

Abstract
Background
Neurodegenerative diseases (NDs) are characterized by the progressive loss of neurons in the human brain. Although the majority of NDs are sporadic, evidence is accumulating that they have a strong genetic component. Therefore, significant efforts have been made in recent years to not only identify disease-causing genes but also genes that modify the severity of NDs, so-called genetic modifiers. To date there exists no compendium that lists and cross-links genetic modifiers of different NDs.
Description
In order to address this need, we present NeuroGeM, the first comprehensive knowledgebase providing integrated information on genetic modifiers of nine different NDs in the model organisms D. melanogaster, C. elegans, and S. cerevisiae. NeuroGeM cross-links curated genetic modifier information from the different NDs and provides details on experimental conditions used for modifier identification, functional annotations, links to homologous proteins and color-coded protein-protein interaction networks to visualize modifier interactions. We demonstrate how this database can be used to generate new understanding through meta-analysis. For instance, we reveal that the Drosophila genes DnaJ-1, thread, Atx2, and mub are generic modifiers that affect multiple if not all NDs.
Conclusion
As the first compendium of genetic modifiers, NeuroGeM will assist experimental and computational scientists in their search for the pathophysiological mechanisms underlying NDs. http://chibi.ubc.ca/neurogem.
Keywords: Neurodegenerative diseases, Genetic modifiers, Database, Knowledgebase, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease
Background
Intracellular protein aggregation is a feature of many late-onset neurodegenerative diseases (NDs), also called proteinopathies. These include Alzheimer’s disease (AD), Parkinson’s disease (PD), and nine polyglutamine expansion diseases exemplified by Huntington’s disease (HD). The pathophysiology of NDs is very complex, which is one of the reasons why there are no effective strategies that slow or prevent neurodegeneration.
In recent years, significant efforts have been made to identify genes that modify the severity of NDs. Altering the activities of these genetic modifier genes on their own may not result in obvious phenotypes in the absence of the conditioning (neurodegeneration-causing) mutation. However, the identified genetic modifiers allow the characterization of biological pathways that modulate the disease and, in some cases, discovery of tractable therapeutic targets. The identification of genetic modifiers has been facilitated due to the development of in vivo models of different proteinopathies in organisms such as D. melanogaster and C. elegans[1-3]. Moreover, genome-wide screens for genetic modifiers have become possible because of high-throughput technologies such as RNA interference [4] or public availability of various transgenic stocks covering most genes such as fly stocks with P-element insertion mutations [5].
Nevertheless, due to the complex nature of the pathological processes underlying proteinopathies, there are large inconsistences in the collected data. Even more importantly, data alone without knowledge or integration into existing databases is bound to remain inaccessible and thus cannot be utilized by the broad scientific community. An integrated database of genetic modifiers of NDs would assist computational and experimental scientists alike in improving their approaches to discover what is common to and distinct for different proteinopathies.
In order to address this need, we assembled the first comprehensive database of genetic modifiers in NDs. NeuroGeM (‘neurodegenerative disease genetic modifiers database’) catalogues and cross-links genetic modifiers of 9 different NDs in three different model organisms, and associates them with information on protein function and other annotations. NeuroGeM contains detailed information on the experimental conditions in which the modifiers were identified, displays the protein-protein interaction sub-network around modifiers, and provides search and display tools to deduce testable hypotheses. Furthermore, in order to demonstrate the broad applicability of the data and tools provided by NeuroGeM, we present the results of a first meta-analysis.
Construction and content
Data collection
NeuroGeM is a comprehensive collection of literature data on genetic modifiers of NDs and associated genetic information from a variety of databases (Figure 1). The ND models include AD subclassified as ADAβ (amyloid-beta models) and ADTau (tau models), HD, PD, Spinocerebellar ataxia type 1, 3 and 7 (SCA1, SCA3, SCA7), Amyotrophic lateral sclerosis (ALS) and generic polyQ-induced disease in D. melanogaster, C. elegans, and S. cerevisiae. At the time of data compilation, all known high-throughput (HT) screens for modifiers carried out in the model organisms and a handful of low-throughput (LT) experimental results were included. Overall, NeuroGeM contains 87,864 experimental records (3,618 for modifiers and 84,246 for non-modifiers) from the 9 different disease models in the three different species (Table 1).
Figure 1.
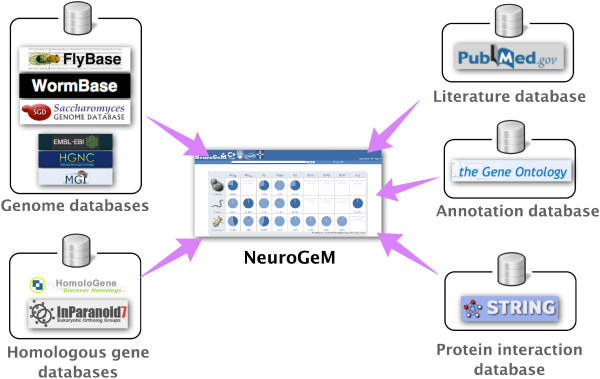
The contents of NeuroGeM. NeuroGeM is a comprehensive collection of genetic modifiers of ND models in D. melanogaster, C. elegans, and S. cerevisiae. In order to provide comprehensive information on genetic modifiers, NeuroGeM also integrates information from genome databases (FlyBase, WormBase, SGD, EBML, HGNC, and MGI), the protein interaction database STRING, GeneOntology, and homologous gene databases (HomoloGene and InParanoid). The statistics of the data currently available in NeuroGeM is shown in Table 1, and all terms used in the database are listed in Table 2.
Table 1.
Statistics of genetic modifiers in NeuroGeM
| Species | Disease models |
Experimental records |
Modifiers1 |
Gene coverage2 (%) | |||
|---|---|---|---|---|---|---|---|
| Positive records | Negative records | Enhancers | Suppressors | Non-modifiers | |||
|
D. melanogaster |
ADTau |
144 |
571 |
65 |
60 |
549 |
4.89 |
| ADAβ |
61 |
6062 |
25 |
22 |
6059 |
44.43 |
|
| PD |
1 |
- |
- |
1 |
- |
0.01 |
|
| HD |
1260 |
8142 |
130 |
90 |
7732 |
57.82 |
|
| SCA1 |
66 |
24 |
16 |
21 |
21 |
0.42 |
|
| SCA3 |
623 |
52 |
55 |
520 |
49 |
4.54 |
|
| SCA7 |
14 |
19 |
8 |
5 |
18 |
0.23 |
|
| PolyQ |
22 |
44 |
7 |
10 |
43 |
0.44 |
|
|
C. elegans |
ADTau |
75 |
15909 |
- |
75 |
15899 |
97.86 |
| ADAβ |
6 |
- |
1 |
5 |
- |
0.04 |
|
| HD |
22 |
2 |
1 |
17 |
2 |
0.12 |
|
| PD |
290 |
18121 |
- |
268 |
15767 |
98.24 |
|
| ALS |
168 |
15909 |
88 |
80 |
15817 |
97.92 |
|
| PolyQ |
459 |
40 |
152 |
195 |
20 |
2.11 |
|
| S. cerevisiae | ADAβ |
106 |
5262 |
18 |
23 |
5248 |
78.83 |
| HD |
82 |
9422 |
28 |
54 |
4670 |
70.83 |
|
| PD |
216 |
4699 |
22 |
181 |
4577 |
71.26 |
|
| PolyQ | 1 | - | - | 1 | - | 0.01 | |
1Certain genes have been identified independently as both an enhancer and suppressor under different experimental conditions.
2With respect to the protein coding genes.
Importantly, we continually update the knowledgebase with newly published results. In addition, users can submit their own data upon request of a login and the uploaded data will be made accessible to all users after curation (see Additional file 1).
In order to provide comprehensive information on genetic modifiers, we integrated relevant data from other sources into NeuroGeM. All entries in NeuroGeM contain information on gene function/annotations and are accompanied by direct links to the relevant information on FlyBase for D. melanogaster (ver Feb 2012) [6], WormBase for C. elegans (ver WS230) [7] and SGD for S. cerevisiae (downloaded in Jan 2012) [8]. Each gene entered in NeuroGeM contains an ID that is identical to the primary ID of the gene in its respective genome database (FlyBase, WormBase, or SGD). These IDs will allow users to easily access other databases and avoid the effort required for ID conversion. NeuroGeM records have a link to the PubMed entry of the original study from which the records stem.
As protein-protein interaction networks allow identification of functionally associated proteins or important functional clusters [9,10], NeuroGeM visualizes the protein interaction sub-network around a queried gene; each protein node in the network is color-coded according to the available experimental results deposited in NeuroGeM. For this feature, NeuroGeM utilizes the protein interaction data from STRING (ver 9.05) [11]. In order to facilitate the identification of genetic modifiers with the same function or involved in the same process, NeuroGeM provides an ontology-based search functionality that searches for genes by GeneOntology (GO) annotations and the hierarchical structure of GO terms [12].
As users might be interested in finding homologs of the genetic modifiers entered in NeuroGeM, we also integrated homologous gene data from NCBI HomoloGene (build 65) [13] and InParanoid (ver7) [14,15]. Orthologs are defined as genes in different species that have evolved from a common ancestral gene, while paralogs are genes related by duplication within the same species that often have different functions. Homologs are either paralogs or orthologs (for details see NCBI HomoloGene (build 65) [13] and InParanoid (ver7) [14,15]). The homology data covers not only the three model organisms but also H. sapiens and M. musculus, though no modifiers from these organisms are deposited in NeuroGeM yet. Cross-linking genes via homology should facilitate the expansion of modifier studies in other organisms; for instance, confirmation of important modifiers in higher organisms. Gene information, GO annotations and protein interaction data for human and mouse genes were also integrated to help users search for modifiers that are homologous to human and mouse genes of interest. (source: EMBL Rel 68 [16], HGNC downloaded in Jan 2013 [17]; EMBL Rel 68 [18]; and MGI downloaded in Dec 2012 [19]). As detailed information on genes and proteins is frequently updated in their source databases, NeuroGeM also provides links to those databases when available.
Database implementation
The database was implemented with a web interface compatible with common web browsers to provide access to researchers. The data is stored in a relational database using a MySQL 5.0.59 server. Data processing and HTML generation for displaying information are carried out using PHP 5.3.3. Javascript and AJAX technology are used to improve search functions. CytoscapeWeb [20] is employed to visualize protein-protein interaction networks. The current database is running on Redhat Linux 5.6 with an Apache server 2.2.3. All the data in NeuroGeM can be downloaded as plain text files.
Utility and discussion
NeuroGeM allows users to access the integrated data in three different ways: (i) a categorical search, (ii) a keyword search, and (iii) an ontology-based search. All search methods take users first to a list of publications or a list of genes that fit the search criteria, from which users can select a gene of interest and consult its modifier information page. Figures 2 and 3 and Additional file 2: Figures S1 and Additional file 3: Figure S2 illustrate the three ways to search genetic modifiers in NeuroGeM and show detailed information on genetic modifiers provided by NeuroGeM.
Figure 2.

Search to access information on genetic modifiers in NeuroGeM. This figure illustrates a keyword-based search to get information of a specific genetic modifier. Other search methods are available in Additional file 2: Figure S1 (categorical search) and Additional file 3: Figure S2 (ontology-based search). The genetic modifier information page includes (a) a report summary of experimental records entered in NeuroGeM, (b) general information on the gene with links to relevant databases, and (c) the protein-protein interaction sub-network around this gene. Details about experimental parameters and results are also shown (Figure 3). The homologous genes section (not shown in this figure) lists genetic information and experimental details of homologous genes.
Figure 3.

Details of the experiments that identified the genetic modifiers. This section of the genetic modifier information page includes the detailed results of the modifier screens (whether the gene is a suppressor, enhancer, or non-modifier), experimental details (mutated genes, method for gene expression modulation, phenotype observation, etc.), and a link to the original article in which the experiment was described (PubMed).
Categorical search for studies that identified modifiers
On the front page of the NeuroGeM web site, research results are organized according to disease and model-organism categories: studies that identified modifiers were categorized according to model organism (n = 3) and diseases (n = 9 including two AD subtypes) (Additional file 2: Figure S1). Clicking on an organism icon or disease-specific icon directs the user to a list of publications that belong to that category. Clicking on a pie chart at the intersection of a specific model organism and disease type directs the user to a list of publications that searched for modifiers in the corresponding disease model and organism. Clicking on a specific publication then directs the user to a list of genes that have been tested in that study.
For instance, clicking on the pie chart at the intersection of D. melanogaster and HD (Additional file 2: Figure S1) lists 24 publications in which genetic modifiers of HD have been identified in D. melanogaster. Clicking on the PubMed ID 17984172 will open a web page that lists the 26 genes that have been examined in this specific study and provides a summary of the experimental records deposited in NeuroGeM for each of these 26 genes (see below for the detailed explanation of the record summary). Clicking on the gene name Nup44A will then direct the user to the genetic modifier information page of Nup44A, which contains the details of the results of different genetic modifier screens for this gene as well as additional information that is discussed in further detail in the “Genetic modifier information” section below.
Keyword-based search for specific genes
NeuroGeM also provides different search methods from a unified search box (Figure 2). As a user starts typing letters in the search box, NeuroGeM suggests matching keywords (gene name, synonyms, IDs, GO terms, etc.). If the user selects one of the suggestions, NeuroGeM will open this gene’s modifier information page (Figure 2, red arrow). If the user presses the ‘Search’ button instead, NeuroGeM will list all genes that contain the keyword in their names, synonyms, or IDs. Then, the user can open a specific genetic information page by clicking on one of the listed genes (Figure 2, blue arrow). For example, when typing ‘nup4’ in the search box, several genes that contain the keyword ‘nup4’ are suggested. Selecting the D. melanogaster Nup44A gene from the suggestions will open this gene’s page directly. On the other hand, entering ‘nup4’ and clicking the ‘Search’ button will list all genes that contain the keyword and show respective experimental record summaries. The user can open the Nup44A gene page by clicking on the gene name Nup44A of the listed genes.
Ontology-based search for related genes
The keyword-based search directs the user to a specific gene, but this feature is not appropriate when searching for functionally related genes, e.g. searching for genes involved in the cell cycle. For such a relation-based search, we provide the ontology-based search. To use the ontology-based search, the user has to type a GO term or GO ID in the search box. Just like the keyword search, suggestions will pop up and the user can select one of the suggested GO terms. Then, NeuroGeM searches for not only genes with the user-specified GO term but also genes with a related (child) term. This ontology-based search will assist users in the identification of modifiers that are associated with specific cellular functions or processes.
For instance, as shown in Additional file 3: Figure S2, the query of ‘cell cycle (GO:0007049)’ in D. melanogaster returns 656 genes that are involved in the cell cycle: 622 out of 656 genes have been evaluated experimentally, and 256 out of 622 genes have been identified as modifiers. Drosophila’s Nup44A gene is also shown in the gene list since Nup44A has GO annotations for regulation of mitotic cell cycle (GO:0007346) and regulation of meiotic cell cycle (GO:0051445), which are child terms of cell cycle. Clicking on the name of Nup44A will direct the user to Nup44A genetic modifier information page.
Genetic modifier information
At the end of each search, NeuroGeM directs users to the genetic modifier information page of a specific gene. As an example, the genetic modifier information page of the Drosophila gene Nup44A (FBgn0033247) is shown in Figures 2 and 3. Terms and definitions used in genetic modifier information pages are listed in Table 2.
Table 2.
Terms used in NeuroGeM
| Terms | Values | Meaning |
|---|---|---|
| Organism |
D. melanogaster |
Three model organisms |
|
C. elegans | ||
|
S. cerevisiae | ||
| Gene ID |
FBgn------ |
Primary IDs used in the respective genome databases (FlyBase, WormBase, and SGD). These IDs are also used as primary IDs in NeuroGeM. |
| W-------- | ||
| S--------- | ||
| Type of modification |
Suppressor |
Suppressors are those genes that alleviate disease pathology or slow disease progression when over-expressed, and those that aggravate disease pathology or accelerate disease progression when down-regulated or deleted. Enhancers are those genes that alleviate disease pathology when down-regulated, and those that aggravate pathology when over-expressed. Non-modifiers have no effect on disease progression. |
| Enhancer | ||
| Non-modifier | ||
| Mode of action |
Toxicity modification |
Toxicity modifiers are those genes that change disease pathology. Aggregation modifiers are those genes that change the size or number of protein aggregates. |
| Aggregation modification | ||
| Disease model |
Alzheimer’s disease (AD) |
ND models compiled in the current version of NeuroGeM. We divided AD into the subtypes ADTau and ADAβ according to the gene used to induce the disease phenotype (mutant Tau protein and Aβ42, respectively). |
| Huntington’s disease (HD) | ||
| Parkinson’s disease (PD) | ||
| Spinocerebellar ataxia type 1 (SCA1) | ||
| Spinocerebellar ataxia type 3 (SCA3) | ||
| Spinocerebellar ataxia type 7 (SCA7) | ||
| Amyotrophic lateral sclerosis (ALS) | ||
| PolyQ disease (PolyQ) | ||
| Disease induction |
Various |
This field contains expression cassette information described in the literature including promoter and disease-causing gene (e.g. polyQ stretch length). |
| Disease-causing mutant proteins compiled in NeuroGeM are Aβ and tau protein for AD, SOD1 for ALS, huntingtin for HD, α-synuclein for PD, Ataxin-1 for SCA1, Ataxin-3 (MJD) for SCA3, Ataxin-7 for SCA7 and polyQ stretches for the PolyQ disease model. | ||
| Modulation method |
Overexpression |
This field describes whether the expression level of the target gene increased (overexpression or gain-of-function) or decreased (knockdown, knockout, or loss-of-function). We adopted the same terms used in the original articles. |
| Gain-of-function | ||
| Knockdown | ||
| Knockout | ||
| Loss-of-function | ||
| Experimental scale |
Primary high-throughput |
This field describes the scale of the experiments. Experiments that were not high-throughput (HT) were assigned as low-throughput (LT). Experiments performed in primary screens were assigned as Primary high-throughput. Experiments to confirm the results obtained from the Primary high-throughput screens were assigned as Secondary high-throughput. |
| Secondary high-throughput | ||
| Low-throughput | ||
| Measurement |
Various |
This field describes how the change of pathology was evaluated. For example, change in the eye phenotype is a common readout in D. melanogaster, and cell growth rate is a common readout in S. cerevisiae. |
| Cell type | Various | This field briefly describes what cell lines and organs were utilized to carry out the experiment. |
At the top of the genetic modifier information page (Figure 2a), the number of experimental records that are available in NeuroGeM for a specific gene and its homologs are reported (record summary) for each experimental scale (“L” stands for LT, “H2” for secondary HT, and “H1” for primary HT). If experimental data that identifies a specific gene as a modifier has been entered in NeuroGeM, this fact is highlighted in red, light red if it is only one HT experiment, and dark red if it is at least two HT experiments or at least one LT experiment. Similarly, records that report that a gene is a non-modifier are colored in light and dark blue. For example, there are currently two records in NeuroGeM for Nup44A indicating that Nup44A (FBgn0033247) is a non-modifier in HD, one based on a LT and one on a primary HT experiment. In addition, there are two records of LT experiments indicating that Nup44A is a modifier in SCA1. By clicking on a specific record in the “record summary” at the top of the page (e.g. “2 0 0” under the column header “L H2 H1” for Nup44A and SCA1), the user is immediately guided to the details of that entry (Figure 3).
Below the report summary, detailed information of the gene is displayed on the left side (Figure 2b), including synonyms, alternative gene names, and IDs used in other databases as well as GO annotations. On the right side (Figure 2c), NeuroGeM displays the protein-protein interaction sub-network around the gene, in which interacting proteins are colored by the type and result of experiments that tested them as modifiers. Specifically, the left half of each node (protein) is colored according to the evidence for it being a modifier. The right half of each node is colored according to the evidence for it being a non-modifier. The same coloring scheme as for the record summary is used. Importantly, the protein-protein interaction network and the coloring are organism- and disease-specific (coloring by different diseases can be selected below the network). Users can navigate to other genetic modifier information pages by clicking on the corresponding nodes. Figure 2c shows the protein-protein interaction sub-network of Nup44A colored according to the results of screens for modifiers of HD in Drosophila. A look at the network immediately reveals that Nup44A, Nup75, and Rae1 were identified as non-modifiers, while Nup107 was identified as modifier of HD in Drosophila. By contrast, SmB, SmD1, Cbp20, and Nup154 were identified as modifiers in some experiments but not in others.
In the next section of the genetic modifier information page, experimental details are displayed (Figure 3). Experiments are categorized by the disease model, and the number of experimental records for each disease model is shown with respect to experimental scale by using the same coloring scheme as for the report summary. The reported experimental details include: (i) type of modification; indicates whether the experiment found the gene to be a suppressor, enhancer, or non-modifier, (ii) mode of action; reports whether the queried gene modified aggregation size/number or changed disease symptoms, (iii) disease induction; denotes which (mutant) gene was used to cause disease symptoms, and shows the mutant gene and its expression cassette information, (iv) modulation method; denotes how the expression of the queried gene was modulated (e.g. over-expressed, knocked out, repressed by RNAi), (v) experimental scale; denotes the scale of performed experiments (LT, primary HT, secondary HT; secondary HT stands for experiments to confirm the results obtained from primary HT experiments), (vi) measurement; denotes what was quantified to identify a modifier and (vii) cell type; denotes the cell lines or stocks utilized in the experiment. For instance, Nup44A was tested as modifier in a Drosophila model of SCA1 that was created by expressing Ataxin-1 with a polyQ expansion of 82 (Figure 3). The impact of the overexpression of Nup44A on the disease model was quantified based on changes in the severity of an eye phenotype. Nup44A was categorized as a suppressor, indicating that the over-expression of the Nup44A gene alleviated the severity of the eye phenotype.
Below the experimental details of a specific gene, its homologous genes are displayed with genetic information, protein interaction sub-network, and experimental details if available.
Search for orthologs of human and mouse genes
The current version of NeuroGeM does not contain any genetic modifiers in human and mouse (to be included at a later stage). Nevertheless, researchers studying genetic modifiers of NDs in the three model organisms are likely to be interested in the homologous genes of the modifiers in higher organisms such as human and mouse. Thus, we also integrated human genome information from the EMBL and HGNC databases and mouse genome information from the EMBL and MGI databases. The user can search for human and mouse genes using their gene names or EMBL IDs, and then obtain not only information on the queried genes but also information on their homologous genes in D. melanogaster, C. elegans, and S. cerevisiae. For example, Drosophila’s Nup44A gene and its homologous genes in other species are listed in Figure 2a and the information on those genes is displayed at the bottom of the Nup44A gene page (omitted in Figures 2 and 3). The user can also search for human and mouse genes homologous to Nup44A by their gene names (Seh1l and SEH1L) or EMBL IDs (ENSMUSG00000079614 and ENSG00000085415) in the unified search box.
Applications of data in NeuroGeM
In order to demonstrate the broad applicability of NeuroGeM and how it can provide new understanding, we performed a variety of meta-analyses using the genetic modifiers data from NeuroGeM. In this section, the results of the meta-analyses are discussed briefly. The detailed methods, results and discussion for the meta-analyses are available in the Additional file 1. Due to the abundance of both HT and LT experimental data from D. melanogaster, mainly results obtained from the meta-analysis of genetic modifiers of D. melanogaster are presented here. Results from the analysis of genetic modifiers in other model organism as well as the comparison of modifiers in all three model organisms are available in Additional file 1.
i) NeuroGeM can be used to identify biological processes that are enriched within genetic modifiers in a specific disease or in groups of diseases. Genes with annotations for these processes can be prioritized for testing in other model organisms or for drug screenings. A meta-analysis of the data deposited in NeuroGeM revealed that modifiers across species are often involved in protein folding (Figure 4a). However, they account for only 3% of all genetic modifiers. The analysis revealed that modifiers are equally often involved in cell cycle and splicing, accounting for 7% and 3% of all genetic modifiers, respectively. This analysis result suggests that researchers expecting to discover more genetic modifiers should focus their efforts also on genes involved in cell cycle and splicing, biological processes that are also often enriched in modifiers. As shown in Figure 4b, a correlation analysis of modifiers between diseases revealed that polyQ diseases (HD, generic PolyQ, SCA1, SCA3, and SCA7 in Figure 4b) share many genetic modifiers and non-modifiers which are not seen in AD models, which is consistent with a previous report [21]. Specifically, a strong anti-correlation is observed when comparing the modifiers and non-modifiers of ADAβ and SCA3. Many SCA3-specific genetic modifiers are involved in protein folding and splicing[22,23], while ADAβ-specific modifiers are involved in protein synthesis[24]. Untested genes involved in modifier-enriched processes can be prioritized in future screens to confirm these trends and elucidate their mechanisms. See Additional file 1.
Figure 4.
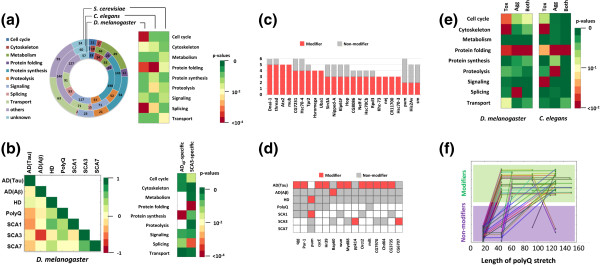
Compilation of meta-analysis results. (a) Functional classification of genetic modifiers and their enrichments represented by p-values. (b) (Left) Correlation analysis of modifiers and non-modifiers between diseases. Pairwise correlations (Matthew’s correlation coefficient) are represented by a color matrix ranging from −1 (inverse correlation, red), via 0 (no correlation, yellow), to 1 (high correlation, green). (Right) Modifiers in SCA3 and ADTau were further analyzed to see which functional categories are enriched among these genetic modifiers. (c) List of genes identified as modifiers of several diseases in D. melanogaster. Red and grey denote the number of diseases in which a gene is identified as a modifier and non-modifier, respectively (see Additional file 1: Figure S7 for full list). (d) List of disease-specific genetic modifiers. The same color annotation as in (c) is used here (see Additional file 1: Figure S8 for full list). (e) Enrichment analysis results of HD modifiers in D. melanogaster and PD modifiers in C. elegans with respect to the mode of action of the modifiers. Tox, toxicity modifiers; Agg, aggregation modifiers; Both, aggregation and toxicity modifiers. (f) Identification of modifiers and non-modifiers depending on the length of the polyQ stretch in HD models in D. melanogaster. Each line refers to one gene and each green dot refers to one experiment with a specific length of the polyQ stretch. If dots are in the purple and green region, it means the gene has been identified as a non-modifier and modifier, respectively. See Additional file 1 for details of meta-analysis.
ii) NeuroGeM allows easy identification (by non computational experts) of genes that modify the neurodegenerative toxicity in several ND models. Hence, cross-disease comparisons can identify potential generic modifiers that then can be tested experimentally in other disease models in other organisms (rodents) or compared to human genetic data. A first search for generic modifiers revealed that the genes DnaJ-1, thread, Atx2, and mub are modifiers in 5 out of 7 ND models in D. melanogaster (Figure 4c). Interestingly, DNAJB4 and BIRC3, the mammalian orthologs of DnaJ-1 and thread, have recently been shown to reduce neuronal cell death when up-regulated in multiple mammalian ND models (see Additional file 1). Moreover, Atx2 has recently also been associated with an increased risk for ALS [25]. Further experiments are necessary to confirm this hypothesis that DnaJ-1, thread, Atx2, and mub are generic modifiers.
iii) Equally, NeuroGeM facilitates the identification of genes that only affect the phenotype of specific NDs. Though all NDs are caused by aggregates, they definitely show different pathophysiology. In this regard, genes capable of modulating disease phenotype only in a specific ND give us hints to understand the difference in disease progression. For instance, modifiers currently confined to ADTau in D. melanogaster include sgg and par-1 (Figure 4d). This finding is consistent with the specific importance of hyper-phosphorylation of the tau protein in AD, which is a process that may be accelerated by par-1 and sgg [26,27]. In S. cerevisiae, modifiers are enriched in protein synthesis in AD (RTG3, TEC1, SPT21, PPR1, MBP1, SRO9, SLF1, and SLS1), protein folding in HD (HSP26, HSP42 and APJ1), and transport in PD (FUN26, YCK3, and GOS1), which is consistent with recent results (Additional file 1).
iv) The database can help get new insights into the mechanism of disease modulation. In NeuroGeM, each modifier is classified as toxicity modifier and/or an aggregation modifier: toxicity modifiers change disease phenotype (eye development, motility, etc.), and aggregation modifiers primarily affect aggregate size or number. As shown in Figure 4e, toxicity modifiers are enriched in cell cycle, cytoskeleton, signaling, and protein folding categories; these modifiers are involved in cellular pathways that modulate the level of tolerance to the stress caused by the aggregates and ultimately lead to phenotypic changes. On the other hand, aggregation modifiers are enriched in splicing, proteolysis, and protein folding, which are the processes directly or indirectly associated with aggregate formation and elimination. Interestingly, modifiers belonging to both groups are only enriched in protein folding. This functional category includes protein quality control, which is a network of cellular processes that in an orchestrated manner works against protein misfolding and aggregation. Therefore, proteins involved in protein folding would be prime therapeutic targets, since they are able to resolve the problem of aggregate formation, and are involved in cellular processes that can increase the tolerance to the cellular stress caused by protein aggregation [28,29]. Moreover, these proteins may play a key role in the pathophysiology of many NDs due to their dual effect. In order to test this hypothesis, we identified Drosophila modifiers that are both aggregation and toxicity modifiers. We found that many of them are indeed able to modulate neurodegeneration in several different disease models (see Table 3 and Additional file 1). Interestingly, the list includes three of the previously identified generic modifiers; DnaJ-1, thread and Atx2. Next, we tested whether this is true across species, i.e. also for higher organisms. We identified human and mouse orthologs of the Drosophila aggregation and toxicity modifiers by using the feature of NeuroGeM. A careful literature search confirmed that for most of the mammalian orthologs of these modifiers there exists experimental evidence that they modify the phenotype of several ND models in mammalian cells (see Table 3 and Additional file 1 for details about the orthologs).
Table 3.
Genes that are toxicity and aggregation modifiers in D. melanogaster and their orthologs in H. sapiens and M. musculus1
| D. melanogaster genes | D. melanogaster ND models2 | Ref | H. sapiens orthologs | M. musculus orthologs | Ref3 |
|---|---|---|---|---|---|
| DnaJ-1 |
ADTau, HD, PolyQ, SCA1, SCA3 |
[30-34] |
DNAJB1, DNAJB4, DNAJB5, DNAJB13 |
Dnajb1, Dnajb4, Dnajb5, Dnajb13 |
[35-40] |
| thread |
ADTau, HD, SCA1, SCA3, SCA7 |
[21,32,41,42] |
BIRC2, BIRC3 |
Birc2, Birc3 |
[43-45] |
| Atx2 |
ADTau, HD, SCA1, SCA3, SCA7 |
[21,41,42,46] |
Atxn2, Atxn2L |
Atxn2, Atxn2L |
[47-52] |
| Hsc70-3 |
HD, SCA1, SCA7 |
[32,42] |
HSPA5 |
Hspa5 |
[53] |
| Hsc70Cb |
ADTau, HD, SCA3 |
[5,54,55] |
HSPH1(HSP110), HSPA4, HSPA4L |
Hsph1 (Hsp110), Hspa4, Hsp4l |
[56,57] |
| Rpd3 |
HD, SCA1, SCA7 |
[34,42,58] |
HDAC1, HDAC2 |
Hdac1, Hdac2 |
[59-65] |
| 14-3-3epsilon |
HD, SCA1 |
[66,67] |
YWHAZ, YWHAB, YWHAE |
Ywhaz, Ywhab, Ywhae |
[67-73] |
| CG5537 |
HD |
[74] |
UPRT |
Uprt |
N/A |
| Hsf |
HD, SCA3 |
[75] |
Hsf2, Hsf4, Hsfx1, Hsfx2, Hsfy1, Hsfy2, Hsf5 |
Hsf2, Hsf3, Hsf4, Hsfy2, Hsf5 |
[76-78] |
| Nipped-A |
HD, SCA3, SCA7 |
[42,54,55] |
TRRAP |
Trrap |
[79,80] |
| Sec61alpha |
HD, SCA3 |
[81] |
Sec61A1, Sec61A2 |
Sec61a1, Sec61a2 |
[82,83] |
| Nup160 |
HD, SCA3 |
[55,74] |
Nup160 |
NUP160 |
[84-86] |
| CG1109 |
HD |
[74] |
WDR33(WDC146) |
Wdr33 (Wdc146) |
N/A |
| Snap |
HD |
[66] |
NAPA, NAPB |
Napa, Napb |
N/A |
| smt3 |
HD |
[74] |
SUMO1, SUMO2, SUMO3, SUMO4 |
Sumo1, Sumo2, Sumo3 |
[87-91] |
| Mef2 |
HD |
[66] |
MEF2A, MEF2B, MEF2BNB, MEF2C, MEF2D |
Mef2a, Mef2b, Mef2c, Mef2d |
[92-97] |
| chic |
HD |
[98] |
PFN4 |
Pfn4 |
[99] |
| Rpt1 |
HD |
[74] |
PSMC2 |
Psmc2 |
[100-102] |
| Sin3A |
HD, SCA1, SCA3 |
[5,32,34] |
Sin3A, Sin3B |
Sin3a, Sin3b |
[103] |
| Rheb | ADTau, HD | [74,104] | Rheb, RhebL1 | Rheb, Rhebl1 | [105,106] |
1Orthologs were obtained from NeuroGeM in which protein homolog groups of NCBI HomoloGene and InParanoid were integrated.
2Disease models in which the genes were identified as modifiers.
3Studies in which the mammalian orthologs were identified as modifiers.
v) NeuroGeM allows assessing the effect of experimental conditions on the consistency and reliability of the identified modifiers. The results of different screens for genetic modifiers are often inconsistent because of the use of different experimental set-ups. NeuroGeM enables the user to infer the best experimental conditions for consistent identification of modifiers. As an example, we investigated the effect of polyQ stretch length on modifier identification in HD models of D. melanogaster. In Figure 4f, each line refers to one gene identified as a modifier or non-modifier in secondary HT or LT experiments in HD models with different polyQ lengths, and each green dot on the line refers to the identification result at a specific polyQ length. Figure 4f suggests some genes were not identified as modifiers in HD models with a polyQ length of 40 (which is above the canonical threshold of 35), but were then identified as modifier in models with a polyQ length of 60. Hence, this analysis suggests that HD models with polyQ > 60 may provide more sensitivity (see Additional file 1 for details).
vi) Most importantly, NeuroGeM facilitates the identification of new, so far untested modifiers. Mapping of genes on the protein interaction networks allows identification of new, untested genetic modifiers based on guilt-by-association. We illustrate this idea on genes involved in anti-apoptosis (GO:0006916). We first obtained 24 anti-apoptotic proteins in D. melanogaster (Figure 5a), and among these genes debcl, Buffy, and thread are interconnected with each other in the protein network (Figure 5b). In order to investigate whether genes interacting with these anti-apoptotic modifiers could also be modifiers, we extended the sub-network by adding proteins that interact with the three proteins. This extension can be easily done, as NeuroGeM allows the user to navigate from one gene to another by clicking on a node in a network. The newly added genes are highly interconnected with each other. Detailed literature surveys of the genes connected to debcl, Buffy, and thread revealed that 5 out of 15 interactors (marked in green in Figure 5b) are modifiers or at least highly related to disease progression. For example, one of the interactors is Ark; inactivation of Ark (FBgn0263864), a key regulator of apoptosis, is known to suppress formation and ubiquitination of polyQ aggregates [107] (Please see Additional file 1 for the details about the other four interactors and potential modifiers). Hence, the search tools of NeuroGeM will facilitate the identification of new modifiers that are involved in specific pathways or cellular processes.
Figure 5.

Ontology-based search results of “anti-apoptosis”. (a) Genes retrieved from NeuroGeM using the Ontology-based search for “anti-apoptosis” (GO:0006916). Genes annotated with anti-apoptosis or its child terms are listed. (b) Protein interaction clusters of the retrieved genes. Proteins that are directly interacting with the discovered modifiers are shown: red nodes, known modifiers; white nodes, untested genes; green nodes, genes with literature evidence (not yet entered into NeuroGeM) for being modifiers.
Conclusion
Here we report, to the best of our knowledge, the first database (NeuroGeM) of genetic modifiers of NDs. NeuroGeM provides a platform for searching modifiers, retrieving experimental conditions used for modifier identification, interpreting the roles of a queried modifier in the context of the protein interaction network, and expanding knowledge in one organism to other organisms through homologous genes. Therefore, NeuroGeM allows users to evaluate their hypotheses and develop new research directions. Furthermore, NeuroGeM provides all information, including gene information, protein interactions, experimental set-ups, etc. in down-loadable files, which will facilitate other computational analyses of modifiers similar to the meta-analysis presented in this work. Consequently, NeuroGeM will assist scientists immensely in their search for the pathophysiological mechanisms underlying NDs by providing the first compendium that catalogues and cross-links their genetic modifiers.
Availability and requirements
NeuroGeM can be accessed from a web browser and is available at http://chibi.ubc.ca/neurogem.
Abbreviations
ND: Neurodegenerative diseases; AD: Alzheimer’s disease; ADTau: Alzheimer’s disease caused by tau; ADAβ: Alzheimer’s disease caused by Aβ; ALS: Amyotrophic lateral sclerosis; PD: Parkinson’s disease; PolyQ: PolyQ disease; SCA1: Spinocerebellar ataxia type 1; SCA3: Spinocerebellar ataxia type 3; SCA7: Spinocerebellar ataxia type 7; HT: High-throughput; LT: Low-throughput; GO: GeneOntology.
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
DN built the database of genetic modifiers with integration of available databases and performed meta-analysis of genetic modifiers. DN, JG, CO, and DR designed the database and web site. MR implemented the web site to access the database of genetic modifiers. JG supervised the project. DN and JG wrote the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
A text with figures addressing the meta-analysis results in detail mentioned in the main text.
A figure to illustrate a categorical search.
A figure to illustrate an ontology-based search.
Contributor Information
Dokyun Na, Email: dkna@chibi.ubc.ca.
Mushfiqur Rouf, Email: nasarouf@cs.ubc.ca.
Cahir J O’Kane, Email: c.okane@gen.cam.ac.uk.
David C Rubinsztein, Email: dcr1000@hermes.cam.ac.uk.
Jörg Gsponer, Email: gsponer@chibi.ubc.ca.
Acknowledgement
Jörg Gsponer, David C. Rubinsztein and Cahir J. O’Kane were supported by the Centres of Excellence in Neurodegeneration Research (CoEN). Jörg Gsponer was supported by the Canadian Institutes of Health Research (CIHR), and David C. Rubinsztein was supported by the Medical Research Council (MRC) and a Wellcome Trust Principal Fellowship.
References
- Marsh JL, Thompson LM. Drosophila in the study of neurodegenerative disease. Neuron. 2006;52:169–178. doi: 10.1016/j.neuron.2006.09.025. [DOI] [PubMed] [Google Scholar]
- Mallik M, Lakhotia SC. Modifiers and mechanisms of multi-system polyglutamine neurodegenerative disorders: lessons from fly models. J Genet. 2010;89:497–526. doi: 10.1007/s12041-010-0072-4. [DOI] [PubMed] [Google Scholar]
- Lu B, Vogel H. Drosophila models of neurodegenerative diseases. Annu Rev Pathol. 2009;4:315–342. doi: 10.1146/annurev.pathol.3.121806.151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nollen EAA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, Plasterk RHA. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci USA. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 2007;3:e177. doi: 10.1371/journal.pgen.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FlyBase. http://flybase.org/
- WormBase. http://www.wormbase.org/
- SGD. http://www.yeastgenome.org/
- Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(Suppl 1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang H-Y, Lee E, Liu Y-T, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol Syst Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRING. http://string-db.org.
- GeneOntology. http://www.geneontology.org/
- NCBI HomoloGene. http://www.ncbi.nlm.nih.gov/homologene/
- Ostlund G, Schmitt T, Forslund K, Köstler T, Messina DN, Roopra S, Frings O, Sonnhammer ELL. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010;38:D196–D203. doi: 10.1093/nar/gkp931. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- InParanoid. http://inparanoid.sbc.su.se/cgi-bin/index.cgi.
- EMBL (Human) ftp://ftp.ensembl.org/pub/release-68/embl/homo_sapiens/
- HGNC. http://www.genenames.org/
- EMBL (Mouse) ftp://ftp.ensembl.org/pub/release-68/embl/mus_musculus/
- MGI. http://www.informatics.jax.org/
- CytoscapeWeb. http://cytoscapeweb.cytoscape.org/
- Shulman JM, Feany MB. Genetic modifiers of tauopathy in Drosophila. Genetics. 2003;165:1233–1242. doi: 10.1093/genetics/165.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick JM, Chan HYE, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- Harris GM, Dodelzon K, Gong L, Gonzalez-Alegre P, Paulson HL. Splice isoforms of the polyglutamine disease protein Ataxin-3 exhibit similar enzymatic yet different aggregation properties. PLoS One. 2010;5:e13695. doi: 10.1371/journal.pone.0013695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, De Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rüb U, Auburger G, Trojanowski JQ, Lee VM-Y, Deerlin VMV, Bonini NM, Gitler AD. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–682. doi: 10.1016/S0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- Folwell J, Cowan CM, Ubhi KK, Shiabh H, Newman TA, Shepherd D, Mudher A. Aβ exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of Alzheimer’s disease. Exp Neurol. 2010;223:401–409. doi: 10.1016/j.expneurol.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Blard O, Feuillette S, Bou J, Chaumette B, Frébourg T, Campion D, Lecourtois M. Cytoskeleton proteins are modulators of mutant tau-induced neurodegeneration in Drosophila. Hum Mol Genet. 2007;16:555–566. doi: 10.1093/hmg/ddm011. [DOI] [PubMed] [Google Scholar]
- Chan HYE, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet. 2000;9:2811–2820. doi: 10.1093/hmg/9.19.2811. [DOI] [PubMed] [Google Scholar]
- Branco J, Al-Ramahi I, Ukani L, Pérez AM, Fernandez-Funez P, Rincón-Limas D, Botas J. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum Mol Genet. 2008;17:376–390. doi: 10.1093/hmg/ddm315. [DOI] [PubMed] [Google Scholar]
- Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–1840. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She W-C, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- Petrakis S, Raskó T, Russ J, Friedrich RP, Stroedicke M, Riechers S-P, Muehlenberg K, Möller A, Reinhardt A, Vinayagam A, Schaefer MH, Boutros M, Tricoire H, Andrade-Navarro MA, Wanker EE. Identification of human proteins that modify misfolding and proteotoxicity of pathogenic Ataxin-1. PLoS Genet. 2012;8:e1002897. doi: 10.1371/journal.pgen.1002897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Bonini NM, Paulson HL. Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J Neurosci. 1999;19:10338–10347. doi: 10.1523/JNEUROSCI.19-23-10338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem. 2000;275:8772–8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- Jana NR, Tanaka M, Wang G, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- Long P, Samnakay P, Jenner P, Rose S. A yeast two-hybrid screen reveals that osteopontin associates with MAP1A and MAP1B in addition to other proteins linked to microtubule stability, apoptosis and protein degradation in the human brain. Eur J Neurosci. 2012;36:2733–2742. doi: 10.1111/j.1460-9568.2012.08189.x. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Feany MB. Comparison of pathways controlling toxicity in the eye and brain in Drosophila models of human neurodegenerative diseases. Hum Mol Genet. 2004;13:2011–2018. doi: 10.1093/hmg/ddh214. [DOI] [PubMed] [Google Scholar]
- Latouche M, Lasbleiz C, Martin E, Monnier V, Debeir T, Mouatt-Prigent A, Muriel M-P, Morel L, Ruberg M, Brice A, Stevanin G, Tricoire H. A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J Neurosci. 2007;27:2483–2492. doi: 10.1523/JNEUROSCI.5453-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugazhenthi S, Wang M, Pham S, Sze C-I, Eckman CB. Downregulation of CREB expression in Alzheimer’s brain and in Aβ-treated rat hippocampal neurons. Mol Neurodegener. 2011;6:60. doi: 10.1186/1750-1326-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio A, Boroni F, Benarese M, Sarnico I, Ghisi V, Bresciani LG, Ferrario M, Borsani G, Spano P, Pizzi M. NF-κB pathway: a target for preventing β-amyloid (Aβ)-induced neuronal damage and Aβ42 production. Eur J Neurosci. 2006;23:1711–1720. doi: 10.1111/j.1460-9568.2006.04722.x. [DOI] [PubMed] [Google Scholar]
- Knight JC, Scharf EL, Mao-Draayer Y. Fas activation increases neural progenitor cell survival. J Neurosci Res. 2010;88:746–757. doi: 10.1002/jnr.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ramahi I, Pérez AM, Lim J, Zhang M, Sorensen R, de Haro M, Branco J, Pulst SM, Zoghbi HY, Botas J. dAtaxin-2 mediates expanded ataxin-1-induced neurodegeneration in a Drosophila model of SCA1. PLoS Genet. 2007;3:e234. doi: 10.1371/journal.pgen.0030234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasumu AW, Hougaard C, Rode F, Jacobsen TA, Sabatier JM, Eriksen BL, Strøbæk D, Liang X, Egorova P, Vorontsova D, Christophersen P, Rønn LCB, Bezprozvanny I. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of Spinocerebellar Ataxia Type 2. Chem Biol. 2012;19:1340–1353. doi: 10.1016/j.chembiol.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damrath E, Heck MV, Gispert S, Azizov M, Nowock J, Seifried C, Rüb U, Walter M, Auburger G. ATXN2-CAG42 sequesters PABPC1 into insolubility and induces FBXW8 in cerebellum of old ataxic knock-in mice. PLoS Genet. 2012;8:e1002920. doi: 10.1371/journal.pgen.1002920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PWJ, Thijs V, Dubois B, Matthijs G, van den Berg LH, Robberecht W. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2 . Neurology. 2011;76:2066–2072. doi: 10.1212/WNL.0b013e31821f445b. [DOI] [PubMed] [Google Scholar]
- Nielsen TT, Svenstrup K, Budtz-Jørgensen E, Eiberg H, Hasholt L, Nielsen JE. ATXN2 with intermediate-length CAG/CAA repeats does not seem to be a risk factor in hereditary spastic paraplegia. J Neurol Sci. 2012;321:100–102. doi: 10.1016/j.jns.2012.07.036. [DOI] [PubMed] [Google Scholar]
- Lee T, Li YR, Ingre C, Weber M, Grehl T, Gredal O, Carvalho de M, Meyer T, Tysnes O-B O-B, Auburger G, Gispert S, Bonini NM, Andersen PM, Gitler AD. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum Mol Genet. 2011;20:1697–1700. doi: 10.1093/hmg/ddr045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross OA, Rutherford NJ, Baker M, Soto-Ortolaza AI, Carrasquillo MM, DeJesus-Hernandez M, Adamson J, Li M, Volkening K, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, Lippa C, Woodruff BK, Knopman DS, White CL, Gerpen JAV, Meschia JF, Mackenzie IR, Boylan K, Boeve BF, Miller BL, Strong MJ, Uitti RJ, Younkin SG, Graff-Radford NR, Petersen RC. et al. Ataxin-2 repeat-length variation and neurodegeneration. Hum Mol Genet. 2011;20:3207–3212. doi: 10.1093/hmg/ddr227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Ling JJ, Yang S, Li X-J, Li S. Neuronal expression of TATA box-binding protein containing expanded polyglutamine in knock-in mice reduces chaperone protein response by impairing the function of nuclear factor-Y transcription factor. Brain. 2011;134(Pt 7):1943–1958. doi: 10.1093/brain/awr146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Binari R, Zhou R, Perrimon N. A genomewide RNA interference screen for modifiers of aggregates formation by mutant huntingtin in Drosophila. Genetics. 2010;184:1165–1179. doi: 10.1534/genetics.109.112516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voßfeldt H, Butzlaff M, Prüßing K, Ní Chárthaigh R-A, Karsten P, Lankes A, Hamm S, Simons M, Adryan B, Schulz JB, Voigt A. Large-scale screen for modifiers of Ataxin-3-derived polyglutamine-induced toxicity in Drosophila. PLoS One. 2012;7:e47452. doi: 10.1371/journal.pone.0047452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K, Yamagishi N, Saito Y, Adachi H, Kobayashi Y, Sobue G, Ohtsuka K, Hatayama T. Hsp105α suppresses the aggregation of truncated androgen receptor with expanded CAG repeats and cell toxicity. J Biol Chem. 2003;278:25143–25150. doi: 10.1074/jbc.M302975200. [DOI] [PubMed] [Google Scholar]
- Eroglu B, Moskophidis D, Mivechi NF. Loss of Hsp110 leads to age-dependent tau hyperphosphorylation and early accumulation of insoluble amyloid beta. Mol Cell Biol. 2010;30:4626–4643. doi: 10.1128/MCB.01493-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM, Marsh JL. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum Mol Genet. 2008;17:3767–3775. doi: 10.1093/hmg/ddn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinti L, Chopra V, Rotili D, Valente S, Amore A, Franci G, Meade S, Valenza M, Altucci L, Maxwell MM, Cattaneo E, Hersch S, Mai A, Kazantsev A. Evaluation of histone deacetylases as drug targets in Huntington’s disease models . PLoS Curr. 2010. Sep 2. doi: 10.1371/currents.RRN1172. [DOI] [PMC free article] [PubMed]
- Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, Geschwind DH, Gottesfeld JM. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci USA. 2008;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PAS, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci USA. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. 2008;283:35402–35409. doi: 10.1074/jbc.M807045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, H. Brown R, Ferrante RJ. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2009;35:870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, Cha G-H, Ukani L, Chepanoske CL, Zhen Y, Sahasrabudhe S, Olson J, Kurschner C, Ellerby LM, Peltier JM, Botas J, Hughes RE. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-K, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EMC, Orr HT, Botas J, Zoghbi HY. Interaction of Akt-phosphorylated Ataxin-1 with 14-3-3 mediates neurodegeneration in Spinocerebellar ataxia type 1. Cell. 2003;113:457–468. doi: 10.1016/S0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- Kaneko K, Hachiya NS. The alternative role of 14-3-3 zeta as a sweeper of misfolded proteins in disease conditions. Med Hypotheses. 2006;67:169–171. doi: 10.1016/j.mehy.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Shirakashi Y, Ihara M, Urushitani M, Oono M, Kawamoto Y, Yamashita H, Shimohama S, Kato S, Hirano A, Tomimoto H, Ito H, Takahashi R. Colocalization of 14-3-3 proteins with SOD1 in Lewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis cases and the animal model. PLoS One. 2011;6:e20427. doi: 10.1371/journal.pone.0020427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiguchi M, Sobue K, Paudel HK. 14-3-3ζ is an effector of tau protein phosphorylation. J Biol Chem. 2000;275:25247–25254. doi: 10.1074/jbc.M003738200. [DOI] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Bai X, Chen Y, Zhao Y, Liu X. Homocysteine induces apoptosis of rat hippocampal neurons by inhibiting 14-3-3ϵ expression and activating calcineurin. PLoS One. 2012;7:e48247. doi: 10.1371/journal.pone.0048247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omi K, Hachiya NS, Tanaka M, Tokunaga K, Kaneko K. 14-3-3zeta is indispensable for aggregate formation of polyglutamine-expanded huntingtin protein. Neurosci Lett. 2008;431:45–50. doi: 10.1016/j.neulet.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Doumanis J, Wada K, Kino Y, Moore AW, Nukina N. RNAi screening in Drosophila cells identifies new modifiers of mutant huntingtin aggregation. PLoS One. 2009;4:e7275. doi: 10.1371/journal.pone.0007275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem. 2008;283:26188–26197. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkawa T, Tan K, Fujimoto M, Hayashida N, Yamamoto K, Takaki E, Takii R, Prakasam R, Inouye S, Mezger V, Nakai A. Heat shock factor 2 is required for maintaining proteostasis against febrile-range thermal stress and polyglutamine aggregation. Mol Biol Cell. 2011;22:3571–3583. doi: 10.1091/mbc.E11-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batulan Z, Shinder GA, Minotti S, He BP, Doroudchi MM, Nalbantoglu J, Strong MJ, Durham HD. High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci. 2003;23:5789–5798. doi: 10.1523/JNEUROSCI.23-13-05789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma S, Jin X, Wang G, Tu N, Min J, Yanasak N, Mivechi NF. Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in hsf1-deficient mice. J Neurosci. 2007;27:7974–7986. doi: 10.1523/JNEUROSCI.0006-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookerjee S, Papanikolaou T, Guyenet SJ, Sampath V, Lin A, Vitelli C, DeGiacomo F, Sopher BL, Chen SF, Spada ARL, Ellerby LM. Posttranslational modification of Ataxin-7 at lysine 257 prevents autophagy-mediated turnover of an N-Terminal caspase-7 cleavage fragment. J Neurosci. 2009;29:15134–15144. doi: 10.1523/JNEUROSCI.4720-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, Gansmüller A, Picaud S, Zoghbi HY, Trottier Y, Tora L, Devys D. Glutamine-expanded Ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006;4:e67. doi: 10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanuka H, Kuranaga E, Hiratou T, Igaki T, Nelson B, Okano H, Miura M. Cytosol-endoplasmic reticulum interplay by Sec61α translocon in polyglutamine-mediated neurotoxicity in Drosophila. Proc Natl Acad Sci. 2003;100:11723–11728. doi: 10.1073/pnas.1934748100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li L, Ye Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J Cell Biol. 2006;174:963–971. doi: 10.1083/jcb.200605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, Toyama BH, Xu T, Yates JR, Hetzer MW. Extremely long-lived nuclear pore proteins in the rat brain. Science. 2012;335:942–942. doi: 10.1126/science.1217421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284–295. doi: 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Hetzer MW. Protein homeostasis: live long, won’t prosper. Nat Rev Mol Cell Biol. 2013;14:55–61. doi: 10.1038/nrm3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janer A, Werner A, Takahashi-Fujigasaki J, Daret A, Fujigasaki H, Takada K, Duyckaerts C, Brice A, Dejean A, Sittler A. SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum Mol Genet. 2010;19:181–195. doi: 10.1093/hmg/ddp478. [DOI] [PubMed] [Google Scholar]
- Poukka H, Karvonen U, Jänne OA, Palvimo JJ. Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (SUMO-1) Proc Natl Acad Sci USA. 2000;97:14145–14150. doi: 10.1073/pnas.97.26.14145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BE, Zoghbi HY, Orr HT. SUMOylation of the polyglutamine repeat protein, Ataxin-1, is dependent on a functional nuclear localization signal. J Biol Chem. 2005;280:21942–21948. doi: 10.1074/jbc.M501677200. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Fishman PS, Thakor NV, Oyler GA. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem. 2003;278:22044–22055. doi: 10.1074/jbc.M212235200. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron. 1999;24:879–892. doi: 10.1016/S0896-6273(00)81035-1. [DOI] [PubMed] [Google Scholar]
- She H, Yang Q, Mao Z. Neurotoxin-induced selective ubiquitination and regulation of MEF2A isoform in neuronal stress response. J Neurochem. 2012;122:1203–1210. doi: 10.1111/j.1471-4159.2012.07860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Mickiewicz AL, Kordower j. α-synuclein aggregation reduces nigral myocyte enhancer Factor-2D in idiopathic and experimental Parkinson’s disease. Neurobiol Dis. 2011;41:71–82. doi: 10.1016/j.nbd.2010.08.022. [DOI] [PubMed] [Google Scholar]
- She H, Mao Z. Regulation of myocyte enhancer factor-2 transcription factors by neurotoxins. Neurotoxicology. 2011;32:563–566. doi: 10.1016/j.neuro.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton TR, Dibrov A, Kashour T, Amara FM. Anti-apoptotic wild-type Alzheimer amyloid precursor protein signaling involves the p38 mitogen-activated protein kinase/MEF2 pathway. Brain Res Mol Brain Res. 2002;108:102–120. doi: 10.1016/S0169-328X(02)00519-3. [DOI] [PubMed] [Google Scholar]
- González P, Álvarez V, Menéndez M, Lahoz CH, Martínez C, Corao AI, Calatayud MT, Peña J, García-Castro M, Coto E. Myocyte enhancing factor-2A in Alzheimer’s disease: genetic analysis and association with MEF2A-polymorphisms. Neurosci Lett. 2007;411:47–51. doi: 10.1016/j.neulet.2006.09.055. [DOI] [PubMed] [Google Scholar]
- Salma J, McDermott JC. Suppression of a MEF2-KLF6 survival pathway by PKA signaling promotes apoptosis in embryonic hippocampal neurons. J Neurosci. 2012;32:2790–2803. doi: 10.1523/JNEUROSCI.3609-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett BG, Andrews J, Ranganathan S, Fischbeck KH, Di Prospero NA. Expression of expanded polyglutamine targets profilin for degradation and alters actin dynamics. Neurobiol Dis. 2008;30:365–374. doi: 10.1016/j.nbd.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso M, Giraudo S, Corpillo D, Bergamasco B, Lopiano L, Fasano M. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteomics. 2004;4:3943–3952. doi: 10.1002/pmic.200400848. [DOI] [PubMed] [Google Scholar]
- Shim SM, Lee WJ, Kim Y, Chang JW, Song S, Jung Y-K. Role of S5b/PSMD5 in proteasome inhibition caused by TNF-α/NFκB in higher eukaryotes. Cell Rep. 2012;2:603–615. doi: 10.1016/j.celrep.2012.07.013. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/S0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]
- Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol. 2006;16:230–241. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Cao M, Tan X, Jin W, Zheng H, Xu W, Rui Y, Li L, Cao J, Wu X, Cui G, Ke K, Gao Y. Upregulation of Ras homolog enriched in the brain (Rheb) in lipopolysaccharide-induced neuroinflammation. Neurochem Int. 2013;62:406–417. doi: 10.1016/j.neuint.2013.01.025. [DOI] [PubMed] [Google Scholar]
- Cheng H-C, Kim SR, Oo TF, Kareva T, Yarygina O, Rzhetskaya M, Wang C, During M, Talloczy Z, Tanaka K, Komatsu M, Kobayashi K, Okano H, Kholodilov N, Burke RE. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci. 2011;31:2125–2135. doi: 10.1523/JNEUROSCI.5519-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang T-K, Li C, Liu W, Rodriguez A, Abrams JM, Zipursky SL, Jackson GR. Inactivation of Drosophila Apaf-1 related killer suppresses formation of polyglutamine aggregates and blocks polyglutamine pathogenesis. Hum Mol Genet. 2005;14:357–372. doi: 10.1093/hmg/ddi032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A text with figures addressing the meta-analysis results in detail mentioned in the main text.
A figure to illustrate a categorical search.
A figure to illustrate an ontology-based search.