

Abstract
Peptide drugs are an exciting class of pharmaceuticals for the treatment of a variety of diseases; however, their short half-life dictates multiple and frequent injections causing undesirable side-effects. Herein, we describe a novel peptide delivery system that seeks to combine the attractive features of prolonged circulation time with a prolonged release formulation. This system consists of glucagon-like peptide-1, a type-2 diabetes drug fused to a thermally responsive, elastin-like-polypeptide (ELP) that undergoes a soluble-insoluble phase transition between room temperature and body temperature, thereby forming an injectable depot. We synthesized a set of GLP-1-ELP fusions and verified their proteolytic stability and potency in vitro. Significantly, a single injection of depot forming GLP-1-ELP fusions reduced blood glucose levels in mice for up to 5 days, 120 times longer than an injection of the native peptide. These findings demonstrate the unique advantages of using ELPs to release peptide-ELP fusions from a depot combined with enhanced systemic circulation to create a tunable peptide delivery system.
Keywords: Peptide, Drug delivery, Elastin-like polypeptide, Glucagon-like peptide-1, subcutaneous depot
1. Introduction
Type-2 diabetes accounts for 90 to 95 percent of all diagnosed cases of adult diabetes, and has been steadily increasing over the past few decades, with an estimated global prevalence of 300 million by 2025 [1]. Glucagon-like peptide-1 (GLP-1) is a 30 amino acid peptide released by gastrointestinal cells upon meal ingestion [2], and stimulates the release of insulin from pancreatic β-cells [3–5]. In addition, GLP-1 has also been shown to inhibit glucagon secretion, reduce appetite, slow gastric emptying, reduce hepatic glucose production [6, 7] and enhance β-cell survival and growth in rodents [8]. Despite these attractive features, native GLP-1 has not been utilized for treatment of type-2 diabetes, as the peptide undergoes rapid deactivation in vivo via N-terminal truncation by dipeptidyl-peptidase IV (DPPIV), leading to a half-life of less than 2 minutes [9]. To circumvent this problem, DPPIV resistant GLP-1 analogs have been designed, but due to the small size of the peptide, renal clearance still limits their half-life to 4–5 min [9].
Significant effort has hence been expended to develop longer acting analogs of GLP-1 with the goal of generating a drug that will, with a single injection, provide a flat concentration versus time profile in the therapeutic range [10]. This will not only reduce the number of injections but also prevent “peak-and-valley” fluctuations associated with bolus injections of short-acting analogs that only provide a therapeutically effective drug concentration for a short time and cause undesired side effects because of their sub-optimal pharmacokinetics [11].
Currently, there are three FDA approved GLP-1R agonists to treat type 2 diabetes. Exenatide, a 39 amino acid peptide with 53% homology to GLP-1, which has been shown to be protease resistant with a modestly increased half-life compared with GLP-1 (2.4 h after s.c. injection in humans [12]), is administered via twice daily injections. Liraglutide is an acylated GLP-1 approved for once-a-day injection (11–15 h half-life after s.c. injection in humans [12]) and Bydureon, a sustained release formulation of exenatide in biodegradable poly(lactic glycolic acid) (PLGA) microspheres, is approved for once-weekly injection [13]. In addition, several groups have proposed the attachment of GLP-1 or exenatide to large carrier molecules such as PEG [9], serum albumin [14], Fc fragments [15] and other recombinant biopolymers [16] as a strategy to increase their in vivo half-life. However, recombinant fusion to Fc fragments or serum albumin requires eukaryotic expression systems that are expensive and difficult to scale-up, while site-specific covalent PEGylation has a low yield and high cost [17]. Similarly, a recent paper describes the generation of a recombinant exenatide-xten fusion wherein the xten moiety is a large recombinant polypeptide engineered to have an unstructured conformation that extends the half-life of exenatide from 0.17 h to 12 h in mice [16]. Nevertheless, to the best of our knowledge, to date GLP-1 delivery systems have been designed for either prolonged release from a drug depot [13, 18] or prolonged circulation.
Herein we describe the design and characterization of an alternative system that seeks to combine the attractive features of a soluble fusion protein, with prolonged circulation time, and a prolonged release formulation, that allows for slow released from a s.c. depot. This system consists of a (GLP-1)-ELP fusion protein in which GLP-1 is fused to a thermally sensitive elastin-like polypeptide (ELP) that is soluble at room temperature but forms a depot – a viscous coacervate – upon subcutaneous (s.c.) injection at body temperature [19]. The central hypothesis that underlies this design is that the ELP domain will drive a thermal transition upon s.c. injection of (GLP-1)-ELP fusions, thereby forming a stable drug depot. Over time, active (GLP-1)-ELP fusion will be released from the depot, providing prolonged release of high MW drug into circulation – which will also enhance the peptide’s half-life. This design combines the ability to titrate the release of the drug from a depot, similar to degradable microsphere delivery systems, with the tunable half-life extension of macromolecular GLP-fusions.
We show that (GLP-1)-ELP fusions can be expressed in E. coli, and that ELP fusion tags enable temperature triggered coacervation that is easily tuned by choice of the ELP fusion protein which also assists in facile, chromatography-free, purification. (GLP-1)-ELP fusions maintain secondary structure, are resistant to degradation by proteolysis, and are capable of activating the GLP-1 receptor. We demonstrate that a single injection of (GLP-1)-ELP forms a s.c. depot that provides superior glucose control as compared with native peptide injections. Finally, we discuss future work and the potential of this fusion technology for the delivery of other peptide therapeutics.
2. Materials and methods
2.1 Recombinant (GLP-1)-ELP synthesis and expression
The gene of modified [G8E22A36]GLP (Table 1) with an alanine-alanine leader, and 5′ NdeI and 3′ HindIII restriction sites was amplified using a [G8E22]GLP synthetic gene template [18] and F primer: “GATATACATATGGCAGCGCACGGTGAAGGCACCTTT” and R primer: “CATAGGAAGCTTGCCCGCACCTTTCACCAGCCACGCGAT”. The PCR reaction mixture consisted of 10 pmol of [G8E22A36]GLP template, 10 pmol each of sense and antisense DNA primers, 50 μL GoTaq green master mix (Promega) and water for a final volume of 100 μL. The PCR reaction conditions were 95 °C for 2 min for initial denaturation, followed by 35 cycles at 95 °C for 30 s, 52 °C for 30 s and 72 °C for 40 s. The resulting PCR product was purified using a PCR purification kit (Qiagen) and visualized on a 1% agarose gel stained with SYBR® Safe DNA stain (Invitrogen). 2 μg PCR products was digested with 2 μl NdeI, 2 μl Hind III, 1x NEB 2 buffer and nuclease free water in total volume of 40 μl at 37 °C for 2 h and PCR purified to remove residual enzymes and buffer. A modified pET25b+ vector containing the ELP tag was modified to include a leader cassette with NdeI and HindIII restriction sites for in-frame ligation of GLP-1 to the ELP. 2 μg vector DNA was digested with 2 μl NdeI, 2 μl Hind III, 1x NEB 2 buffer and nuclease free water in total volume of 40 μl at 37 °C for 2 h and PCR purified to remove residual enzymes and buffer. The restriction product was ligated to the vector using 4 units of T4 DNA ligase, 1X ligation buffer, ~250 ng of the annealed product, ~250 ng of digested vector, and nuclease-free water in a total volume of 20 μl. The ligation mixture was incubated at room temperature for 1 h, and BL21(DE3) cells were then transformed with 7 μl of the ligation mixture for 15 min in an ice-water bath, heat-shocked at 42 °C for 30 s, and returned to the ice-water mixture for another 2 min. The cells were recovered in SOC media while horizontally shaking at 200 rpm at 37 °C for 40–60 min, and were then plated on TB agarose plates containing 1 mg/mL ampicillin. Correct clones were identified by colony PCR and verified by DNA sequencing. GLP-ELP constructs were expressed and purified using a modified Inverse Transition Cycling (ITC) protocol [20] detailed in the Supplementary Information section.
Table 1.
Sequences used for generation of (GLP-1)-ELP fusion proteins. By convention, the first amino acid in the GLP-1 sequence (Histidine (H)) is numbered 7. Mutations in native GLP-1 sequence are underlined. The Alanine (A) to Glycine (G) mutation in the 8th position confers DPPIV resistance, The Glycine (G) to Glutamic acid (E) mutation in the 22nd position stabilizes the alpha helical structure of GLP-1 and the Arginine (R) to Alanine (A) mutation in the 36th position prevents cleavage by Arginine-targeting proteases in the s.c. space. ELPs were chosen to have a Tt that is lower (ELPDepot) or higher (ELPSol) than body temperature which was modulated by the choice of guest residue composition. More hydrophobic amino acid residues (e.g., Valine) reduce the Tt whereas more hydrophilic amino acids (Alanine and Glycine) increase it. A DPPIV cleavable Alanine-Alanine (AA) dipeptide was added to the N-terminus of each construct to facilitate removal of any extra Methionine residues and the linker between the GLP and ELP moieties was dictated by the choice of restriction endonucleases in the assembly of the fusion gene.
| Native GLP-1 | HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR |
| [G8E22A36]GLP-1 | AAHGEGTFTSDVSSYLEEQAAKEFIAWLVKGA |
| ELPSol | (GAGVPGGGVP)60GY |
| ELPDepot | (GVGVP)120GWP |
| Leader | AA |
| Linker | KLAM |
2.2 Phase transition and secondary structure analysis
The secondary structure was studied by circular dichroism (CD) using an Aviv Model 202 instrument and 1 mm quartz cells (Hellma) by scanning from 280 nm to 180 nm with 1 nm steps and a 3 second averaging time at 19 °C. Purified constructs were diluted to 7.5 μM in water. Data were considered for analysis whenever the Dynode voltage was below 500 V [21].
To characterize the inverse transition temperature of (GLP-1)-ELP constructs, the optical density of a 200 μM – 10 μM solution in PBS was monitored at a wavelength of 350 nm as a function of temperature, with heating and cooling performed at a rate of 1 °C min-1 on a Cary 300 UV-visible spectrophotometer equipped with a multicell thermoelectric temperature controller (Varian Instruments, Walnut Creek, CA).
2.3 Degradation by Neutral Endopeptidase (NEP)
(GLP-1)-ELPDepot and native GLP-1 (30 μM) were incubated at 20 or 37 °C for up to 18 h with 0.46 μg NEP protease (Enzo). Following incubation, fractions were separated on a 10–20% tris-tricine SDS gel (Bio-Rad) and stained with Coomassie brilliant blue.
2.4 In vitro assay for GLP-1 activity
The ability of (GLP-1)-ELPDepot and (GLP-1)-ELPSol to activate the GLP-1 receptor (GLP-1R) in vitro after incubation for 20 h with 0.5 μg DPPIV (ProSpec) at 20 or 37 °C to remove the AA leader was assessed using Baby Hamster Kidney (BHK) cells that are stably transfected with rat GLP-1R (a gift of Prof. Drucker, University of Toronto [22]). Intracellular cAMP concentrations were measured using a competitive binding assay according to the manufacturer’s instructions (Assay Designs).
2.5 Animal Studies
5–6 week old male C57BL/6J mice [23] (stock number 000664) were purchased from Jackson Laboratories (Bar Harbor, Me., USA). All experimental procedures were approved by the Duke Institutional Animal Care & Use Committee
2.6 In vivo near infra-red fluorescence tomography
For in vivo tomography of depot formation, (GLP-1)-ELPDepot and (GLP-1)-ELPSol were labeled with IRDye® 800CW NHS Ester (LICOR) by conjugation to lysine residues in GLP-1. All mice were anesthetized with 2% isoflurane throughout the imaging procedures. The torsos of mice (7 weeks old, n=3 per group) were shaved, and labeled constructs were injected s.c. (175 or 700 nmol/kg). Images were collected immediately after injection and at 5, 24, 48, 72, 96, 120 and 144 h post-injection. Imaging was performed with an FMT2500LX fluorescence tomography in vivo imaging system (PerkinElmer) and the images were acquired and analyzed with TrueQuant 3D software (PerkinElmer). When depicted in the same panel, all image intensities were set to the maximum intensity scale of the first (0 h) image.
2.7 Fed blood glucose measurements
The effect of (GLP-1)-ELPDepot and (GLP-1)-ELPSol on fed blood glucose levels was measured following a single s.c. injection. Before blood glucose measurement, the tail was wiped with a sterilizing alcohol solution, and wiped dry. A tiny incision of the mouse tail vain was made and the first 1 μl drop of blood was wiped. The second 1–2 μl blood drop was measured via the glucose oxidase test, using a hand-held glucometer (AlphaTrack, Abbott, set to code 7). Blood glucose levels were measured at 3 and 1 days prior to initiating the experiment. On the day of injection, animals were weighed, blood glucose was measured and (GLP-1)-ELPDepot solution (14, 7 and 3.5 nmol/mouse, or 700, 350 and 175 nmol/kg at 3.5 μl/g, 13–17 °C) or PBS (n=7–11) was injected s.c. Immediately following injection, mice were placed back in the cage, with free access to food and water and blood glucose was measured at 3, 6, 24, 48, 72, 96, 120, 144 and 168 h after the injection. Blood glucose levels were normalized by the average glucose levels taken on the days before and immediately prior to injection to reflect the percent change in glucose levels and to correct for transient variations in glucose.
2.8 Intraperitoneal Glucose Tolerance Test (IPGTT)
At the beginning of the experiment, mice were randomly divided into 4 groups (n=5) based on two previous measurements of blood glucose. At 9 am on the first day, all mice were injected s.c. with (GLP-1)-ELPDepot (700 nmol/kg) or PBS. In the morning of the first IPGTT experiment (9 AM, 48 h following injection), 2 groups of mice (injected with either (GLP-1)-ELPDepot or PBS control) were fasted by placement in a fresh cage and removal of food for 5 h. At the end of the fast period (52 h following injection), mice were given 1 g glucose/kg (10% sterile glucose solution, Sigma) via intraperitoneal injection. Blood was drawn from the tail vein and glucose levels were measured using a glucometer (Alpha-Track, Abbott) at 0, 20, 40, 60, 90 and 120 min after glucose administration. In the morning of the second IPGTT experiment (9 AM, 97 h following injection) the 2 remaining groups of mice (injected with either (GLP-1)-ELPDepot or PBS control) were subjected to the same protocol and an IPGTT was similarly performed (102 h following injection).
2.8 Data Analysis
Data are presented as means and standard errors. Treatment effects on fed glucose levels were first analyzed using repeated measures ANOVA for all groups. When the initial analysis indicated a significant difference, lower-order ANOVAs were conducted to determine main effects of each treatment, followed by post-hoc evaluations of individual differences at each time point for treatment and PBS using Fisher’s Protected Least Significant Difference. For AUC of IPGTT evaluation, treatment and PBS were compared using a one-tailed heteroschedastic t-test.
3. Results
3.1 Design of (GLP-1)-ELP fusion protein
The wild-type (WT) GLP-1 peptide, comprised of amino acids 7–37 of the pro-GLP-1 sequences [24] (by convention the first residue is labeled residue 7), was mutated at three residues to create a DPPIV resistant, helically stabilized GLP-1 analog. This GLP-1 mutant, named [G8E22A36]GLP-1 (Table 1), contains an Alanine (A) to Glycine (G) substitution at residue 8 that confers resistance to DPPIV deactivation, a Glycine (G) to Glutamic Acid (E) substitution at residue 22 that stabilizes the α-helix [9], and a C-terminal Arginine (R) to Alanine (A) substitution at residue 30 that prevents s.c. cleavage of the GLP-1 from the ELP fusion by s.c. proteases [18]. In addition, a dipeptide (Alanine-Alanine, AA) was added to the N-terminus of the fusion protein to ensure efficient removal of the extraneous first methionine (Met) residue [25] and is designed to be cleaved in vivo by DPPIV (Figure 1A). Ensuring removal of the extraneous Met is essential, as the N-terminal sequence of GLP-1 is crucial for receptor binding and activation [26].
Figure 1.

Design and characterization of (GLP-1)-ELP depots. (A) Modified amino acid sequence of GLP-1: Amino acids highlighted in red are essential for GLP-1 binding; modified amino acids are highlighted in blue (modified after [33]), arrow marks site of activation by DPPIV. (B) ITC purification of (GLP-1)-ELPDepot: lanes (1) sonicated cell lysate, (2) soluble cell lysate, (3) re-suspended cold spin pellet, (4) purified (GLP-1)-ELPDepot construct after 2 ITC cycles. (C) Transition temperature of (GLP-1)-ELPDepot and (GLP-1)-ELPSol. (D) CD spectra of monomer native GLP-1, ELPDepot and (GLP-1)-ELPDepot (all at 7.5 μM).
We chose ELPs as the depot forming segment of the fusion because they display a tunable, predictable thermally responsive behavior. ELPs are a family of peptide polymers derived from a recurring VPGVG pentapeptide that is found in tropoelastin [27]. They are soluble in aqueous solution below their inverse transition temperature (Tt), but undergo a sharp (~2 °C range) phase transition when the temperature is raised above their Tt, leading to the formation of an ELP-rich coacervate phase. To modulate the thermally responsive behavior of (GLP-1)-ELP, two ELPs were designed by exploiting the two molecular parameters that control their phase transition behavior – their composition and molecular weight. In our design, we fixed the ELP MW at 50 kDa, because we have shown in previous studies that ELPs with this MW show a reasonably long half-life of 8–12 h in circulation, but are still cleared by renal filtration. Having fixed the MW, we designed two ELPs with the appropriate phase transition behavior based on the correlation between guest residue composition and Tt from our previous studies [27, 28]. The first ELP was designed to form a depot in vivo, as it has a Tt below body temperature (ELPDepot), by selecting Valine (V) as 4th guest residues in the pentapeptide repeat. To create a soluble control, a second ELP (ELPSol) was designed to have a Tt that is above body temperature (Table 1) by alternating Glycine (G) and Alanine (A) in the guest residue position.
Beyond their ability to undergo a thermally triggered phase transition, ELPs have other useful attributes for the delivery of biologics. Because ELPs are genetically encoded, their sequence and molecular weight can be precisely controlled, and modulation of these parameters allows the ELP transition behavior to be precisely tuned, which in turn controls its in vivo half-life [27]. Importantly, protein-ELP fusions have been shown to maintain temperature responsive behavior [29] and the ELPs themselves are biodegradable [30] and non-immunogenic [31]. In addition, ELP fusions can be purified to very high purity without the need for chromatography by a batch purification process –termed inverse transition cycling (ITC)– that takes advantage of their reversible soluble-insoluble phase transition behavior [32]. [33]
3.2 Production and characterization of (GLP-1)-ELP
The (GLP-1)-ELP fusion proteins were successfully expressed in E. coli with IPTG induction. Chromatography-free purification by ITC (Figure 1B) [20] typically yielded 30–50 mg of fusion protein per liter of culture. SDS-PAGE showed that the purified construct had the expected MW, as seen by a single band at approximately 50 kDa, and sequencing of (GLP-1)-ELPDepot confirmed that the N-terminal methionine was correctly removed. Temperature triggered coacervation was controlled, as predicted, by varying the ELP hydrophobicity, generating (GLP-1)-ELP fusions with transition temperatures below body temperature (depot forming, named (GLP-1)-ELPDepot) or above body temperature (soluble control, named (GLP-1)-ELPSol), with the same molecular weight. Both fusion proteins displayed a sharp, reversible transition profile at their Tt (Figure 1C) which increases with decreasing ELP fusion protein concentration. We hypothesize that this concentration dependent increase in Tt will mediate dissolution of (GLP-1)-ELP from the depot boundaries (Supplementary informaiton Figure S1). The α-helical structure of GLP-1 in (GLP-1)-ELPDepot fusions appeared to be preserved, as measured by circular dichroism by negative 208 and 222 nm peaks and slightly positive 195 nm peak, compared with the highly disordered structure observed for ELP alone [9] (Figure 1D).
3.3 Assessment of depot viability
As the (GLP-1)-ELP depot was designed to reside in the s.c. space for a prolonged period of time, we were interested in testing its stability against neutral endopeptidase (NEP), a protease that is known to degrade GLP-1 in vivo. Although considerably slower than DPPIV-mediated deactivation of GLP-1, NEP has been previously implicated in GLP-1 degradation [34, 35], which is of some concern, as susceptibility of (GLP-1)-ELP fusions to NEP would greatly reduce the available GLP-1 in the depot. Remarkably, both soluble and depot forming fusion proteins were not degraded even after 18 h incubation with NEP at 20 or 37 C, whereas native GLP-1 was almost completely degraded (Figure 2A, Supplementary information Figure S2). This suggests that (GLP-1)-ELP fusions are proteolytically more stable than GLP-1 monomer, presumably because the high MW ELP in the fusion impedes access of the enzyme to the cleavage site. This suggests that a GLP-1-ELP depot is a viable candidate for long-term delivery of the (GLP-1)-ELP fusion as a drug for type-2 diabetes.
Figure 2.
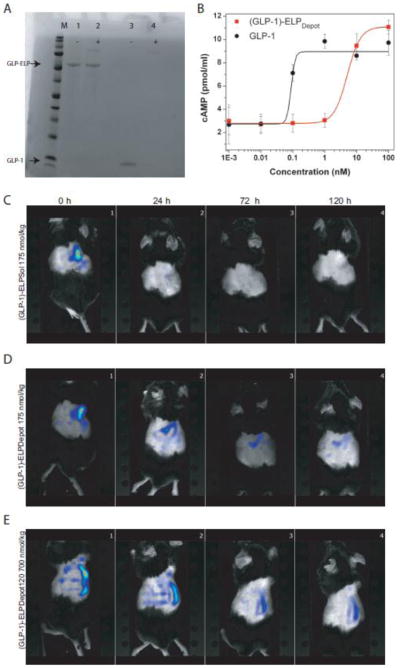
(A) Degradation of (GLP-1)-ELPDepot or GLP-1 by NEP after 18 h incubation (lane 1: (GLP-1)-ELPDepot lane 2: (GLP-1)-ELPDepot+NEP, lane 3: GLP-1, lane 4: GLP-1+NEP. NEP may be seen as a faint band at ~85kDa. (B) cAMP response of native GLP vs. (GLP-1)-ELPDepot in BHK cells expressing the GLP-1R. (C-E) NIR tomography images after a single s.c. injection of (C) (GLP-1)-ELPSol, 175 nmol/kg (D) (GLP-1)-ELPDepot, 175 nmol/kg and (E) (GLP-1)-ELPDepot, 700 nmol/kg immediately following injection and 24, 72 and 120 h after s.c. injection.
Next, we examined the ability of the fusions to activate the GLP-1 receptor (GLP-1R) in a receptor activation assay using baby hamster kidney (BHK) cells that express rat GLP-1R. GLP-1)-ELPDepot produced dose-related cAMP accumulation, albeit less effectively than native GLP-1, with a 57-fold increase in EC50 (Figure 2B, 0.092 vs. ~5.2 nmol/L for GLP-1 vs. (GLP-1)-ELPDepot respectively). Likewise, (GLP-1)-ELPSol, which is attached to a soluble ELP of the same MW as ELPDepot, produced dose-related cAMP accumulation, with a similar EC50 increase as (GLP-1)-ELPDepot, (Supplementary information Figure S3). The observed increase in EC50 is neither unexpected nor unprecedented as fusion of GLP-1 to BSA (Albugon) or of exendin to BSA resulted in 30 and 20 fold reduction in bioactivity respectively [11, 36] and a GLP-1–PEG fusion reduced receptor binding affinity by 30–500 fold [9, 37].
To visualize s.c. depot formation and persistence in vivo, NIR labeled (GLP-1)-ELPDepot (175 and 700 nmol/kg) and (GLP-1)-ELPSol (175 nmol/kg) soluble control were injected s.c. and imaged for up to 1 week post injection using an NIR fluorescence tomography system. Tomography images confirmed the presence of a (GLP-1)-ELPDepot s.c. depot, which could be visualized for 1 week following a single injection. Furthermore, it was visually apparent that, while some small amount of (GLP-1)-ELPSol soluble control persisted in the injection site (Figure 2C, Supplementary information Figure S4), the size of depots created by both doses of (GLP-1)-ELPDepot was substantially larger, thus confirming the role of in vivo transition of the ELP in depot formation (Figure 2D and 2E, Supplementary information Figure S5 and S6).
3.4 In vivo activity of (GLP-1)-ELPDepot
We next determined whether a single injection of (GLP-1)-ELPDepot can provide extended temporal reduction of glucose levels compared with monomer GLP-1, and whether this release profile is affected by the Tt of the ELP. A single s.c. injection of (GLP-1)-ELPDepot was administered, using a 27 gauge needle, at a temperature below the Tt. Fed glucose levels were monitored throughout the first day, and every morning subsequently for up to 1 week post-injection. (GLP-1)-ELPDepot reduced fed blood glucose levels significantly in a dose-dependent manner: 175, 350 and 700 nmol/kg reduced blood glucose levels for 24, 72 and 144 h respectively (ANOVA p<0.0001 for all treatment groups). Differences were also observed in the magnitude of glucose reduction which increased from ~13% to 30% with the increase in dose (Figure 3A–C). In contrast, a single injection of GLP-1 monomer caused a modest reduction in blood glucose that vanished 1.5 h post injection (Figure 3C inset).
Figure 3.

Fed glucose reduction following a single injection of (GLP-1)-ELPs. Daily fed blood glucose levels normalized by baseline glucose levels after a single s.c. injection of PBS control or (A) (GLP-1)-ELPDepot (175 nmol/kg, ANOVA p<0.001), (B) (GLP-1)-ELPDepot (350 nmol/kg, ANOVA p<0.0001) (C) (GLP-1)-ELPDepot (700 nmol/kg, ANOVA p<0.0001) with inset showing the effect of a single injection of GLP-1 (50 nmol/kg) (D) a separate experiment comparing the effects of (GLP-1)-ELPSol (175 nmol/kg) with (GLP-1)-ELPDepot (175 and 700 nmol/kg) (ANOVA p<0.0001). Injection was administered at the 0 h time point. (*** p<0.05, ** p<0.01, * p<0.001). (E-F) IPGTT following a single injection of (GLP-1)-ELPDepot. Blood glucose levels in 7 week old mice during IPGTT performed (E) 52 h and (F) 102 h after a single injection of (GLP-1)-ELPDepot (700 nmol/kg, *** p<0.001).
To examine the influence of depot formation on glucose reduction, a second experiment was conducted to compare a single injection of the soluble fusion, (GLP-1)-ELPSol (175 nmol/kg), with that of (GLP-1)-ELPDepot (175 nmol/kg and 700 nmol/kg). Upon injection of (GLP-1)-ELPSol, which was designed to remain soluble at body temperature, blood glucose levels dropped sharply due to the rapid dissemination of the soluble fusion protein from the s.c. compartment into systemic circulation. At peak activity, the soluble construct reduced glucose levels by 60% compared with PBS treated controls, whereas the depot forming (GLP-1)-ELPDepot administered at the same dose (175 nmol/kg) caused a smaller ~20% reduction in glucose levels. These results clearly show that the soluble construct releases most of its payload from the s.c. space in a short duration of time, resulting in a bolus that is released into systemic circulation that causes a peak-and valley response with a steep transient drop in blood glucose. In contrast, (GLP-1)-ELPDepot showed a more damped response that persists for a longer duration. As seen in (Figure 3D), upon increasing the dose of (GLP-1)-ELPDepot to 700 nmol/kg, we again observed a steady ~30% reduction in fed glucose levels that persisted for over 5 days post injection. Hence, these results show that the depot is capable of controlling both the degree of blood glucose reduction and its duration, simply by adjusting the injected dose.
To validate these results, an intraperitoneal glucose tolerance test (IPGTT) was performed 54 h and 102 h (between the 2nd and 3rd day and between the 4th and 5th day post-injection, respectively) after a single injection of (GLP-1)-ELPDepot (700 nmol/kg). IPGTT results confirmed the presence of active GLP-1 fusion in circulation and its significant effect on glucose clearance. At 54 h post-injection, the area under the curve (AUC) is reduced by 40% (p<0.001, Figure 3E) for treated mice compared with PBS treated controls. Similarly, at 102 h post-injection, AUC is reduced by 32% for treated mice compared with PBS treated controls (p<0.001, Figure 3F) thus confirming the prolonged effects of the depot. IPGTT tests done after the 5th day did not show a consistent reduction in AUC (data not shown) despite the fact that fed glucose levels remained below baseline at the 6th day. This may be attributed to residual low levels of circulating (GLP-1)-ELPDepot that are unable to effect a significant reduction upon glucose challenge.
4. Discussion and conclusions
We have shown that the (GLP-1)-ELP drug delivery system is a viable and attractive method for the sustained delivery of GLP-1 for up to 5 days following a single subcutaneous injection. While GLP-1 has been touted as a potential exciting new drug to treat type-2 diabetes [10], its very short half necessitates frequent injections leading to a bolus effect that can cause undesirable side-effects [11] and reduce patient compliance [12]. Constant, prolonged administration of GLP-1 via continuous subcutaneous infusion for 6 weeks has been shown to reduce fasting glucose, hemoglobin A1c, body weight and appetite [38]. Concurrently, it has been shown that there is no attenuation of the GLP-1 receptor (GLP-1R) in vivo in response to constant GLP-1R activation [39]. Furthermore, high GLP-1 levels do not pose a risk for hypoglycemia, as the insulinotropic effects of GLP-1 are glucose dependent and vanish when glucose levels drop below 60 mg/dL [40]. Therefore, a sustained delivery system of GLP-1 is safe and effective and minimizing injection frequency is likely to increase patient compliance.
While several groups attempted to increase GLP-1 circulation time by attachment to large polymers or proteins [9, 14–16, 41] none of these systems create a s.c. drug depot of a high molecular weight fusion protein or protein-polymer conjugate, which can control the rate of release from a depot and increase the duration of peptide action by enhancing its plasma half-life. The (GLP-1)-ELPDepot fusion protein described here is entirely genetically encoded, allowing for complete control over the overall fusion size and composition – which also controls the construct’s Tt and depot properties. Production of (GLP-1)-ELP fusion proteins is not only molecularly precise but also economical, as they successfully express in E. coli expression systems with good yield, and can be purified by ITC, allowing for high-throughput screening of multiple constructs and easy scale-up of protein purification. Most importantly, the ELP allows for extended GLP-1 circulation by two independent parameters that we can control at the molecular design level: (1) s.c. depot formation, in which the GLP-1 fusion proteins are stored and released over time; and (2) increased MW of the fusion protein, which increases the plasma half-life of the circulating drug once released from the depot. The significance of these two parameters in providing sustained efficacy is illustrated by the comparison of a single injection of the depot-forming fusion and an equimolar soluble fusion (Figure 3D). The depot-forming construct reduces fed glucose levels that persisted for over 5 days post injection and correlated with decreased AUC in IPGTT. In contrast the high molecular weight of the soluble fusion (50 kDa compared with 3.2 kDa for native GLP-1) allows this fusion to reduce glucose levels for only 24 h, demonstrating the beneficial effect of prolonged circulation, as monomer GLP-1 reduces glucose levels for only 1.5h.
A comparison with other GLP-1 delivery systems that are in the preclinical and clinical pipeline is useful to benchmark the translational potential of the (GLP-1)-ELP depot delivery system. Because our system shares features with both fusions/conjugates and depots, we briefly discuss both approaches. One approach to increasing peptide circulation time is to design binding sites for albumin, thus generating non-covalant peptide-albumin fusions post-injection. Liraglutide™, an acylated GLP-1 variant designed to bind Albumin in vivo after injection, is FDA approved for once-daily injection in humans. Similarly, Semaglutide is a more sophisticated and complex design consisting of a GLP-1 analog that is acylated via a PEG linker. Semaglutide is currently in pre-clinical evaluation for once-weekly injections [42]. The synthesis of both Liragludie and Semaglutide, however, require chemical modifications of GLP-1 that adds complexity and cost to the synthesis. In the similar category of covalent GLP-1 macromolecule fusions, Albugon™ is a GLP-1–albumin fusion that is the only GLP-1 fusion that is close to FDA approval. Albugon. however, requires expression in an engineered yeast strain [43], and Albugon™ has only been shown to reduce blood glucose levels for ~24 h in db/db mice [44]. Pre-clinical data on GLP-1 fused to an Fc domain has been reported, but this too requires complex and expensive expression in a mammalian expression system that will likely make its use expensive in the chronic, long-term setting of diabetes management. In contrast, production of (GLP-1)-ELPDepot does not require expensive eukaryotic expression hosts or post-expression covalent conjugation.
The only clinically approved depot system, Bydureon™, is a sustained release formulation of exenatide entrapped in biodegradable poly(lactic glycolic acid) (PLGA) microspheres, and was recently approved for once-weekly injection in humans [13]. Adaptation of microsphere encapsulation formulations to peptide drugs is attractive, as PLGA microspheres are FDA approved, and a broad release profile of days to months can be achieved with different formulations. However, despite their commercial success and clinical utility, PLGA microspheres suffer from significant limitations. First, the dose required by Bydureon is 14–28 fold larger compared to exenatide [45], exemplifying the fact that microsphere optimization is difficult and in vivo behavior can be unpredictable. Second, their preparation requires separate synthesis of the peptide and polymer and co-formulation into microspheres of a precise size, a complex process that must be carried out in a sterile environment [46]. Third, sustained release polymer microsphere suspensions are viscous, and hence require painful injections with large gauge needles. Fourth, degradation of PLGA can cause fibrosis [47]. Finally, peptide bioavailability may also be significantly reduced due to peptide adsorption to the polymer matrix and denaturation of the peptide [48].
To circumvent some of the issues with microsphere-based delivery systems, we recently developed protease operated depots (PODs) - an entirely genetically encoded, ELP-based prolonged release formulation designed to release monomer GLP-1 from a s.c. depot [18]. By design, PODs are constructed for prolonged release of monomer GLP-1 peptide and are the first molecularly engineered analog of- and alternative to- polymer microsphere sustained release formulations. As PODs release the native GLP-1 peptide without an attached fusion protein, similar to polymer microsphere formulations such as Bydureon™, drug potency is higher compared to (GLP-1)-ELP fusions, but the peptide is rapidly cleared upon release from the depot, necessitating injection of a higher dose. The (GLP-1)-ELPSol construct, does not create a s.c. depot, but extends peptide circulation solely by increasing its MW. (GLP-1)-ELPSol is therefore analogous to peptide-PEG conjugates and requires a lower dose to cause a sharp drop in blood glucose level, but this reduction is temporally quite limited (~hours) compared with prolonged release formulations that can control glucose levels for days to a week. In contrast to these two orthogonal approaches, the depot forming, (GLP-1)-ELPDepot construct combines the two mechanisms, and provides prolonged glucose control after injection of an intermediate GLP-1 dose. It does so by first forming a depot upon s.c. injection, from which intact and soluble (GLP-1)-ELP fusion is released, which has a much longer plasma half-life than the native peptide.
These different designs demonstrate the utility and flexibility of ELPs as vehicles for drug delivery and constitute a peptide-delivery “toolbox” for GLP-1 as well as other peptide drugs. We further plan to expand this “toolbox” by integrating albumin binding motifs in the ELP domain to further extend the circulation time of the released fusions. Going forward, the appropriate system can be selected and adapted for other peptides according to their biological characteristics and optimal pharmacokinetic profile. This ELP-based drug-delivery “toolbox” is entirely genetically encoded with the following attractive features: (1) fusion proteins are easy to produce at high level by recombinant expression of a single polypeptide chain in E. coli; (2), they do not require co-formulation with a matrix or excipients, and can be injected s.c. as a highly concentrated solution through a narrow gauge needle; (3) they spontaneously undergo a phase transition at body temperature upon s.c. injection, forming a stable, long-lasting depot; (4) they release bioactive peptide or peptide fusions over a sustained duration from the depot and can reduce glucose levels for up to 5 days following a single s.c. injection in mice. We believe these features augur well for clinical translation of ELP-based delivery systems for GLP-1 and other peptide drugs.
Supplementary Material
Acknowledgments
A.C. acknowledges the financial support of NIH grant R01DK091789, and M.A. acknowledges the support of a graduate fellowship from the Center for Biologically Inspired Materials and Material Systems at Duke University. We thank the Duke University School of Medicine for the use of the Duke Proteomics Core Facility, which provided MALDI-TOF and protein sequencing services.
Footnotes
Author contributions:
A.C. designed experiments, analyzed data and prepared the manuscript. M.A. designed and performed experiments, analyzed data and prepared the manuscript. X.L. assisted with animal experiments, K.M.L performed experiments, and M.N.F analyzed data.
Competing financial interests:
A.C. is co-founder of a start-up company, PhaseBio Pharmaceuticals, in Malvern, PA, USA that is commercializing elastin-like polypeptides for applications in biotechnology and medicine.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hays NP, Galassetti PR, Coker RH. Prevention and treatment of type 2 diabetes: current role of lifestyle, natural product, and pharmacological interventions. Pharmacol Ther. 2008;118:181–191. doi: 10.1016/j.pharmthera.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brubaker PL. Minireview: update on incretin biology: focus on glucagon-like peptide-1. Endocrinology. 2010;151:1984–1989. doi: 10.1210/en.2010-0115. [DOI] [PubMed] [Google Scholar]
- 3.Nauck MA. Glucagon-like peptide 1 (GLP-1): a potent gut hormone with a possible therapeutic perspective. Acta Diabetologica. 1998;35:117–129. doi: 10.1007/s005920050116. [DOI] [PubMed] [Google Scholar]
- 4.Hui HX, Zhao HN, Perfetti R. Structure and function studies of glucagon-like peptide-1 (GLP-1): the designing of a novel pharmacotogical agent for the treatment of diabetes. Diabetes-Metabolism Research and Reviews. 2005;21:313–331. doi: 10.1002/dmrr.553. [DOI] [PubMed] [Google Scholar]
- 5.Nauck MA. Glucagon-like peptide 1 (GLP-1): a potent gut hormone with a possible therapeutic perspective. Acta Diabetol. 1998;35:117–129. doi: 10.1007/s005920050116. [DOI] [PubMed] [Google Scholar]
- 6.DeFronzo RA. From the Triumvirate to the Ominous Octet: A New Paradigm for the Treatment of Type 2 Diabetes Mellitus. Diabetes. 2009;58:773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holst JJ, Vilsboll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol. 2009;297:127–136. doi: 10.1016/j.mce.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Brubaker PL, Drucker DJ. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology. 2004;145:2653–2659. doi: 10.1210/en.2004-0015. [DOI] [PubMed] [Google Scholar]
- 9.Miranda LP, Winters KA, Gegg CV, Patel A, Aral J, Long J, Zhang J, Diamond S, Guido M, Stanislaus S, Ma M, Li H, Rose MJ, Poppe L, Veniant MM. Design and synthesis of conformationally constrained glucagon-like peptide-1 derivatives with increased plasma stability and prolonged in vivo activity. J Med Chem. 2008;51:2758–2765. doi: 10.1021/jm701522b. [DOI] [PubMed] [Google Scholar]
- 10.Lovshin JA, Drucker DJ. Incretin-based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5:262–269. doi: 10.1038/nrendo.2009.48. [DOI] [PubMed] [Google Scholar]
- 11.Madsbad S, Kielgast U, Asmar M, Deacon CF, Torekov SS, Holst JJ. An overview of once-weekly glucagon-like peptide-1 receptor agonists--available efficacy and safety data and perspectives for the future. Diabetes Obes Metab. 2011;13:394–407. doi: 10.1111/j.1463-1326.2011.01357.x. [DOI] [PubMed] [Google Scholar]
- 12.Fineman MS, Cirincione BB, Maggs D, Diamant M. GLP-1 Based Therapies: Differential Effects on Fasting and Postprandial Glucose. Diabetes Obes Metab. 2012 doi: 10.1111/j.1463-1326.2012.01560.x. [DOI] [PubMed] [Google Scholar]
- 13.Diamant M, Van Gaal L, Stranks S, Northrup J, Cao D, Taylor K, Trautmann M. Once weekly exenatide compared with insulin glargine titrated to target in patients with type 2 diabetes (DURATION-3): an open-label randomised trial. Lancet. 2010;375:2234–2243. doi: 10.1016/S0140-6736(10)60406-0. [DOI] [PubMed] [Google Scholar]
- 14.Kim JG, Baggio LL, Bridon DP, Castaigne JP, Robitaille MF, Jette L, Benquet C, Drucker DJ. Development and characterization of a glucagon-like peptide 1-albumin conjugate - The ability to activate the glucagon-like peptide 1 receptor in vivo. Diabetes. 2003;52:751–759. doi: 10.2337/diabetes.52.3.751. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Chen K, Liu R, Zhao F, Gupta S, Zhang N, Prud’homme GJ. Novel GLP-1 fusion chimera as potent long acting GLP-1 receptor agonist. PLoS One. 2010;5:e12734. doi: 10.1371/journal.pone.0012734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schellenberger V, Wang CW, Geething NC, Spink BJ, Campbell A, To W, Scholle MD, Yin Y, Yao Y, Bogin O, Cleland JL, Silverman J, Stemmer WPC. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nature Biotechnology. 2009;27:1186–U1155. doi: 10.1038/nbt.1588. [DOI] [PubMed] [Google Scholar]
- 17.Banga AK. Therapeutic Peptides and Proteins. CRC Press; 2005. Parenteral Controlled Delivery and Pharmacokinetics of Therapeutic Peptides and Proteins; pp. 177–227. [Google Scholar]
- 18.Amiram M, Luginbuhl KM, Li X, Feinglos MN, Chilkoti A. Injectable protease-operated depots of glucagon-like peptide-1 provide extended and tunable glucose control. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2792–2797. doi: 10.1073/pnas.1214518110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer DE, Chilkoti A. Quantification of the effects of chain length and concentration on the thermal behavior of elastin-like polypeptides. Biomacromolecules. 2004;5:846–851. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- 20.Christensen T, Amiram M, Dagher S, Trabbic-Carlson K, Shamji MF, Setton LA, Chilkoti A. Fusion order controls expression level and activity of elastin-like polypeptide fusion proteins. Protein Sci. 2009;18:1377–1387. doi: 10.1002/pro.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baggio LL, Huang Q, Brown TJ, Drucker DJ. A recombinant human glucagon-like peptide (GLP)-1-albumin protein (albugon) mimics peptidergic activation of GLP-1 receptor-dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis. Diabetes. 2004;53:2492–2500. doi: 10.2337/diabetes.53.9.2492. [DOI] [PubMed] [Google Scholar]
- 23.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 24.Drucker DJ. Glucagon-like peptides: regulators of cell proliferation, differentiation, and apoptosis. Molecular endocrinology. 2003;17:161–171. doi: 10.1210/me.2002-0306. [DOI] [PubMed] [Google Scholar]
- 25.Hirel PH, Schmitter JM, Dessen P, Fayat G, Blanquet S. Extent of N-Terminal Methionine Excision from Escherichia-Coli Proteins Is Governed by the Side-Chain Length of the Penultimate AminoAcid. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:8247–8251. doi: 10.1073/pnas.86.21.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegel EG, Gallwitz B, Scharf G, Mentlein R, Morys-Wortmann C, Folsch UR, Schrezenmeir J, Drescher K, Schmidt WE. Biological activity of GLP-1-analogues with N-terminal modifications. Regulatory peptides. 1999;79:93–102. doi: 10.1016/s0167-0115(98)00155-4. [DOI] [PubMed] [Google Scholar]
- 27.Meyer DE, Chilkoti A. Genetically encoded synthesis of protein-based polymers with precisely specified molecular weight and sequence by recursive directional ligation: Examples from the elastin-like polypeptide system. Biomacromolecules. 2002;3:357–367. doi: 10.1021/bm015630n. [DOI] [PubMed] [Google Scholar]
- 28.Urry DW, Luan CH, Parker TM, Gowda DC, Prasad KU, Reid MC, Safavy A. Temperature of Polypeptide Inverse Temperature Transition Depends on Mean Residue Hydrophobicity. Journal of the American Chemical Society. 1991;113:4346–4348. [Google Scholar]
- 29.Trabbic-Carlson K, Liu L, Kim B, Chilkoti A. Expression and purification of recombinant proteins from Escherichia coli: Comparison of an elastin-like polypeptide fusion with an oligohistidine fusion. Protein Sci. 2004;13:3274–3284. doi: 10.1110/ps.04931604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Urry DW. Elastic molecular machines in metabolism and soft-tissue restoration. Trends Biotechnol. 1999;17:249–257. doi: 10.1016/s0167-7799(99)01306-2. [DOI] [PubMed] [Google Scholar]
- 31.Ong SR, Trabbic-Carlson KA, Nettles DL, Lim DW, Chilkoti A, Setton LA. Epitope tagging for tracking elastin-like polypeptides. Biomaterials. 2006;27:1930–1935. doi: 10.1016/j.biomaterials.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 32.Meyer DE, Chilkoti A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nature Biotechnology. 1999;17:1112–1115. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 33.Knudsen LB. Glucagon-like peptide-1: The basis of a new class of treatment for type 2 diabetes. Journal of Medicinal Chemistry. 2004;47:4128–4134. doi: 10.1021/jm030630m. [DOI] [PubMed] [Google Scholar]
- 34.Hupe-Sodmann K, McGregor GP, Bridenbaugh R, Goke R, Goke B, Thole H, Zimmermann B, Voigt K. Characterisation of the processing by human neutral endopeptidase 24.11 of GLP-1(7-36) amide and comparison of the substrate specificity of the enzyme for other glucagon-like peptides. Regul Pept. 1995;58:149–156. doi: 10.1016/0167-0115(95)00063-h. [DOI] [PubMed] [Google Scholar]
- 35.Malm-Erjefalt M, Bjornsdottir I, Vanggaard J, Helleberg H, Larsen U, Oosterhuis B, van Lier JJ, Zdravkovic M, Olsen AK. Metabolism and excretion of the once-daily human glucagon-like peptide-1 analog liraglutide in healthy male subjects and its in vitro degradation by dipeptidyl peptidase IV and neutral endopeptidase. Drug Metab Dispos. 2010;38:1944–1953. doi: 10.1124/dmd.110.034066. [DOI] [PubMed] [Google Scholar]
- 36.Huang YS, Chen Z, Chen YQ, Ma GC, Shan JF, Liu W, Zhou LF. Preparation and characterization of a novel exendin-4 human serum albumin fusion protein expressed in Pichia pastoris. J Pept Sci. 2008;14:588–595. doi: 10.1002/psc.942. [DOI] [PubMed] [Google Scholar]
- 37.Chae SY, Chun YG, Lee S, Jin CH, Lee ES, Lee KC, Youn YS. Pharmacokinetic and pharmacodynamic evaluation of site-specific PEGylated glucagon-like peptide-1 analogs as flexible postprandial-glucose controllers. J Pharm Sci. 2009;98:1556–1567. doi: 10.1002/jps.21532. [DOI] [PubMed] [Google Scholar]
- 38.Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359:824–830. doi: 10.1016/S0140-6736(02)07952-7. [DOI] [PubMed] [Google Scholar]
- 39.Baggio LL, Kim JG, Drucker DJ. Chronic exposure to GLP-IR agonists promotes homologous GLP-1 receptor desensitization in vitro but does not attenuate GLP-1R-dependent glucose homeostasis in vivo. Diabetes. 2004;53:S205–S214. doi: 10.2337/diabetes.53.suppl_3.s205. [DOI] [PubMed] [Google Scholar]
- 40.LeRoith D, Taylor SI, Olefsky JM. Diabetes mellitus: a fundamental and clinical text. 3. Lippincott Williams & Wilkins; Philadelphia: 2004. [Google Scholar]
- 41.Wang QH, Chen K, Liu R, Zhao F, Gupta S, Zhang NN, Prud’homme GJ. Novel GLP-1 Fusion Chimera as Potent Long Acting GLP-1 Receptor Agonist. Plos One. 2010;5 doi: 10.1371/journal.pone.0012734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug discovery today. 2013 doi: 10.1016/j.drudis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 43.Chuang VT, Kragh-Hansen U, Otagiri M. Pharmaceutical strategies utilizing recombinant human serum albumin. Pharmaceutical research. 2002;19:569–577. doi: 10.1023/a:1015396825274. [DOI] [PubMed] [Google Scholar]
- 44.Chee JME, Chia W. Biology and therapeutic potential of GLP-1 in the treatment of diabetes. Drug Discovery Today: Disease Mechanisms. 2005;2:295–301. [Google Scholar]
- 45.Kwak HH, Shim WS, Hwang S, Son MK, Kim YJ, Kim TH, Yoon ZH, Youn HJ, Lee GI, Kang SH, Shim CK. Pharmacokinetics and Efficacy of a Biweekly Dosage Formulation of Exenatide in Zucker Diabetic Fatty (ZDF) Rats. Pharm Res-Dord. 2009;26:2504–2512. doi: 10.1007/s11095-009-9966-3. [DOI] [PubMed] [Google Scholar]
- 46.Niu CH, Chiu YY. FDA perspective on peptide formulation and stability issues. J Pharm Sci. 1998;87:1331–1334. doi: 10.1021/js9800782. [DOI] [PubMed] [Google Scholar]
- 47.Shive MS, Anderson JM. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 48.Pai SS, Tilton RD, Przybycien TM. Poly(ethylene glycol)-modified proteins: implications for poly(lactide-co-glycolide)-based microsphere delivery. AAPS J. 2009;11:88–98. doi: 10.1208/s12248-009-9081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.