state ensemble are 10s to 100s of microseconds, while the time to equilibrate the unfolded state is of the order of 100 ns. These times are to be compared with the folding time for Trp-cage, which is 5.5 microseconds.
An estimate of the time required to equilibrate the protein unfolded state is also needed to understand the implications of the recently introduced kinetic hub model of protein folding.29,31,32 In this model, the folded state F acts as a hub, so that most paths, which connect pairs of unfolded states U1 and U2 pass through F.33,34 Hub like behavior appears to imply that the unfolded state partitions into subspaces, which largely fold along different pathways, but we have shown that this is not the case for Trp-cage.28 Furthermore, when the time to equilibrate within the unfolded state ensemble is much faster than the folding time, the hub like behavior simply reflects the fact that the F state has sufficient population to have a high probability of being on most paths between typical points U1 and U2 within the unfolded state ensemble. It has recently become clear that hub like behavior is consistent with a smooth folding funnel.28
We use the integral of the time correlation function, which quantifies how the population fluctuations within the unfolded state relax to equilibrium as the measure of the time to equilibrate the unfolded state.35 There are two contributions to the relaxation of population fluctuations within the unfolded state ensemble of a protein, or equivalently the equilibration of the unfolded state. The first corresponds to relaxation of fluctuations, which originate and propagate entirely within the U state and the second to relaxation within U, which arises from the equilibration between the unfolded and folded states. When the former relaxation process is much faster than the later, the protein folding is two-state. In this communication we mostly focus on the fast relaxation processes entirely within U. For our analysis we use a discrete master equation model of Trp-cage with 20 states parameterized on a 208 microseconds all atom molecular dynamics simulation of this mini-protein in water provided by the D.E. Shaw group.23 The kinetics is characterized by the implied timescale spectrum of the transition matrix, which contains all the information about the relaxation times of the states within the discrete time Markov State Model (MSM). The Trp-cage implied timescale spectrum has a substantial gap between the longest implied timescale, which is associated with folding and the others, therefore the intra U state fluctuations can be separated from the folding and the mini-protein folds in a two-state manner with single exponential kinetics. That the remaining eigenmodes correspond to intra U-state relaxation can be verified by comparing the spectrum with the corresponding implied timescale spectrum obtained using reflecting boundary conditions at F, as we do in the following section.
Results and Discussion
We use a master equation to study the timescales over which the unfolded state equilibrates. The formal solution to the master equation is:
| (1) |
where P is a vector of state probabilities and the transition matrix T (also called the propagator) contains all the information about the kinetics of the system (see Supporting Information). The propagator matrix element Tij(t) is the probability that the system is in state j at time t given that it was in state i at time zero. All observables of the system can be calculated in terms of functions of the Tij(t). The Tij(t) in turn can be expressed in terms of the eigenvalues and eigenvectors of T. Figure 1 shows the spectrum of implied timescales for the Trp-cage transition matrix constructed from the Shaw trajectory and for a modified transition matrix with a reflecting boundary added at F. Imposing the reflecting boundary condition here provides a model for the dynamics of the unfolded state alone. It can be seen that the spectrum is very similar except that the largest nonzero eigenvalue is missing from the spectrum with reflecting boundary condition at F; this eigenmode corresponds to the relaxation between the unfolded (U) state ensemble and the folded (F) state. The large gap between the largest implied timescale and the others means that the folding is two-state and the implied timescale (∼1.2 μs) is the inverse of the sum of the folding plus unfolding rates.
Figure 1.
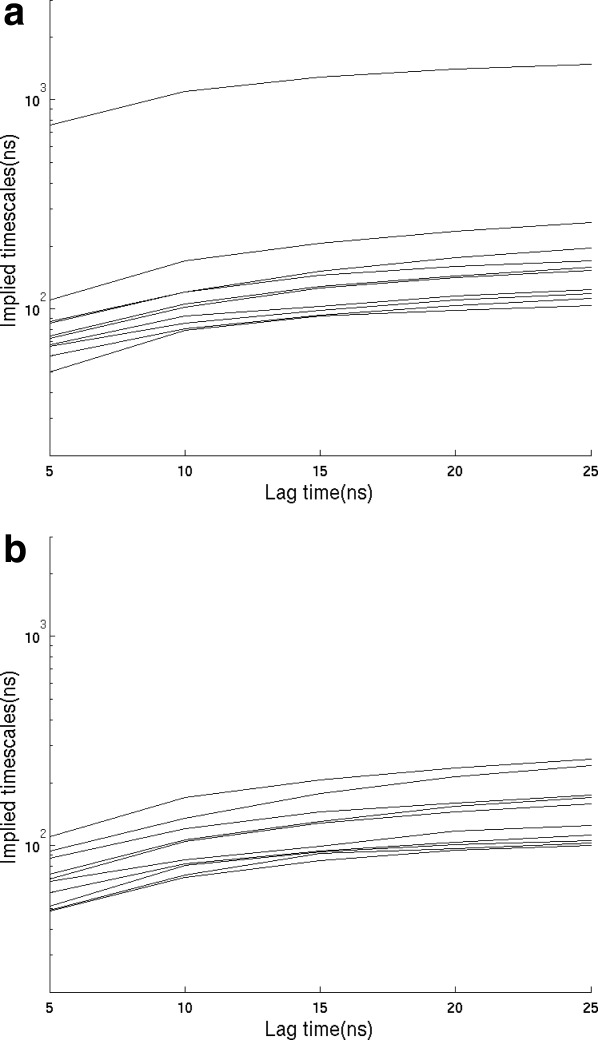
(a) The implied timescales corresponding to the 10 slowest decaying eigenmodes using transition matrices T(τ), with different boundary conditions, (a) unmodified equilibrium and (b) reflecting at F and I states. The optimal lag time 10 ns is chosen for further analysis based on the tradeoff between the network being Markovian and the resolution being sufficient for studying folding mechanism.
In Figure 2 we show a typical propagator matrix element Tij(t) from state i to state j, both within U, calculated three ways; using absorbing, unmodified equilibrium, and reflecting boundary conditions at F. The time dependence of Tij(t) describes the relaxation process following an initial point perturbation at state i. On a timescale of a few hundred nanoseconds they look very similar. Each rises rapidly to a plateau value which “overshoots” the equilibrium population of state j by a small amount. When added up over all the states in U, the excess corresponds to the equilibrium population of F that folds from U to F on the slower timescale of ∼5 µs. After a few hundred nanoseconds, the Tij(t) matrix elements shown in Figure 2 have the following longer time behavior. Under reflecting boundary conditions Tij(t) is approximately constant, the unmodified transition matrix Tij(t) relaxes to the equilibrium population with a relaxation time ∼1.2 µs, while under absorbing boundary condition the matrix elements relax to zero with a relaxation time ∼5 µs. The results shown in Figure 2 are suggestive as to the timescales for equilibrating the unfolded state, but the full relaxation involves all the elements Tij(t) of the propagator. We consider the full expression for the relaxation now.
Figure 2.
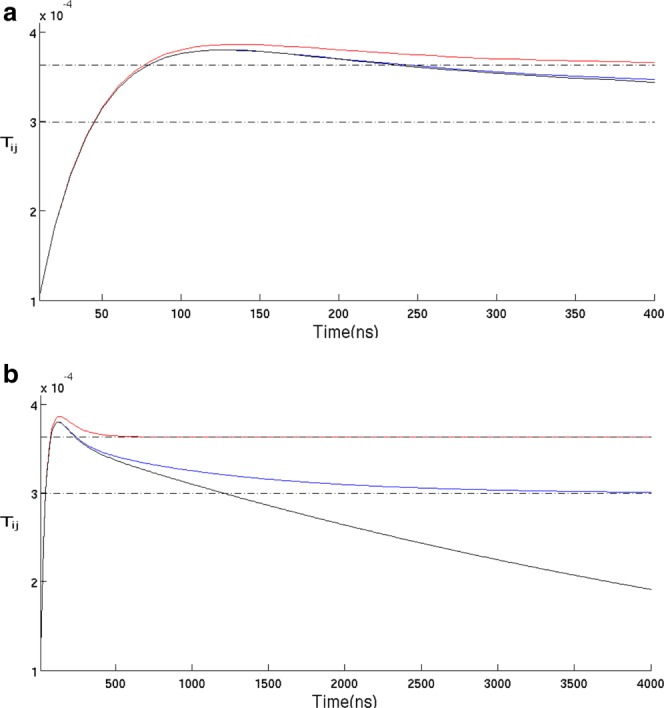
(a) A typical propagator matrix element Tij(t) from state i to state j, both within U, calculated three ways; using absorbing at F (black), unmodified equilibrium (blue) and reflecting boundary conditions at F (red). The upper and lower dash and point lines in each subplot are correspondingly the equilibrium population of state j under reflecting and unmodified equilibrium boundary conditions. All the propagator elements are calculated from spectral decomposition using all the 20 eigenmodes at lag time of 10 ns. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
The way to estimate the time it takes to equilibrate a system from equilibrium statistical mechanics is to calculate an integral of the appropriate time correlation function.35 The correlation function of interest here corresponds to the decay of the population fluctuations in the unfolded state. After some manipulation (see Supporting Information), this correlation function can be expressed as:
| (2a) |
| (2b) |
| (2c) |
where ψnR(i) and ψnL(i) are the ith element of the nth right and left eigenvectors of the T matrix. λn is the nth eigenvalue of the T matrix. ΔPi(t) = Pi(t) − Peq(i). Peq(i) is the equilibrium population of state i. Pi(t) is an indicator function, which is 1 when the trajectory is on state i and 0 otherwise at time t.
In Figure 3 we show the unfolded state population fluctuation correlation function. When the motions are restricted to the unfolded state, the time to equilibrate the unfolded state is estimated from the time integral of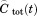 to be ∼100 ns; when the additional relaxation of U due to the much slower equilibration between U and F is also considered, the time to equilibrate the unfolded state is increased to ∼540 ns. The separation of timescales between the equilibration within U and the folding is implicit in the folding funnel model of protein folding.3,5 While folding on a flat “golf-course” landscape,11 which lacks the energy bias can also produce a separation of timescales, the very fast equilibration (∼100 ns) within the unfolded state is a feature of the funneled landscape.
to be ∼100 ns; when the additional relaxation of U due to the much slower equilibration between U and F is also considered, the time to equilibrate the unfolded state is increased to ∼540 ns. The separation of timescales between the equilibration within U and the folding is implicit in the folding funnel model of protein folding.3,5 While folding on a flat “golf-course” landscape,11 which lacks the energy bias can also produce a separation of timescales, the very fast equilibration (∼100 ns) within the unfolded state is a feature of the funneled landscape.
Figure 3.
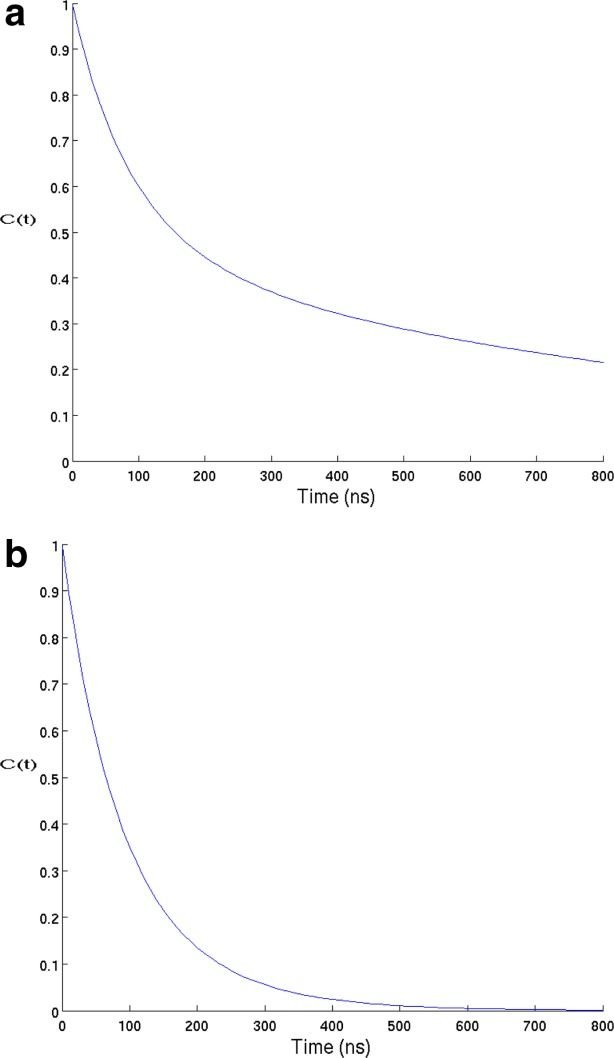
(a) The population fluctuation relaxation functions [Eq. (2)] of the 20-node network at a lag time of 10 ns, using two different boundary conditions, (a) unmodified equilibrium and (b) reflecting on F. The integrals of the functions are the relaxation times, which are 543 and 100 ns correspondingly. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Our estimate of the time to equilibrate the protein unfolded state based on the decay of fluctuations of the U state population (eq. 2b) is independent of the kind of experiment chosen to monitor the system. Any particular experiment will measure the time evolution of the population fluctuations reweighted by how sensitive that particular probe is to the different modes by which the population fluctuations relax. If for example, the experiment is sensitive to the fluctuations of some property f, then the experimental relaxation time measured for that probe of the unfolded state dynamics would be:
| (3) |
where f(i), f(j) are the values of the experimental observables in state i and j.
A common choice of the experimental observable f is the FRET efficiency, which is a nonlinear function of the distance between two particular residues within the protein. The relaxation time thus determined depends on the choice of those residues.25
We turn now to an analysis of the MFPTs between different states within the unfolded state ensemble. From MSMs, the MFPTs between unfolded states have been reported to be tens of microseconds or longer.29,31,32 For the Trp-cage model we studied it extends to ∼200 microseconds. The MFPT to an unfolded state i can be obtained from the formula:
| (4a) |
| (4b) |
where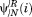 and
and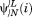 are the ith element of the nth right and left eigenvectors of the transition matrix with an absorbing boundary at i Tabs→i. μn is its the nth implied timescale (see Supporting Information).
are the ith element of the nth right and left eigenvectors of the transition matrix with an absorbing boundary at i Tabs→i. μn is its the nth implied timescale (see Supporting Information).
The average shown in eq. 4a is taken over all the other states j in U and includes a sum over all the eigenmodes n. In Figure 4(a) we show the implied timescale spectrum of the transition matrix with absorbing boundary at a typical unfolded state i. The large gap between the largest implied timescale and the rest is the signature of the exponential distribution of first passage times to unfolded state i. The longest implied timescale is of the order of ∼100 microseconds. Because the unfolded state ensemble relaxes on a timescale a hundred to a thousand times faster than the time it takes on average to reach state i, the MFPT to state i does not depend on the starting point within U. The kinetics involving the transitions between any specific state i and all the other states taken collectively is then effectively two state and the MFPT to state i can be written as:
Figure 4.
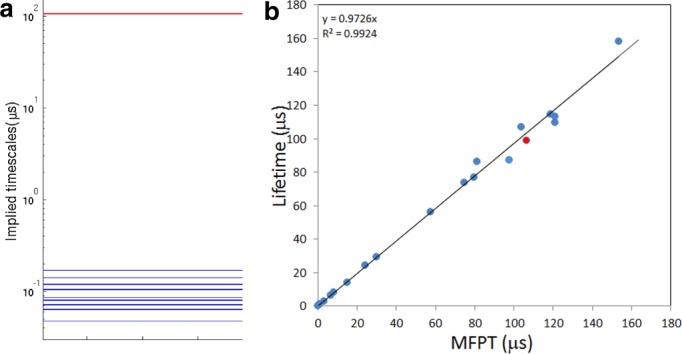
(a) The implied timescale spectrum to the state, which is highlighted as red in Figure 4(b). (b) The average lifetime of the collective state (U-i), which excludes the state i versus the MFPT to state i using Eq. (4b). [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
| (5) |
The MFPT to the unfolded state i chosen for the example shown in Figure 4(a) is found to be 106 microseconds.
To understand why the MFPTs to states within U are so long, we consider the relationship between the average lifetime of a state i within U and the average lifetime of the collective state consisting of the remainder of U excluding state i:
| (6) |
where ti is the average lifetime of state i and tU-i is the average lifetime of the collective state U-i consisting of the remainder of U excluding state i. Here we define the lifetime distribution of a state as the distribution of times recorded upon entering a state when the clock starts and then leaving it when the clock stops, during a single very long trajectory when the state is visited many times [see Supporting Information for the derivation of Eq. 6].
In Figure 4(b) we plot the MFPT to state i [Eq. (4b)] against the average lifetime of the collective state, tU-i [Eq. 6] for each of the unfolded states in the 20-state model. It can be seen that these times are almost equal. This is true when the time to equilibrate within the unfolded state (U-i) is much shorter than the average lifetime of (U-i). Under these circumstances, the MFPT to any unfolded state i is proportional to the average lifetime of the state ti divided by the population, and there is an equality involving Eqs 4, 5 and 6. Because the average lifetimes of the unfolded states decay on the same timescale as the decay of the population fluctuations, we find that the MFPT to any state within U is approximately equal to the time to equilibrate U divided by the population of the target state. Importantly, the MFPTs depend on the resolution of the model for the unfolded state, the more fine grained the model, the longer the MFPTs to an individual state. On the other hand, the time to equilibrate the unfolded state is a characteristic of the macrostate, which depends only weakly on the resolution. For the 20-state model of Trp-cage studied here, the longest MFPT (∼200 µs) is to the state with the smallest population 0.003, while the average lifetime of that state is 48 ns, comparable to the time to equilibrate the unfolded state.
In this communication we have resolved a paradox about kinetics within the unfolded state of proteins, which leads to a better understanding of why most small proteins fold with two-state kinetics. When the equilibration of the unfolded state ensemble is very fast as it is for most small proteins, the protein will fold with single exponential kinetics. While it seems paradoxical that the time to equilibrate the unfolded state can be orders of magnitude shorter than MFPTs within U, we have shown there is no inconsistency. Using a time-correlation function approach, we have presented a general formula for the timescale of population relaxation within U [Eq. (2c)]. Applying this formula to the folding of the two-state mini-protein Trp-cage, we found that the folding follows a two-step process: starting from an arbitrary nonequilibrium conformational distribution within the unfolded region the protein population will quickly relax to a pre-equilibrium within the unfolded state on timescales (∼100 ns for Trp-cage) much faster than folding. From this time forward, while the relative populations of all the unfolded microstates remain constant, the “excess” population within U, which will populate the folded state at equilibrium, folds with single exponential kinetics (rate ∼1/5.5 μs). It should be noted that as we reported in a recent article, an individual Trp-cage folding trajectory only visits a fraction (e.g., ∼25%) of the unfolded state space.28 The key to reconciling this with the rapid equilibration in the U-state is to realize that while any one trajectory explores only a small part of U before folding, an ensemble of such trajectories starting from the same initial condition within U will explore all of the U states with a probability that is close to the equilibrium population of that state before folding.11,28,30 The methodology developed in this study is also well suited for studying the kinetics of larger and more complex proteins where the timescales to equilibrate within U and to fold may overlap and the folding is no longer two state.
Materials and Methods
A MD trajectory of Trp-cage, which contains 1 million snapshots and saved at every 200 ps, was obtained from D.E. Shaw Research.23 The simulation length is 208 µs using a modified CHARMM22 all-atom force field in the TIP3P explicit solvent. A 25000-node fine-grained network and a 20-node coarse-grained network were generated from the trajectory (see Supporting Information for detailed descriptions of how the fine-grained network was generated).
Acknowledgments
Some of the calculations were performed using the XSEDE allocation TG-MCB100145. The authors thank Dr. Attila Szabo for very helpful discussions. ND would like to thank Dr. Kyle Beauchamp from Dr. Vijay Pande group for help with the MSMBuilder2.36
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Creighton TE. Toward a better understanding of protein folding pathways. Proc Natl Acad Sci U S A. 1988;85:5082–5086. doi: 10.1073/pnas.85.14.5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson SE, Fersht AR. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry (Mosc.) 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 3.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 4.Eaton WA, Thompson PA, Chan C-K, Hage SJ, Hofrichter J. Fast events in protein folding. Structure. 1996;4:1133–1139. doi: 10.1016/s0969-2126(96)00121-9. [DOI] [PubMed] [Google Scholar]
- 5.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 6.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 7.Zwanzig R. Two-state models of protein folding kinetics. Proc Natl Acad Aci U S A. 1997;94:148–150. doi: 10.1073/pnas.94.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perl D, Welker C, Schindler T, Schröder K, Marahiel MA, Jaenicke R, Schmid FX. Conservation of rapid two-state folding in mesophilic, thermophilic and hyperthermophilic cold shock proteins. Nat Struct Biol. 1998;5:229–235. doi: 10.1038/nsb0398-229. [DOI] [PubMed] [Google Scholar]
- 9.Jackson SE. How do small single-domain proteins fold? Fold Des. 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 10.Cieplak M, Henkel M, Karbowski J, Banavar J. Master equation approach to protein folding and kinetic traps. Phys Rev Lett. 1998;80:3654–3657. [Google Scholar]
- 11.Bicout DJ, Szabo A. Entropic barriers, transition states, funnels, and exponential protein folding kinetics: a simple model. Protein Sci. 2000;9:452–465. doi: 10.1110/ps.9.3.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinner AR, Šali A, Smith LJ, Dobson CM, Karplus M. Understanding protein folding via free-energy surfaces from theory and experiment. Trends Biochem Sci. 2000;25:331–339. doi: 10.1016/s0968-0004(00)01610-8. [DOI] [PubMed] [Google Scholar]
- 13.Mirny L, Shakhnovich E. Protein folding theory: From lattice to all-atom models. Annu Rev Biophys Biomol Struct. 2001;30:361–396. doi: 10.1146/annurev.biophys.30.1.361. [DOI] [PubMed] [Google Scholar]
- 14.Makarov DE. How the folding rate constant of simple, single-domain proteins depends on the number of native contacts. Proc Natl Acad Sci U S A. 2002;99:3535–3539. doi: 10.1073/pnas.052713599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang WY, Gruebele M. Folding at the speed limit. Nature. 2003;423:193–197. doi: 10.1038/nature01609. [DOI] [PubMed] [Google Scholar]
- 16.Kaya H, Chan HS. Simple two-state protein folding kinetics requires near-levinthal thermodynamic cooperativity. Proteins. 2003;52:510–523. doi: 10.1002/prot.10506. [DOI] [PubMed] [Google Scholar]
- 17.Weikl TR. Cooperativity in two-state protein folding kinetics. Protein Sci. 2004;13:822–829. doi: 10.1110/ps.03403604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhoades E, Cohen M, Schuler B, Haran G. Two-state folding observed in individual protein molecules. J Am Chem Soc. 2004;126:14686–14687. doi: 10.1021/ja046209k. [DOI] [PubMed] [Google Scholar]
- 19.Ellison PA, Cavagnero S. Role of unfolded state heterogeneity and en-route ruggedness in protein folding kinetics. Protein Sci. 2006;15:564–582. doi: 10.1110/ps.051758206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrick D. What have we learned from the studies of two-state folders, and what are the unanswered questions about two-state protein folding? Phys Biol. 2009;6:015001. doi: 10.1088/1478-3975/6/1/015001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng W, Andrec M, Gallicchio E, Levy RM. Recovering kinetics from a simplified protein folding model using replica exchange simulations: A kinetic network and effective stochastic dynamics. J Phys Chem B. 2009;113:11702–11709. doi: 10.1021/jp900445t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Best RB, Hummer G. Coordinate-dependent diffusion in protein folding. Proc Natl Acad Sci U S A. 2009;107:1088–1093. doi: 10.1073/pnas.0910390107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How fast-folding proteins fold. Science. 2011;334:517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 24.Karplus M. Behind the folding funnel diagram. Nat Chem Biol. 2011;7:401–404. doi: 10.1038/nchembio.565. [DOI] [PubMed] [Google Scholar]
- 25.Soranno A, Buchli B, Nettels D, Cheng RR, Muller-Spath S, Pfeil SH, Hoffmann A, Lipman EA, Makarov DE, Schuler B. Quantifying internal friction in unfolded and intrinsically disordered proteins with single-molecule spectroscopy. Proc Natl Acad Sci U S A. 2012;109:17800–17806. doi: 10.1073/pnas.1117368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Chan HS. Transition paths, diffusive processes, and preequilibria of protein folding. Proc Natl Acad Sci U S A. 2012;109:20919–20924. doi: 10.1073/pnas.1209891109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Sancho D, Mittal J, Best RB. Folding kinetics and unfolded state dynamics of the GB1 hairpin from molecular simulation. J Chem Theory Comput. 2013;9:1743–1753. doi: 10.1021/ct301033r. [DOI] [PubMed] [Google Scholar]
- 28.Deng N, Dai W, Levy RM. How kinetics within the unfolded state affects protein folding: An analysis based on Markov state models and an ultra-long MD trajectory. J Phys Chem B. 2013:130524112618003. doi: 10.1021/jp401962k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowman GR, Pande VS. Protein folded states are kinetic hubs. Proc Natl Acad Sci U S A. 2010;107:10890–10895. doi: 10.1073/pnas.1003962107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lane TJ, Schwantes CR, Beauchamp KA, Pande VS. Probing the origins of two-state folding. Physicsbio-Ph. doi: 10.1063/1.4823502. Arxiv13050963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voelz VA, Bowman GR, Beauchamp K, Pande VS. Molecular simulation of ab initio protein folding for a millisecond folder NTL9(1−39) J Am Chem Soc. 2010;132:1526–1528. doi: 10.1021/ja9090353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowman GR, Voelz VA, Pande VS. Taming the complexity of protein folding. Curr Opin Struct Biol. 2011;21:4–11. doi: 10.1016/j.sbi.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dickson A, Brooks CL. Quantifying hub-like behavior in protein folding networks. J Chem Theory Comput. 2012;8:3044–3052. doi: 10.1021/ct300537s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dickson A, Brooks CL. Native states of fast-folding proteins are kinetic traps. J Am Chem Soc. 2013;135:4729–4734. doi: 10.1021/ja311077u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandler D. Introduction to modern statistical mechanics. New York: Oxford University Press; 1987. [Google Scholar]
- 36.Beauchamp KA, Bowman GR, Lane TJ, Maibaum L, Haque IS, Pande VS. MSMBuilder2: Modeling conformational dynamics on the picosecond to millisecond scale. J Chem Theory Comput. 2011;7:3412–3419. doi: 10.1021/ct200463m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.