

Abstract
Tissue trauma in the peritoneal and pelvic cavities following surgery or bacterial infection results in adhesions that are a debilitating cause of intestinal obstruction, chronic pelvic pain, and infertility in women. We recently demonstrated that CD4+ αβ T cells are essential for development of this process. Using a murine model of experimental adhesion formation, we now demonstrate that adhesion formation is characterized by the selective recruitment of Tim-3+, CCR5+, CXCR3+, IFN-γ+ cells, indicating the presence of a Th1 phenotype. We further demonstrate that adhesion formation is critically dependent on the function of Th1 cells because mice genetically deficient for IFN-γ, T-bet, or treated with Abs to the Th1-selective chemoattractant IL-16 show significantly less adhesion formation than wild-type mice. In addition, disrupting the interaction of the Th1-specific regulatory molecule Tim-3, with its ligand, significantly exacerbates adhesion formation. This enhanced response is associated with increases in the level of neutrophil-attracting chemokines KC and MIP-2, known to play a role in adhesiogenesis. These data demonstrate that the CD4+ T cells orchestrating adhesion formation are of the Th1 phenotype and delineate the central role of T-bet, Tim-3, IFN-γ, and IL-16 in mediating this pathogenic tissue response.
Timely resolution of the inflammatory response to tissue trauma is necessary to avoid unchecked adhesion formation that is a hallmark of chronic infectious, allergic, and autoimmune tissue disorders. During the response to surgical trauma, unchecked fibrosis leads to the development of fibrous adhesions that bind apposing tissues together in an inappropriate manner. Surgical adhesions are a major complication of gynecologic and abdominal surgery, with an incidence as high as 50–90% (1–3). Adhesions that develop after gynecologic surgery are a source of chronic pelvic pain and infertility (3). In addition, when most severe, adhesions in the abdominal cavity can cause potentially fatal small bowel obstruction and organ failure (4–6). Current treatments to prevent or reduce the severity of adhesions rely on the use of barrier devices or bioresorbable gels, but the mechanisms involved in the development of adhesions are not fully understood.
Little is known about the cellular host response that leads to adhesion formation. A few reports describe a role for neutrophils and macrophages in this process (7–9); however, information on the role of T cells in this process is lacking. We have shown that CD4+ αβ T cells are required for surgical and postinfectious adhesion formation because following cecal abrasion, activated CD4+ T cells enter the peritoneal cavity, orchestrate chemokine production and leukocyte trafficking, and then home to the site of developing adhesions (10, 11). Modulating the activity of these T cells by blockade of costimulatory and coinhibitory molecules significantly affects the severity of adhesions that eventually develop.
Our initial experiments suggested that the CD4+ T cells mediating adhesion formation are of the Th1 phenotype (10). In this study, we confirm that the Th1 cell is central to adhesion development and, furthermore, identify the requirement of two key regulators of Th1 cell differentiation and activation, T-bet and Tim-3, in this process (12–17). Our studies further demonstrate the requirement for IFN-γ, a prototypical Th1 cytokine, and IL-16 (18), a chemoattractant for Th1 cells (19), in this host response. Importantly, CD4+ T cells that home to sites of adhesion formation express Tim-3 and IFN-γ, as well as CCR5 and CXCR3, chemokine receptors preferentially associated with Th1 cells. Our data strongly suggest that the CD4+ T cells associated with surgical adhesions are Th1 effector cells and identify chronic adhesion formation as a dysregulated type I immune response.
Materials and Methods
Animals
IFN-γ-deficient (B6.129S7-Ifngtm1Ts/J), CCR5-deficient (B6;129P2-Ccr5tm1Kuz/J), C57BL/6J, and DO11.10 mice were purchased from The Jackson Laboratory. T-bet-deficient mice (16) on the C57BL/6J background (backcrossed >10 generations) were bred at the Channing Laboratory Animal Facility. All animals were provided with food and water ad libitum and housed under specific pathogen-free conditions. The mice were maintained according to the Harvard Medical School animal management program, which is accredited by the American Association for the Accreditation of Laboratory Animal Care.
Rodent model of surgical adhesion formation
Abdominal surgery was performed following guidelines approved by the Harvard Medical School animal management program. Mice were anesthetized with a single injection i.p. of 0.2 ml of pentobarbital sodium (50 mg/ml; Abbott Laboratories) diluted 1/5 v/v in PBS (10 mg/ml). Abdominal adhesions were induced by abrasion of the cecum and the abdominal wall as described previously (10). Briefly, sterile surgical gauze was used to abrade the cecum with seven to eight strokes of a consistent medium pressure. This resulted in the development of petechiae on the surface of the cecum. Using this technique, the resultant adhesions were predominately classified as low to moderate according to the adhesion classification score described below. Where indicated, the degree of abrasion was modified to induce adhesions of increased severity. Severe abrasions were induced with 15 strokes of medium pressure. The procedures used induced adhesions that were reproducible in terms of number and intensity in control animals and found to be similar in reproducibility as those obtained using a regulated mechanical device. Animals were euthanized and examined for adhesion formation 6 days later by an observer blinded to the identity of the experimental groups. The severity of adhesions in each animal was evaluated according to the following scoring system: 0, no adhesions; 1, one thin filmy adhesion; 2, more than one thin adhesion; 3, thick adhesion with focal point; 4, thick adhesion with planar attachment, or more than one thick adhesion with focal point; and 5, very thick vascularized adhesion or more than one planar adhesion. To ensure reproducibility, all experiments were conducted on at least two different groups of mice on separate occasions. The median adhesion scores for the various experimental groups were compared by the Mann-Whitney U test. Differences between groups were considered significant at p < 0.05.
For the Tim-3Ig Ab studies, the mice received s.c. injections of either Tim-3Ig or control human IgG1 (250 μg each) at −24, 0, and +24 h relative to cecal abrasion surgery. For in vivo neutralization of IL-16, mice were injected i.p. with 100 μg of anti-IL-16 mAb (clone 14.1, IgG2a) or isotype control (R&D Systems) at 6 h, 24 h, 3 days, and 5 days following surgery.
Antibodies
Anti-Tim-3 mAb 8B.2C12, anti-IL-16 mAb (clone 14.1), and Tim-3Ig, a chimeric protein in which the extracellular portion of Tim-3 is fused to the Fc portion of human IgG1, have been described previously (12, 14, 18, 31). Control human IgG1 and mouse IgG were purchased from Sigma-Aldrich. Polyclonal goat Abs specific for mouse CCR5 and CXCR3, as well as control goat IgG, were purchased from Santa Cruz Biotechnology. For the confocal microscopy studies, Abs were conjugated with Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647 using Alexa Fluor mAb Labeling Kits (Molecular Probes). The anti-mouse CD4 mAb RM4-5 and control rat IgG2a Ab conjugated with Alexa Fluor 488 were purchased from Caltag Laboratories.
Measurement of chemokines and cytokines by ELISA or ELISPOT assay
Levels of MIP-2, KC, and IL-16 in peritoneal lavage fluid and IL-16 production in Th1/Th2 cell cultures were determined by specific ELISA kits purchased from R&D Systems. IFN-γ production by purified CD4+ T cells was determined by ELISPOT assay. Briefly, single-cell suspensions from mesenteric lymph nodes (LN)4 or peritoneal lavage fluid were depleted of RBC by centrifugation on Histopaque (ρ = 1.119) at 700 × g (Sigma-Aldrich). The buoyant leukocyte populations were passed over CD4 Cell Enrichment Columns (R&D Systems). Purified CD4+ T cells were cultured in polyvinylidene difluoride-backed 96-well microplates at either 0.5 or 1 × 105 cells/well, with or without 1 × 105 syngeneic 3000 rad irradiated spleen cells as a source of APCs, and with or without 2 μg/ml Con A. Culture medium consisted of RPMI 1640 containing 10% FBS, 1 mM sodium pyruvate, 100 μM nonessential amino acids, 2 mM l-glutamine, 100 U/ml penicillin plus 100 μg/ml streptomycin, and 10 mM HEPES buffer (Invitrogen Life Technologies). After a 36-h incubation, the number of IFN-γ-producing cells was determined by a specific ELISPOT assay kit (MabTech). The values are expressed as the averages ± SDs and the asterisk denotes significantly different from control wells at the 5% confidence level.
Histology
Samples of the abdominal wall comprising the skin, muscle mass, peritoneum, and any tissue associated by adhesions were harvested and flash frozen in Tissue-Tek OCT compound (Sakura). Sections (6- to 10-μm thick) were cut and one representative section was stained with H&E (Richard-Allan Scientific) to confirm the integrity of the adhesions. For analysis by confocal microscopy, samples were fixed in ice-cold acetone for 10 min, followed by extensive washing in PBS. For analysis of cytokines, the samples were incubated in 0.05% saponin in PBS for 30 min, followed by washing in PBS. The samples were blocked with 10% goat or rat serum for 20 min, followed by incubation with primary Ab (directly conjugated with Alexa Fluor dyes; see above) for 1 h at room temperature. Samples were then washed in PBS and mounted with SlowFade Antifade mounting reagent (Molecular Probes) and analyzed with a Zeiss Axiovert 200M microscope equipped with helium/neon and argon lasers. The images were generated and analyzed with Pascal software (Borland).
T cell skewing and peritoneal macrophage isolation
To generate Th1 and Th2 cell populations, DO11.10 CD4+ T cells were isolated from LN and positively selected using a density gradient centrifugation technique (Stem Cell Technologies) as previously described (19). CD4+ T cell purity was assessed by flow cytometry. APCs were generated from syngeneic T cell-depleted splenocytes prepared by positive selection using a magnetic bead pan-T cell Ab to Thy-1.2 (Dynal). Ag-presenting splenocytes were treated with mitomycin C (50 μg/ml; Sigma Aldrich) for 20 min on ice before washing twice in PBS. Th1 and Th2 cells were generated by adding 2 × 106 CD4+ T cells/ml with 2 × 106 APCs/ml and 1 μg/ml pOVA323–339 (Invitrogen). The Th1 or Th2 cell skewing mixture was then added to the cell cultures. Th1 cell mixture included IL-12 (5 ng/ml) and anti-IL-4 (2.5 μg/ml; both R&D Systems). The Th2 cell mixture included IL-4 (5 ng/ml), anti-IL-12 (10 μg/ml; R&D Systems), and anti-IFN-γ (2.5 μg/ml) (Sigma Aldrich). Purity of the skewed populations were determined by assessing IL-4, IL-5, and IFN-γ production in an aliquot from each cell culture following Con A (5 μg/ml) stimulation.
Peritoneal macrophages were obtained from C57BL/6 mice following peritoneal lavage with 1 ml of PBS. The macrophages were isolated using plastic adherence and purity assessed by labeling with a fluorescently conjugated anti-CD14 Ab as detected by flow cytometry.
Results
CD4+ T Th1 cells are present at the site of surgical adhesion formation
Histological analysis was performed on tissue 6 days after low to moderate cecal abrasion in C57BL/6J mice. Fig. 1 shows a representative adhesion that developed distal to the site of abrasion and that involved the small intestine. H&E staining of a section of this adhesion revealed a marked inflammatory cell infiltrate surrounded by a matrix of collagen-like fibrils binding the smooth muscle of the small intestine to the remains of the abdominal wall (Fig. 1). As reported previously, these infiltrates consist largely of macrophages, some neutrophils, and, CD4+ T cells (11). To further investigate the phenotype of the recruited T cells to adhesion sites, histological sections were stained with Abs to CD4 as well as counterstained with Abs to Tim-3, CCR5, and CXCR3, all specific for Th1 cells, or with the Th2 marker CCR3. Tim-3 is a recently characterized marker for differentiated Th1 cells, up-regulated on Th1-polarized CD4+ T cells (13). When adhesions were stained with fluorescence-labeled Abs and examined by confocal microscopy, CD4+ cells expressing Tim-3, CCR5, and CXCR3 were detected (Fig. 2). Cells expressing these markers were not detected in surrounding tissue, indicating selective recruitment to the inflamed site. CCR3+ cells were not detected at the sites of adhesions nor in surrounding tissue, suggesting that Th2 cells are not involved in the inflammatory precess (Fig. 2).
FIGURE 1.
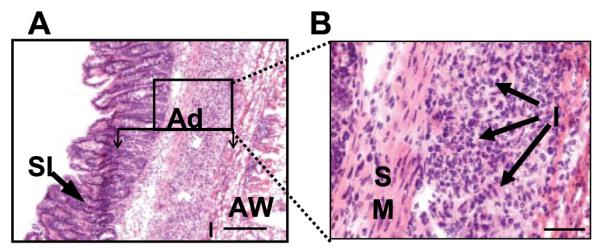
Histologic examination of a representative surgical adhesion in which a portion of the small intestine has become attached to the abdominal wall. A, H&E staining. Original magnification, ×100. The adhesion (Ad) comprises the small intestine (SI), inflammatory infiltrate (I), and the remains of the abdominal wall (AW), which are indicated. The box indicates the field under higher magnification in B. B, The same adhesion under higher magnification. Bar, 100 μm. Original magnification, ×400. The smooth muscle (SM) of the intestine and the inflammatory infiltrate (I) are indicated. These results are representative of surgical adhesions obtained from the intestine, cecum, and intestinal wall, examined for each animal, from 10 different C57BL/6J mice.
FIGURE 2.

CD4+ T cells expressing Tim-3, CCR5, and CXCR3 are present at the site of surgical adhesions. Sections were stained with anti-CD4 conjugated with Alexa Fluor 488 (green) and anti-CCR5 (A), anti-CXCR3 (B), anti-Tim-3 (C), and anti-CCR3 (D), all conjugated with Alexa Fluor 594 (red). Adhesions were examined under confocal microscopy. Bars, 20 μm. Original magnification, ×670. Cells costaining green and red are evident when the two channels are merged. These are representative images from five different mice for each receptor.
Formation of severe adhesions requires the presence of IFN-γ-producing Th1 cells
Having shown that Th1 cells are selectively recruited to sites of adhesions, we next wanted to determine whether these cells were required for adhesion formation. Th1 cells are positive for the transcription factor T-bet and produce IFN-γ. We predicted that, if surgical adhesion formation is mediated by CD4+ Th1 cells, T-bet-deficient mice should develop significantly reduced adhesions in response to cecal abrasion. As shown in Fig. 3A, mice subjected to a moderate cecal abrasion protocol developed adhesions with a median score of 2.5. In contrast, the majority of T-bet-deficient mice developed very few detectable adhesions, resulting in a median score of 0 ( p = 0.0422; Mann-Whitney U test).
FIGURE 3.
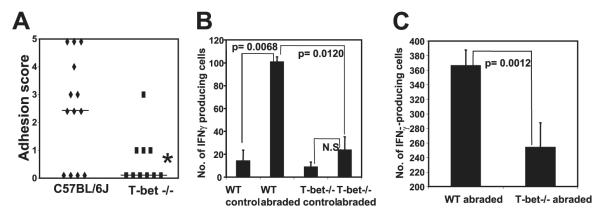
Mice deficient in T-bet expression have reduced adhesion formation due to reduced homing of IFN-γ-producing CD4+ T cells. A, C57BL/6J and T-bet-deficient mice were subjected to cecal abrasion surgery and adhesion formation was assessed 6 days later. *, p = 0.0422; Mann-Whitney U test. The data are combined from three independent experiments. The bars indicate median adhesion scores. B and C, CD4+ T cells were purified from mesenteric LN (B) or peritoneal lavage cells (C) of T-bet−/− or C57BL/6J mice 6 days after cecal abrasion surgery, and the number of IFN-γ-producing cells was determined by ELISPOT assay. The number of IFN-γ-producing cells per 1 × 105 (B) or 5 × 104 (C) input CD4+ T cells is indicated. No CD4+ T cells were present in peritoneal lavage samples of control mice and therefore could not be tested. The data were generated using 10 T-bet−/− mice and 13 control mice.
To begin to establish a potential mechanism by which T-bet+ cells contributed to adhesion formation, we used an ELISPOT assay to quantitate the number of IFN-γ-producing CD4+ T cells in T-bet−/− and wild-type (WT) mice. Following cecal abrasion surgery, there was a marked increase in the number of IFN-γ-producing CD4+ T cells in the draining mesenteric LN of WT mice at the 6-day time point ( p = 0.0068; unpaired t test) (Fig. 3B). In contrast, there was no significant increase in the number of these cells in the T-bet-deficient mice. More important, the number of IFN-γ-producing CD4+ T cells in abraded WT mice was considerably higher than in abraded T-bet−/− mice, whether purified from mesenteric LN ( p = 0.0120; Fig. 3B) or from peritoneal lavage cells ( p = 0.0012; Fig. 3C). Thus, the reduced adhesion formation observed in T-bet−/− mice correlated with fewer IFN-γ-producing CD4+ T cells in the draining mesenteric LN and peritoneal cavity, an indication that trafficking of Th1 cells to the site of injury and to the draining LN was profoundly reduced in these mice, consistent with other reports indicating reduced T cells at inflammatory sites in T-bet-deficient mice (20–22). It is interesting to note that in T-bet−/− mice a low number of IFN-γ-producing cells are present and that this correlates with detectable, but less severe, adhesion formation, suggesting a role of IFN-γ in adhesion formation.
IFN-γ is required for adhesion formation
To investigate the requirement of IFN-γ for adhesion formation, we subjected IFN-γ-deficient mice to the cecal abration model. As shown in Fig. 4, these mice had no evidence of adhesions following moderate cecal abrasion surgery since their peritoneal cavities appeared indistinguishable from those of sham-treated mice. WT C57BL/6J mice, in contrast, had a median adhesion score of 3 ( p < 0.0025; Mann-Whitney U test). Taken together, these data indicate that adhesion formation requires the presence of IFN-γ-producing CD4+ Th1 cells.
FIGURE 4.
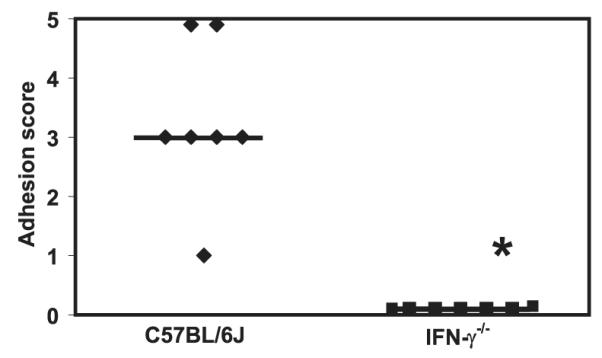
IFN-γ-deficient mice do not form detectable adhesions. C57BL/6J mice and IFN-γ-deficient mice were subjected to cecal abrasion surgery and examined and scored 6 days later for adhesions. *, p < 0.0025; Mann-Whitney U test. The bars indicate median adhesion scores. The data were generated from seven IFN-γ−/− and control mice, respectively.
Blockade of the Th1-associated protein Tim-3 exacerbates adhesion formation
We have demonstrated that IFN-γ production by Th1 cells recruited to sites of adhesion formations function as a proinflammatory mediator. The presence of Tim-3+ cells at these sites presents the possibility that the Th1 cells are regulating the inflammatory process. Because Tim-3 has been shown to inhibit effector (Th1) cells during immune responses, we hypothesized that blockade of Tim-3, by Tim-3 Ig, should lead to exacerbation of adhesion formation. To investigate this, we subjected C57BL/6J mice to the cecal abrasion model and treated them with Tim-3Ig. To determine alteration of adhesion formation following Tim-3 Ig treatment, the surgical protocol was moderated to generate low adhesion scores in the control group (seven strokes per site). As shown in Fig. 5, mice treated with a control human IgG1 Ab (250 μg at −24, 0, and 24 h) had a median adhesion score of 1.5. In contrast, treatment with Tim-3 Ig (250 μg at −24, 0, and 24 h) markedly exacerbated adhesion formation, median score of 5 ( p < 0.0001; Mann-Whitney U test).
FIGURE 5.
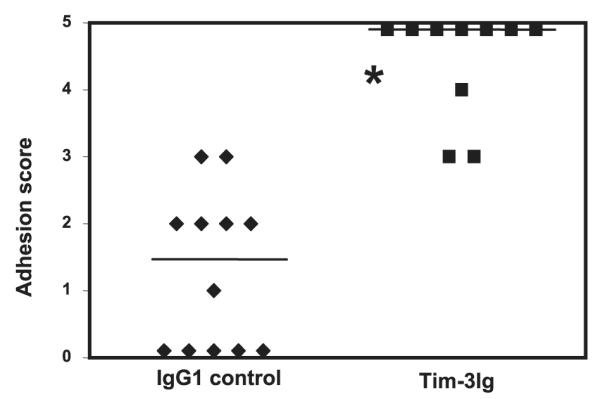
Tim-3 blockade exacerbates adhesion formation in the peritoneal cavity of WT mice. C57BL/6J mice were treated with Tim-3Ig or a control human IgG1, 250 μg s.c. at −24, 0, and 24 h relative to cecal abrasion surgery. Six days after surgery, the mice were euthanized and scored for adhesions. *, p < 0.0001; Mann-Whitney U test. The bars indicate median adhesion scores. For these experiments, 10 mice received Tim-3 Ig and 12 mice received IgG control Ab.
In addition to CD4+ Th1 cells, the other major immune cell recruited to sites of adhesion formation are neutrophils, which generate oxygen free radicals (23). We have reported previously that the neutrophil-attracting chemokines MIP-2 and KC play a critical role in the acute inflammatory response that leads to adhesion formation (10). Therefore, peritoneal lavage fluid taken from IgG1-and Tim-3 Ig-treated mice was assessed for MIP-2 and KC levels (Fig. 6). We have previously reported that following surgery neutrophilic infiltrates can be detected as early as 6 h, peak at 24 h, and are significantly diminished by 72 h (10). Therefore, we assessed MIP-2 and KC levels at both 6 and 24 h. Tim-3 Ig treatment significantly increased expression of MIP-2 at both 6 and 24 h after surgery and KC at 24 h after surgery. These data suggest that treatment with Tim-3 Ig affects adhesion formation at the earliest stages of this host response by increasing expression of these inflammatory mediators. Neither MIP-2 nor KC were detected in peritoneal lavage fluid from nonabraded mice (data not shown).
FIGURE 6.
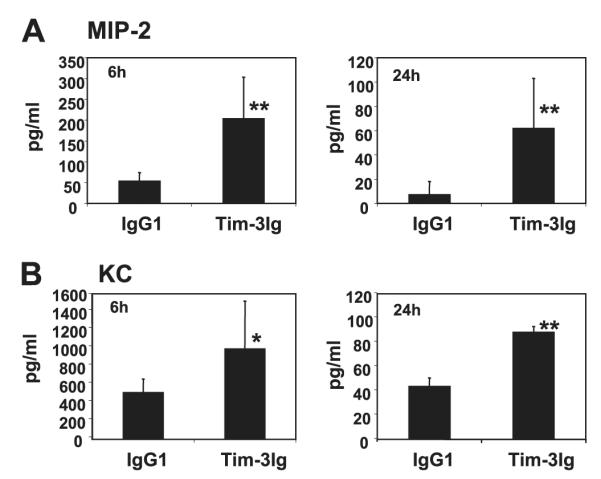
Tim-3 blockade increases chemokine production in the peritoneal cavity of mice following cecal abrasion. C57BL/6J mice were treated with Tim-3Ig or a control human IgG1, 250 μg s.c., once at −24 h and once at the time of cecal abrasion surgery. The mice were euthanized 6 or 24 h after surgery. MIP-2 (A) and KC (B) levels in peritoneal lavage fluid were assessed by ELISAs specific for these chemokines. The data are expressed as average values ± SD and **, p < 0.05; Student’s unpaired t test; *, p < 0.10. The averages were obtained from four different mice for each group.
The peritoneal fluid was also assessed for the presence of the Th1 cell chemoattractant IL-16 (19). Mice receiving Tim-3 Ig treatment demonstrated an increase in IL-16 expression at the 24-h time point, which was 3-fold greater than that detected in the isotype control-treated mice (Fig. 7). Our previous work (10) also demonstrated that following surgery, unlike neutrophils, T cell numbers continued to increase beyond 48 h; therefore, IL-16 levels were also assessed on days 3 and 6. By day 3, the amount of detectable IL-16 in the peritoneal fluid was 106 ± 14 pg/ml, with the day 6 measurement indicating a further rise to 125 ± 19 pg/ml (four mice per group were used for the 3- and 6-day time points). IL-16 was not detected in mice that were nonabraded (data not shown).
FIGURE 7.
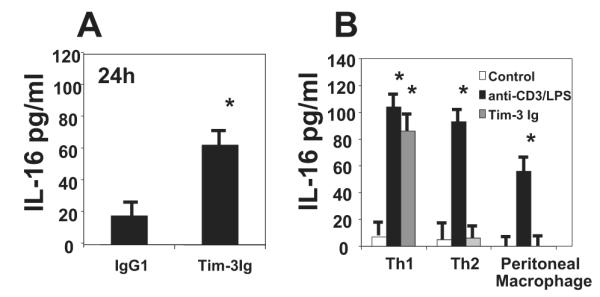
Tim-3 blockade increases IL-16 production in the peritoneal cavity of mice following cecal abrasion. A, Ten C57BL/6J mice were treated with either Tim-3Ig or a control human IgG1, 250 μg s.c., once at −24 h and once at the time of cecal abrasion surgery. The mice were euthanized 24 h after surgery and IL-16 levels were assessed in the peritoneal lavage fluid by ELISA. B, Skewed murine Th1 or Th2 cells along with isolated peritoneal macrophages were cultured at 1 × 106 cells/ml for 24 h in the absence or presence of 5 μg of Tim-3 Ig, 1 μg of immobilized anti-CD3 Ab, or 1 μg of LPS (for the macrophage cultures only), at which time the cell supernatants were assessed for IL-16 protein by ELISA. The data are expressed as average values ± SD and *, p < 0.05; Student’s unpaired t test.
To investigate the potential cell source for the IL-16, skewed murine Th1 or Th2 cells, as well as freshly isolated peritoneal macrophages, were cultured with 5 μg of Tim-3 Ig, control Ab or positive control stimulants anti-CD3 Ab (1 μg/ml plate bound) for T cells or 1 μg of LPS for the macrophage culture. The supernatants were assessed for IL-16 production following a 24-h culture period. Tim-3 Ig stimulation induced IL-16 production only in supernatants from the Th1 cells, although all cell types were capable of producing IL-16 when stimulated with the positive control stimulants (Fig. 7).
Surgical adhesion formation requires the activity of the Th1 chemoattractant cytokine IL-16
Based on our findings that Th1 cell recruitment and activation are required for adhesion formation and that Th1 cells can secrete a Th1-specific chemoattractant, IL-16, we hypothesized that neutralization of IL-16 would reduce adhesion formation in mice by inhibiting the trafficking of CD4+ Th1 cells to the site of tissue damage. Accordingly, we subjected C57BL/6J mice to severe cecal abrasion surgery, treating them with a control Ab or a neutralizing Ab to IL-16. Anti-IL-16 treatment markedly reduced adhesion formation in these mice (median adhesion score of 0), compared with mice receiving a control Ab (median adhesion score of 4; p = 0.0022) (Fig. 8). Taken together, these data indicate that Th1 cell-generated IL-16 may function as a positive feedback mechanism for further recruitment of Th1 cells to sites of adhesion formation.
FIGURE 8.
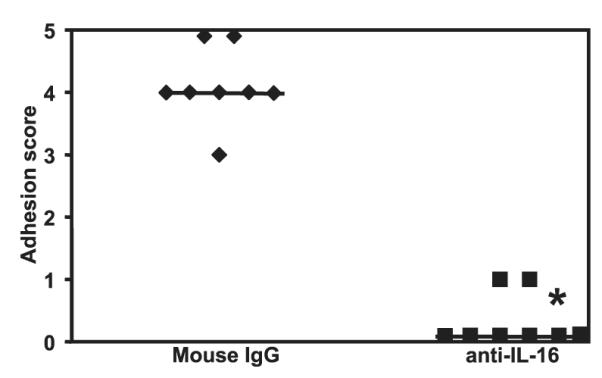
Adhesion formation requires expression of IL-16. Sixteen C57BL/6 mice were subjected to cecal abrasion surgery and treated with 100 μg of either anti-IL-16 or control Ab at 6 h, 24 h, 3 days, and 5 days after surgery. Six days after surgery, the mice were euthanized and scored for adhesions. *, p = 0.0022; Mann-Whitney U test. The bars indicate the median adhesion score.
Discussion
Inappropriately regulated inflammation leading to unchecked fibrosis is a hallmark of infectious, allergic, and autoimmune disorders. In the case of abdominal and gynecologic surgical procedures, unresolved fibrosis leads to the formation of adhesions. Little is known about the cellular mechanisms by which adhesions form, but the fact that they regenerate after secondary surgical lysis and form distally to the initial sites of tissue trauma (5, 24) underscores their dynamic character and suggests that they could result from a chronic inflammatory response.
Using a rodent model of experimental adhesion formation, we have investigated cellular and molecular regulators that could potentially mediate the inflammatory response to abdominal surgery. Using a cecal abrasion model, we have previously shown that CD4+ T cells home to the site of adhesion formation and function to control chemokine expression (10). Furthermore, mice genetically deficient for Stat4, which mediates IL-12 signaling, have significantly less adhesion formation than WT mice, while Stat6−/− mice deficient in IL-4 signaling developed adhesions as severe as those of WT mice (10). These results indicated that IL-12 signaling and development of a Th1 response is important for adhesion formation.
To address the role of Th1 cells in adhesion formation, we first identified whether there was preferential recruitment of a CD4+ T cell subset. Using relatively specific Th1 surface markers, Tim-3, CCR5, and CXCR3, it was noted that these cells were present, while CCR3+ Th2 cells were not detected. Interestingly, Th1+ cells were not detected in tissue adjacent to adhesion formation, suggesting a selective recruitment of Th1 cells to those sites.
We next investigated functional contributions of Th1 cells to the formation of surgical adhesions. Studies have identified the transcription factor T-bet as a key regulator of CD4+ Th1 cell differentiation (15–17). T-bet is up-regulated in CD4+ T cells by IFN-γ produced by APCs. T-bet then stimulates IFN-γ production by the T cell, thus completing a positive feedback loop. The central role played by T-bet in the development of Th1-mediated autoimmune diseases strongly suggested that it could also be a key regulator of adhesion formation. As predicted, T-bet-deficient mice showed significantly reduced adhesion formation following cecal abrasion surgery, which correlated with fewer IFN-γ-producing CD4+ T cells in the peritoneal cavity and draining mesenteric LN. Along those lines, Lord et al. (25) have recently shown that T-bet-deficient CD4+ T cells have reduced binding to P-selectin, as well as markedly reduced expression of CXCR3 and responsiveness to two of its cognate chemokines, CXCL10 (IFN-γ-inducible protein 10) and CXCL11 (IFN-inducible T cell α chemoattractant). Because we observed CXCR3 expression on CD4+ T cells at the site of adhesions, as well as CXCL10 expression in the peritoneal cavity of WT mice following cecal abrasion (11), the results of Lord et al. (25) suggest a plausible mechanism for the reduced trafficking of CD4+ T cells observed in our experiments. The ability of T-bet-deficient mice to develop mild adhesions could be attributed to residual IFN-γ-producing CD4+ T cells in these animals, a concept supported by our observation that adhesion development was undetectable in IFN-γ-deficient mice. It is important to note that both T-bet and IFN-γ are expressed by a variety of cell types. Thus, the effects we observed in T-bet−/− and IFN-γ−/− mice must be interpreted in light of the fact that both T cells and critical APCs are affected in these mice.
Tim-3+ cells were also detected at adhesion sites. Tim-3 is a discrete cell surface marker for differentiated Th1 cells, which regulates Th1 immune responses and induction of peripheral tolerance. Treatment of mice with Tim-3Ig, a chimeric recombinant protein comprising the extracellular portion of mouse Tim-3 fused to a human IgG1 Fc tail, accelerates the development of experimental autoimmune encephalitis and is accompanied by hyperproliferation of T cells producing Th1 cytokines (14). Therefore, Tim-3 along with its ligand galectin-9, is involved in negative regulation of effector function in Th1 cells (26, 27). Our data indicate that treatment of mice with Tim-3Ig markedly exacerbated adhesion formation, which correlated with increased production of neutrophil-attracting chemokines. The specific attraction of neutrophils by MIP-2 and KC implies that blockade of Tim-3 with its ligand affects trafficking of leukocytes to the peritoneal cavity. This appears to be a rapid event as both cytokines were produced within 24 h and is consistent with an early neutrophilic infiltrate (10). Expression of both Tim-3 and galectin-9 is restricted mostly to T cells, suggesting that Tim-3Ig inhibits a critical T cell-T cell interaction (12, 14).
Tim-3 also appears to be involved in regulating recruitment of Th1 T cells. Tim-3Ig stimulation of Th1 cells, but not Th2 or peritoneal macrophages, resulted in the production of IL-16. These studies required that the T cells undergo artificial skewing into either a Th1 or Th2 subset; however, since neither the Th2 subset nor the macrophage population produced IL-16 in response to Tim-3 Ig, it is likely that the Th1 subset also represents the major source in vivo. Recruitment of Th1 cells to sites of inflammation requires either a selective expression of chemoattractant receptors or selective responsiveness by the Th1 cells to certain chemoattractants (28–30). The CD4 ligand-specific chemoattractant IL-16 has previously been reported to have a preferential migratory effect for Th1 cells, likely involving cosignaling through CCR5 (19). In models of Th1-mediated inflammation, experimental autoimmune encephalitis (31), inflammatory bowel disease (32), and contact hypersensitivity (33), treatment with anti-IL-16 results in a reduction of Th1+ cells and attenuation of inflammation. We now demonstrate that neutralization of IL-16 significantly reduced the formation of adhesions, demonstrating that it plays a major role in this process. These findings also suggest a positive regulatory mechanism whereby activated Th1 cells at sites of cecal abration can selectively recruit additional Th1 cells for development of adhesion formation. As with most inflammatory models, the number of cells required to regulate the response is difficult to assess. Additional studies are needed to more fully elucidate the numbers, and location relative to adhesion formation, of Th1 cells that are required for initiation and perpetuation of the inflammatory response.
The obvious goal in adhesion prevention is to decrease newly forming postoperative adhesions in the abdominal cavity without compromising dermal or peritoneal wound healing. However, despite reducing adhesion formation, many experimental adhesion prevention modalities have failed due to their negative impact on wound healing. A good example is the systemic or i.p. administration of corticosteroids, which reduced adhesion formation but severely compromised wound healing and caused immunosuppression in postsurgical patients. In our studies, there was no gross evidence of a loss of wound integrity or dermal wound dehiscence in any of the groups. Interestingly, similar adhesion studies (34) have shown that colonic anastomotic healing correlates well with dermal wound healing, further suggesting that monitoring the healing of the primary incision is a good indicator of overall wound healing. It was evident from our study that the lack of functional Th-1 cells did not interfere with the healing of the primary incision. Although there are mixed reports linking IFN-γ deficiency to impaired wound healing (35), a recent study by Ishida et al. (36) showed that compared with WT mice, IFN-γ −/− mice show signs of accelerated wound healing.
In summary, we have demonstrated that CD4+ T cells, instrumental in controlling experimental postsurgical adhesion formations, are under the control of the Th1 master switch T-bet and the Th1 cell regulator Tim-3. Tim-3 activation appears to be critical for induction of neutrophil and Th1-specific cell recruitment, by IL-16, with subsequent elaboration of IFN-γ. Our data illustrate for the first time the regulatory role of Th1 cells in adhesion formation. And, more specifically, that Tim-3, a newly described surface marker of Th1 cells, is expressed on T cells during an active inflammatory process in vivo. The formation of abdominal surgical adhesions is the result of the accumulative effect of many contributory factors and cell types. We have previously identified the involvement of IL-17 and CXCL8 in adhesion formation and found that neutralization of these cytokines partially attenuates adhesion formation (10). In further delineation of the model, we now report a central role for Th1 cells in the pathogenesis and identify that specifically targeting the activity of either Tim-3 or IL-16 could have significant therapeutic effects following abdominal surgery in humans.
Acknowledgment
We thank Dr. Karen Reed for critical review of this manuscript and insightful comments.
Footnotes
This work was supported by research grants from the National Institutes of Health (RM64805-01, HL32802, and CA112663 to L.H.G.).
Abbreviations used in this paper; LN, lymph node; WT, wild type.
Disclosures W.W.C. is listed as co-inventor on patents related to anti-IL-16 Ab.
References
- 1.Trimbos-Kemper T, Trimbos J, van Hall E. Adhesion formation after tubal surgery: results of the eighth-day laparoscopy in 188 patients. Fertil. Steril. 1985;43:395–400. doi: 10.1016/s0015-0282(16)48438-4. [DOI] [PubMed] [Google Scholar]
- 2.Monk BJ, Berman M, Montz F. Adhesions after extensive gynecologic surgery: clinical significance, etiology, and prevention. Am. J. Obstet. Gynecol. 1994;170:1396–1403. doi: 10.1016/s0002-9378(94)70170-9. [DOI] [PubMed] [Google Scholar]
- 3.Lower A, Hawthorn RJ, Ellis H, O’Brien F, Buchan S, Crowe AM. The impact of adhesions on hospital readmissions over ten years after 8849 open gynaecological operations: an assessment from the Surgical and Clinical Adhesions Research Study. Br. J. Obstet. Gynaecol. 2000;107:855–862. doi: 10.1111/j.1471-0528.2000.tb11083.x. [DOI] [PubMed] [Google Scholar]
- 4.Ellis H. The clinical significance of adhesions: focus on intestinal obstruction. Eur. J. Surg. 1997;(Suppl. 577):5–9. [PubMed] [Google Scholar]
- 5.Wilson M, Hawkswell J, McCloy RF. Natural history of adhesional small bowel obstruction: counting the cost. Br. J. Surg. 1998;85:1294–1298. doi: 10.1046/j.1365-2168.1998.00822.x. [DOI] [PubMed] [Google Scholar]
- 6.Diamond MP, Freeman ML. Clinical implications of postsurgical adhesions. Hum. Reprod. Update. 2001;7:567–576. doi: 10.1093/humupd/7.6.567. [DOI] [PubMed] [Google Scholar]
- 7.Ar’Rajab A, Mileski W, Sentementes JT, Sikes P, Harris R, Dawidson IJ. The role of neutrophils in peritoneal adhesion formation. J. Surg. Res. 1996;61:143–146. doi: 10.1006/jsre.1996.0095. [DOI] [PubMed] [Google Scholar]
- 8.Haney AF. Serious complications of uterine artery embolization for conservative treatment of fibroids. Fertil. Steril. 2003;79:128–131. doi: 10.1016/s0015-0282(02)04398-4. [DOI] [PubMed] [Google Scholar]
- 9.Vural B, Cantürk NZ, Esen N, Solakoglu S, Cantürk Z, Kirkali G, Sökmensüer C. The role of neutrophils in the formation of peritoneal adhesions. Hum. Reprod. 1999;14:49–54. doi: 10.1093/humrep/14.1.49. [DOI] [PubMed] [Google Scholar]
- 10.Chung D, Chitnis T, Panzo RJ, Kasper D, Sayegh M, Tzianabos A. CD4+ T cells regulate surgical and postinfectious adhesion formation. J. Exp. Med. 2002;195:1471–1478. doi: 10.1084/jem.20020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holsti MA, Chitnis T, Panzo RJ, Bronson RT, Yagita H, Sayegh MH, Tzianabos AO. Regulation of postsurgical fibrosis by the programmed death-1 inhibitory pathway. J. Immunol. 2004;172:5774–5781. doi: 10.4049/jimmunol.172.9.5774. [DOI] [PubMed] [Google Scholar]
- 12.Sánchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos C, Manlongat N, Bender O, Kamradt T, Kuchroo VK, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003;4:1093–1101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- 13.Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 14.Sabatos CA, Chakravarti S, Cha E, Schubart A, Sánchez-Fueyo A, Zheng XX, Coyle AJ, Strom TB, Freeman GJ, Kuchroo VK. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 2003;4:1102–1110. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- 15.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 16.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 17.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu. Rev. Immunol. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 18.Cruikshank WW, Center DM, Nisar N, Wu M, Natke B, Theodore AC, Kornfeld H. Molecular and functional analysis of a lymphocyte chemoattractant factor: association of biologic function with CD4 expression. Proc. Natl. Acad. Sci. USA. 1994;91:5109–5113. doi: 10.1073/pnas.91.11.5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lynch EA, Heijens CA, Horst N, Center DM, Cruikshank W. Cutting edge: IL-16/CD4 preferentially induces Th1 cell migration: requirement of CCR5. J. Immunol. 2003;171:4965–4968. doi: 10.4049/jimmunol.171.10.4965. [DOI] [PubMed] [Google Scholar]
- 20.Bettelli E, Sullivan BM, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J. Exp. Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juedes A, Rodrigo E, Togher L, Glimcher LH, von Herrath M. T-bet controls autoaggressive CD8 lymphocyte responses in type 1 diabetes. J. Exp. Med. 2004;199:1153–1162. doi: 10.1084/jem.20031873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudter J, Galle PR, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J. Exp. Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.ten Raa S, van den Tol MP, Sluiter W, Hofland LJ, van Eijck CH, Jeekel H. The role of neutrophils and oxygen free radicals in post-operative adhesions. J. Surg. Res. 2006;136:45–52. doi: 10.1016/j.jss.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Diamond MP, Daniell JF, Feste J, Surrey MW, McLaughlin DS, Friedman S, Vaughn WK, Martin DC. Adhesion reformation and de novo adhesion formation after reproductive pelvic surgery. Fertil. Steril. 1987;47:864–866. doi: 10.1016/s0015-0282(16)59181-x. [DOI] [PubMed] [Google Scholar]
- 25.Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106:3432–3439. doi: 10.1182/blood-2005-04-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van de Weyer PS, Muchlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem. Biophys. Res. Commun. 2006;351:571–576. doi: 10.1016/j.bbrc.2006.10.079. [DOI] [PubMed] [Google Scholar]
- 27.Zhu C, Anderson AC, Schubart Z, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 28.Siveke JT, Hamann A. T helper 1 and T helper 2 cells respond differentially to chemokines. J. Immunol. 1998;160:550–554. [PubMed] [Google Scholar]
- 29.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. A Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 30.Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigagli F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skundric DS, Cai J, Cruikshank W, Gveric D. Production of IL-16 correlates with CD4+ Th1 inflammation and phosphorylation of axonal cytoskeleton in multiple sclerosis lesions. J. Neuroinflammation. 2006;3:13–27. doi: 10.1186/1742-2094-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keates AC, Castagliuolo I, Cruikshank W, Qui B, Arseneau KO, Brazer W, Kelly CP. Interleukin 16 is up-regulated in Crohn’s disease and participates in TNBS colitis in mice. Gastroenterology. 2000;119:972–982. doi: 10.1053/gast.2000.18164. [DOI] [PubMed] [Google Scholar]
- 33.Masuda K, Katoh N, Soga F, Kishimoto S. The role of interleukin-16 in murine contact hypersensitivity. Clin. Exp. Immunol. 2005;140:213–219. doi: 10.1111/j.1365-2249.2005.02752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen PA, Aarons CB, Gower AC, Stucchi AF, Leeman SE, Becker JM, Reed KL. The effectiveness of a single intraperitoneal infusion of a neurokinin-1 receptor antagonist in reducing postoperative adhesion formation is time dependent. Surgery. 2007;141:368–375. doi: 10.1016/j.surg.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 35.Schäffer M, Bongartz M, Hoffmann W, Viebahn R. Regulation of nitric oxide synthesis in wounds by IFN-γ depends on TNF-α. J. Invest. Surg. 2006;19:371–379. doi: 10.1080/08941930600985710. [DOI] [PubMed] [Google Scholar]
- 36.Ishida Y, Kondo T, Takayasu T, Iwakura Y, Mukaida N. The essential involvement of cross-talk between IFN-γ and TGF-β in the skin wound-healing process. J. Immunol. 2004;172:1848–1855. doi: 10.4049/jimmunol.172.3.1848. [DOI] [PubMed] [Google Scholar]