

Abstract
Homology modeling and molecular dynamics simulations have been carried out to model the detailed structures of human neonatal Fc receptor (FcRn) binding with wild-type Fc of human immunoglobulin G1 (IgG1) and its various mutants. Based on the modeled human FcRn-Fc binding structures, the protein-protein binding interface is composed of three subsites. The first subsite is a hydrophobic core where residue I39 of human Fc can be accommodated very well, and the other two subsites are all composed of critical salt bridges between human FcRn and human Fc. All of the modeled structures and the calculated binding energies are qualitatively consistent with the available experimental data, suggesting that the modeled human FcRn-Fc binding structures are reasonable. The modeled human FcRn-Fc binding structure may be valuable for future rational design of novel mutants of human Fc and Fc-fused therapeutic proteins with a potentially higher binding affinity for human FcRn and, thus, a longer in vivo half-life in human.
Introduction
Human neonatal Fc receptor (FcRn) for immunoglobulin G (IgG) is a 52-kDa heterodimeric glycoprotein bound on the membrane of endosome. It is composed of a heavy chain and a light chain named as β2-microglobulin (β2m).1,2,3,4 FcRn is expressed in human placenta and can transfer maternal IgG to the fetus or the newborn, providing humoral immunity for the first weeks of mammalian life.1,5,6 Further studies7,8 found that FcRn is expressed in the vascular endothelial cells, epithelial cells, hepatocytes, intestinal macrophages, peripheral blood monocytes, and dendritic cells. FcRn expression was also demonstrated at vascular endothelial cells in brain.9 The primary function of FcRn is to maintain the long half-life of IgG in the serum through binding with the Fc portion of IgG.3,4.10 IgG is a class of antibody predominantly present in the normal human serum, and additional amount of IgG could be generated from the secondary response under continuous stimulation of an external pathogen.10,11 Without the binding with FcRn, IgG is circulated and degraded quickly through lysosomal degradation pathway, e.g. the half-life of IgG is reduced from 6–8 days to about 1 day in the FcRn-deficient mice.12 IgG is internalized into FcRn-expressing cells most likely by fluid phase pinocytosis as the steady-state location of FcRn is endosomal.13 IgG binds with FcRn in the acidic environment of endosome, and is later transported to the cell surface where upon exposure to a neutral pH IgG is released back to the main bloodstream. Such pH dependence of FcRn-IgG binding ensures that IgG binds to FcRn in the intracellular acidic compartments but be released rapidly upon encountering the slightly basic pH 7.4 of the extracellular milieu.10,12,13
Seeking a longer half-life for a specifically designed therapeutic protein is of great significance and is a grand challenge in the area of protein drug design, discovery, and development.14,15 With a growing number of protein drugs being developed, the in vivo half-life extension strategies have attracted more and more attention by the biotech and pharmaceutical industries.16 Compared to other in vivo half-life prolonging methods, Fc fusion, i.e. genetically fusing the Fc portion of IgG to a protein drug, has become the most clinically and commercially successful strategy with possibly enhanced efficacy, greater safety, and reduced immunogenicity or improved delivery.15,16 The Fc part of an Fc-fused protein can bind with FcRn like the Fc part of IgG1 binding with FcRn. So, the Fc-fused protein drug is expected to have a longer in vivo half-life compared to the corresponding unfused protein drug. Currently, there are a number of marketed and clinical candidate antibodies and Fc fusion proteins, including Alefacept, that have successfully taken advantage of the FcRn-Fc binding.17,18,19
The X-ray crystal structures20,21,22 of FcRn at various pH and from different species (human and rat) revealed that the overall conformation of FcRn is persistent, indicating that the pH-dependence of FcRn-Fc binding is not mediated by the conformational change of FcRn, but possibly by the electrostatic interactions involving histidine amino acids. The X-ray crystal structures23,24,25,26 of rat FcRn bound with rat Fc revealed that the hinge region of Cγ2-Cγ3 domain of Fc binds to the top of α1 and α2 helices of FcRn. The hinge region of Fc was demonstrated to be a consensus site of recognition by a series of proteins associated with Fc.25 The X-ray crystal structures27,28,29,30,31 of IgG1 Fc and the M38Y/S40T/T42E mutant32 under acidic pH condition demonstrated that the overall shape of Fc is similar to that of a “horseshoe”, and most of the internal space is filled with oligosaccharide chains through residue N83 (we renumbered Fc residues and ignored other portion of IgG1 for convenience). The IgG1 Fc is a homodimer (with two equal subunits) linked by disulfide bridges in the N-terminal region and non-covalent interactions between the C-terminal regions of the two subunits, and each subunit is comprised of two immunoglobulin domains known as Cγ2 and Cγ3. The Cγ2 domain of Fc can take large extent of rigid body motion, leading to a “closed” conformation of Fc when residue N83 is totally unglycosylated and an “open” conformation when residue N83 is fully glycosylated. The distance between the end points of the Cγ2 domains of the two subunits of Fc varies from 10 Å for the “closed” conformation to 14 Å in the “open” conformation.
A large number of Fc mutants3,4,10,11,19,32,33,34,35,36,37,38,39,40,41,42,43,44 have been generated and fused with different effector proteins in order to explore the relationship between the FcRn-Fc binding affinity and the in vivo half-life of a Fc-fused protein. These mutational studies also aimed to understand how the change in the FcRn-Fc binding affinity affects the pharmacokinetics of the Fc-fused effector protein,36,39,42 and how to control the safety profile such as antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDCC).19,35,40,41 It is also known45,46 that there is a dramatic cross-species difference in the affinity of FcRn-Fc binding. At acidic pH, mouse FcRn has a high affinity with either mouse IgG1 or human IgG1, whereas human FcRn has a very low affinity with mouse IgG1. At physiological pH 7.4, mouse FcRn keeps micromolar (μM) range of binding affinity with human IgG1, but no detectable binding with mouse IgG1. These studies established that the Fc mutants with a higher FcRn affinity under acidic pH condition usually have a longer in vivo half-life when fused with an effector protein, and a lower cytotoxicity if the binding affinity of Fc with Fc gama receptors type II and type III (FcγRIIs, FcγRIIIs) are not enhanced simultaneously.
As the structures of rat FcRn-Fc complex, human FcRn, and human Fc are available, it is now possible to explore the structural difference at atomic level between the human FcRn-Fc binding and non-human FcRn-Fc binding. It is also interesting to explore the structural determinants for the reported relationships between the binding affinity of Fc mutants with FcRn and the experimentally measured in vivo half-life and safety properties of Fc-fused effector proteins. Understanding of these structural determinants is fundamentally important for rational design of novel Fc mutants that can be fused with a protein drug in order to prolong the in vivo half-life of the protein drug and produce satisfactory pharmacological properties and good safety profile.
In the present study, the binding structure of human FcRn-Fc complex was modeled and structurally optimized through molecular dynamics (MD) simulations. Binding structure of human FcRn in complex with each of 30 human Fc mutants was also modeled and energy-minimized. Binding energies were calculated for all these optimized binding structures by using a ranking score function47,48 that was specifically designed to study protein-protein interactions. Our modeling and simulation results together with the data of calculated binding energies have provided novel insights into the molecular mechanism for FcRn-Fc binding across different species. The modeled binding structures can be used as a starting point to rationally design novel Fc mutants that can more potently bind with human FcRn. By fusing a novel, high-affinity Fc mutant with a protein drug, it would be possible to develop a truly effective Fc-fused therapeutic protein which could have not only a sufficiently high in vivo potency and stability, but also a sufficiently long in vivo half-life for practical medical uses.
Methods
Homology modeling and MD simulations of human FcRn-Fc binding structure
In order to explore the mode of human FcRn binding with human Fc and to identify the structural difference between the human FcRn-Fc binding and non-human FcRn-Fc binding, we first constructed a binding structure of human FcRn-Fc complex through homology modeling and molecular structure optimization by using the Protein Modeling module of Discovery Studio (version 2.5.5, Accelrys, Inc., San Diego, CA). The X-ray crystal structure of rat FcRn-Fc complex (PDB entry code 1I1A with resolution of 2.80 Å)26 determined at pH 5.4 was used as a template to model human FcRn-Fc complex, as the resolution of this template is the highest within the available X-ray crystal structures including that determined at pH 5.6 (PDB code entry 1FRT with resolution of 4.50Å).23 The amino acid sequence of human FcRn was directly extracted from the PubMed protein database (access number (NP_001129491). For convenience of the sequence and structure comparison, the amino acid sequences of FcRn from other species (monkey, access number AAL92101; mouse, access number NP_034319) were also extracted from the same PubMed protein database and were aligned together. The sequence alignment for all these amino acid sequences was generated by using ClusterW with the Blosum scoring function.49,50 The best alignment was selected according to both the alignment score and the reciprocal positions of the conserved residues among all these FcRn proteins from different species, particularly the conserved residues on the binding interface as revealed in the template structure. Considering the fact that X-ray crystal structures of ligand-free FcRn are also available, we selected the structure of lignad-free human FcRn with PDB entry code 3M17 at resolution of 2.60 Å22 as a template to model the regions composed of non-conserved residues between human FcRn and rat FcRn. We did not select the ligand-free human FcRn structure corresponding to PDB code 1EXU21 (determined at pH 8.2 with resolution of 2.70 Å), because the human FcRn can bind tightly with human Fc only under acidic pH condition. The coordinates of the conserved residues of human FcRn were directly copied from the template rat FcRn-Fc complex structure, and the coordinates of non-conserved residues were copied from the X-ray crystal structure of the ligand-free human FcRn corresponding to PDB code 3M17.22
Modeling of human Fc in the human FcRn-Fc complex was performed in a similar way as described above. The amino acid sequences of Fc from human, monkey, rat, and mouse were aligned together using the ClusterW program.49,50 The coordinates of the conserved residues of human Fc were copied directly from the template rat Fc in the rat FcRn-Fc complex, whereas the coordinates of the non-conserved residues between human Fc and rat Fc were mutated from the template to the corresponding ones of human Fc. The side chains of the non-conserved residues of human Fc were relaxed during the process of homology modeling to remove possible steric overlap or hindrance with the neighboring conserved residues.
After the initial human FcRn-Fc complex structure was built, all the ionizable residues were set to the standard protonated or deprotonated states, and all the histidine residues were set as protonated. The whole complex structure was then solvated in an orthorhombic box of 38,126 TIP3P water molecules, with a minimal distance of 10 Å from the protein to the box boundary. The whole system was neutralized by adding 18 chloride counterions, and reached the size as 113 Å × 124 Å × 105 Å. After the whole system was set up, a series of energy minimizations were carried out by using the Sander module of Amber 11 program51 with a non-bonded cutoff of 10 Å and a conjugate gradient energy-minimization method. First, the atomic positions of water molecules and counterions were energy-minimized for 40,000 steps. Next, 40,000 steps of the energy minimization on the side chains of the complex along with water molecules and counterions were performed. Finally, the entire system was energy-minimized to reach a convergence criterion of 0.001 kcal/(mol Å).
In order to further relax the energy-minimized structure of human FcRn-Fc complex, MD simulations were performed by using the Sander module of Amber 11.51 First, 0.5 ns MD simulations were performed on the water molecules and counterions with the NTV ensemble at T = 300 K in order to get the complex better solvated. The whole system was energy-minimized again and the same convergence criterion was reached. Then, the whole system was gradually heated to 300 K by a weak-coupling method52 and equilibrated for 1.0 ns. In order to avoid the relative motion of the Cγ2 and Cγ3 domains of human Fc in the complex during the simulations and, meanwhile, keep some degrees of freedom of the residues, a weak harmonic restraint with the force constant of 5.0 kcal/(mol Å) was imposed on the backbone atoms of human Fc, and the distance between the mass center of the Cγ2 domain of one subunit and the mass center of the Cγ2 domain of the other subunit was fixed. Throughout the MD simulations, a 10 Å non-bonded interaction cutoff was used and the non-bonded list was updated every 25 steps. The particle mesh Ewald (PME) method53 was applied to treat long-range electrostatic interactions. The lengths of covalent bonds involving hydrogen atoms were fixed with the SHAKE algorithm,54 enabling the use of a 2-fs time step to numerically integrate the equations of motion. Finally, the production MD was kept running for about 5.0 ns with a periodic boundary condition (PBC) in the NTP ensemble at T = 300 K with Berendsen temperature coupling52 and P = 1 atm with anisotropic molecule-based scaling.
Mutational modeling of human Fc mutants and binding energy calculations
We collected a total of 30 mutants of human Fc reported in literature33,39 to explore the structural determinants for the relationship between the change in human FcRn-Fc binding affinity and the prolonged in vivo half-life. The mutational modeling of these collected Fc mutants started from an energy-minimized human FcRn-Fc binding structure after the MD simulations. The protein-protein binding structure for each of the mutants was generated automatically by using a script developed in house at University of Kentucky and the X-leap module of Amber 11 program51. In order to more accurately reflect the structural perturbation of the mutation to the binding interface of human FcRn-Fc complex, and for convenience of the rapid geometry refinement, the Cβ atom and the backbone atoms of each mutant were retained from the energy-minimized starting structure. Once the protein-protein binding structure for a mutant was generated, it was subject to further energy minimization in order to optimize the interactions between human FcRn and the mutant of human Fc. The energy minimization was performed by using the Sander module of Amber 1151 via a combined use of the steepest descent/conjugate gradient algorithms, with a convergence criterion of 0.1 kcal/(mol.Å), and the non-bonded cutoff distance was set to 10.0 Å. The energy minimization was performed first on the mutated residue, and then on those residues which were within 5 Å distance around the mutated residue, and finally on the entire complex structure. After the energy minimization, we visually checked each of the mutant-type complex structures to make sure that there was no any unexpected structural distortion.
The energy-minimized binding structures for wild-type human FcRn with wild-type human Fc and its mutants were evaluated for their binding energies by using the ZRANK score function47,48,55 which was developed by Weng et al. to score optimized protein-protein binding complexes. The ZRANK score function is a linear weighted sum of van der Waals attractive and repulsive energies, electrostatic short- and long-range attractive and repulsive energies, desolvation energy, and a pairwise interface statistical potential energy.55
Results and Discussion
Binding interface of human FcRn-Fc complex
Depicted in Figure 1 are the aligned sequences for the heavy chain of FcRn from human, monkey, mouse, and rat. It is not surprising to note in Figure 1 that the overall homology between human and monkey FcRn proteins (group 1) is high, and that the overall homology between mouse and rat FcRn proteins (group 2) is also high. The overall homology between the group 1 and the group 2 proteins is relatively low, such as ~64% between human and rat FcRn proteins. The high homology between different members of the same group and the relatively bigger difference between the two groups are also reflected in the glycosylation sites: only one glycosylation site (N102) in human/monkey FcRn versus four glycosylaton sites (N87, N104, N141, and N225) in mouse/rat FcRn. The glycosylation sites N141 and N225 of rat/mouse FcRn are all replaced by glutamines (Q139 and Q223) of human/monkey FcRn. The residue in human/monkey FcRn corresponding to the glycosylation site N87 of rat/mouse FcRn is totally depleted during the evolution process. The difference at residue #139 of human/monkey FcRn and residue #141 of rat/mouse FcRn (Q139 versus N141) is expected to have a significant impact on the binding with Fc. As observed from the template structure of rat FcRn-Fc complex,26 the glycans attached to residue N141 of rat FcRn bridged the binding interface between rat FcRn and rat Fc, and these glycans at N141 of rat FcRn have direct interactions with residues of rat Fc (see below).
Figure 1.

Sequence alignment of the heavy chain of FcRn from human (H), monkey (MK), rat (R), and mouse (M). The position of the signal peptide is also indicated by arrows and labeled. Residues in red color are at the binding interface with human Fc, and residues in blue color are the sites for glycosylation. The overall homology between human FcRn and monkey FcRn is 96.0%, 89% between rat FcRn and mouse FcRn, and 64% between human FcRn and rat FcRn.
Despite of the difference in glycosylation sites, the residues of human FcRn on the binding interface of human FcRn-Fc complex are generally similar to those of rat FcRn in the rat FcRn-Fc complex (red colored ones in Figure 1). One difference exists in residue #88 of human FcRn (Y88) that corresponds to residue F90 of rat FcRn. Another difference exists in the hydrophobic residue L135 of human FcRn corresponding to a negatively charged residue D137 of rat FcRn. This difference on the binding interface could affect the affinity of the FcRn-Fc binding. As observed in the X-ray crystal structure of rat FcRn-Fc,26 the negatively charged residue D137 could form a salt bridge with residue H436 (the numbering used in the crystal structure) of rat Fc when H436 is protonated.
As seen in Figure 2, the light chain of human FcRn (β2m) shares an overall high homology (~85%) with the light chain of FcRn from another species (monkey, mouse, or rat). Only the first two residues of the light chain are involved in the binding of FcRn. It has been demonstrated that the proper association with light chain is important for the normal biological function of FcRn, i.e. protecting IgG1 from the lysosomal degradation pathway.3,4,10,11
Figure 2.

Sequence alignment of the light chain (β2m) of FcRn from human (H), monkey (MK), rat (R), and mouse (M). Red colored residues are at the binding interface with Fc. The overall homology is ~85% for the light chain of Fc from these different species.
Shown in Figure 3 are the aligned sequences for Fc fragment of IgG1 from human, monkey, rat, and mouse. Except the obvious different amino acid sequence in the beginning of the Cγ2 domain, most of the remaining sequence of human Fc is similar to these from other species, particularly for human Fc amino acids (red colored ones in Figure 3) located on the interface of binding with FcRn. Fc from all these species share the same site of glycosylation corresponding to residue #83 (N83, blue colored in Figure 3) of human Fc. At residue #74, human Fc bears a positively charged lysine (K74) corresponding to a polar residue glutamine (Q288) of rat Fc. The Q288 of rat Fc is located nearby residue Q2 from the light chain of rat FcRn as shown in the X-ray crystal structure of rat FcRn-Fc complex.26
Figure 3.
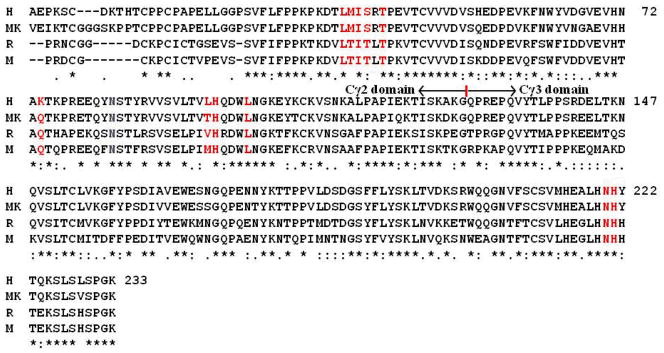
Sequence alignment of human Fc from human (H), monkey (MK), rat (R), and mouse (M). Residues in red color are on the binding interface with human FcRn. The blue colored residue is the common glycosylation site. Indicated is the rough division position (red straight line and black arrows) between the Cγ2 and Cγ3 domains of Fc.
Figure 4 represents the energy-minimized structure of human FcRn-Fc binding complex, in comparison with the template rat FcRn-Fc complex, ligand-free human FcRn complex, and ligand-free human Fc structures reported in literature.22,26,27,28 The overall structure of human FcRn-Fc complex (Figure 4A) resembles that of the rat FcRn-Fc complex26 with the positional root-mean-square deviation (RMSD) for Cα atoms being only 0.958 Å, suggesting the high fidelity for the human FcRn-Fc binding structure obtained from the current modeling and MD simulations. The FcRn structure (Figure 4A) in the human FcRn-Fc complex is also very close to the reported ligand-free human FcRn structure22 with the RMSD value for Cα atoms being only 0.79 Å (Figure 4B). The structural similarity between the human FcRn bound with human Fc and the ligand-free human FcRn is consistent with the observation that the rat FcRn structure kept a very similar conformation in both pH 6.5 and pH 8.0.20 The heavy chain of human FcRn (Figure 4A) in the FcRn-Fc complex is composed of eight-stranded anti-parallel β-sheet which is topped by α1 and α2 helices. The light chain of human FcRn in the FcRn-Fc complex interacts with the heavy chain by contacting the underside of the α1 and α2 helices. However, the conformation of human Fc structure in the human FcRn-Fc complex is quite different from ligand-free human Fc structures. When superimposed with the reported ligand-free Fc structures (Figure 4C),27,28 significant conformational difference can be observed for the Cγ2 domain of each subunit of human Fc with positional RMSD values being above 1.50 Å, although the difference is smaller for the hinge region between the Cγ2 and Cγ3 domains. The overall structural flexibility of human Fc suggests that human Fc may always adjust its conformation when it associates with either partner protein like human FcRn or is glycosylated differentially under different physiological stimulus.
Figure 4.
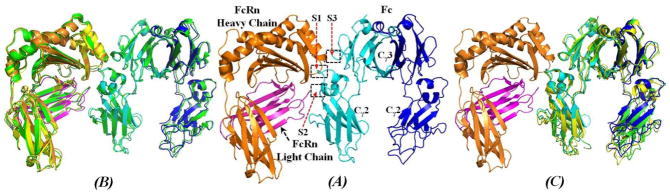
The energy-minimized structure of human FcRn-Fc binding complex in comparison with reported X-ray crystal structures of lignd-free human FcRn and human Fc. (A) The heavy chain of human FcRn is represented as orange ribbon, and magenta ribbon for the light chain (β2m) of human FcRn. Human Fc is also represented as ribbon colored in cyan for one subunit and blue for another subunit. The binding interface between human FcRn and human Fc is roughly represented as three subsites (S1, S2, and S3) as labeled. (B) Human FcRn-Fc complex superimposed with that of rat FcRn-Fc complex26 (PDB code 1I1A, green ribbon), ligand-free human FcRn22 (PDB code 3M17, yellow ribbon); only Cα atoms were used in the superimposition. The positional RMSD for Cα atoms between human FcRn-Fc structure and the rat FcRn-Fc structure is 0.96 Å, and the RMSD between the human FcRn in human FcRn-Fc complex and the ligand-free human FcRn is 0.79 Å. (C) Human FcRn-Fc binding complex superimposed with two typical ligand-free Fc structures. One ligand-free human Fc structure corresponding to PDB code 3AVE28 (green ribbon) has a positional RMSD value of 1.55 Å for the Cα atoms. Another ligand-free human Fc structure corresponding to PDB code 1H3Y27 (yellow ribbon) has an RMSD value of 1.85 Å. Obvious differences appeared at the relative position of the Cγ2 domain of each subunit of human Fc dimer.
A detailed inspection of the binding interface for the human FcRn-Fc complex reveals that the binding site spans a large surface area and is highly complementary. The binding site of human FcRn for Fc is composed of the top portion of α1 and α2 helices of the heavy chain and the first two residues of the light chain. On the human Fc side, the binding site for human FcRn mainly consists of the hinge region between the Cγ2 and Cγ3 domains. The whole binding interface can be roughly divided into three subsites: S1, S2, and S3 (Figure 4A). Depicted in Figure 5 are the structural features for all of the subsites (Figure 5A) and the overall intermolecular interactions between human FcRn and human Fc (Figure 5B). Summarized in Table 1 are typical inter-molecular interactions including hydrophobic contacts and hydrogen bonding as observed in the energy-minimized human FcRn-Fc binding structure. Figure 6 shows the tracked distances for several typical inter-molecular interactions derived from the MD trajectory. As shown in Figure 5A, the S1 subsite is the only one hydrophobic site composed by residues L112, F117, and W131 of FcRn. The hydrophobic residue I39 of human Fc is embedded very well inside this pocket. This can be demonstrated by three important distances tracked from the MD trajectory, i.e. the distance between the center of I39 side chain from Fc and the center of L112 side chain from FcRn; the distance between the center of I39 side chain of Fc and the center of the aromatic side chain of F117 of FcRn; and the distance between the center of I39 side chain of Fc and the center of aromatic side chain of W131 of FcRn (lower panel of Figure 6). Other residues such as L37, M38, S40, and T42 of Fc are anchored around the inside wall of this hydrophobic well, indicating that this hydrophobic core is critical for the association between human FcRn and Fc. The subsite S2 is located right under subsite S1, and is formed by the salt bridge between negatively charged E115 from the heavy chain of FcRn and the protonated H96 of Fc (Figure 5A, upper panel of Figure 6). The protonated H96 of Fc is also hydrogen-bonded with the carbonyl oxygen atom on the backbone of K41. E115 from the heavy chain of FcRn is also salt-bridged with the positively charged N-terminal group of residue I1 from the light chain of FcRn, and is hydrogen-bonded with the –NH group on the backbone of H96 of Fc (Figure 5B). The subsite S3 is situated just above subsite S1. It is formed by a salt bridge between the negatively charged residue D130 of FcRn and the protonated H221 of Fc (Figure 5, upper panel of Figure 6). The protonated H221 of Fc also closely contacts with the aromatic side chain of residue W131 of FcRn through cation-π interactions (Figure 5B, upper panel of Figure 6). As the protonated H96 and H221 play such important roles in human FcRn-Fc binding by forming critical salt bridges and cation-π interactions under the acidic condition, it is reasonable to assume that human FcRn-Fc binding at the physiological pH 7.4 can be weakened dramatically due to the break of these critical inter-molecular interactions. Other complementary intermolecular interactions include the hydrophobic contacts between residue W131 of FcRn and L100 of Fc, hydrophobic contacts between residue P132 of FcRn and L37 of Fc, hydrophobic contacts between residue I1 of the light chain of FcRn and residue L95 of Fc (Figure 5B, Table 1). In addition, the negatively charged E133 side chain and the hydroxyl group on the Y88 side chain of FcRn are both hydrogen-bonded with residue S40 of Fc. As shown in Figure 5B and listed in Table 1, N220 side chain of Fc is hydrogen-bonded with the carbonyl oxygen atoms on the backbone of G129 and W131 of FcRn. The positively charged side chain of K74 of Fc is hydrogen-bonded with the carbonyl oxygen on the side chain of residue Q2 from the light chain of FcRn (Figure 5B, Table 1).
Figure 5.
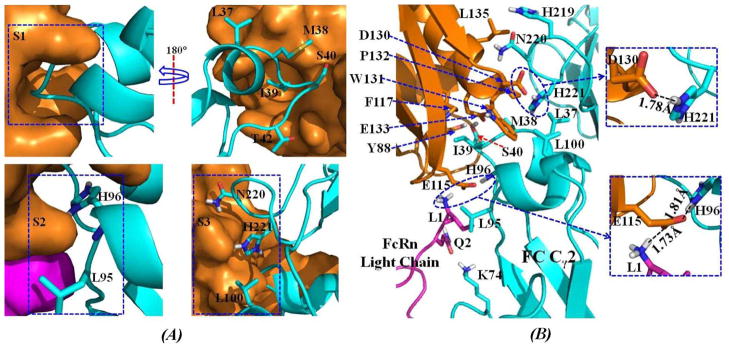
(A) Structural features of the subsites S1, S2, and S3 on the binding interface of human FcRn-Fc complex. The interface on the FcRn side is represented by molecular surface, while residues of Fc are show as sticks. The color scheme is the same as that shown in Figure 4. (B) Atomic details of the inter-molecular interactions on the binding interface of human FcRn-Fc complex. The proteins are represented as colored ribbon. The residues on the binding interface are shown as sticks and the hydrogen bonding interactions are represented as dashed lines with the distances labeled in Å. The color scheme is also the same as that in Figure 4.
Table 1.
Key inter-molecular interactions in the optimized human FcRn-Fc binding structure.
| FcRn | Distance (Å) | Fc | ||
|---|---|---|---|---|
| Hydrophobic contacts (listed as shortest distance, cutoff of 5 Å) | ||||
| residue | Atom | atom | residue | |
| W131 | Cε2 | 4.43 | Cγ2 | I39 |
| L112 | Cδ2 | 3.88 | Cδ1 | I39 |
| F117 | Cβ | 3.75 | Cδ1 | I39 |
| P132 | Cγ | 4.12 | Cδ2 | L37 |
| I1 (β2m) | Cβ | 4.08 | Cδ1 | L95 |
| W131 | Cζ2 | 3.67 | Cδ2 | L100 |
| Salt bridge and hydrogen bonding | ||||
| D130 | Oδ2 | 1.78(salt bridge) | Hε2 | H221 |
| E115 | Oε2 | 1.81(salt bridge) | Hδ1 | H96 |
| E115 | Oε2 | 1.74 (Hydrogen bonding) | Backbone NH | H96 |
| E133 | Oε1 | 1.90 (Hydrogen bonding) | Backbone NH | S40 |
| Y88 | Hydroxyl hydrogen | 2.00 (Hydrogen bonding) | Oγ | S40 |
| G129 | O=C | 2.13(Hydrogen bonding) | Hδ21 | N220 |
| W131 | O=C | 2.02 (Hydrogen bonding) | Hδ22 | N220 |
| Q2 (β2m) | Oε1 | 1.77 (Hydrogen bonding) | Hζ | K74 |
Figure 6.
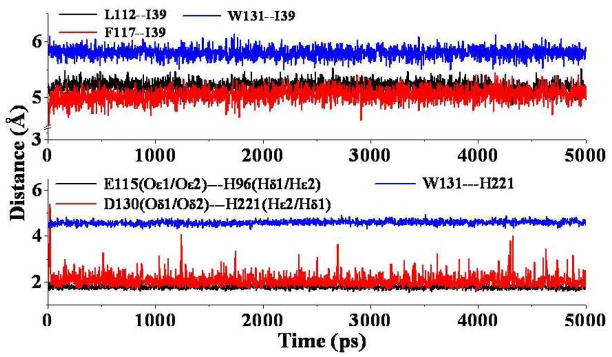
Tracked key internuclear distances in the MD-simulated human FcRn-Fc binding structure. Upper panel: L112—I39 represents the distance between the center of L112 side chain of FcRn and the center of I39 side chain of Fc; F117--I39 refers to the distance between the center of F117 side chain of FcRn and the center of I39 side chain of Fc; and W131—I39 refers to the distance between the center of W131 side chain of FcRn and the center of I39 side chain of Fc. Lower panel: E115(Oε1/Oε2)—H96(Hδ1/Hε2) represents the shortest distance between the charged atoms (Oε1 or Oε2) on E115 side chain of FcRn and the hydrogen atoms on the protonated H96 side chain of Fc; D130(Oδ1/Oδ2)--H221(Hε2/Hδ1) refers to the shortest distance between the negatively charged atoms (Oδ1 or Oδ2) on D130 side chain of FcRn and the hydrogen atoms on the protonated H221 side chain of Fc; and W131—H221 refers to the cation-π interaction distance between the center of W131 side chain of FcRn and the center of H221 side chain of Fc.
Comparison with available experimental data
As a large number of Fc mutants have been tested and reproted,10,11,19,33,36,39 it is interesting for validation of the modeling to appropriately compare our modeled human FcRn-Fc binding structure with the available experimental data. As discussed below, the modeled human FcRn-Fc binding structure is qualitatively consistent with all of the experimental observations and data.
All of the reported mutants can be categorized roughly into three groups. One group of mutants were focused on subsite S1 (Figure 5A) around residues #38, #40, and #42. Another group of mutants were focused on residue #220, which is nearby subsite S2 (Figure 5B). The third group is associated with mutations on residues that are far away from human FcRn-Fc binding interface. As shown in our modeled human FcRn-Fc binding structure, residue I39 of Fc is orientated inside the hydrophobic pocket formed by residues L112, F117, and W131 of FcRn. If I39 side chain of Fc is replaced with a smaller or bigger one, it will definitely reduce the hydrophobic packing between FcRn and Fc. The observation that the I39A mutant of Fc showed no detectable affinity with FcRn33 is consistent with the structural features around subsite S1 of our modeled FcRn-Fc binding structure. According to our modeled human FcRn-Fc binding structure, residue M38 of Fc is close to R41 side chain of Fc on the surface, and has no direct contact with the surrounding polar or charged residues like S40 of Fc and E133 of FcRn. Our modeled FcRn-Fc binding structure suggests that the binding affinity is expected to improve when residues M38, S40, and/or T42 of Fc are mutated into bigger, polar, or charged residues. Specifically, if M38 is mutated into a polar or aromatic residue, it could improve the local intermolecular or intra-molecular interactions, e.g. better packing with positively charged side chain of residue R41 of Fc would enhance the intra-molecular interaction of Fc. As observed in our modeled M38-related mutant-type FcRn-Fc structures, the M38Y, M38W, M38F/T42D, and M38Y/S40T/T42E mutations do have cation-π interactions with residue R41 of Fc (Figure 7A). Accordingly, these improvements around subsite S1 on residues S40 and T42 together with M38 of Fc are beneficial to the FcRn-Fc association. These observations around residue M38 of Fc from our modeled human FcRn-Fc binding structure and the mutant structures agree well with the results of experimentally measured Kd values (Table 2) and the increased in vivo half-life of these mutants focused on M38, S40, and T42.19,32,33,35
Figure 7.
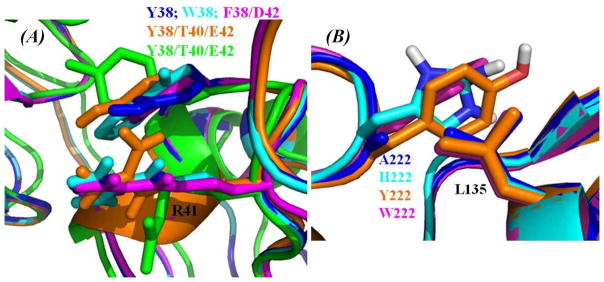
(A) Representative of the intra-molecular interactions between the aromatic side chain of residue #38 and the positively charged side chain of residue R41 from human Fc after superimposing with the mutant-type human FcRn-Fc binding structures. M38Y mutant is colored blue, M38W mutant is colored cyan, M38F/T42D mutant is colored magenta, M38Y/S40T/T42E mutant of Fc in the modeled human FcRn-Fc complex is colored orange, and the ligand-free M38Y/S40T/T42E mutant Fc structure from X-ray diffraction22 is colored green. (B) The intermolecular packing between residue L135 of human FcRn and the mutated residues at #222 of human Fc in the modeled mutant-type FcRn-Fc binding structures. N222A mutant of Fc is colored blue, N222H mutant is colored cyan, N222Y mutant is colored orange, and N222W mutant is colored magenta.
Table 2.
Calculated Fc mutation-caused shifts (ΔΔEcalc, kcal/mol) of human FcRn-Fc binding energy in comparison with available experimental data (ΔΔGexpt, kcal/mol).
| Fc and its mutants | ΔΔEcalca | ΔΔGexptb | Kd (nM)c | Ref. |
|---|---|---|---|---|
| Human IgG1-Fc | 0.00 | 0.00 | 2527±117 | 33 |
| I39A | +33.88 | N.D.d | 33 | |
| M38Y | −21.45 | −0.92 | 532±37 | 33 |
| M38W | −20.72 | −1.08 | 408±24 | 33 |
| M38F/T42D | −32.45 | −0.59 | 933±170 | 33 |
| M38Y/T42Q | −5.28 | −0.89 | 560±102 | 33 |
| N220F/Y222H | −67.35 | −0.97 | 493±7 | 33 |
| M38Y/S40T/T42E | −13.03 | −1.43 | 225±10 | 33 |
| V94T/L95P/Q97S | −11.18 | −0.15 | 1964±84 | 33 |
| G171D/Q172P/N175S | −30.57 | −0.09 | 2164±331 | 33 |
| H219R/N220Y/Y222H | −54.46 | −0.99 | 472±61 | 33 |
| H219K/N220F/Y222H | −61.84 | −1.09 | 399±47 | 33 |
| G171R/Q172T/P173R/N175P | −13.11 | −0.26 | 1620±61 | 33 |
| M38Y/S40T/T42E/H219K/N220F/Y222H | −45.23 | −2.40 | 44±3 | 33 |
| M38Y/S40T/T42E/G171R/Q172T/P173R/N175P | −11.54 | −1.39 | 243±48 | 33 |
| Human Fc | 0.00 | 0.00 | 1700±20 | 39 |
| N220A | −23.11 | −0.65 | 570±30 | 39 |
| N220H | −6.43 | −0.90 | 370±30 | 39 |
| N220Y | −66.97 | −1.83 | 78±12 | 39 |
| N220W | −57.24 | −2.16 | 44±5 | 39 |
| A164V/N220A | −26.04 | −0.87 | 390±40 | 39 |
| N220A/Y222I | −14.98 | −0.95 | 340±80 | 39 |
| M38Y/N220A | −18.22 | −1.21 | 220±20 | 39 |
| M214L/N220A | −17.93 | −1.03 | 300±40 | 39 |
| V94P/N220Y | −65.17 | −3.03 | 10.2±1.8 | 39 |
| T93Q/N220A | −8.67 | −1.17 | 235±20 | 39 |
| T36Q/M214L | −28.63 | −1.17 | 234±22 | 39 |
| V94P/N220A | −18.04 | −1.84 | 76±10 | 39 |
| T93Q/E166A/N220A | −5.41 | −1.07 | 280±10 | 39 |
| M38Y/V94P/N220Y | −60.76 | −3.63 | 3.7±1.0 | 39 |
| Human Fc | 0.00 | 0.00 | 37±5 | 36 |
| D162V/N220H | −28.31 | −1.60 | 2.5±1.4 | 36 |
| P43I/Q97I | −5.47 | −1.76 | 1.9±0.4 | 36 |
| P43I/N220H | −11.22 | −1.65 | 2.3±0.2 | 36 |
Directly calculated binding free energy shift from human FcRn binding with wild-type human Fc to human FcRn binding with the mutant of human Fc.
The binding free energy shift (from human FcRn binding with wild-type human Fc to human FcRn binding with the mutant of human Fc) derived from the experimentally measured dissociation constant (Kd) change as according to equation ΔΔG=RTlnKd(mutant)/Kd(wild-type).
The experimental dissociation constant for the FcRn-Fc binding reported in literature cited.
No detectable binding.
The results of sequence alignment (Figures 1 and 3) and our modeled human FcRn-Fc binding structure (Figures 4 and 5) suggest that, residue N220 of human Fc is not compatible with the neighboring hydrophobic residue L135 of human FcRn. Although residue N220 is conserved among all the species (human, monkey, rat, and mouse), the counter partner on the FcRn side for residue N220 of Fc is changed from the negatively charged residue (D137 of rat Fc or E137 of mouse Fc) to a hydrophobic leucine (L135 of human FcRn or monkey FcRn). Structurally, this change from D/E to L on the FcRn side from mouse/rat to human/monkey does certainly decrease the binding affinity of human FcRn-Fc binding. Accordingly, the binding affinity of rat FcRn associated with rat Fc under acidic condition should be higher than that of human FcRn associated with human Fc. This structural difference on the binding interface of FcRn-Fc complex is the reason why the experimentally measured affinity of wild-type rat/mouse FcRn-Fc binding is higher than that of the wild-type human FcRn-Fc binding.33 This structural difference between the L135-N220 neighborhood in human FcRn-Fc complex and the D137-N448 neighborhood in rat FcRn-Fc complex might be one of the factors contributing to the dramatic difference across species for the affinity of FcRn-Fc binding.45,46 Hence, the wild-type mouse FcRn can still bind with wild-type human Fc at physiological pH 7.4 with the affinity at μM level. Based on our modeled structure of human FcRn-Fc complex (Figures 4 and 5), we would propose that residue N220 of human Fc be mutated into a smaller hydrophobic residue such as alanine or a larger aromatic residues such as tyrosine or tryptophan in order to enhance the human FcRn-Fc association. Our modeled mutant-type human FcRn-Fc complexes generated from the mutations on residue N220 of human Fc also indicate that N220A, N220Y, N220H, and N220W are all good choices to enhance the affinity of human FcRn-Fc binding (Figure 7B). The reported affinity changes (see Table 2)19,34,36,39 of human FcRn-Fc binding from the mutations involving residue N220 are consistent with the observable structural features around N220 residue in our modeled structure of human FcRn-Fc complex.
Based on our modeled human FcRn-Fc binding structure, the T36Q mutation may enhance the interactions between the Cγ2 and Cγ3 domains of Fc, thus further stabilizing the FcRn-Fc complex. According to the Fc structure in our modeled human FcRn-Fc complex, residue P43 packs against the protonated side chain of residue H96 and, thus, the P43I mutation should improve the intra-molecular packing of Fc. The T93Q mutation may help to increase the opportunity of hydrogen bonding with residue Q2 from the light chain of FcRn and, thus, it can be expected that the T93Q mutation is beneficial to FcRn-Fc binding. The V94T mutation of Fc may enhance the intra-molecular hydrogen bonding with its neighboring backbone carbonyl atoms of Fc. The L95P, Q97S, and Q97I mutations would not benefit the inter-molecular interactions between human Fc and the light chain of human FcRn. In the human Fc structure, residue D162 is neighbored with residues P33, L37, and W99 so that the D162V mutation may make a better intra-molecular packing and, thus, indirectly enhance the FcRn-Fc association. As residue A164 of Fc is situated between the Cγ2 and Cγ3 domains, and it is close to residue P33 of human Fc, the A164V mutation may improve its hydrophobic interaction with residue P33 and, thus, is helpful for human FcRn-Fc binding. Residues G171, G172, P173, and N175 of human Fc are located far away from the binding interface in human FcRn-Fc complex and, hence, these residues should have no direct impact on the affinity of FcRn-Fc binding. As observed in the Fc structure in human FcRn-Fc complex, residue M214 is close to M38, but not directly involved in the inter-molecular interaction. It can be expected that the M214L mutation has no significant effect on the human FcRn-Fc binding.
In order to better understand the structure-binding affinity correlation based on the experimental binding affinity data available in literature,33,36,39 we performed binding energy calculations by using the ZRANK score function based on the energy-minimized mutant-type human FcRn-Fc binding structures. By using the calculated binding energies, we evaluated the binding energy difference ΔΔEcalc, i.e. the mutational-caused shift of the binding energy from the wild-type FcRn-Fc binding to the mutant-type FcRn-Fc binding (ΔΔEcalc = ΔEmutant –ΔEwild-type). Summarized in Table 2 are the calculated ΔΔEcalc values, along with the reported dissociation constant (Kd) values and the corresponding binding free energy differences derived from the use of experimental Kd values and equation ΔΔG = RTlnKd(mutant)/Kd(wild-type). Based on the calculated results, a negative ΔΔEcalc value suggest that the particular Fc mutation should enhance to the human FcRn-Fc binding, whereas a positive ΔΔEcalc value will be definitely detrimental to the FcRn-Fc association. According to the calculated ΔΔEcalc values listed in Table 2, ΔΔEcalc is a plus value for the I39A mutant, but a negative value for each of the remaining mutants of human IgG1-Fc, suggesting that all these mutants of human Fc should enhance the affinity of human FcRn-Fc binding except the I39A mutant.
It should be noted that the above-discussed binding energy calculations using the ZRANK score function were all performed on the human FcRn-Fc binding structures that were energy-minimized with explicit solvent water molecules. Further, we also energy-minimized the wild-type and mutant-type human FcRn-Fc binding structures by using an implicit solvent model, i.e. Generalized Born (GB) model,56,57 and repeated the binding energy calculations (using the same ZRANK score function) with the energy-minimized human FcRn-Fc binding structures. It turned out that the ΔΔEcalc values (provided as Supplementary Information) calculated with the human FcRn-Fc binding structures based on the implicit solvent model are close to the corresponding the ΔΔEcalc values (Table 2) calculated with the human FcRn-Fc binding structures based on the explicit solvent water molecules, with a high linear correlation coefficient (R = 0.99). Both the explicit and implicit solvent models led to similar ΔΔEcalc values, with the signs of the calculated ΔΔEcalc values being consistent with those of the corresponding experimental data.
On the other hand, one may also note in Table 2 that the directly calculated ΔΔEcalc value is always either much more negative or much more positive compared to the experimentally derived binding free energy difference (ΔΔGexpt). In other words, the calculations using the ZRANK score function systematically estimated the binding energy shifts from the wild-type Fc to the mutants. Nevertheless, based on the data collected in Table 2, there is a reasonable (though not perfect) linear correlation relationship between the calculated ΔΔEcalc values and the experimentally derived ΔΔGexpt values, with a correlation coefficient of 0.54 and the standard deviation of 0.68 kcal/mol. The reasonable agreement between the calculated and experimental binding energies further supports the conclusion that our modeled human FcRn-Fc binding structure is reasonable. Based on the modeled human FcRn-Fc binding structure, in order to predict the binding energy more accurately, one might have to perform a potential of mean force (PMF) simulation (with a sufficiently large number of windows) on the binding/unbinding process of each FcRn-Fc binding complex (with the wild-type or a mutant), as we did for other protein-ligand binding complexes.58,59,60 On the other hand, the MD sampling-based PMF simulations on all of the FcRn-Fc binding complexes (with the wild-type and mutants) concerned in the current study would be very time-consuming. In any situation, it can be expected that our modeled human FcRn-Fc binding structure will help to design novel human Fc mutants that may be used to significantly prolong the in vivo half-lives of Fc-fused therapeutic proteins.
Conclusion
The detail analysis of the FcRn and IgG1 Fc sequences from various species (human, monkey, rat, and mouse) has revealed the interesting similarity and differences between different species. Based on the aligned amino acid sequences, the structure of human FcRn binding with human Fc has been constructed by performing homology modeling and molecular dynamics simulations. According to the modeled human FcRn-Fc binding structure, the binding interface of human FcRn with human Fc is composed of three subsites denoted as S1 to S3. The first subsite S1 is a hydrophobic core where residue I39 of human Fc can be accommodated very well. The second subsite S2 and the third subsite S3 are all composed of critical salt bridges between human FcRn and human Fc. The modeled structures of human FcRn binding with wild-type human Fc and its mutants, as well as the calculated binding free energies, are qualitatively consistent with the available experimental data, suggesting that the modeled human FcRn-Fc binding structure is reasonable. The modeled human FcRn-Fc binding structure may be valuable for rational design of novel mutants of human Fc and Fc-fused therapeutic proteins that can more potently bind with human FcRn to prolong their in vivo half-lives in human.
Supplementary Material
Acknowledgments
This work was supported by the NIH grants to Zhan (R01 DA035552, R01 DA032910, R01 DA013930, and R01 DA025100). The authors also acknowledge the Computer Center at University of Kentucky for supercomputing time on a Dell Supercomputer Cluster consisting of 388 nodes or 4,816 processors.
Footnotes
Supplementatry Information Available. A table (Table S1) for the calculated Fc-mutation-caused shifts of binding energy based on the implicit solvent model; a figure (Figure S1) for the plots of the calculated shifts of binding energy vs the experimentally-derived shifts of binding free energy. These materials are free via the Internet http://pubs.rsc.org.
References
- 1.Simister NE, Mostov KE. Nature. 1989;337:184–187. doi: 10.1038/337184a0. [DOI] [PubMed] [Google Scholar]
- 2.Garg A, Balthasar JP. The AAPS J. 2009;11(3):553–557. doi: 10.1208/s12248-009-9129-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wines BD, Trist HM, Farrugia W, Ngo C, Trowsdale J, Areschoug T, Lindahl G, Fraser JD, Ramsland PA. Curr Topics Innate Immu II. Adv Expt Med Biol. 2012;946:87–112. doi: 10.1007/978-1-4614-0106-3_6. [DOI] [PubMed] [Google Scholar]
- 4.Jefferis R. Arch Biochem Biophys. 2012;526(2):159–166. doi: 10.1016/j.abb.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 5.Kristoffersen EK, Matre R. Eur J Immunol. 1996;26(2):505–507. doi: 10.1002/eji.1830260234. [DOI] [PubMed] [Google Scholar]
- 6.Leach JL, Sedmak DD, Osborne JM, Rahill B, Lairmore MD, Anderson CL. J Immunol. 1996;157(8):3317–3322. [PubMed] [Google Scholar]
- 7.Blumberg RS, Koss T, Story CM, Barisani D, Polischuk J, Lipin A, Pablo L, Green R, Simister NE. J Clin Invest. 1995;95(5):2397–402. doi: 10.1172/JCI117934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Israel EJ, Taylor S, Wu Z, Mizoguchi E, Blumberg RS, Bhan A, Simister NE. Immunology. 1997;92(1):69–74. doi: 10.1046/j.1365-2567.1997.00326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlachetzki F, Zhu C, Pardridge WM. J Neurochem. 2002;81(1):203–206. doi: 10.1046/j.1471-4159.2002.00840.x. [DOI] [PubMed] [Google Scholar]
- 10.Woof JM, Burton DR. Nature Rev Immunol. 2004;4(2):89–99. doi: 10.1038/nri1266. [DOI] [PubMed] [Google Scholar]
- 11.Roopenian DC, Akilesh S. Nature Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 12.Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, Eden PA, Anderson CL. J Immunol. 2003;170:3528–3533. doi: 10.4049/jimmunol.170.7.3528. [DOI] [PubMed] [Google Scholar]
- 13.Goebl NA, Babbey CM, Datta-Mannan A, Witcher DR, Wroblewski VJ, Dunn KW. Mol Biol Cell. 2008;19(12):5490–5505. doi: 10.1091/mbc.E07-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beck A, Wurch T, Bailly C, Corvaia N. Nature Rev Immunol. 2010;10:345–352. doi: 10.1038/nri2747. [DOI] [PubMed] [Google Scholar]
- 15.Huang C. Curr Opin Biotech. 2009;20:692–699. doi: 10.1016/j.copbio.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Carter PJ. Expt Cell Res. 2011;317:1261–1269. doi: 10.1016/j.yexcr.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Dustin ML, Starr T, Coombs D, Majeau GR, Meier W, Hochman PS, Douglass A, Vale R, Goldstein B. J Biol Chem. 2007;282(48):34748–34757. doi: 10.1074/jbc.M705616200. [DOI] [PubMed] [Google Scholar]
- 18.Davis PM, Abraham R, Xu L, Nadler SG, Suchard SJ. J Rheumatol. 2007;34:2204–2210. [PubMed] [Google Scholar]
- 19.Strohl W. Curr Opin Biotech. 2009;20:685–691. doi: 10.1016/j.copbio.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Vaughn DE, Bjorkman PJ. Structure. 1998;6:63–73. doi: 10.1016/s0969-2126(98)00008-2. [DOI] [PubMed] [Google Scholar]
- 21.West AP, Bjorkman PJ. Biochem. 2000;39:9698–9708. doi: 10.1021/bi000749m. [DOI] [PubMed] [Google Scholar]
- 22.Mezo AR, Sridhar V, Badger J, Sakorafas P, Nienaber V. J Biol Chem. 2010;285(36):27694–27701. doi: 10.1074/jbc.M110.120667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burmeister WP, Huber AH, Bjorkman PJ. Nature. 1994;372:379–383. doi: 10.1038/372379a0. [DOI] [PubMed] [Google Scholar]
- 24.Martin WL, Bjorkman PJ. Biochem. 1999;38:12639–12647. doi: 10.1021/bi9913505. [DOI] [PubMed] [Google Scholar]
- 25.DeLano WL, Ultsch MH, de Vos AM, Wells JA. Science. 2000;287:1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- 26.Martin WL, West AP, Jr, Gan L, Bjorkman PJ. Mol Cell. 2001;7:867–877. doi: 10.1016/s1097-2765(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 27.Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. J Mol Biol. 2003;325:979–989. doi: 10.1016/s0022-2836(02)01250-0. [DOI] [PubMed] [Google Scholar]
- 28.Matsumiya S, Yamaguchi Y, Saito JI, Nagano M, Sasakawa H, Otaki S, Satoh M, Shitara M, Kato K. J Mol Biol. 2007;368:767–779. doi: 10.1016/j.jmb.2007.02.034. [DOI] [PubMed] [Google Scholar]
- 29.Feige MJ, Nath S, Catharino SR, Weinfurtner D, Steinbacher S, Buchner J. J Mol Biol. 2009;391:599–608. doi: 10.1016/j.jmb.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 30.Crispin M, Bowden TA, Coles CH, Harlos K, Aricescu AR, Harvey DJ, Stuart DI, Jones EY. J Mol Biol. 2009;387:1061–1066. doi: 10.1016/j.jmb.2009.02.033. [DOI] [PubMed] [Google Scholar]
- 31.Baruah K, Bowden TA, Krishna BA, Dwek RA, Crispin M, Scanlan CN. J Mol Biol. 2012;420(1–2):1–7. doi: 10.1016/j.jmb.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oganesyan V, Damschroder MM, Woods RM, Cook KE, Wu H, Dall’Acqua WF. Mol Immunol. 2009;46:1750–1755. doi: 10.1016/j.molimm.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 33.Dall’sAcqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. J Immunol. 2002;169:5171–5180. doi: 10.4049/jimmunol.169.9.5171. [DOI] [PubMed] [Google Scholar]
- 34.Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Khabbaz HA, Brown AC, Presta LG, Meng YG, Roopenian DC. Int Immu. 2006;18(12):1759–1769. doi: 10.1093/intimm/dxl110. [DOI] [PubMed] [Google Scholar]
- 35.Dall’Acqua WF, Kiener PA, Wu H. J Biol Chem. 2006;281(33):23514–23524. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- 36.Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Jiang W. Drug Metab Dispo. 2007;35:86–94. doi: 10.1124/dmd.106.011734. [DOI] [PubMed] [Google Scholar]
- 37.Ying T, Chen W, Gong R, Feng Y, Dimitrov DS. J Biol Chem. 2012;287(23):19399–19408. doi: 10.1074/jbc.M112.368647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis PM, Abraham R, Xu L, Nadler SG, Suchard SJ. J Rheumatol. 2007;34:2204–2210. [PubMed] [Google Scholar]
- 39.Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, Starovasnik MA, Lowman J Immunol. 2009;182:7663–7671. doi: 10.4049/jimmunol.0804182. [DOI] [PubMed] [Google Scholar]
- 40.Heider KH, Kiefer K, Zenz T, Volden M, Stilgenbauer S, Ostermann E, Baum A, Lamche H, Küpcü Z, Jacobi A, Müller S, Hirt U, Adolf GR, Borges E. Blood. 2011;118:4159–4168. doi: 10.1182/blood-2011-04-351932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuo TT, Baker K, Yoshida M, Qiao SW, Aveson VG, Lencer WL, Blumberg RS. J Clin Immunol. 2010;30:777–789. doi: 10.1007/s10875-010-9468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Drug Metab Dispo. 2011;39:1469–1477. doi: 10.1124/dmd.111.039453. [DOI] [PubMed] [Google Scholar]
- 43.Dumont JA, Liu T, Low SC, Zhang X, Kamphaus G, Sakorafas P, Fraley C, Drager D, Reidy T, McCue J, Franck HWG, Merricks EP, Nichols TC, Bitonti AJ, Pierce GF, Jiang H. Blood. 2012;119:3024–3030. doi: 10.1182/blood-2011-08-367813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wozniak-Knopp G, Stadlmann J, Rüker F. PLoS One. 2012;7(1):e30083. doi: 10.1371/journal.pone.0030083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andersoen JT, Daba MB, Berntzen G, Michaelsen TE, Sandlie I. J Biol Chem. 2010;285(7):4826–4836. doi: 10.1074/jbc.M109.081828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andersen JT, Foss S, Kenanova VE, Olafsen T, Leikfoss IS, Roopenian D, Wu AM, Sandlie I. J Biol Chem. 2012;287(27):22927–22937. doi: 10.1074/jbc.M112.355131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pierce B, Weng Z. Proteins Struct Funct Bioinfo. 2007;67:1078–1086. doi: 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- 48.Pierce B, Weng Z. Proteins Struct Funct Bioinfo. 2008;72:270–279. doi: 10.1002/prot.21920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henikoff S, Henikoff JG. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman P. AMBER 11. University of California; San Francisco: 2010. [Google Scholar]
- 52.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 53.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 54.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 55.Mintseris J, Pierce B, Wiehe K, Anderson R, Chen R, Weng Z. Proteins Struct Funct Bioinfo. 2007;69:511–520. doi: 10.1002/prot.21502. [DOI] [PubMed] [Google Scholar]
- 56.Hawkins GD, Cramer CJ, Truhlar DG. Chem Phys Lett. 1995;246:122–129. [Google Scholar]
- 57.Hawkins D, Cramer CJ, Truhlar DG. J Phys Chem. 1996;100:19824–19839. [Google Scholar]
- 58.Huang X, Zheng F, Zhan CG. J Phys Chem B. 2011;115:11254–11260. doi: 10.1021/jp2047807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang X, Zhao X, Zheng F, Zhan CG. J Phys Chem B. 2012;116:3361–3368. doi: 10.1021/jp2111605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hao GF, Tan Y, Wang ZF, Yang SG, Zhan CG, Xi Z, Yang GF. PLoS One. 2013;8:e69198. doi: 10.1371/journal.pone.0069198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.