

Abstract
A growing body of evidence suggests that the efficacy of cytokines in cancer therapy can be increased by targeting strategies based on conjugation with ligands that recognize receptors expressed by tumor cells or elements of the tumor microenvironment, including the tumor vasculature. The targeting approach is generally conceived to permit administration of low, yet pharmacologically active, doses of drugs, thereby avoiding toxic reactions. However, it is becoming clear that, in the case of cytokines, this strategy has another inherent advantage, i.e. the possibility of administering extremely low doses that do not activate systemic counter-regulatory mechanisms, which may limit their potential therapeutic effects. This review is focused on the use of tumor vasculature-homing peptides as vehicles for targeted delivery of cytokines to tumor blood vessel. In particular, we provide an overview of peptide-cytokine conjugates made with peptides containing the NGR, RGD, isoDGR or RGR sequences and describe, in more details, the biological and pharmacological properties of NGR-hTNF, a peptide-tumor necrosis factor-α conjugate that is currently being tested in phase II and III clinical studies. The results of preclinical and clinical studies performed with these products suggest that peptide-mediated vascular-targeting is indeed a viable strategy for delivering bioactive amounts of cytokines to tumor endothelial cells without causing the activation of counter-regulatory mechanisms and toxic reactions.
Introduction
The efficacy of cytokines in cancer therapy is often limited by systemic toxicity and counter-regulatory mechanisms. Recent studies suggest that these limitations can be overcome by targeting strategies based on the conjugation of these proteins with ligands capable of delivering them to the tumor site, thereby allowing administration of lower doses and reducing systemic effects [1, 2]. Among the various approaches that have been developed, cytokine conjugation or fusion with antibodies or peptide ligands capable of recognizing specific receptors in tumor tissues are the most advanced. These ligands typically recognize receptors expressed by tumor cells or elements of the tumor microenvironment, including the tumor vasculature [1]. A comprehensive database of most tumor-homing peptides so far developed and their receptors has been recently reported [3]. Remarkably, a large proportion of these peptides have some common motifs, such as NGR and RGD [3]. This review is focused on the application of peptides containing these or other motifs as ligands for targeting tumor vessels with cytokines capable of altering the physiology of endothelial cells and tumor microenvironment, and, consequently, capable of promoting the tumor penetration of antitumor drugs, enhancing the infiltration of immune cells and inhibiting tumor growth. As a prototypic example of this class of molecules, we describe, in more detail, the biological and pharmacological properties of NGR-hTNF, a peptide-tumor necrosis factor (TNF)-α conjugate originally developed by our group, which is currently being tested in phase II and III clinical studies. In addition, we provide an overview of the various peptide-based delivery systems containing the NGR, RGD, isoDGR or RGR sequences that have been exploited for the delivery of TNF and other cytokines, such as interferon (IFN)-γ and IFNα2a, to tumor vessels.
The NGR-Mediated Targeting of Cytokines to Tumor Vasculature
The NGR Motif
The NGR motif was discovered in the 1990s by in vivo selection of peptide-phage libraries in tumor-bearing mice [4]. Systemic administration of a phage library into nude mice bearing human breast carcinoma xenografts led to the selection of tumor vasculature-homing phages carrying various peptide sequences containing this motif. Mechanistic studies showed that NGR can specifically recognize vessels expressing aminopeptidase N (CD13), a membrane-bound metalloproteinase that is barely or not at all expressed by normal blood vessels, but is up-regulated in angiogenic blood vessels [5–8]. This protease has a role in protein degradation, cytokine regulation, antigen presentation, cell proliferation, cell migration, and angiogenesis [9–11]. In tumor tissues, CD13 is expressed by endothelial cells and pericytes, and, in some cases, by tumor cells and fibroblasts. CD13 is also expressed by many cells of normal tissues, including epithelial cells from the small intestine, proximal renal tubules, prostate, bile duct canaliculi, keratinocytes, mast cells, myeloid cells, and antigen-presenting cells [12–15]. Immunohistochemical and biodistribution studies showed that CNGRC-containing compounds bind CD13-positive tumor blood vessels, but not other CD13-rich tissues [6, 16]. The structural basis of this NGR selectivity is still unknown. The recognition of angiogenic blood vessels by NGR has also been demonstrated with cyclic-NGR-labeled paramagnetic quantum dots and quantitative molecular magnetic resonance imaging (MRI) in tumor mouse models [17]. Ex vivo two-photon laser scanning microscopy showed that these particles bind primarily to the endothelial lining of tumor vessels.
Peptides containing the NGR sequence have been used by several investigators for delivering a variety of different compounds to tumor blood vessels, including chemotherapeutic drugs, liposomes, anti-angiogenic compounds, DNA complexes, viral particles, fluorescent compounds, contrast agents, and other biological response modifiers. The stability and immunogenicity of NGR peptides and the pharmacological properties of these compounds have recently been reviewed [18]. NGR peptides have also been fused to cytokines, such as TNF, IFNγ and IFNα2a, in an attempt to improve their therapeutic index. These compounds will be described with more detail in the following sections.
The NGR-TNF Example
Historical Background and Rationale for the TNF Vascular Targeting Approach
The discovery of TNF in 1975 as an endotoxin-induced serum protein capable of causing hemorrhagic necrosis of tumors in mice [19, 20] led to an explosion of basic and translational research aimed at exploiting its potential anti-tumor activity in cancer patients. This research led, in the mid-1980s, to the identification of the TNF gene and to the production of recombinant TNF in Escherichia coli cells for clinical trials. Unfortunately, despite the impressive anti-tumor effects observed in animal models, phase I–II clinical trials, carried out few years later, showed that TNF induces toxic effects and no, or very low, anti-tumor responses when administered systemically to patients [21, 22]. Attempts were made, therefore, to prepare less toxic TNF mutants and to increase its efficacy by combining it with other drugs or cytokines or by manipulating its half-life in the circulation [23, 24]. Although these strategies did not meet initial expectations, later studies, reported in the early 1990s, showed that the loco-regional administration of high-dose TNF in combination with chemotherapeutic drugs induces high response rates in patients with melanoma or sarcoma of the extremities [25–28]. These results, which led to the registration of TNF as a drug for loco-regional treatments of sarcomas confined to the limbs, are of outstanding relevance because they show that the anti-tumor effects of TNF can be successfully exploited in patients if sufficient dose levels are attained locally. These findings prompted further studies aimed at developing ligand-directed delivery systems, in an attempt to achieve high local concentrations of TNF through systemic administration of low, non-toxic doses of targeted TNF. Using an antibody-avidin-based targeting system, we demonstrated, in the mid 1990s, that a significant increase of anti-tumor activity of TNF can be achieved in animal models by pre-targeting tumors with a biotin-antibody conjugate specific for a tumor cell surface antigen, followed by administration of avidin and biotinylated TNF [29, 30]. Although this ‘three-step’ targeting approach was not further developed for clinical studies, owing to its complexity, the good results obtained in animal models suggested that targeted delivery of TNF to tumors might indeed represent a valuable strategy for increasing its therapeutic index. Notably, in these studies, targeted TNF could affect well established and vascularized tumors, but not avascular, freshly transplanted tumors [29]. Furthermore, we showed that bioactive TNF could dissociate from the targeted tumor cells and reach endothelial cells in the tumor microenvironment, thereby causing vascular damage and tumor tissue necrosis [31].
These findings provided the rationale for developing other targeting systems capable of directly delivering TNF to tumor vessels. For this purpose, TNF was fused to CNGRCG, a peptide capable of selectively recognizing targets expressed by angiogenic vessels and therefore to ‘home’ to the tumor vasculature [4, 16]. Based on promising results obtained in preclinical studies, this fusion protein, which has been generated and studied at the San Raffaele Scientific Institute and called NGR-TNF, was out-licensed to Molmed SpA. The biochemical properties of this drug and the results of preclinical and clinical phase I, II, and III studies are reviewed in the following sections.
The NGR-TNF Receptors
As the NGR-TNF fusion protein is made by two functional domains (NGR peptide and TNF), the receptor system of this conjugate includes receptors for both ligands, i.e. CD13 and TNF receptors. The role of CD13 as a receptor for NGR peptides has been discussed above. Regarding TNF receptors, it is important to keep in mind that soluble bioactive TNF is a homotrimeric protein capable of interacting with two distinct cell surface receptors of 55–60 and 75–80 kDa, respectively (TNF-R1 and TNF-R2) [32]. TNF-R1 is thought to mediate most TNF effects, whereas TNF-R2, owing to its higher affinity and rapid dissociation rate, plays a role in increasing the local concentration of TNF and passing it to TNF-R1 [33–36]. TNF-R2 also contributes to several cellular responses by direct signaling upon TNF-mediated clustering [37–40]. Notably, human TNF can efficiently bind murine TNF-R1, but it cannot bind murine TNF-R2, because of species-specificity of this interaction [41]. For this reason, murine NGR-TNF (NGR-mTNF) has been used in preclinical studies in mice and human NGR-TNF (NGR-hTNF) in clinical studies in patients. Structure-activity and receptor-binding studies performed with both products, prepared by recombinant DNA technology, showed that CNGRCG peptide does not prevent folding, oligomerization, and binding of the TNF domain to TNF-R1 and TNF-R2 [16]. On the other hand, the TNF subunits do not prevent the interaction of CNGRCG with the CD13 receptor. As a consequence, NGR-TNF may undertake multivalent high-avidity interactions with both TNF and CD13 receptors on cells that express both receptor types [16]. While TNF receptors are unlikely to provide selectivity for tumor vessels, being expressed by most normal cells, CD13 binding is likely the major mechanism for tumor vessel selectivity when low doses of NGR-TNF are administered. In fact, tumor vessels, but not other CD13-expressing tissues, express a functionally active form of CD13 that binds NGR-TNF [6].
Anti-Tumor Activity of NGR-TNF in Animal Models
Early preclinical studies performed in murine RMA lymphoma or B16F1 melanoma models showed that systemically administered NGR-TNF was more active and less toxic than TNF [16]. The dose-response curve of NGR-TNF was wider and more complex than that of TNF [42]: while microgram doses of TNF were necessary to induce anti-tumor effects, a wider range of NGR-TNF doses (from picograms to micrograms) exerted anti-tumor effects in these models (see Fig. 1a). Interestingly, intermediate doses of NGR-TNF (3–10 ng) were paradoxically less active than picogram doses. Mechanistic studies showed that NGR-TNF and TNF, when injected at doses greater than 1 ng, could induce the shedding in circulation of soluble TNF receptors, which can act as TNF inhibitors [42]. This counter-regulatory mechanism can be overwhelmed by the administration of microgram doses, leading to anti-tumor effects and, for the same reason, also to systemic toxicity. Interestingly, low doses of NGR-TNF or TNF (in the picogram range) did not activate soluble receptor shedding (Fig. 1b), likely because of insufficient interaction with endothelial cells in normal vessels [42]. However, in this case, only NGR-TNF could exert pharmacological effects, likely owing to high-avidity interaction with TNF-Rs and CD13 on tumor vessels [6, 15] (see Fig. 1c for a schematic representation of this concept). Thus, the vascular targeting strategy can be exploited, in the case of TNF, to avoid not only toxic reactions but also negative feedback mechanisms, provided that very low doses of targeted TNF are used.
Fig. 1.
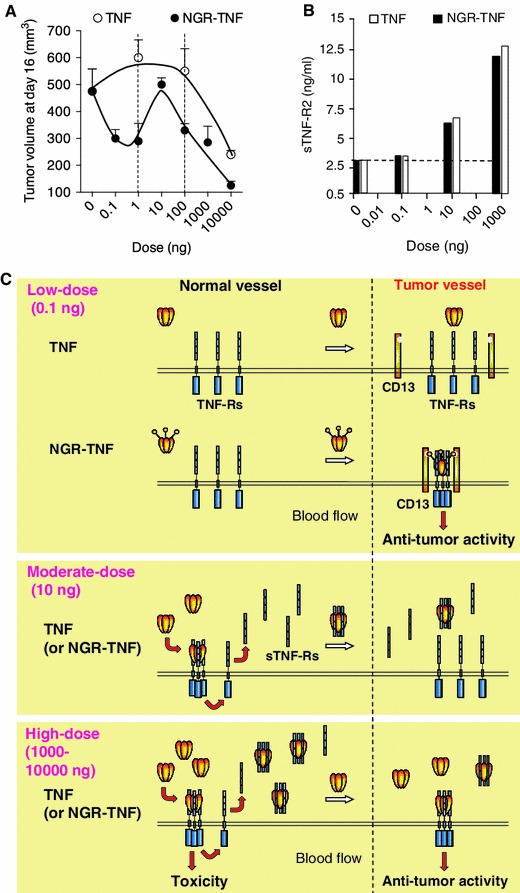
Effect of NGR-TNF and TNF on tumor growth and soluble TNF receptor shedding, an important counter-regulatory mechanism. a Effect on the growth of RMA-T lymphomas implanted subcutaneously in mice (tumor volumes, 4 days after treatment, showing different dose-response curves for NGR-TNF and TNF) [42]. b Effect of low (0.1 ng/mouse), moderate (10 ng), and high (1,000 ng) doses of NGR-TNF or TNF on the shedding of soluble TNF-R2 (serum levels, 1 h after treatment). Strong activation of this counter-regulatory mechanism occurs only with moderate and high doses of both cytokines. c Schematic representation of the hypothetical interactions of low, moderate, and high doses of NGR-TNF or TNF with soluble and membrane TNF receptors in normal (CD13-negative) and tumor-associated vessels (CD13-positive). This model can explain the different anti-tumor dose-response curves observed (adapted from [42]). According to this model, moderate doses of NGR-TNF or TNF (e.g. 10 ng/mouse), can efficiently interact with membrane TNF receptors throughout the body and trigger the systemic release of sTNF-Rs, which act as potent TNF inhibitors. High doses of both cytokines (e.g. 1,000–10,000 ng) can saturate the circulating sTNF-Rs, thereby leaving a significant amount of bioactive TNF free to interact with membrane receptors in neoplastic and normal tissues and to induce, therefore, anti-tumor as well as toxic effects. In contrast, low-dose NGR-TNF or TNF (e.g. 0.1 ng) cannot induce massive soluble receptor shedding, likely because of insufficient interaction with endothelial cells in normal vessels. However, in this case, NGR-TNF can still induce anti-tumor effects owing to high-avidity interaction with membrane TNF receptors and CD13 on tumor vessels. TNF tumour necrosis factor
Studies on the mechanism of action showed that low-dose NGR-TNF (0.1 ng/mouse) is sufficient to induce apoptosis of endothelial cells and, at later time points, tumor cell apoptosis, likely as a consequence of vascular damage [43]. This effect was markedly increased when NGR-TNF was combined with low doses of endothelial-monocyte-activating polypeptide II (EMAP-II), a tumor-derived anti-angiogenic cytokine that sensitizes the tumor vasculature to the damaging activity of TNF [43]. However, when NGR-TNF was used alone, marked vascular occlusion was observed only with high doses (e.g. 10 μg) but not, or much less, with the low dose (0.1 ng), suggesting that different doses can trigger different anti-tumor mechanisms [44]. Thus, other mechanisms are likely brought into play by low-dose NGR-TNF to delay tumor growth. A recent study showed that administration of low-dose NGR-TNF to tumor-bearing mice induces the up-regulation VCAM-1 and ICAM-2 in endothelial cells and the release of several cytokines/chemokines involved in T-cell activation and migration, such as MCP-1/CCL-2, MCP-3/CCL-7, MIP-2, oncostatin-M, and stem cell factor (SCF), in tumors [45], suggesting that inflammatory/immune responses might contribute to the activity of NGR-TNF.
Anti-Tumor Activity of NGR-TNF in Combination with Chemotherapy in Animal Models
High-dose TNF exerts synergistic effects with melphalan or doxorubicin in patients treated by regional isolated limb perfusion [25–28]. Remarkably, low-dose NGR-TNF (0.1 ng/mouse, six orders of magnitude lower than the LD50), but not an equivalent dose of TNF, was sufficient to enhance the anti-tumor activity of melphalan and doxorubicin in murine models, with no evidence of toxicity [42]. Low-dose NGR-TNF could also enhance the response to other drugs, including cisplatin, gemcitabine, and paclitaxel in various transplantable tumor models and in the orthotopic TRAMP model of prostate cancer, with no evidence of increased toxicity [44, 46]. The optimal administration schedule for maximal synergism required a 2-h delay between the administration of NGR-TNF and chemotherapeutic drugs, irrespective of drug and tumor model used. It is well known that TNF can rapidly increase endothelial permeability and decrease the interstitial fluid pressure in tumors, both representing important barriers that limit drug penetration [26, 47–54]. These mechanisms may increase the convective transport of drugs through tumor vessel walls and interstitium, eventually leading to increased drug uptake by tumor cells. MRI studies performed with mice bearing subcutaneous lymphomas showed a greater leakage of ultra-small iron oxide particles (USPIO) from the vasculature to the interstitial area after treatment with NGR-TNF, than with TNF [55]. Furthermore, pre-administration of low-dose NGR-TNF to mice increased the tumor uptake of doxorubicin (administered 2 h later) in murine melanoma and lymphoma models [42]. These findings support the concept that NGR-TNF can indeed increase drug uptake by increasing the leakiness of tumor vessels, at least in those areas characterized by poor perfusion and low permeability. It appears, therefore, that low-dose NGR-TNF has distinct vascular effects, causing early activation of endothelial cells (after 1–2 h) and consequent barrier alteration and expression of leukocyte adhesion molecules, followed by apoptosis of endothelial cells 8–24 h later [43]. Interestingly, chromogranin A, a protein capable of inhibiting the vascular leakage induced by TNF [56], could inhibit the synergism between NGR-TNF and chemotherapy in murine models [57]. Synergism between low-dose NGR-TNF and doxorubicin was observed in immunocompetent mice but not in nude or IFNγ-knockout mice, pointing to a crucial role for locally produced IFNγ in mediating the overall antitumor effects [58].
Anti-Tumor Activity of NGR-TNF in Combination with Immunotherapy in Animal Models
A recent study showed that low-dose NGR-TNF (0.1 ng) promotes lymphocyte extravasation in tumors [45]. Mechanistic studies showed that NGR-TNF, besides up-regulating leukocyte adhesion molecules on tumor vessels, can induce the release of various chemokines in tumor tissues (mentioned above). These mechanisms associate with increased tumor infiltration of endogenous or adoptively transferred cytotoxic T lymphocytes in transplantable models of melanoma and in the TRAMP model of spontaneous prostate cancer [45]. Remarkably, NGR-TNF did not modify T-cell distribution in the blood, spleen, or kidney of tumor-bearing mice. The combination of NGR-TNF and adoptive immunotherapy increased the overall survival of tumor-bearing mice, with no evidence of toxic reactions. NGR-TNF could also increase the efficacy of active immunotherapy (vaccination) either alone or in combination with chemotherapy [45]. In all the experimental conditions tested, a comparable dose of TNF was marginally or not active, supporting the hypothesis that peptide-mediated targeted delivery of TNF to tumor vessels was crucial for the activity.
Clinical Studies with NGR-hTNF in Patients with Solid Tumors
Various phase I and II studies have been performed with NGR-hTNF in patients with solid tumors, both as a single agent and in combination with chemotherapy.
NGR-hTNF as a single agent The first phase I study (EORTC 16041) was aimed at investigating dose-limiting toxicities, maximum tolerated dose (MTD), pharmacokinetics, pharmacodynamics, and vascular response in solid tumors [59]. Escalating doses (0.2–60 μg/m2) of NGR-hTNF were administered to 69 patients once every 3 weeks via a 20 or 60 min infusion rate (intravenously). NGR-TNF was well tolerated (MTD: 45 μg/m2 administered in 1 h). Chills and fever were the most frequently observed toxicities. The mean apparent terminal half-life ranged from about 1 to 2 h. Dynamic contrast-enhanced MRI (DCE-MRI) showed a vascular response to NGR-hTNF [59]. No objective responses were observed, but 39 % of patients had stable disease, with a median duration of 3 months [59]. In another phase I study, performed in 31 patients with advanced solid cancer, DCE-MRI showed larger anti-vascular effects of NGR-hTNF in smaller tumors, which have less mature neovasculature [60]. No patients developed anti-NGR-TNF antibodies during treatment. The circulating levels of soluble TNF receptors increased in a dose-proportional manner, but only at doses greater than 1.3 μg/m2. As preclinical studies showed that soluble receptors shedding in circulation can lead to a bell-shaped dose-response curve, another phase I study was designed to define the optimal biological dose of NGR-hTNF in the low-dose range (0.2–1.6 μg/m2, 1 h infusion) [61]. Based on soluble receptor kinetics, tolerability, anti-vascular effects, and disease control (44 % stable diseases for a median time of 5.9 months were observed), the dose of 0.8 μg/m2 of NGR-hTNF was chosen for subsequent studies, either alone or with standard chemotherapy.
Single-agent phase II studies with low-dose NGR-TNF (0.8 μg/m2, 1 h infusion, every 3 weeks or weekly) were conducted in malignant pleural mesothelioma (MPM), hepatocellular carcinoma (HCC), and colorectal cancer (CRC) [62–64]. These studies showed radiological anti-vascular effects and significant disease control with low-dose NGR-hTNF. The study in MPM patients showed disease control in about half of 57 previously treated patients, which was maintained in the tri-weekly cohort for 4.4 months and for 9.1 months in the weekly cohort [62]. In the overall study population, the observed median survival was 12.1 months. NGR-hTNF was well tolerated in both cohorts. In the three-weekly cohort (n = 43), common grades 1 and 2 short-lived chills (71 %) and one grade 3 drug-related toxicity were observed [62]. These results are remarkable considering that, currently, there are no standard options for patients with MPM who are failing a front-line pemetrexed-based regimen and considering the easily manageable toxicity profile of NGR-TNF.
In the study in patients with HCC, NGR-hTNF was administered to 27 previously treated patients with advanced-stage disease [63]. One complete response, maintained after 20 months in a sorafenib-refractory patient, one partial response in a Child–Pugh class B patient, and six stable diseases were observed. The disease control rate was 30 %, median progression-free survival (PFS) time was 4.3 months, and median survival time was 8.9 months [63]. NGR-hTNF was considered well tolerated, as no grade 3–4 treatment-related toxicities were noted in this study. Similar results were observed in the patients enrolled in the weekly cohort.
In metastatic CRC, NGR-hTNF, which was administered to 33 patients after failure of standard therapy, was well tolerated, and the median PFS and overall survival were 2.5 and 13.1 months, respectively [64]. The disease control rate was 39.4 %, including one partial response and 12 stable diseases. Based on these observations, it was concluded that NGR-hTNF deserves further evaluation in MPM, HCC, and CRC.
Combination studies A phase Ib study of NGR-hTNF in combination with doxorubicin explored cytokine doses of 0.2, 0.4, 0.8, and 1.6 μg/m2 [65]. This combination was well tolerated and a high disease control rate (73 %) was achieved in a population of heavily chemotherapy pre-treated patients, including anthracycline-pretreated patients. No shedding of soluble TNF receptors occurred with 0.8 μg/m2 or lower doses. The dose of 0.8 μg/m2 NGR-hTNF was selected for a phase II study of NGR-hTNF in combination with doxorubicin in relapsed ovarian cancer patients (n = 37) [66]. This study showed that the drug combination has interesting clinical activity and a safe toxicity profile: 23 % of patients had a partial response and 43 % had stable disease. Median PFS and overall survival were 5 and 17 months, respectively. Considering that about two thirds of these patients were platinum-refractory/resistant, these results have been considered clinically relevant. Furthermore, most commonly reported adverse events were those typically expected when each drug is given alone, suggesting that toxicity profiles of the two drugs do not overlap or increase. Interestingly, a strong association between baseline lymphocyte counts and outcomes was observed, especially in refractory/resistant subsets [66].
Another phase I trial of 0.8 μg/m2 of NGR-TNF in combination with cisplatin, for patients with refractory solid tumors, showed that this combination was also well tolerated and induced objective response and disease stabilization even in patients pre-treated with cisplatin [67].
Regarding combination studies, another phase II study evaluated the safety of low-dose (0.8 μg/m2, optimal biological dose) and higher-dose (45 μg/m2, MTD) NGR-hTNF in combination with fixed doses of oxaliplatin plus capecitabine (XELOX) in CRC patients who had previously failed standard treatment [68]. Both doses of NGR-hTNF were safely combined with XELOX in these patients. However, a hint of activity was apparent only with the lower dose. Remarkably, induction of sTNF-R1 and sTNF-R2 was observed with the higher dose. Furthermore, plasma levels of sTNF-Rs tended to correlate inversely with progression-free intervals. These data are consistent with preclinical studies in mice showing that a dose of 10 ng/mouse of NGR-mTNF, which is 100-fold higher than the optimal biological dose (0.1 ng/mouse) leads to sTNF-Rs shedding in circulation and, consequently, to inhibition of its potential antitumor activity [42] (as discussed in the above sections; see also Fig. 1). It appears therefore that NGR-hTNF has a complex dose-response curve also in humans, likely owing to the activation of counter-regulatory mechanisms when moderate-high doses are used.
High-dose NGR-hTNF in patients with refractory solid tumors A recent phase I study was undertaken in 48 patients with refractory solid tumors to explore doses (60–325 μg/m2) higher than the previously established MTD (45 μg/m2), using a more protracted infusion length (2 h) and mild paracetamol premedication [69]. The MTD was not reached in this study and common related toxicity included grade 1–2 chills (58 %). Changes in sTNF-R shedding were not observed with the different doses, suggesting a plateau effect in shedding kinetics. As best response, 29 % of patients had stable disease. Lower levels of sTNF-R2 and greater K trans values, by DCE-MRI, after the first cycle, were associated with improved survival [69]. These results suggest that NGR-hTNF can be safely escalated to high doses. The authors concluded that, based on the observed tolerability profile and biological effects, further testing of doses higher than 200 μg/m2 of NGR-hTNF combined with chemotherapy and comparison with a low dose of 0.8 μg/m2 is warranted [69].
Ongoing phase III study Based on the encouraging results obtained with low-dose NGR-hTNF in MPM patients, a randomized, double-blind, phase III study of NGR-hTNF plus best investigator’s choice (BIC) versus placebo plus BIC in previously treated patients with advanced MPM started in 2010 (http://www.clinicaltrialsfeeds.org/clinical-trials/show/NCT01098266). The main objective of this trial is to document the efficacy of low-dose weekly NGR-hTNF in combination with BIC in advanced MPM patients previously treated with a pemetrexed-based chemotherapy regimen. The results obtained in the phase II study in MPM and HCC patients enabled NGR-TNF to be granted ‘Orphan Drug’ designation for these indications in the EU and the USA (http://www.molmed.com/eng/pipeline_arenegyr.asp).
Other NGR-Cytokine Fusion Products
NGR-peptides have also been used for delivering, in addition to TNF, IFNγ and IFN-α2a to the tumor neovasculature. The rationale for this approach relies on the observation that IFNγ can inhibit tumor angiogenesis [70, 71] and augment the expression of MHC-I and II on cancer and endothelial cells [72, 73]. Within tumor stroma IFNγ can induce cytokine and chemokine secretion, including IP-10, an angiostatic protein and a chemoattractant factor for lymphocytes and monocytes [71, 74]. Evidence has been obtained to suggest that IFNγ produced by tumor-infiltrating macrophages plays a role in tumor blood vessel destruction [75]. Combined treatment of endothelial cells with IFNγ and TNF results in synergistic cytotoxic effects, likely important for tumor vasculature destruction [76]. In addition, IFNγ induces anti-proliferative and pro-apoptotic effects on many tumor cell types and activate natural killer cells and macrophages to kill a variety of tumor cell targets [75]. IFNγ is also an important regulator of CD4+ T-helper cells and is the major physiological macrophage-activating factor [77, 78]. As a consequence of these effects on tumor vasculature and on cells of the immune system, IFNγ can activate inflammatory/immune responses against established tumors and inhibit tumor growth. Because of its immunomodulatory and anticancer activities, IFNγ has been used as an anticancer drug in several clinical studies, unfortunately with modest results [79, 80]. Attempts to increase the response by increasing the dose or by repeated injections often resulted in lower efficacy, likely due to counter-regulatory mechanisms [81, 82]. Experiments in murine models showed that targeted delivery of minute amounts (picograms) of an IFNγ-GCNGRC fusion product (called IFNγ-NGR) to tumor vasculature could be a valuable strategy for uncoupling antitumor activity and counter-regulatory mechanisms [83]. However, the dose-response curve observed in animal models with this conjugate was bell-shaped, suggesting that counter-regulatory mechanisms can still be activated with high-dose IFNγ-NGR. Furthermore, although the therapeutic efficacy of this protein in animal models is greater than that of IFNγ, frequent administrations of IFNγ-NGR result in lower efficacy and tumor resistance [83]. Mechanistic studies of tumor resistance to repeated treatments with IFNγ-NGR suggested a crucial role of indoleamine 2,3-dioxygenase (IDO), an IFNγ-inducible enzyme that may down-regulate T cells by affecting local tryptophan catabolism. Coadministration of IFNγ-NGR with 1-methyltryptophan, an inhibitor of IDO, increased tumor responses to multiple treatments in lymphoma, melanoma, and fibrosarcoma models [84].
Other investigators showed that the anti-tumor activity of IFNα2a can also be increased by coupling this protein with an NGR peptide [85]. Studies performed in animal models showed that the IFNα2a-NGR conjugate, but not IFNα2a, accumulates and target tumor vessels [86–88].
Notably, a fusion protein consisting of the extracellular domain of tissue factor (truncated tissue factor [tTF]) and peptide GNGRAHA (tTF-NGR), but not untargeted tTF, induced thrombosis of blood vessels in solid tumors in mice [89]. Clinical application of low dosages of this targeted coagulation factor revealed good tolerability and decreased tumor perfusion. These observations further highlight the utility of NGR peptides as targeting ligands in patients.
RGD-, isoDGR-, and RGR-Mediated Targeting of Cytokines to Tumor Vasculature
Besides the NGR-cytokine fusion products, other peptide-cytokine conjugates based on the RGD, isoDGR, or RGR sequences have been developed. Their biochemical and biological properties are described in the following sections.
Cytokine Targeting via Peptides Containing the RGD Motif
Peptides containing the RGD sequence have been widely used, like NGR-peptides, as ligands for the delivery of a variety of drugs, nanoparticles, and diagnostic agents to tumors, including cytokines [90–93]. The RGD sequence was originally discovered as a binding site of a number of extracellular matrix, blood, and cell surface proteins for integrins, a large class of αβ heterodimeric membrane receptors involved in cell adhesion [94, 95]. Various αβ heterodimers can be recognized by RGD, including αvβ1-, αvβ3-, αvβ5-, αvβ6-, αvβ8-, α5β1-, α8β1-, and αIIbβ3-integrins [94, 96]. These integrins can be recognized in a differential manner by different RGD peptides, as the molecular scaffold in which RGD is inserted may contribute to control its conformation and therefore its integrin-binding affinity and specificity [97]. Thus, a variety of different peptides with different binding properties, in the low nanomolar and subnanomolar range, have been developed [97]. Interestingly, because the αvβ3 heterodimer is overexpressed in the tumor vasculature, this receptor is an attractive pharmacological target for ligand-directed delivery of cytokines to tumor vessels [98, 99]. To assess this hypothesis, TNF has been fused, by recombinant DNA technology, to ACDCRGDCFCG, a peptide ligand of αv integrins discovered by in vivo panning of peptide-phage libraries in tumor-bearing mice [4, 93]. Subnanogram doses of this conjugate, but not of TNF, were sufficient to induce antitumor effects in tumor-bearing mice when combined with melphalan, suggesting that these integrins are indeed good targets for cytokine delivery to tumors. Unfortunately, this peptide contains four Cys residues. Thus, the trimeric RGD-TNF fusion product was difficult to fold in a homogeneous manner and, for this reason, NGR-TNF was preferred for clinical development. However, intramuscular administration of plasmid DNA encoding ACDCRGDCFCG–TNF or CNGRCG-TNF, but not plasmids encoding TNF, inhibited to a similar extent the growth of murine melanomas and lymphomas implanted subcutaneously at sites distant from the site of plasmid injection, suggesting that both fusion products are valid for a gene therapy approach [100].
Cytokine Targeting via Peptides Containing the isoDGR Motif
IsoDGR is a tripeptide sequence, closely related to RGD, which was originally discovered in aged fibronectin, a protein of the extracellular matrix that contains four NGR sites [101]. This motif was later found to also represent an important degradation product of NGR-drug conjugates. The isoDGR sequence can arise in fibronectin and NGR-peptides as a consequence of asparagine deamidation [101, 102]. Experimental evidence suggests that the NGR-to-isoDGR transition, which occurs spontaneously and in a relatively short time in fibronectin fragments and in cyclic NGR peptides (half life 4–5 h), can work as a biological switch for the regulation of cell adhesion, isoDGR being an integrin-recognition motif [101–103]. Biochemical and functional studies have shown that NGR flanking residues and molecular scaffold markedly contribute to peptide stability [104]. Studies performed with synthetic peptides showed that isoDGR, like RGD, can recognize RGD-dependent integrins (such as αvβ3, αvβ5, αvβ6, αvβ8, and α5β1) with different affinity and selectivity, depending on the molecular scaffold in which isoDGR is inserted [104–107]. Biochemical studies, NMR structure analysis and αvβ3-docking experiments performed with a disulphide-bridged CisoDGRC peptide showed that isoDGR can fit into the RGD-binding pocket of this integrin, recapitulating not only the canonical RGD/αvβ3 contacts but also establishing additional polar interactions [108]. Interestingly, a recent study showed that isoDGR, but not RGD, is a pure integrin antagonist [109]. The structural and functional similarity with RGD suggests that isoDGR can also be exploited, like RGD, as a ligand for targeted delivery of drugs, imaging agents, or other compounds to tumors. Consistent with this, fluorescent nanoparticles coupled to a cyclic CisoDGRC peptide could bind αvβ3-integrin and colocalize with antibodies against this integrin in vessels of human renal cell carcinoma [110]. Furthermore, extremely low doses (1–10 pg) of a recombinant protein made up of CisoDGRC fused to TNF induced anti-tumor effects in tumor-bearing mice through specifically targeting TNF to tumor sites (Curnis et al. [110]). Another peptide, consisting of head-to-tail cyclized c(CGisoDGRG), recognizes αvβ3 with very good selectivity after chemical conjugation to human serum albumin via thiol group [111]. IsoDGR-tagged albumin has been exploited for the preparation of TNF-bearing gold nanoparticles that efficiently home to tumors [111]. Colloidal gold is a well tolerated nanomaterial currently exploited for several applications in the field of nanomedicine, including TNF delivery [112–115]. Notably, gold nanoparticles coated with both isoDGR-tagged albumin and TNF have improved anti-tumor activity compared with TNF and gold-TNF nanoparticles [111]. This suggests that other TNF-based nanoparticles and polymer conjugates, which are currently being developed by different laboratories [115], might also be improved by tagging with isoDGR peptides.
RGR-Mediated Targeting of Cytokines to Tumor Vasculature
Another ligand that has been used for the delivery of cytokines, such as TNF and IFNγ, to the tumor vasculature is the RGR peptide [116]. This peptide has also been identified by phage display against pancreatic tumors and been shown to specifically bind to highly angiogenic vessels in murine insulinomas [116]. Studies in animal models showed that recombinant TNF-RGR and IFNγ-RGR selectively home to tumors and are retained in the microenvironment [117]. Interestingly, IFNγ-RGR predominantly acted as an anti-vascular agent in RIP1-Tag5 transgenic mice, which express the SV40 Large T antigen under control of the rat insulin gene promoter and develop pancreatic neuroendocrine tumors over time. In contrast, TNF-RGR (2 μg over 2 weeks) did not destroy angiogenic vessels but instead induced a more regular and less leaky vascular network with small vessel calibers and mural stabilization, thus improving tumor perfusion. TNF-RGR enhanced T-cell infiltration and overall survival, an effect that was exclusively mediated by CD8+ effector cells. Vessel remodeling induced by TNF-RGR substantially improved antitumor vaccination or adoptive T-cell therapy [118]. These findings, and the observation that NGR-TNF can also be exploited to enhance T-cell infiltration in tumor tissues [45], suggest that vascular targeting with peptide-TNF conjugates is a good strategy for improving immunotherapy.
Conclusion
The results of preclinical and clinical studies performed so far with peptide-cytokine conjugates suggest that the peptide-based tumor vascular targeting approach is a viable strategy for improving their therapeutic index, at least for certain cytokines, such as TNF, IFNγ, and IFNα2a. Although the targeting approach is generally conceived to permit administration of low, yet pharmacologically active, doses of drugs in order to avoid toxic reactions, it is becoming clear that this strategy has another inherent advantage, i.e., the possibility to administer extremely low doses that do not activate systemic counter-regulatory mechanisms, which tend to block the potential therapeutic effects of cytokines. This is made possible by the good accessibility of the target (endothelial cells) and by the high affinity for targeting receptors that can be achieved with peptide-cytokine conjugates, likely owing to multiple receptor interactions. In principle, the same goals can be achieved with antibodies and, indeed, many studies have demonstrated the utility of this class of targeting ligands for cytokine delivery, including TNF and several other cytokines [1, 2, 119]. However, the low molecular weight and the poor immunogenicity observed with peptides may represent an important advantage, particularly for therapies that need prolonged and repeated treatments. Considering the large number of tumor vasculature-homing peptides discovered in the last decade [3, 120] and the promising results obtained so far with the prototypic peptide-cytokine conjugates described here, the development of other conjugates made with different peptides and synergistic cytokines might lead to the development of new therapeutic options for cancer patients.
Acknowledgments
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC 9965 and 9180) of Italy.
Disclosure
The authors have no conflicts of interest that are directly relevant to the content of this article.
References
- 1.Pasche N, Neri D. Immunocytokines: a novel class of potent armed antibodies. Drug Discov Today. 2012;17(11–12):583–590. doi: 10.1016/j.drudis.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Kontermann RE. Antibody-cytokine fusion proteins. Arch Biochem Biophys. 2012;526(2):194–205. doi: 10.1016/j.abb.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Kapoor P, Singh H, Gautam A, Chaudhary K, Kumar R, Raghava GP. TumorHoPe: a database of tumor homing peptides. PLoS One. 2012;7(4):e35187. doi: 10.1371/journal.pone.0035187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279(5349):377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 5.Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60(3):722–727. [PMC free article] [PubMed] [Google Scholar]
- 6.Curnis F, Arrigoni G, Sacchi A, Fischetti L, Arap W, Pasqualini R, et al. Differential binding of drugs containing the NGR motif to CD13 isoforms in tumor vessels, epithelia, and myeloid cells. Cancer Res. 2002;62(3):867–874. [PubMed] [Google Scholar]
- 7.Lahdenranta J, Sidman RL, Pasqualini R, Arap W. Treatment of hypoxia-induced retinopathy with targeted proapoptotic peptidomimetic in a mouse model of disease. Faseb J. 2007;21(12):3272–3278. doi: 10.1096/fj.07-8273com. [DOI] [PubMed] [Google Scholar]
- 8.Buehler A, van Zandvoort MA, Stelt BJ, Hackeng TM, Schrans-Stassen BH, Bennaghmouch A, et al. cNGR: a novel homing sequence for CD13/APN targeted molecular imaging of murine cardiac angiogenesis in vivo. Arterioscler Thromb Vasc Biol. 2006;26(12):2681–2687. doi: 10.1161/01.ATV.0000245807.65714.0b. [DOI] [PubMed] [Google Scholar]
- 9.Mina-Osorio P. The moonlighting enzyme CD13: old and new functions to target. Trends Mol Med. 2008;14(8):361–371. doi: 10.1016/j.molmed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luan Y, Xu W. The structure and main functions of aminopeptidase N. Curr Med Chem. 2007;14(6):639–647. doi: 10.2174/092986707780059571. [DOI] [PubMed] [Google Scholar]
- 11.Bhagwat SV, Lahdenranta J, Giordano R, Arap W, Pasqualini R, Shapiro LH. CD13/APN is activated by angiogenic signals and is essential for capillary tube formation. Blood. 2001;97(3):652–659. doi: 10.1182/blood.v97.3.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor A. Aminopeptidases: structure and function. FASEB J. 1993;7(2):290–298. doi: 10.1096/fasebj.7.2.8440407. [DOI] [PubMed] [Google Scholar]
- 13.Shipp MA, Look AT. Hematopoietic differentiation antigens that are membrane-associated enzymes: cutting is the key. Blood. 1993;82(4):1052–1070. [PubMed] [Google Scholar]
- 14.Dixon J, Kaklamanis L, Turley H, Hickson ID, Leek RD, Harris AL, et al. Expression of aminopeptidase-n (CD 13) in normal tissues and malignant neoplasms of epithelial and lymphoid origin. J Clin Pathol. 1994;47(1):43–47. doi: 10.1136/jcp.47.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Matteo P, Arrigoni GL, Alberici L, Corti A, Gallo-Stampino C, Traversari C, et al. Enhanced expression of CD13 in vessels of inflammatory and neoplastic tissues. J Histochem Cytochem. 2011;59(1):47–59. [DOI] [PMC free article] [PubMed]
- 16.Curnis F, Sacchi A, Borgna L, Magni F, Gasparri A, Corti A. Enhancement of tumor necrosis factor alpha antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13) Nat Biotechnol. 2000;18(11):1185–1190. doi: 10.1038/81183. [DOI] [PubMed] [Google Scholar]
- 17.Oostendorp M, Douma K, Hackeng TM, Dirksen A, Post MJ, van Zandvoort MA, et al. Quantitative molecular magnetic resonance imaging of tumor angiogenesis using cNGR-labeled paramagnetic quantum dots. Cancer Res. 2008;68(18):7676–7683. doi: 10.1158/0008-5472.CAN-08-0689. [DOI] [PubMed] [Google Scholar]
- 18.Corti A, Curnis F. Tumor vasculature targeting through NGR peptide-based drug delivery systems. Curr Pharm Biotechnol. 2011;12(8):1128–1134. doi: 10.2174/138920111796117373. [DOI] [PubMed] [Google Scholar]
- 19.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. 1975;72(9):3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helson L, Green S, Carswell E, Old LJ. Effect of tumor necrosis factor on cultured human melanoma cells. Nature. 1975;258:731–732. doi: 10.1038/258731a0. [DOI] [PubMed] [Google Scholar]
- 21.Lejeune FJ, Lienard D, Matter M, Ruegg C. Efficiency of recombinant human TNF in human cancer therapy. Cancer Immun. 2006;6:6. [PubMed] [Google Scholar]
- 22.Fraker DL, Alexander HR, Pass HI. Biologic therapy with TNF: systemic administration and isolation-perfusion. In: De Vita V, Hellman S, Rosenberg S, editors. Biologic therapy of cancer: principles and practice. Philadelphia: J.B. Lippincott Company; 1995. pp. 329–345. [Google Scholar]
- 23.Corti A, Marcucci F. Tumour necrosis factor: strategies for improving the therapeutic index. J Drug Target. 1998;5:403–413. doi: 10.3109/10611869808997869. [DOI] [PubMed] [Google Scholar]
- 24.Gerspach J, Pfizenmaier K, Wajant H. Improving TNF as a cancer therapeutic: tailor-made TNF fusion proteins with conserved antitumor activity and reduced systemic side effects. Biofactors. 2009;35(4):364–72. [DOI] [PubMed]
- 25.Lienard D, Ewalenko P, Delmotte JJ, Renard N, Lejeune FJ. High-dose recombinant tumor necrosis factor alpha in combination with interferon gamma and melphalan in isolation perfusion of the limbs for melanoma and sarcoma. J Clin Oncol. 1992;10(1):52–60. doi: 10.1200/JCO.1992.10.1.52. [DOI] [PubMed] [Google Scholar]
- 26.Eggermont AM, Schraffordt Koops H, Lienard D, Kroon BB, van Geel AN, Hoekstra HJ, et al. Isolated limb perfusion with high-dose tumor necrosis factor-alpha in combination with interferon-gamma and melphalan for nonresectable extremity soft tissue sarcomas: a multicenter trial. J Clin Oncol. 1996;14(10):2653–2665. doi: 10.1200/JCO.1996.14.10.2653. [DOI] [PubMed] [Google Scholar]
- 27.Fraker DL, Alexander HR, Andrich M, Rosenberg SA. Treatment of patients with melanoma of the extremity using hyperthermic isolated limb perfusion with melphalan, tumor necrosis factor, and interferon gamma: results of a tumor necrosis factor dose-escalation study. J Clin Oncol. 1996;14(2):479–489. doi: 10.1200/JCO.1996.14.2.479. [DOI] [PubMed] [Google Scholar]
- 28.Eggermont AM, de Wilt JH, ten Hagen TL. Current uses of isolated limb perfusion in the clinic and a model system for new strategies. Lancet Oncol. 2003;4(7):429–437. doi: 10.1016/s1470-2045(03)01141-0. [DOI] [PubMed] [Google Scholar]
- 29.Gasparri A, Moro M, Curnis F, Sacchi A, Pagano S, Veglia F, et al. Tumor pretargeting with avidin improves the therapeutic index of biotinylated tumor necrosis factor alpha in mouse models. Cancer Res. 1999;59(12):2917–2923. [PubMed] [Google Scholar]
- 30.Moro M, Pelagi M, Fulci G, Paganelli G, Dellabona P, Casorati G, et al. Tumor cell targeting with antibody-avidin complexes and biotinylated tumor necrosis factor alpha. Cancer Res. 1997;57(10):1922–1928. [PubMed] [Google Scholar]
- 31.Corti A, Gasparri A, Sacchi A, Curnis F, Sangregorio R, Colombo B, et al. Tumor targeting with biotinylated tumor necrosis factor alpha: structure-activity relationships and mechanism of action on avidin pretargeted tumor cells. Cancer Res. 1998;58:3866–3872. [PubMed] [Google Scholar]
- 32.Corti A, Fassina G, Marcucci F, Barbanti E, Cassani G. Oligomeric tumour necrosis factor alpha slowly converts into inactive forms at bioactive levels. Biochem J. 1992;284(Pt 3):905–910. doi: 10.1042/bj2840905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Ostade X, Vandenabeele P, Everaerdt B, Loetscher H, Gentz R, Brockhaus M, et al. Human TNF mutants with selective activity on the p55 receptor. Nature. 1993;361(6409):266–269. doi: 10.1038/361266a0. [DOI] [PubMed] [Google Scholar]
- 34.Bigda J, Beletsky I, Brakebusch C, Varfolomeev Y, Engelmann H, Bigda J, et al. Dual role of the p75 tumor necrosis factor (TNF) receptor in TNF cytotoxicity. J Exp Med. 1994;180(2):445–460. doi: 10.1084/jem.180.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tartaglia LA, Pennica D, Goeddel DV. Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J Biol Chem. 1993;268(25):18542–18548. [PubMed] [Google Scholar]
- 36.Espevik T, Brockhaus M, Loetscher H, Nonstad U, Shalaby R. Characterization of binding and biological effects of monoclonal antibodies against a human tumor necrosis factor receptor. J Exp Med. 1990;171(2):415–426. doi: 10.1084/jem.171.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grell M, Douni E, Wajant H, Lohden M, Clauss M, Georgopulos S, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 38.Vandenabeele P, Declercq W, Vercammen D, Van de Craen M, Grooten J, Loetscher H, et al. Functional characterization of the human tumor necrosis factor receptor p75 in a transfected rat/mouse T cell hybridoma. J Exp Med. 1992;176(4):1015–1024. doi: 10.1084/jem.176.4.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naume B, Shalaby R, Lesslauer W, Espevik T. Involvement of the 55- and 75-kDa tumor necrosis factor receptors in the generation of lymphokine-activated killer cell activity and proliferation of natural killer cells. J Immunol. 1991;146(9):3045–3048. [PubMed] [Google Scholar]
- 40.Pelagi M, Curnis F, Colombo B, Rovere P, Sacchi A, Manfredi AA, et al. Caspase inhibition reveals functional cooperation between p55- and p75-TNF receptors in cell necrosis. Eur Cytokine Netw. 2000;11(4):580–588. [PubMed] [Google Scholar]
- 41.Lewis M, Tartaglia LA, Lee A, Bennet LG, Rice GR, Wong GHW, et al. Cloning and expression of cDNA for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc Natl Acad Sci USA. 1991;88:2830–2834. doi: 10.1073/pnas.88.7.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Curnis F, Sacchi A, Corti A. Improving chemotherapeutic drug penetration in tumors by vascular targeting and barrier alteration. J Clin Invest. 2002;110(4):475–482. doi: 10.1172/JCI15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crippa L, Gasparri A, Sacchi A, Ferrero E, Curnis F, Corti A. Synergistic damage of tumor vessels with ultra low-dose endothelial-monocyte activating polypeptide-II and neovasculature-targeted tumor necrosis factor-alpha. Cancer Res. 2008;68(4):1154–1161. doi: 10.1158/0008-5472.CAN-07-2085. [DOI] [PubMed] [Google Scholar]
- 44.Sacchi A, Gasparri A, Gallo-Stampino C, Toma S, Curnis F, Corti A. Synergistic antitumor activity of cisplatin, paclitaxel, and gemcitabine with tumor vasculature-targeted tumor necrosis factor-alpha. Clin Cancer Res. 2006;12(1):175–182. doi: 10.1158/1078-0432.CCR-05-1147. [DOI] [PubMed] [Google Scholar]
- 45.Calcinotto A, Grioni M, Jachetti E, Curnis F, Mondino A, Parmiani G, et al. Targeting TNF-alpha to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J Immunol. 2012;188(6):2687–94. [DOI] [PubMed]
- 46.Bertilaccio MT, Grioni M, Sutherland BW, Degl’Innocenti E, Freschi M, Jachetti E, et al. Vasculature-targeted tumor necrosis factor-alpha increases the therapeutic index of doxorubicin against prostate cancer. Prostate. 2008;68(10):1105–1115. doi: 10.1002/pros.20775. [DOI] [PubMed] [Google Scholar]
- 47.Brett J, Gerlach H, Nawroth P, Steinberg S, Godman G, Stern D. Tumor necrosis factor/cachectin increases permeability of endothelial cell monolayers by a mechanism involving regulatory G proteins. J Exp Med. 1989;169(6):1977–1991. doi: 10.1084/jem.169.6.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldblum SE, Sun WL. Tumor necrosis factor-alpha augments pulmonary arterial transendothelial albumin flux in vitro. Am J Physiol. 1990;258(2 Pt 1):L57–L67. doi: 10.1152/ajplung.1990.258.2.L57. [DOI] [PubMed] [Google Scholar]
- 49.van der Veen AH, de Wilt JH, Eggermont AM, van Tiel ST, Seynhaeve AL, ten Hagen TL. TNF-alpha augments intratumoural concentrations of doxorubicin in TNF-alpha-based isolated limb perfusion in rat sarcoma models and enhances anti-tumour effects. Br J Cancer. 2000;82(4):973–980. doi: 10.1054/bjoc.1999.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lejeune FJ. High dose recombinant tumour necrosis factor (rTNF alpha) administered by isolation perfusion for advanced tumours of the limbs: a model for biochemotherapy of cancer. Eur J Cancer. 1995;31A(6):1009–1016. doi: 10.1016/0959-8049(94)00512-5. [DOI] [PubMed] [Google Scholar]
- 51.Kristensen CA, Nozue M, Boucher Y, Jain RK. Reduction of interstitial fluid pressure after TNF-alpha treatment of three human melanoma xenografts. Br J Cancer. 1996;74(4):533–536. doi: 10.1038/bjc.1996.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suzuki S, Ohta S, Takashio K, Nitanai H, Hashimoto Y. Augmentation for intratumoral accumulation and anti-tumor activity of liposome-encapsulated adriamycin by tumor necrosis factor-alpha in mice. Int J Cancer. 1990;46(6):1095–1100. doi: 10.1002/ijc.2910460624. [DOI] [PubMed] [Google Scholar]
- 53.de Wilt JH, ten Hagen TL, de Boeck G, van Tiel ST, de Bruijn EA, Eggermont AM. Tumour necrosis factor alpha increases melphalan concentration in tumour tissue after isolated limb perfusion. Br J Cancer. 2000;82(5):1000–1003. doi: 10.1054/bjoc.1999.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271(1):58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 55.van Laarhoven H, Gambarota G, Heerschap A, Lok J, Verhagen I, Corti A, et al. Early response measurements of NGR-TNF efficacy in murine lymphomas using magnetic resonance and immunohistochemical methods. Invest New Drugs. 2006;24(1):27–3. [DOI] [PubMed]
- 56.Ferrero E, Scabini S, Magni E, Foglieni C, Belloni D, Colombo B, et al. Chromogranin A protects vessels against tumor necrosis factor alpha-induced vascular leakage. FASEB J. 2004;18(3):554–555. doi: 10.1096/fj.03-0922fje. [DOI] [PubMed] [Google Scholar]
- 57.Dondossola E, Gasparri AM, Colombo B, Sacchi A, Curnis F, Corti A. Chromogranin A restricts drug penetration and limits the ability of NGR-TNF to enhance chemotherapeutic efficacy. Cancer Res. 2011;71(17):5881–5890. doi: 10.1158/0008-5472.CAN-11-1273. [DOI] [PubMed] [Google Scholar]
- 58.Sacchi A, Gasparri A, Curnis F, Bellone M, Corti A. Crucial role for interferon-gamma in the synergism between tumor vasculature-targeted tumor necrosis factor alpha (NGR-TNF) and doxorubicin. Cancer Res. 2004;64(19):7150–7155. doi: 10.1158/0008-5472.CAN-04-1445. [DOI] [PubMed] [Google Scholar]
- 59.van Laarhoven HW, Fiedler W, Desar IM, van Asten JJ, Marreaud S, Lacombe D, et al. Phase I clinical and magnetic resonance imaging study of the vascular agent NGR-hTNF in patients with advanced cancers (European Organization for Research and Treatment of Cancer Study 16041) Clin Cancer Res. 2010;16(4):1315–1323. doi: 10.1158/1078-0432.CCR-09-1621. [DOI] [PubMed] [Google Scholar]
- 60.Desar IM, van Herpen CM, van Asten JJ, Fiedler W, Marreaud S, Timmer-Bonte JN, et al. Factors affecting the unexpected failure of DCE-MRI to determine the optimal biological dose of the vascular targeting agent NGR-hTNF in solid cancer patients. Eur J Radiol. 2011;80(3):655–661. doi: 10.1016/j.ejrad.2010.08.045. [DOI] [PubMed] [Google Scholar]
- 61.Gregorc V, Citterio G, Vitali G, Spreafico A, Scifo P, Borri A, et al. Defining the optimal biological dose of NGR-hTNF, a selective vascular targeting agent, in advanced solid tumours. Eur J Cancer. 2010;46(1):198–206. doi: 10.1016/j.ejca.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 62.Gregorc V, Zucali PA, Santoro A, Ceresoli GL, Citterio G, De Pas TM, et al. Phase II study of asparagine–glycine–arginine-human tumor necrosis factor alpha, a selective vascular targeting agent, in previously treated patients with malignant pleural mesothelioma. J Clin Oncol. 2010;28(15):2604–2611. doi: 10.1200/JCO.2009.27.3649. [DOI] [PubMed] [Google Scholar]
- 63.Santoro A, Pressiani T, Citterio G, Rossoni G, Donadoni G, Pozzi F, et al. Activity and safety of NGR-hTNF, a selective vascular-targeting agent, in previously treated patients with advanced hepatocellular carcinoma. Br J Cancer. 2010;103(6):837–844. doi: 10.1038/sj.bjc.6605858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Santoro A, Rimassa L, Sobrero AF, Citterio G, Sclafani F, Carnaghi C, et al. Phase II study of NGR-hTNF, a selective vascular targeting agent, in patients with metastatic colorectal cancer after failure of standard therapy. Eur J Cancer. 2010;46(15):2746–2752. doi: 10.1016/j.ejca.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 65.Gregorc V, Santoro A, Bennicelli E, Punt C, Citterio G, Timmer-Bonte J, et al. Phase Ib study of NGR-hTNF, a selective vascular targeting agent, administered at low doses in combination with doxorubicin to patients with advanced solid tumours. Br J Cancer. 2009;101:219–224. doi: 10.1038/sj.bjc.6605162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lorusso D, Scambia G, Amadio G, di Legge A, Pietragalla A, De Vincenzo R, et al. Phase II study of NGR-hTNF in combination with doxorubicin in relapsed ovarian cancer patients. Br J Cancer. 2012;107(1):37–42. doi: 10.1038/bjc.2012.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gregorc V, De Braud FG, De Pas TM, Scalamogna R, Citterio G, Milani A, et al. Phase I study of NGR-hTNF, a selective vascular targeting agent, in combination with cisplatin in refractory solid tumors. Clin Cancer Res. 2011;17(7):1964–1972. doi: 10.1158/1078-0432.CCR-10-1376. [DOI] [PubMed] [Google Scholar]
- 68.Mammoliti S, Andretta V, Bennicelli E, Caprioni F, Comandini D, Fornarini G, et al. Two doses of NGR-hTNF in combination with capecitabine plus oxaliplatin in colorectal cancer patients failing standard therapies. Ann Oncol. 2011;22(4):973–978. doi: 10.1093/annonc/mdq436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zucali PA, Simonelli M, De Vincenzo F, Lorenzi E, Perrino M, Bertossi M, et al. Phase I and pharmacodynamic study of high-dose NGR-hTNF in patients with refractory solid tumours. Br J Cancer. 2013;108(1):58–63. doi: 10.1038/bjc.2012.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qin Z, Blankenstein T. CD4+ T cell-mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity. 2000;12(6):677–686. doi: 10.1016/s1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]
- 71.Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshwari S, et al. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med. 1995;182(1):155–162. doi: 10.1084/jem.182.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 73.Basham TY, Merigan TC. Recombinant interferon-gamma increases HLA-DR synthesis and expression. J Immunol. 1983;130(4):1492–1494. [PubMed] [Google Scholar]
- 74.Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13(2):95–109. doi: 10.1016/s1359-6101(01)00038-7. [DOI] [PubMed] [Google Scholar]
- 75.Ibe S, Qin Z, Schuler T, Preiss S, Blankenstein T. Tumor rejection by disturbing tumor stroma cell interactions. J Exp Med. 2001;194(11):1549–1559. doi: 10.1084/jem.194.11.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ruegg C, Yilmaz A, Bieler G, Bamat J, Chaubert P, Chaubert P, et al. Evidence for the involvement of endothelial cell integrin alpha v beta 3 in the disruption of the tumor vasculature induced by TNF and IFN-gamma. Nat Med. 1998;4:408–414. doi: 10.1038/nm0498-408. [DOI] [PubMed] [Google Scholar]
- 77.Gajewski TF, Schell SR, Nau G, Fitch FW. Regulation of T-cell activation: differences among T-cell subsets. Immunol Rev. 1989;111:79–110. doi: 10.1111/j.1600-065x.1989.tb00543.x. [DOI] [PubMed] [Google Scholar]
- 78.Schreiber RD, Hicks LJ, Celada A, Buchmeier NA, Gray PW. Monoclonal antibodies to murine gamma-interferon which differentially modulate macrophage activation and antiviral activity. J Immunol. 1985;134(3):1609–1618. [PubMed] [Google Scholar]
- 79.Gleave ME, Elhilali M, Fradet Y, Davis I, Venner P, Saad F, et al. Interferon gamma-1b compared with placebo in metastatic renal-cell carcinoma. Canadian Urologic Oncology Group. N Engl J Med. 1998;338(18):1265–1271. doi: 10.1056/NEJM199804303381804. [DOI] [PubMed] [Google Scholar]
- 80.Windbichler GH, Hausmaninger H, Stummvoll W, Graf AH, Kainz C, Lahodny J, et al. Interferon-gamma in the first-line therapy of ovarian cancer: a randomized phase III trial. Br J Cancer. 2000;82(6):1138–1144. doi: 10.1054/bjoc.1999.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, et al. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin Cancer Res. 2003;9(1):84–92. [PubMed] [Google Scholar]
- 82.Talmadge JE, Black PL, Tribble H, Pennington R, Bowersox O, Schneider M, et al. Preclinical approaches to the treatment of metastatic disease: therapeutic properties of rH TNF, rM IFN-gamma, and rH IL-2. Drugs Exp Clin Res. 1987;13(6):327–337. [PubMed] [Google Scholar]
- 83.Curnis F, Gasparri A, Sacchi A, Cattaneo A, Magni F, Corti A. Targeted delivery of IFN-gamma to tumor vessels uncouples anti-tumor from counter-regulatory mechanisms. Cancer Res. 2005;65:2906–13. [DOI] [PubMed]
- 84.Gasparri AM, Jachetti E, Colombo B, Sacchi A, Curnis F, Rizzardi GP, et al. Critical role of indoleamine 2,3-dioxygenase in tumor resistance to repeated treatments with targeted IFNgamma. Mol Cancer Ther. 2008;7(12):3859–3866. doi: 10.1158/1535-7163.MCT-08-0538. [DOI] [PubMed] [Google Scholar]
- 85.Zhang B, Gao B, Dong S, Zhang Y, Wu Y. Anti-tumor efficacy and pre-clinical immunogenicity of IFNalpha2a-NGR. Regul Toxicol Pharmacol. 2011;60(1):73–78. doi: 10.1016/j.yrtph.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 86.Meng J, Yan Z, Wu Y, Gao M, Li W, Gao F, et al. Preclinical safety evaluation of IFNalpha2a-NGR. Regul Toxicol Pharmacol. 2008;50(3):294–302. doi: 10.1016/j.yrtph.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 87.Meng J, Yan Z, Wu J, Li L, Xue X, Li M, et al. High-yield expression, purification and characterization of tumor-targeted IFN-alpha2a. Cytotherapy. 2007;9(1):60–68. doi: 10.1080/14653240601094322. [DOI] [PubMed] [Google Scholar]
- 88.Wang XX, Lu L, Song CL, Qian WN, Zhang SY, Zhang YQ, et al. Comparative pharmacokinetics of a tumour-targeting therapy candidate rh-IFNalpha2a-NGR with rh-IFNalpha2a administered intravenously in mice and rats. J Pharmacy Pharmacol. 2013;65(4):574–581. doi: 10.1111/jphp.12022. [DOI] [PubMed] [Google Scholar]
- 89.Bieker R, Kessler T, Schwoppe C, Padro T, Persigehl T, Bremer C, et al. Infarction of tumor vessels by NGR-peptide-directed targeting of tissue factor: experimental results and first-in-man experience. Blood. 2009;113(20):5019–5027. doi: 10.1182/blood-2008-04-150318. [DOI] [PubMed] [Google Scholar]
- 90.Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188(6):759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gaertner FC, Kessler H, Wester HJ, Schwaiger M, Beer AJ. Radiolabelled RGD peptides for imaging and therapy. Eur J Nucl Med Mol Imaging. 2012;39(Suppl 1):S126–S138. doi: 10.1007/s00259-011-2028-1. [DOI] [PubMed] [Google Scholar]
- 92.Mas-Moruno C, Rechenmacher F, Kessler H. Cilengitide: the first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anti-Cancer Agents Med Chem. 2010;10(10):753–768. doi: 10.2174/187152010794728639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Curnis F, Gasparri A, Sacchi A, Longhi R, Corti A. Coupling tumor necrosis factor-alpha with alphaV integrin ligands improves its antineoplastic activity. Cancer Res. 2004;64(2):565–571. doi: 10.1158/0008-5472.can-03-1753. [DOI] [PubMed] [Google Scholar]
- 94.Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 95.Ruoslahti E, Pierschbacher D. Arg-Gly-Asp: a versatile cell recognition signal. Cell. 1986;44:517–518. doi: 10.1016/0092-8674(86)90259-x. [DOI] [PubMed] [Google Scholar]
- 96.Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115(Pt 20):3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 97.Heckmann D, Kessler H. Design and chemical synthesis of integrin ligands. Methods Enzymol. 2007;426:463–503. doi: 10.1016/S0076-6879(07)26020-3. [DOI] [PubMed] [Google Scholar]
- 98.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8(8):604–617. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zarovni N, Monaco L, Corti A. Inhibition of tumor growth by intramuscular injection of cDNA encoding tumor necrosis factor alpha coupled to NGR and RGD tumor-homing peptides. Hum Gene Ther. 2004;15(4):373–382. doi: 10.1089/104303404322959524. [DOI] [PubMed] [Google Scholar]
- 101.Curnis F, Longhi R, Crippa L, Cattaneo A, Dondossola E, Bachi A, et al. Spontaneous formation of l-isoaspartate and gain of function in fibronectin. J Biol Chem. 2006;281(47):36466–36476. doi: 10.1074/jbc.M604812200. [DOI] [PubMed] [Google Scholar]
- 102.Corti A, Curnis F. Isoaspartate-dependent molecular switches for integrin-ligand recognition. J Cell Sci. 2011;124(Pt 4):515–522. doi: 10.1242/jcs.077172. [DOI] [PubMed] [Google Scholar]
- 103.Corti A, Curnis F, Arap W, Pasqualini R. The neovasculature homing motif NGR: more than meets the eye. Blood. 2008;112(7):2628–2635. doi: 10.1182/blood-2008-04-150862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Curnis F, Cattaneo A, Longhi R, Sacchi A, Gasparri AM, Pastorino F, et al. Critical role of flanking residues in NGR-to-isoDGR transition and CD13/Integrin receptor switching. J Biol Chem. 2010;285:9114–9123. doi: 10.1074/jbc.M109.044297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bochen A, Marelli UK, Otto E, Pallarola D, Mas-Moruno C, Di Leva FS, et al. Biselectivity of isoDGR peptides for fibronectin binding integrin subtypes alpha5beta1 and alphavbeta6: conformational control through flanking amino acids. J Med Chem. 2013;56(4):1509–19. [DOI] [PubMed]
- 106.Frank AO, Otto E, Mas-Moruno C, Schiller HB, Marinelli L, Cosconati S, et al. Conformational control of integrin-subtype selectivity in isoDGR peptide motifs: a biological switch. Angewandte Chemie. 2010;49(48):9278–9281. doi: 10.1002/anie.201004363. [DOI] [PubMed] [Google Scholar]
- 107.Mingozzi M, Dal Corso A, Marchini M, Guzzetti I, Civera M, Piarulli U, et al. Cyclic isoDGR peptidomimetics as low-nanomolar alphav beta3 integrin ligands. Chemistry. 2013;19(11):3563–3567. doi: 10.1002/chem.201204639. [DOI] [PubMed] [Google Scholar]
- 108.Spitaleri A, Mari S, Curnis F, Traversari C, Longhi R, Bordignon C, et al. Structural basis for the interaction of isoDGR with the RGD-binding site of avbeta 3 integrin. J Biol Chem. 2008;283(28):19757–68. [DOI] [PubMed]
- 109.Ghitti M, Spitaleri A, Valentinis B, Mari S, Asperti C, Traversari C, et al. Molecular dynamics reveal that isoDGR-containing cyclopeptides are true alphavbeta3 antagonists unable to promote integrin allostery and activation. Angewandte Chemie. 2012;51(31):7702–7705. doi: 10.1002/anie.201202032. [DOI] [PubMed] [Google Scholar]
- 110.Curnis F, Sacchi A, Gasparri A, Longhi R, Bachi A, Doglioni C, et al. Isoaspartate–glycine–arginine: a new tumor vasculature-targeting motif. Cancer Res. 2008;68(17):7073–7082. doi: 10.1158/0008-5472.CAN-08-1272. [DOI] [PubMed] [Google Scholar]
- 111.Curnis F, Sacchi A, Longhi R, Colombo B, Gasparri A, Corti A. IsoDGR-tagged albumin: a new avb3 selective carrier for nanodrug delivery to tumors. Small. 2013;9(5):673–8. [DOI] [PubMed]
- 112.Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Gold nanoparticles for biology and medicine. Angewandte Chemie. 2010;49(19):3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, et al. Colloidal gold: a novel nanoparticle vector for tumor directed drug delivery. Drug Deliv. 2004;11(3):169–83. [DOI] [PubMed]
- 114.Goel R, Shah N, Visaria R, Paciotti GF, Bischof JC. Biodistribution of TNF-alpha-coated gold nanoparticles in an in vivo model system. Nanomedicine (Lond) 2009;4(4):401–410. doi: 10.2217/nnm.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shenoi MM, Iltis I, Choi J, Koonce NA, Metzger GJ, Griffin RJ, et al. Nanoparticle delivered vascular disrupting agents (VDAs): use of TNF-alpha conjugated gold nanoparticles for multimodal cancer therapy. Mol Pharm. 2013;10(5):1683–94. [DOI] [PMC free article] [PubMed]
- 116.Joyce JA, Laakkonen P, Bernasconi M, Bergers G, Ruoslahti E, Hanahan D. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2003;4(5):393–403. doi: 10.1016/s1535-6108(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 117.Johansson A, Hamzah J, Payne CJ, Ganss R. Tumor-targeted TNFalpha stabilizes tumor vessels and enhances active immunotherapy. Proc Natl Acad Sci USA. 2012;109(20):7841–7846. doi: 10.1073/pnas.1118296109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Johansson A, Hamzah J, Ganss R. Intratumoral TNFalpha improves immunotherapy. Oncoimmunology. 2012;1(8):1395–1397. doi: 10.4161/onci.20981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Balza E, Mortara L, Sassi F, Monteghirfo S, Carnemolla B, Castellani P, et al. Targeted delivery of tumor necrosis factor-alpha to tumor vessels induces a therapeutic T cell-mediated immune response that protects the host against syngeneic tumors of different histologic origin. Clin Cancer Res. 2006;12(8):2575–2582. doi: 10.1158/1078-0432.CCR-05-2448. [DOI] [PubMed] [Google Scholar]
- 120.Aina OH, Sroka TC, Chen M-L, Lam KS. Therapeutic cancer targeting peptides. Biopolymers. 2002;66:184–199. doi: 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]