

Abstract
Background and Purpose
Drug interference with normal hERG protein trafficking substantially reduces the channel density in the plasma membrane and thereby poses an arrhythmic threat. The chemical substructures important for hERG trafficking inhibition were investigated using pentamidine as a model drug. Furthermore, the relationship between acute ion channel block and correction of trafficking by dofetilide was studied.
Experimental Approach
hERG and KIR2.1 trafficking in HEK293 cells was evaluated by Western blot and immunofluorescence microscopy after treatment with pentamidine and six pentamidine analogues, and correction with dofetilide and four dofetilide analogues that displayed different abilities to inhibit IKr. Molecular dynamics simulations were used to address mode, number and type of interactions between hERG and dofetilide analogues.
Key Results
Structural modifications of pentamidine differentially affected plasma membrane levels of hERG and KIR2.1. Modification of the phenyl ring or substituents directly attached to it had the largest effect, affirming the importance of these chemical residues in ion channel binding. PA-4 had the mildest effects on both ion channels. Dofetilide corrected pentamidine-induced hERG, but not KIR2.1 trafficking defects. Dofetilide analogues that displayed high channel affinity, mediated by pi-pi stacks and hydrophobic interactions, also restored hERG protein levels, whereas analogues with low affinity were ineffective.
Conclusions and Implications
Drug-induced trafficking defects can be minimized if certain chemical features are avoided or ‘synthesized out’; this could influence the design and development of future drugs. Further analysis of such features in hERG trafficking correctors may facilitate the design of a non-blocking corrector for trafficking defective hERG proteins in both congenital and acquired LQTS.
Keywords: ion channel trafficking, pentamidine, dofetilide, drug-induced trafficking defects, hERG, KIR2.1, chemical substructures, Long QT syndrome, safety pharmacology
Introduction
The rapid component of the delayed rectifier K+ current (IKr) plays a major role in cardiomyocyte repolarization. The current is carried by pore-forming α-subunits encoded by the human ether-a-go-go-related gene (hERG or KCNH2) (Warmke and Ganetzky, 1994; Sanguinetti and Tristani-Firouzi, 2006). Gene mutations and accompanying hERG (Kv11.1) channel current reduction (Curran et al., 1995), or a gain of function mechanism leading to loss of K+ selectivity and inward rectification (Lees-Miller et al., 2000b) have been linked to human hereditary long QT syndrome (LQTS), which is characterized by a prolonged QT interval on the ECG and an increased risk of sudden cardiac death due to potentially life-threatening Torsade de Pointes (TdP) arrhythmias (Dessertenne, 1966). The acquired form of LQTS can arise as a result of antiarrhythmic drug therapy or as a rare unintended side effect of structurally diverse medications, such as pentamidine, cisapride, terfenadine or sertindole (Thomsen et al., 2003; Kannankeril et al., 2010). Consequently, hERG blockade, QT prolongation and pro-arrhythmia have become an important component of cardiac safety screening of new pharmacological entities (Haverkamp et al., 2000; Fenichel et al., 2004). The list of (potentially) pro-arrhythmic compounds is still growing and consists mainly of IKr blockers (Haverkamp et al., 2000; Roden and Viswanathan, 2005) (up-to-date list: http://www.torsades.org).
For years, malfunctioning of the hERG channel was presumed to be mainly caused by mutations or drugs that interfered with normal ion conduction, leading to a loss of ion channel function. However, more recently, an entirely different mechanism has been postulated; disruption of normal ion channel trafficking can lead to severely reduced functional protein levels at the plasma membrane, thereby posing an arrhythmic threat. The process of trafficking comprises both anterograde (forward transport towards the plasma membrane) and retrograde (internalization from the plasma membrane) transport of ion channel proteins. Loss of hERG channel function in type 2 LQTS (LQTS2) has been shown to be caused by different mechanisms; mutations result in channels that either gate abnormally, are retained in the endoplasmic reticulum (ER) due to abnormal intracellular processing, or are non-functional (Zhou et al., 1998). Remarkably, it has been shown more recently that the majority of the mutations found in LQTS2 lead to a trafficking defective hERG channel that is trapped in the ER (Anderson et al., 2006). Similarly, a growing number of drugs have been shown to interfere with normal hERG protein trafficking, thereby reducing the number of channels in the cell membrane (van der Heyden et al., 2008). Of note, this chronic drug effect is not detected in conventional cardiac safety screening, and can thus pose a potential arrhythmic threat.
The antiprotozoal agent pentamidine has been known to cause QT prolongation and TdP when used clinically (Jha, 1983; Wharton et al., 1987; Bibler et al., 1988; Girgis et al., 1997). It severely reduces hERG cell surface expression without interfering with ion conduction (Cordes et al., 2005; Kuryshev et al., 2005), acutely blocks the IK1 current, and interferes with normal KIR2.1 protein expression (de Boer et al., 2010; Nalos et al., 2011). Recently, it was shown that pentamidine inhibits hERG ER exit and consequently maturation; an effect that can be corrected by application of the class III agent astemizole (Dennis et al., 2012). Interestingly, several trafficking deficient LQTS2 mutants can also be rescued by application of class III agents (Balijepalli et al., 2010).
Currently, it is not known which chemical substructures are responsible for drug-induced hERG trafficking inhibition or the restoration of both acquired and inherited LQTS. Therefore, we aimed to (i) determine the chemical features within pentamidine that are important for acquired trafficking defects, (ii) determine whether dofetilide can restore the acquired trafficking defects of hERG and KIR2.1, and (iii) determine the relationship between acute ion channel block and correction of trafficking. We found that structural modifications of pentamidine differentially affect hERG and/or KIR2.1 trafficking. Dofetilide and its analogues corrected pentamidine-induced hERG trafficking defects and this effect was found to be dependent on their affinity for the channel.
Methods
Animal experiments
Animal care and handling was performed in accordance with the ‘European Directive for the Protection of Vertebrate animals used for Experimental and Scientific Purpose, European Community Directive 86/609/CEE’. All experiments were approved by the Committee for Experiments on Animals of the Utrecht University, The Netherlands. Three adult purpose-bred mongrel dogs (Marshall, North Rose, NY, USA) were used. General anaesthesia was induced by pentobarbital (25 mg·kg−1 i.v.) and maintained by isoflurane (1.5% in O2 : NO2 1:2). Atrioventricular block was induced by radiofrequency ablation in all dogs, and ventricular activation was controlled using a DDDR pacemaker (Vitatron, Arnhem, The Netherlands). Detailed descriptions of procedures, pacing protocols and data analysis were published previously (Winckels et al., 2007). After a minimum of 4 weeks (4–6 weeks), pentamidine (10 mg·kg−1 for 60 min, i.v) was administered under full anaesthesia. Standard six-lead ECG recordings were made regularly over a period of 6 weeks to determine the long-term electrophysiological effects of pentamidine. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Cell culture
HEK-293 cells stably expressing GFP-tagged murine wildtype KIR2.1 (HEK-KWGF) were generated as described previously (de Boer et al., 2006). HEK-hERG cells were obtained from C. January (Zhou et al., 1998). Cells were maintained in DMEM supplemented with 10% fetal calf serum, 2 mM l-glutamine, 50 U·mL−1 penicillin, and 50 μg·mL−1 streptomycin (Verviers, Belgium). For experiments cells were seeded and incubated in complete DMEM containing different drugs at various concentrations as indicated. Variable incubation times were achieved by replacing control medium with drug-containing medium at appropriate time points. During the experiments, drug-containing medium was refreshed every 24 h. The amount of DMSO added to the cells was kept below 0.01%.
Chemical compounds
Pentamidine-isethionate (Pentacrit® 300, Sanofi Aventis, Gouda, The Netherlands) was dissolved in water at a concentration of 0.1 M, sterilized by filtration (22 μM), aliquoted and stored at −20°C until further use. Pentamidine analogues (PAs; see Table 1, PA-1 to PA-7) were synthesized as described previously (Jones et al., 1990; Tidwell et al., 1990; Bakunova et al., 2009a,b), and were dissolved in water (PA-1 to PA-5) or DMSO (PA-7) at the highest concentration possible (≤25 mM). Dofetilide and its analogues were dissolved in DMSO at a concentration of 10 mM. Dofetilide analogues (DA) were synthesized as described previously (Shagufta et al., 2009).
Table 1.
Chemical structures of pentamidine (P) and dofetilide (D) and their derivatives used in this study
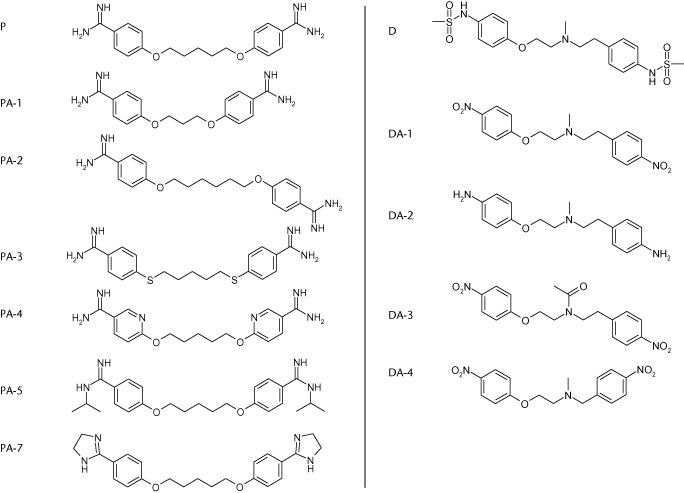 |
Western blot
Cell lysates were prepared in buffer D (20 mM HEPES, 125 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 10% glycerol, and 1% Triton X-100) supplemented with 1 mM PMSF and 6.8 μg·mL−1 aprotinin (Sigma-Aldrich, Zwijndrecht, The Netherlands). Fifteen micrograms of protein lysate was mixed with Laemmli sample buffer, separated by 7% (hERG) or 10% (KIR2.1) SDS-PAGE and subsequently electroblotted onto a nitrocellulose membrane (Biorad, Veenendaal, The Netherlands). Reversible Ponceau staining was used to reveal equal protein loading and subsequent quantification. KIR2.1-GFP was detected by monoclonal anti-GFP (cat. no. Sc-9996; Santa Cruz Biotechnology, Santa Cruz, CA, USA), hERG protein was detected by polyclonal anti-hKv11.1 primary antibody (cat. no. APC-062; Alomone Labs, Jerusalem, Israel). Peroxidase-conjugated secondary antibodies were used to facilitate final detection with a standard ECL procedure (Santa Cruz Biotechnology).
Immunofluorescence microscopy
HEK-hERG cells were cultured on poly-l-lysine (Sigma)-coated 15 Ø glass cover slips (Smethwick, Warley, UK), fixated with 3% paraformaldehyde dissolved in PBS. Cells were quenched with 50 mM glycine-PBS after permeabilization with 0.5% Triton X-100, and subsequently blocked with NET-gel (150 mM NaCl, 5 mM EDTA, 50 mM Tris-HCl, pH 7.4, 0.05% igepal, 0.25% gelatin, 0.02% NaN3). Cells were then incubated overnight with polyclonal anti-hKv11.1 primary antibody (cat. no. APC-062; Alomone Labs), followed by incubation with anti-rabbit FITC-conjugated secondary antibody (Jackson ImmunoResearch) for 2 h. Covers slips were mounted with Vectashield (Vector Laboratories Inc., Burlingame, CA, USA), and confocal images were obtained using a Zeiss Axiovert 200 M confocal microscope (Carl Zeiss Microscopy GmbH, Jena, Germany) equipped with a ×40 water immersion objective (NA 1.2) plus ×2 digital zoom. Excitation was performed with an air-cooled argon ion laser (LASOS, RMC 7812Z, 488 nm).
Drug docking and molecular dynamics (MD) simulations
Docking was performed with the program Gold 4.0.1 (GOLD, 2008) using the Gold scoring functions. The recently published hERG homology model termed ‘model 6’ (Stary et al., 2010) after 50 ns MD simulations was used as a template for docking. Coordinates of the geometric centre calculated among the Y652 and F656 residues were taken as binding site. The binding site radius was set to 10 Å. The 10 best-ranked poses of each docking run were used for visual analysis. The most frequent binding mode was used as a starting conformation for MD simulations.
MD simulations were performed with Gromacs v. 4.5.4. (Hess et al., 2008) as described previously (Knape et al., 2011). Briefly, the hERG model was embedded in an equilibrated membrane consisting of 280 dioleolylphosphatidylcholine (DOPC) lipids by making use of the g_membed tool (Wolf et al., 2010). The amber99sb force field (Hornak et al., 2006) with lipid parameters taken from Siu et al. (2008), and the TIP3P water model (Jorgensen et al., 1983) were utilized. Geometry optimization and topology generation of the drugs was carried out with HF/3-21G, implemented in Gaussian09 (Frisch et al., 2009) and antechamber (Case et al., 2010) respectively. Prior to simulations, 1000 conjugate gradient energy-minimization steps were performed. Subsequently, the systems were equilibrated for 1 ns for each docked compound. During equilibration, the protein and the ligand were restrained with a force constant of 1000 kJ·mol·nm−2, while the lipids, the ions, and the water were allowed to move freely. Each drug was simulated for 50 ns.
Statistics
Group averages are presented as mean ± SEM, unless indicated otherwise. Differences between groups were tested using one-way ANOVA with a Bonferroni post hoc test. A statistically significant difference was considered if P < 0.05. All analyses were carried out using SPSS (SPSS 19.0 IBM, Armonk, NY, USA).
Drugs and molecular target nomenclature conform to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2011).
Results
A single dose of pentamidine lengthens the QT interval
The time course of QT interval changes after a single dose of pentamidine is summarized in Figure 1. The QT interval did not change during and directly after pentamidine administration in any of the three dogs. After 7 days, the QT interval prolongs progressively, which persists until day 21, demonstrating the typical chronic effect of pentamidine on repolarization.
Figure 1.

Electrophysiological effects of 10 mg·kg−1 pentamidine over 60 min in AV-blocked dogs paced from the high septum. A delayed effect of pentamidine on the QT interval was observed. QT prolongation became apparent 4 days after i.v. pentamidine administration that persisted for 17 days, after which the prolongation dissipated gradually. Data are presented as mean ± SD (n = 3).
Pentamidine and its analogues differentially inhibit hERG and/or KIR2.1 trafficking
Pentamidine inhibited anterograde hERG trafficking, as can be seen in Figure 2A. The mature protein level (upper band) was severely reduced compared with control levels after 48 h treatment with 10 μM pentamidine (0.2 ± 0.02 vs. 1.0 ± 0.0, P < 0.05). In addition, KIR2.1 protein levels were also analysed. As expected, 10 μM pentamidine for 48 h significantly decreased KIR2.1 protein levels compared with control (Figure 2B). At present, the chemical characteristics and substructures within trafficking inhibiting drugs that specifically interfere with the normal trafficking process of these potassium ion channels have not been identified. To identify potentially trafficking inhibiting substructures, pentamidine was used as a model drug, and several analogues were tested for ion channel trafficking inhibiting capacity. The molecular structure of pentamidine is depicted in Table 1. Compared with the lead compound, PA-1, 2 and 3 contain modifications in the linker between the benzamidine groups, PA-4 has pyridine instead of phenyl rings, and PA-5 and 7 contain substituted benzamidine moieties (Table 1).
Figure 2.
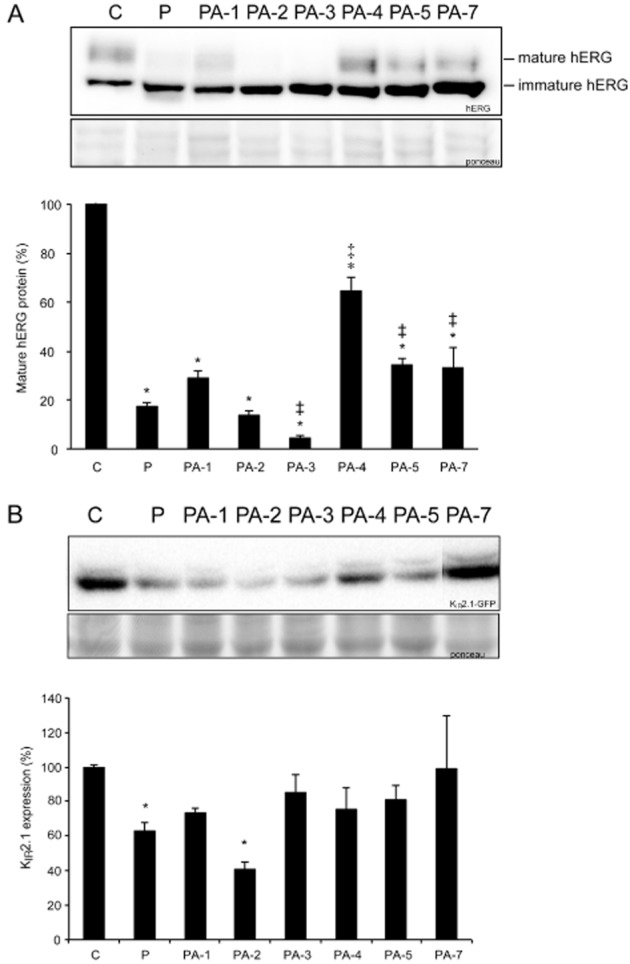
Pentamidine and its analogues differentially affect hERG and KIR2.1 protein levels. (A) Western blot showing the effects of pentamidine or its structural analogues (10 μM, 48 h) on hERG forward trafficking (n = 12 for control and pentamidine, n = 3 for PA-4, n = 4 for all other analogues). (B) Western blot showing the effects of pentamidine or its structural analogues (10 μM, 48 h) on KIR2.1 protein expression (n = 3 for PA-4 and 7, n = 4 for control, pentamidine, and all other analogues). Total protein staining (Ponceau) was used as a loading control. Control protein levels (untreated cells) were designated as 100% after correction; *indicates P < 0.05 versus control, ‡indicates P < 0.05 versus pentamidine treatment.
Both HEK-hERG and HEK-KWGF cells were treated with 10 μM pentamidine or its analogues for 48 h (Figure 2). Compared with pentamidine, either shortening or lengthening the carbon linker between the aromatic rings had slight effects on hERG protein maturation. PA-1 was a slightly less effective trafficking inhibitor (0.30 ± 0.03 vs. P 0.20 ± 0.02, ns), while PA-2 was slightly more potent than pentamidine (0.15 ± 0.02 vs. P 0.20 ± 0.02, ns). Interestingly, substitution of the oxygen directly attached to the aromatic residues by sulphur considerably increased hERG trafficking inhibition (PA-3 0.04 ± 0.02 vs. P, PA-1, PA-4, PA-5, or PA-7, P < 0.05). In contrast, PA-5 (P < 0.05 vs. P, and PA-2, 3, 4) and PA-7 (P < 0.05 vs. P, and PA-2, 3, 4) were significantly less potent than pentamidine, although protein levels comparable with control were not reached. PA-4 – in which the phenyl rings are replaced by pyridine – was the least potent hERG trafficking inhibiting compound (P < 0.05 for PA-4 vs. P and PA-1 to 7). Although control protein levels were not reached, mature hERG protein levels were substantially increased when PA-4 and pentamidine treatment were compared (Figure 2A). All these data indicate that an unaltered amidine substituent and the nature of the aromatic rings are important determinants of the interaction of pentamidine and the hERG channel during the trafficking process. It should be kept in mind though, that the compounds were tested at one concentration only, and that full concentration–response curves would allow a more definitive and quantitative SAR analysis.
In accordance with their effect on hERG trafficking, the effect of PA-1 and 2 on KIR2.1 protein levels differed slightly when compared with pentamidine (Figure 2B). In contrast, PA-4, 5 and 7 hardy affected KIR2.1 protein levels. Interestingly, PA-4 also slightly affected hERG trafficking. When comparing the effects of the pentamidine analogues on hERG and KIR2.1, the differential effect of PA-3 on these proteins represents an obvious discrepancy. While hERG trafficking was severely inhibited, KIR2.1 protein levels were hardly affected (0.90 ± 0.2 vs. control 1.0 ± 0.0, ns), which could imply that the mechanism by which pentamidine inhibits trafficking differs between these ion channels.
Dofetilide rescues pentamidine-induced hERG, but not KIR2.1, trafficking defects
In order to reduce the pentamidine-induced hERG channel trafficking block, we applied the high affinity class III agent dofetilide in different concentrations to HEK-hERG cells in the continuous presence of 10 μM pentamidine. Application of even 0.03 μM dofetilide significantly increased mature hERG levels compared with pentamidine (0.7 ± 0.1 vs. 0.3 ± 0.03, P < 0.05, Figure 3A), and at a concentration of 1 μM, dofetilide completely restored anterograde hERG trafficking. Interestingly, higher concentrations of dofetilide seemed to increase hERG trafficking efficiency, since significantly higher mature protein levels were reached compared with control levels.
Figure 3.
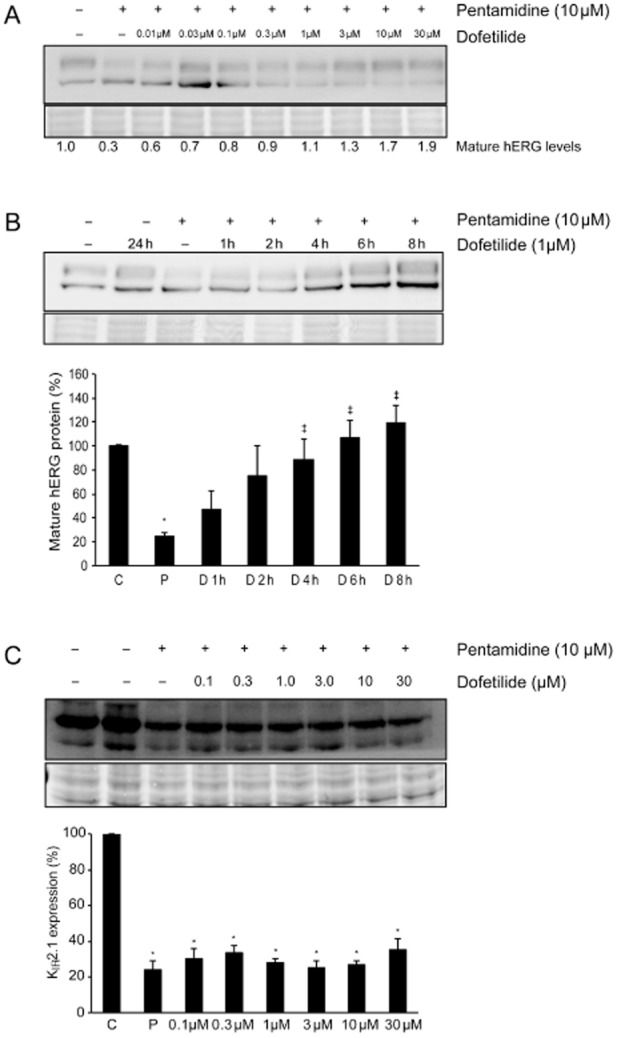
Dofetilide corrects pentamidine-induced hERG trafficking defects. (A) Western blot showing the concentration-dependent correction of hERG protein levels in the presence of pentamidine. Dofetilide 1 μM completely restored mature hERG protein levels, higher concentrations increased mature hERG protein levels even further (n = 24 for control and pentamidine, n = 6 for all dofetilide concentrations). (B) Dofetilide time-dependently corrects mature hERG protein levels. Treatment of pentamidine-exposed (10 μM, 48 h) HEK-hERG cells with 1 μM dofetilide restored mature hERG levels within 4–6 h (n = 24 for control and pentamidine, n = 3 dofetilide). (C) In contrast to the beneficial effect on hERG trafficking, pentamidine-mediated downregulation of KIR2.1 could not be corrected by application of dofetilide (n = 3 for all conditions). Total protein staining (Ponceau) was used as a loading control. Control protein levels (untreated cells) were designated as 100% after correction; *indicates P < 0.05 versus control, ‡indicates P < 0.05 versus pentamidine treatment.
The timeframe in which the pharmacological correction of drug-induced trafficking was taking place is unknown. Therefore, HEK-hERG cells were first exposed to pentamidine (10 μM) for 48 h; subsequently, dofetilide (1 μM) was added in the continued presence of pentamidine and mature protein levels were determined after different incubation times. Mature protein levels increased time- dependently, and were completely restored after 4–6 h of dofetilide treatment (Figure 3B). This timeframe strongly suggests that the correction of pentamidine-induced trafficking defects occurred post-transcriptionally, most likely via the relief of ER retention.
Since dofetilide is a high affinity class III agent, we hypothesized that its correction of ion channel trafficking defects could be ion channel-specific, and the pharmacological chaperone used has to bind directly to the particular ion channel involved. To test this hypothesis, we evaluated whether pentamidine-affected KIR2.1 protein levels could be corrected by application of dofetilide. As expected, treatment with dofetilide up to 30 μM did not restore KIR2.1 protein levels (0.4 ± 0.06 vs. P 0.25 ± 0.05, ns, n = 3, Figure 3C).
Correction of pentamidine-induced hERG trafficking defects depends on an affinity for the channel
Since dofetilide has a high affinity for the hERG channel and specifically restores drug-induced hERG trafficking defects, we wondered which substructures that affect channel affinity are important in the molecule. The structure of dofetilide is depicted in Table 1; its protonated tertiary nitrogen, phenyl rings and methanesulphonamide substituents are known to be important for high affinity binding (Shagufta et al., 2009). Furthermore, the spacing between the central basic nitrogen and the aromatic residues might also be of significance for efficient binding (Pearlstein et al., 2003). In all the dofetilide analogues (DA) used in this study, the methanesulphonamide substituent was replaced; in DA-2 by –NH2, in DA-1, 3 and 4 by NO2 groups. In addition, DA-3 was differently substituted at the central nitrogen, and in DA-4, the length of the chain connecting the two phenyl groups was modified. DA-1 was used as a reference compound for synthesis of DA-3 and 4. Compared with dofetilide (Ki 4.9 ± 0.9 nM), DA-1 had a comparable affinity (Ki 5.7 ± 3.0 nM) for the hERG channel, DA-3 and 4 had moderate affinity (Ki 278 ± 73 nM and Ki 202 ± 4.9 nM), and DA-2 had a very low affinity (Ki > 10 μM) for the channel (Shagufta et al., 2009).
Dofetilide and the four analogues were tested for their ability to correct drug-induced trafficking defects (Figure 4A). DA-1, in which a nitro-substituent replaces the methanesulphonamide group, was almost as effective as dofetilide. In contrast, substitution with a polar NH2 group (DA-2) completely abolished the trafficking correction capacity (30 μM DA-2 0.40 ± 0.04 vs. control 1.0 ± 0.0, P < 0.05; 30 μM DA-2 0.40 ± 0.04 vs. 30 μM dofetilide 1.9 ± 0.4, P < 0.05) in association with a substantial decrease in hERG binding affinity (Shagufta et al., 2009). In DA-3, the basicity of the central nitrogen was abolished by acylation; as a consequence, not only was hERG affinity decreased considerably, but also hERG trafficking defects were only partially corrected (30 μM DA-3 0.60 ± 0.1 vs. control 1.0 ± 0.0, P < 0.05; 30 μM DA-3 0.60 ± 0.1 vs. 30 μM dofetilide 1.9 ± 0.4, P < 0.05). DA-4, which had a moderate hERG affinity due to a variation in chain length, also partially corrected the pentamidine-induced trafficking defect. It remains puzzling, however, why higher concentrations of the lower-affinity compounds DA-3 and DA-4 do not fully restore mature protein levels.
Figure 4.

Correction of pentamidine-induced hERG trafficking defects is influenced by structural modifications of dofetilide (A). Compared with dofetilide, the high affinity analogue DA-1 was an equally effective corrector, while the other analogues were significantly less effective when tested at 1 μM (n = 24 for control and pentamidine, n = 6 for dofetilide, n = 3 for DA-1 and 2, n = 5 for DA-3 and 4). Total protein staining (Ponceau) was used as a loading control. Control protein levels (untreated cells) were designated as 100% after correction; *indicates P < 0.05 versus control, ‡indicates P < 0.05 versus pentamidine treatment. (B) In control conditions, the hERG protein is present in the cytoplasm and at the plasma membrane. A similar staining pattern was seen after only dofetilide treatment. After pentamidine treatment, only cytoplasmic staining was apparent. Staining patterns after correction of these effects with dofetilide or its structural analogues were in agreement with the Western blot results; effective correction showed hERG staining both at the plasma membrane and intracellularly, while ineffective correctors showed only intracellular staining. The hERG protein is shown in green.
The subcellular localization of the hERG protein after trafficking inhibition with pentamidine and correction thereof with dofetilide and its analogues (1 μM) was studied by immunofluorescence staining of hERG (Figure 4B). In untreated control cells, hERG was distributed throughout the cell and clear membrane staining was detected, as was the case after only dofetilide treatment. As expected, membrane staining was lost after pentamidine treatment, and intracellular staining had a more restricted perinuclear pattern. The localization of the hERG protein after application of the correctors correlated with their efficacy and was in agreement with the results of the Western blots. Correction of pentamidine-induced trafficking defects with dofetilide or DA-1 revealed a pattern comparable with control cells, as both compounds completely restored mature hERG protein levels. In contrast, DA-2- treated cells displayed a staining similar to cells treated with pentamidine alone. After treatment with DA-3 or DA-4, which are both partial correctors, the hERG protein was scattered throughout the cell but membrane staining was considerably less when compared to control cells.
Finally, to gain further insight in the relationship between drug binding affinity and correction of hERG trafficking defects, drug docking and molecular dynamic simulations were performed. All dofetilide derivatives in our hERG model displayed considerable stability during the course of 50 ns MD simulations. Key interactions, involving Y652 and F656, are shown in Figure 5. The numbers of hydrophobic and aromatic interactions a compound has (Table 2) are in good agreement with its ability to correct pentamidine-induced trafficking defects. While the compounds dofetilide and DA-1 (Figure 5A–D) had a large number of favourable interactions, DA-2, DA-3 and DA-4 had very few interactions with the hERG model. Our modelling studies suggest that dofetilide and DA-1 stabilize the hERG structure via pi-pi stacking, while the low affinity compound DA-2 cannot provide sufficient stabilizing interactions to correct pentamidine trafficking defects. DA-3 and DA-4 take an intermediate position between D and DA-1 on the one, and DA-2 on the other hand.
Figure 5.
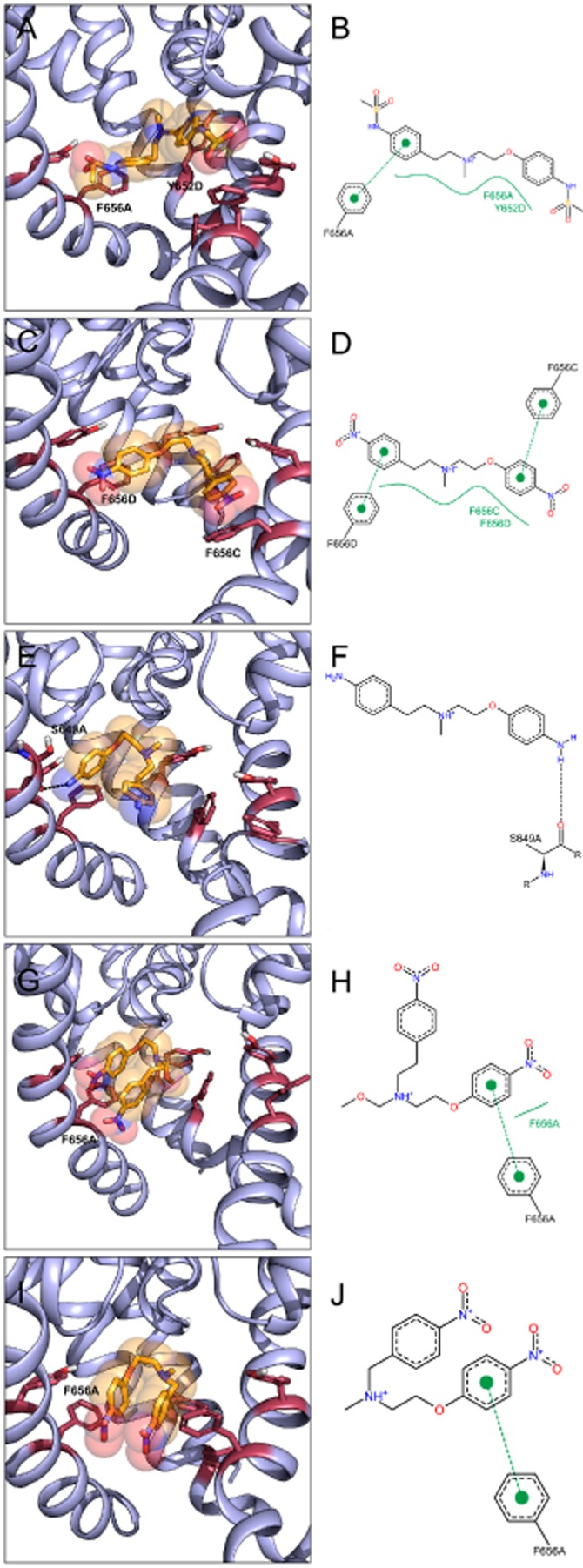
MD poses and 2D interaction profiles of dofetilide (A,B), DA-1 (C,D), DA-2 (E,F), DA-3 (G,H) and DA-4 (I,J). Snapshots after 50 ns are shown. Binding determinants Y652 and F656 are shown as raspberry sticks. Drug molecules are coloured orange, with nitrogen atoms coloured blue, oxygen atoms coloured red and sulfur groups coloured yellow. 2D interaction profiles were generated with PoseView (poseview.zbh.uni-hamburg.de). Green dotted lines indicate pi-pi interactions between aromatic rings, green solid lines represent hydrophobic interactions. Hydrogen bonds are shown as black dotted lines.
Table 2.
Number of interactions for dofetilide derivatives
| Compound | Σ of hydrophobic IAs (pi-pi stacking) | HB IAs |
|---|---|---|
| D | 3 (1) | – |
| DA-1 | 4 (2) | – |
| DA-2 | – | 1 |
| DA-3 | 2 (1) | – |
| DA-4 | 1 (1) | – |
HB, hydrogen bonds; IAs, interactions.
Discussion
Detrimental drug interactions with the hERG potassium channel constitute a major pharmacological safety concern during drug development. In recent years, it has become recognized that, in addition to hERG current inhibition, chronic drug effects should also be considered as a potential safety hazard (van der Heyden et al., 2008), as the dangerous effects of trafficking inhibiting agents like pentamidine and arsenic trioxide are not always detected in conventional hERG safety screening assays (Katchman et al., 2006). Although ion channel trafficking assays are available (HERG-Lite® and CHAN-Lite®, ChanTest™, Cleveland, Ohio), they have not been adopted by the current guidelines. In order to avoid toxicity, it is imperative to understand the structural requirements needed not only for hERG binding, but also for hERG trafficking inhibition, and this would be of great benefit for the drug design and development process.
At present, it is not known which chemical substructures in pentamidine are important for selective disruption of hERG ion channel trafficking. Our results indicate that the aromatic residues, in particular, are essential, as modifications of these groups (PA-3 and 4) have a large effect on hERG ion channel trafficking. PA-4, which had the most favourable trafficking safety profile, was evaluated in a mouse model of trypanosomiasis. Unfortunately, the compound was not effective, although it was well tolerated (Bakunova et al., 2009b). Interestingly, both PA-3 and 4 mildly affected KIR2.1 density, while PA-3 severely affected hERG maturation. PA-2 had a large effect on both KIR2.1 and hERG density. The differential effect of these compounds suggests that the mechanism by which pentamidine exerts protein downregulation differs between these potassium channels. In line with this, we found that the specific IKr inhibitor dofetilide corrected hERG channel but not KIR2.1 channel density after pentamidine treatment. Previously, we showed that inhibition of lysosomal degradation restores KIR2.1 protein levels in pentamidine-treated HEK-KWGF cells, which strongly suggests that pentamidine increases KIR2.1 retrograde trafficking and degradation (Nalos et al., 2011). In addition, pharmacological interference at different levels in the clathrin-dependent internalization route and thus retrograde trafficking, increase pentamidine-affected KIR2.1 protein levels (Varkevisser et al., 2013).
In the present study, we showed that dofetilide also corrects pentamidine-induced hERG anterograde trafficking defects and the correction efficacy depends on the compound's affinity for the channel. A fair agreement between affinity and correction was observed; a high affinity analogue (DA-1) completely restored mature hERG protein levels in the presence of pentamidine while analogues with moderate (DA-3 and 4) or low affinity (DA-2) restored mature protein levels partially or not at all. Our results favour the hypothesis that pentamidine and its correctors compete for the same binding site within the hERG channel. While pentamidine supposedly binds to an emerging hERG drug binding site in a conformation folding intermediate leading to an arrest in channel maturation (Dennis et al., 2012), displacement by a high affinity corrector stabilizes the channel, thereby alleviating the block in maturation. However, based on their chemical structure, it seems unlikely that dofetilide and pentamidine share a common binding site. Alternatively, pentamidine could bind to a different binding site in the immature hERG protein, which is not available in the mature protein. When performing MD simulations with pentamidine, no consistent interactions between the drug and the hERG model were found, in contrast to dofetilide and its analogues (Figure 5). This finding would be consistent with pentamidine binding to a channel intermediate instead of the mature channel. Presumably, pentamidine binding destabilizes the immature hERG protein, leading to its retention in the ER by quality control mechanisms; the binding of a high affinity class III agent at an allosteric site will then stabilize the immature protein and promote maturation. If this hypothesis is correct, dofetilide would operate similarly in the presence of other destabilizing agents. Mutation of amino acid residues F656 and Y652 that affect pentamidine sensitivity (Dennis et al., 2012) could, therefore, also have an indirect effect, as a mutation of these residues can affect the characteristics of the current (Lees-Miller et al., 2000a; Fernandez et al., 2004; Kamiya et al., 2006) possibly by small conformational changes. This could either directly obscure the binding site or reduce pentamidine binding because of steric hindrance. The direct hERG block and trafficking inhibition induced by several other drugs were found to be mediated by the drugs binding to different sites. Ketoconazole, fluconazole and fluoxetine, which are all direct IKr blockers, as well as hERG trafficking inhibitors, do not seem to bind to the canonical drug binding site in the immature hERG protein, as a mutation of residues Phe656 and Tyr652 abolished the direct IKr block but had no effect on hERG trafficking inhibition (Rajamani et al., 2006; Takemasa et al., 2008; Han et al., 2011). Furthermore, application of the high affinity IKr blocker E-4031 did not restore the hERG trafficking inhibition induced by either fluoxetine or norfluoxetine (Rajamani et al., 2006).
The mechanism by which pentamidine arrests hERG channel maturation is not known. It has been suggested that disruption of the channel–chaperone interactions could play a role. Wild-type hERG proteins have been shown to interact with the cystosolic chaperones Hsp70 and Hsp90 and the ER chaperone calnexin (Ficker et al., 2003; Gong et al., 2006). Not surprisingly, drugs like geldanamycin and As2O3 that inhibit Hsp70 and 90, arrested hERG maturation (Ficker et al., 2003; 2004). However, it should be noted that these drugs have complex actions and could potentially inhibit the correct folding of other proteins, including other (cardiac) ion channels. On the other hand, it has been shown that pentamidine does not interfere with the actions of Hsp70 or Hsp90 (Dennis et al., 2012), but it could disrupt the association of hERG with other chaperones, like calnexin. Calnexin associates transiently with wild-type immature hERG channels for up to 8 h, whereas its association with the trafficking deficient LQTS2 mutant N470D was prolonged (Gong et al., 2006). Association times between calnexin and this mutant channel approached the normal time frame in the presence of E-4031 and accordingly the protein levels of this mutant hERG were completely restored within 8 h (Zhou et al., 1999). In agreement with these findings, we showed that dofetilide completely restored hERG protein levels within 8 h in the presence of pentamidine. Further research is needed to elucidate the precise mechanism by which pentamidine arrests hERG maturation.
In several human diseases mutant proteins fold or assemble improperly, resulting in defective protein trafficking (Cohen and Kelly, 2003). Correction of defective trafficking by protein-specific modulators or drug substrates was first shown for misprocessed human P-glycoprotein mutants (Loo and Clarke, 1997). Using this approach as potential clinical therapy, great progress has been achieved in the treatment of cystic fibrosis. A promising small molecule (VX-809) that corrects anterograde trafficking of ΔF508-CFTR mutant proteins has entered clinical trials (Clancy et al., 2012). Pharmacological correctors of either mutant or drug-induced trafficking deficient hERG channels are typically also direct hERG channel blockers, and their efficacy appears to be related to the domain in which the mutation is located and the affinity of the class III agent used (Ficker et al., 2002; Balijepalli et al., 2010; Dennis et al., 2012). In agreement with earlier results in LQTS2 mutants (Ficker et al., 2002), in the present study, we showed that correction of drug-induced hERG trafficking defects is dependent on channel affinity and is channel specific. In particular, modifications near a compound's aromatic residues greatly affect its ability to restore the drug-induced hERG trafficking. Further research into the difference between direct hERG channel blockade and correction of defective trafficking is necessary and may be of benefit for the treatment of defective hERG trafficking.
In conclusion, structural modifications of pentamidine differentially affected plasma membrane levels of hERG and KIR2.1. Modification of the phenyl ring or substituents directly attached to it had the largest effect, highlighting the importance of these features in ion channel binding. Thus, if certain residues are avoided or ‘synthesized out’, the ability to inhibit trafficking can be abolished, as was shown with PA-4, which had mild effects on both ion channels. Correction of defective protein trafficking is ion channel-specific and requires high affinity binding. Further analysis of important features in other hERG channel trafficking correctors may facilitate the design of a non-blocking corrector for trafficking defective hERG proteins in both congenital and acquired LQTS.
Acknowledgments
We would like to thank Dr C. January (University of Wisconsin) for providing HEK-hERG cells. Part of the research leading to the results has received funding from the European Community's Seventh Framework Programme FP7/2007–2013 under grant agreement No. HEALTH-F2-2009-241526, EUTrigTreat. Part of this work was supported by The Austrian Science Fund (FWF; Grants P22395, W1232). T. L. was supported by a research fellowship 2013 from the University of Vienna.
Glossary
- ER
endoplasmic reticulum
- hERG
human ether-a-go-go-related gene
- IK1
cardiac inward rectifying K+ current
- IKr
rapid component of the delayed rectifier K+ current
- LQTS
long QT syndrome
- MD
molecular dynamics
- TdP
Torsade de Pointes
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- Bakunova SM, Bakunov SA, Patrick DA, Kumar EV, Ohemeng KA, Bridges AS, et al. Structure-activity study of pentamidine analogues as antiprotozoal agents. J Med Chem. 2009a;52:2016–2035. doi: 10.1021/jm801547t. [DOI] [PubMed] [Google Scholar]
- Bakunova SM, Bakunov SA, Wenzler T, Barszcz T, Werbovetz KA, Brun R, et al. Synthesis and antiprotozoal activity of pyridyl analogues of pentamidine. J Med Chem. 2009b;52:4657–4667. doi: 10.1021/jm900805v. [DOI] [PubMed] [Google Scholar]
- Balijepalli SY, Anderson CL, Lin EC, January CT. Rescue of mutated cardiac ion channels in inherited arrhythmia syndromes. J Cardiovasc Pharmacol. 2010;56:113–122. doi: 10.1097/FJC.0b013e3181dab014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibler MR, Chou TC, Toltzis RJ, Wade PA. Recurrent ventricular tachycardia due to pentamidine-induced cardiotoxicity. Chest. 1988;94:1303–1306. doi: 10.1378/chest.94.6.1303. [DOI] [PubMed] [Google Scholar]
- de Boer TP, van Veen TA, Houtman MJ, Jansen JA, van Amersfoorth SC, Doevendans PA, et al. Inhibition of cardiomyocyte automaticity by electrotonic application of inward rectifier current from Kir2.1 expressing cells. Med Biol Eng Comput. 2006;44:537–542. doi: 10.1007/s11517-006-0059-8. [DOI] [PubMed] [Google Scholar]
- de Boer TP, Nalos L, Stary A, Kok B, Houtman MJ, Antoons G, et al. The anti-protozoal drug pentamidine blocks KIR2.x-mediated inward rectifier current by entering the cytoplasmic pore region of the channel. Br J Pharmacol. 2010;159:1532–1541. doi: 10.1111/j.1476-5381.2010.00658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, et al. AMBER 11. San Francisco, CA: University of California; 2010. [Google Scholar]
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67:12–18. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426:905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- Cordes JS, Sun Z, Lloyd DB, Bradley JA, Opsahl AC, Tengowski MW, et al. Pentamidine reduces hERG expression to prolong the QT interval. Br J Pharmacol. 2005;145:15–23. doi: 10.1038/sj.bjp.0706140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- Dennis AT, Wang L, Wan H, Nassal D, Deschenes I, Ficker E. Molecular determinants of pentamidine-induced hERG trafficking inhibition. Mol Pharmacol. 2012;81:198–209. doi: 10.1124/mol.111.075135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessertenne F. [Ventricular tachycardia with 2 variable opposing foci] Arch Mal Coeur Vaiss. 1966;59:263–272. [PubMed] [Google Scholar]
- Fenichel RR, Malik M, Antzelevitch C, Sanguinetti M, Roden DM, Priori SG, et al. Drug-induced torsades de pointes and implications for drug development. J Cardiovasc Electrophysiol. 2004;15:475–495. doi: 10.1046/j.1540-8167.2004.03534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D, Ghanta A, Kauffman GW, Sanguinetti MC. Physicochemical features of the HERG channel drug binding site. J Biol Chem. 2004;279:10120–10127. doi: 10.1074/jbc.M310683200. [DOI] [PubMed] [Google Scholar]
- Ficker E, Obejero-Paz CA, Zhao S, Brown AM. The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether-a-gogo-related gene (HERG) mutations. J Biol Chem. 2002;277:4989–4998. doi: 10.1074/jbc.M107345200. [DOI] [PubMed] [Google Scholar]
- Ficker E, Dennis AT, Wang L, Brown AM. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res. 2003;92:e87–100. doi: 10.1161/01.RES.0000079028.31393.15. [DOI] [PubMed] [Google Scholar]
- Ficker E, Kuryshev YA, Dennis AT, Obejero-Paz C, Wang L, Hawryluk P, et al. Mechanisms of arsenic-induced prolongation of cardiac repolarization. Mol Pharmacol. 2004;66:33–44. doi: 10.1124/mol.66.1.33. [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks CW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 09. Wallingford, CT: Gaussian, Inc; 2009. [Google Scholar]
- Girgis I, Gualberti J, Langan L, Malek S, Mustaciuolo V, Costantino T, et al. A prospective study of the effect of I.V. pentamidine therapy on ventricular arrhythmias and QTc prolongation in HIV-infected patients. Chest. 1997;112:646–653. doi: 10.1378/chest.112.3.646. [DOI] [PubMed] [Google Scholar]
- GOLD. 2008. GOLD, version 4.0: Cambridge Crystallographic Data Centre; Cambridge, UK.
- Gong Q, Jones MA, Zhou Z. Mechanisms of pharmacological rescue of trafficking-defective hERG mutant channels in human long QT syndrome. J Biol Chem. 2006;281:4069–4074. doi: 10.1074/jbc.M511765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Zhang Y, Chen Q, Duan Y, Zheng T, Hu X, et al. Fluconazole inhibits hERG K(+) channel by direct block and disruption of protein trafficking. Eur J Pharmacol. 2011;650:138–144. doi: 10.1016/j.ejphar.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C, et al. The potential for QT prolongation and pro-arrhythmia by non-anti-arrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Cardiovasc Res. 2000;47:219–233. doi: 10.1016/s0008-6363(00)00119-x. [DOI] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- van der Heyden MA, Smits ME, Vos MA. Drugs and trafficking of ion channels: a new pro-arrhythmic threat on the horizon? Br J Pharmacol. 2008;153:406–409. doi: 10.1038/sj.bjp.0707618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha TK. Evaluation of diamidine compound (pentamidine isethionate) in the treatment resistant cases of kala-azar occurring in North Bihar, India. Trans R Soc Trop Med Hyg. 1983;77:167–170. doi: 10.1016/0035-9203(83)90058-5. [DOI] [PubMed] [Google Scholar]
- Jones SK, Hall JE, Allen MA, Morrison SD, Ohemeng KA, Reddy VV, et al. Novel pentamidine analogs in the treatment of experimental Pneumocystis carinii pneumonia. Antimicrob Agents Chemother. 1990;34:1026–1030. doi: 10.1128/aac.34.6.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of HERG channel block. Mol Pharmacol. 2006;69:1709–1716. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- Kannankeril P, Roden DM, Darbar D. Drug-induced long QT syndrome. Pharmacol Rev. 2010;62:760–781. doi: 10.1124/pr.110.003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katchman AN, Koerner J, Tosaka T, Woosley RL, Ebert SN. Comparative evaluation of HERG currents and QT intervals following challenge with suspected torsadogenic and nontorsadogenic drugs. J Pharmacol Exp Ther. 2006;316:1098–1106. doi: 10.1124/jpet.105.093393. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knape K, Linder T, Wolschann P, Beyer A, Stary-Weinzinger A. In silico analysis of conformational changes induced by mutation of aromatic binding residues: consequences for drug binding in the hERG K+ channel. Plos ONE. 2011;6:e28778. doi: 10.1371/journal.pone.0028778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryshev YA, Ficker E, Wang L, Hawryluk P, Dennis AT, Wible BA, et al. Pentamidine-induced long QT syndrome and block of hERG trafficking. J Pharmacol Exp Ther. 2005;312:316–323. doi: 10.1124/jpet.104.073692. [DOI] [PubMed] [Google Scholar]
- Lees-Miller JP, Duan Y, Teng GQ, Duff HJ. Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol Pharmacol. 2000a;57:367–374. [PubMed] [Google Scholar]
- Lees-Miller JP, Duan Y, Teng GQ, Thorstad K, Duff HJ. Novel gain-of-function mechanism in K(+) channel-related long-QT syndrome: altered gating and selectivity in the HERG1 N629D mutant. Circ Res. 2000b;86:507–513. doi: 10.1161/01.res.86.5.507. [DOI] [PubMed] [Google Scholar]
- Loo TW, Clarke DM. Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J Biol Chem. 1997;272:709–712. doi: 10.1074/jbc.272.2.709. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalos L, de Boer TP, Houtman MJ, Rook MB, Vos MA, van der Heyden MA. Inhibition of lysosomal degradation rescues pentamidine-mediated decreases of K(IR)2.1 ion channel expression but not that of K(v)11.1. Eur J Pharmacol. 2011;652:96–103. doi: 10.1016/j.ejphar.2010.10.093. [DOI] [PubMed] [Google Scholar]
- Pearlstein R, Vaz R, Rampe D. Understanding the structure-activity relationship of the human ether-a-go-go-related gene cardiac K+ channel. A model for bad behavior. J Med Chem. 2003;46:2017–2022. doi: 10.1021/jm0205651. [DOI] [PubMed] [Google Scholar]
- Rajamani S, Eckhardt LL, Valdivia CR, Klemens CA, Gillman BM, Anderson CL, et al. Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br J Pharmacol. 2006;149:481–489. doi: 10.1038/sj.bjp.0706892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest. 2005;115:2025–2032. doi: 10.1172/JCI25539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Shagufta GD, Guo D, Klaasse E, de Vries H, Brussee J, Nalos L, et al. Exploring chemical substructures essential for HERG k(+) channel blockade by synthesis and biological evaluation of dofetilide analogues. ChemMedChem. 2009;4:1722–1732. doi: 10.1002/cmdc.200900203. [DOI] [PubMed] [Google Scholar]
- Siu SW, Vacha R, Jungwirth P, Bockmann RA. Biomolecular simulations of membranes: physical properties from different force fields. J Chem Phys. 2008;128:125103. doi: 10.1063/1.2897760. [DOI] [PubMed] [Google Scholar]
- Stary A, Wacker SJ, Boukharta L, Zachariae U, Karimi-Nejad Y, Aqvist J, et al. Toward a consensus model of the hERG potassium channel. ChemMedChem. 2010;5:455–467. doi: 10.1002/cmdc.200900461. [DOI] [PubMed] [Google Scholar]
- Takemasa H, Nagatomo T, Abe H, Kawakami K, Igarashi T, Tsurugi T, et al. Coexistence of hERG current block and disruption of protein trafficking in ketoconazole-induced long QT syndrome. Br J Pharmacol. 2008;153:439–447. doi: 10.1038/sj.bjp.0707537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MB, Volders PG, Stengl M, Spatjens RL, Beekman JD, Bischoff U, et al. Electrophysiological safety of sertindole in dogs with normal and remodeled hearts. J Pharmacol Exp Ther. 2003;307:776–784. doi: 10.1124/jpet.103.052753. [DOI] [PubMed] [Google Scholar]
- Tidwell RR, Jones SK, Geratz JD, Ohemeng KA, Cory M, Hall JE. Analogues of 1,5-bis(4-amidinophenoxy)pentane (pentamidine) in the treatment of experimental Pneumocystis carinii pneumonia. J Med Chem. 1990;33:1252–1257. doi: 10.1021/jm00166a026. [DOI] [PubMed] [Google Scholar]
- Varkevisser R, Houtman MJ, Waasdorp M, Man JC, Heukers R, Takanari H, et al. Inhibiting the clathrin-mediated endocytosis pathway rescues K(IR)2.1 downregulation by pentamidine. Pflugers Arch. 2013;465:247–259. doi: 10.1007/s00424-012-1189-5. [DOI] [PubMed] [Google Scholar]
- Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci U S A. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton JM, Demopulos PA, Goldschlager N. Torsade de pointes during administration of pentamidine isethionate. Am J Med. 1987;83:571–576. doi: 10.1016/0002-9343(87)90774-1. [DOI] [PubMed] [Google Scholar]
- Winckels SK, Thomsen MB, Oosterhoff P, Oros A, Beekman JD, Attevelt NJ, et al. High-septal pacing reduces ventricular electrical remodeling and proarrhythmia in chronic atrioventricular block dogs. J Am Coll Cardiol. 2007;50:906–913. doi: 10.1016/j.jacc.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Wolf MG, Hoefling M, Aponte-Santamaria C, Grubmuller H, Groenhof G. g_membed: efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J Comput Chem. 2010;31:2169–2174. doi: 10.1002/jcc.21507. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem. 1998;273:21061–21066. doi: 10.1074/jbc.273.33.21061. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, January CT. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J Biol Chem. 1999;274:31123–31126. doi: 10.1074/jbc.274.44.31123. [DOI] [PubMed] [Google Scholar]