


Abstract
Breakage-fusion-bridge (BFB) cycle is a series of chromosome breaks and duplications that could lead to the increased copy number of a genomic segment (gene amplification). A critical step of BFB cycles leading to gene amplification is a palindromic fusion of sister chromatids following the rupture of a dicentric chromosome during mitosis. It is currently unknown how sister chromatid fusion is produced from a mitotic break. To delineate the process, we took an integrated genomic, cytogenetic and molecular approach for the recurrent MCL1 amplicon at chromosome 1 in human tumor cells. A newly developed next-generation sequencing-based approach identified a cluster of palindromic fusions within the amplicon at ∼50-kb intervals, indicating a series of breaks and fusions by BFB cycles. The physical location of the amplicon (at the end of a broken chromosome) further indicated BFB cycles as underlying processes. Three palindromic fusions were mediated by the homologies between two nearby inverted Alu repeats, whereas the other two fusions exhibited microhomology-mediated events. Such breakpoint sequences indicate that homology-mediated fold-back capping of broken ends followed by DNA replication is an underlying mechanism of sister chromatid fusion. Our results elucidate nucleotide-level events during BFB cycles and end processing for naturally occurring mitotic breaks.
INTRODUCTION
Gene amplification, a selective copy-number increase of genomic segments through DNA rearrangements, is a clinically important form of genome instability in cancer, as gene amplification causes advanced tumors and acquired therapy resistance (1–4). Thus, a better understanding of the underlying mechanisms of gene amplification could improve prognosis of cancer patients. Cytogenetically, amplified genomic segments reside either in small chromosomes (double minute chromosomes, DM) or in intrachromosomal, homogenously staining regions (HSR) (5–9). Continuous DNA breaks and rearrangements through breakage-fusion-bridge (BFB) cycles have been implicated as an underlying mechanism for intrachromosomal gene amplification (10–12).
The BFB cycle was originally described by Barbara McClintock in 1939 as a fate of a dicentric chromosome during meiotic mitosis and endosperm development in maize (13). She observed the following: (i) ‘breakage’ of a dicentric chromosome in meiotic anaphase when the two centromeres pass to opposite poles, (ii) ‘fusion’ at the breakage site between two sister halves of the broken chromatid resulting in a duplicated chromatid with two centromeres, and (iii) the formation of a chromatid ‘bridge’ in the following mitotic anaphase. The bridge eventually ruptures, and broken chromatids enter into each daughter nuclei. Because the rupture can occur at any site between the two centromeres, the broken chromatids can inherit unequal amounts of genetic material: a partial inverted (palindromic) duplication in one chromatid and a partial deletion in the other (Figure 1a). The resulting broken chromatids repeat the cycle in following mitotic divisions, and, as a result, a segment between two centromeres is amplified in some cell descendants. This was observed phenotypically in kernels with extremely dark color because the gene required for pigment production was located between two centromeres. These observations inform about key steps leading to gene amplification: DNA double-strand breaks (DSBs) in mitosis and the subsequent sister chromatid fusion.
Figure 1.
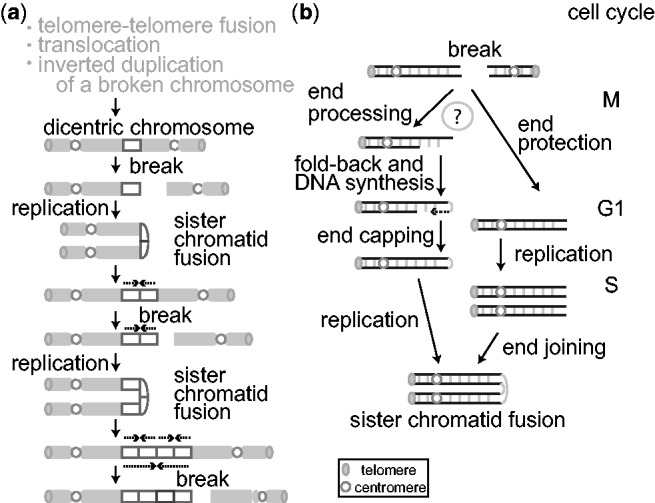
(a) Sister chromatid fusion during the BFB cycle promotes gene amplification. Only the fate of one broken chromatid is shown. Three events that can create a dicentric chromosome are listed on the top (see in the ‘Introduction’ section of the main text). Following a chromosome rupture and DNA replication, a broken chromatid undergoes inverted duplication (dotted arrows) with a fusion at the end (sister chromatid fusion), which results in the formation of a dicentric chromosome. Repeated occurrence of the cycle leads to the unequal distribution of chromsomal regions (rectangle). (b) Two models for the nucleotide-level mechanisms of sister chromatid fusion: end processing–capping-replication model (left) and NHEJ-dependent model (right).
In cancer cells, dicentric chromosomes can arise from a variety of events: the fusion of two chromosomes with extremely short telomeres (14,15), the fusion between two centromere-bearing broken non-homologous chromosomes (translocation) (16–18) and the inverted duplications of centromere-bearing broken chromosomes (19,20). Once formed, dicentric chromosomes can enter into BFB cycles and initiate gene amplification through sister chromatid fusion, as McClintock described for maize. If the resulting amplified segments harbor genes that promote cell proliferation, the cells can become dominant in a cancer cell population and confer aggressive tumor phenotypes. Despite the clinical importance, how sister chromatids fuse in tumor cells remains elusive. Cytogenetic studies provide ample evidence for the occurrence of BFB cycles in tumor cells (21–24) but do not provide base-pair resolution on palindromic junctions. Base-pair resolution can be obtained by recent next-generation sequencing (NGS)-based breakpoint analyses (25,26), but breakpoint sequences themselves are not direct evidence of sister chromatid fusion and can be produced by other mechanisms, such as replication fork stalling and template switching (27,28).
Two plausible mechanisms have been proposed for sister chromatid fusion (Figure 1b) (25,26,29,30). A broken end in mitosis would undergo end resection and leave a 3′ single-stranded DNA (ssDNA) tail. The ssDNA would fold back and anneal using homologies. DNA synthesis would fill the gap and complete the end capping. The entire chromosome would duplicate in the S-phase of next cell cycle to produce chromatids fused at the broken end. This intra-strand fold-back annealing of ssDNA between homologous sequences has been shown for endonuclease-induced DSBs in both mammalian cells and yeast (29,30). Alternatively, a non-homologous end joining (NHEJ)-based mechanism predicts that a broken end in mitosis would be protected in G1 phase and would enter into S phase. After DNA replication, the two ends of sister chromatids would fuse to each other by NHEJ.
Distinguishing these possibilities requires (i) identifying the amplicon that is created by BFB cycles and (ii) defining the DNA sequences at the breakpoints. A homology-dependent end-capping mechanism could exhibit homologies at breakpoints, whereas a NHEJ-dependent process could predict breakpoints either without homologies or with the insertion of non-templated sequences. In this manuscript, we address this issue for the recurrent chromosome 1q21 amplicon in human cancer cells using an integrated NGS-based genomic, cytogenetic and molecular approach.
MATERIALS AND METHODS
Genome-wide analysis of palindrome formation (GAPF-seq) and array-comparative genomic hybridization
The colorectal cancer cell line Colo320DM and primary fibroblast IMR90 were obtained from ATCC. Genomic DNA was processed for genome-wide analysis of palindrome formation (GAPF) as previously described with minor modifications (31). Two micrograms of genomic DNA was used as starting material, of which 1 μg was digested with KpnI and 1 μg was digested with SbfI for 16 h (20 μl of reaction with 10 U of each enzyme). After the heat inactivation of the enzyme (65°C for 20 min), digests were combined for denaturation (7 min in boiling water) and rapid renaturation in the presence of 100 mM NaCl and 50% formamide (90 μl of reaction mix consisting of 40 μl of DNA digests, 3 μl of 3 M NaCl, 45 μl of formamide and 2 μl of water). Following the digestion with nuclease S1(Invitrogen) (120 μl of reaction consisting of 90 μl of DNA, 8 μl of 3 M NaCl, 12 μl of S1 nuclease buffer, 2 μl of 100 U/μl S1 nuclease and 8 μl of water), DNA was purified using the ChargeSwitch PCR Clean-up Kit.
After fragmentation using the M220 focused ultrasonicator (Covaris), fragments between 100 and 250 bp were purified for NGS library construction using 5500 SOLiD™ Fragment Library Core Kit (Applied Biosystems). Each DNA sample was ligated to linkers with unique barcodes. Libraries were then pooled together and sequenced in a single flowcell run using a SOLiD 5500 × l sequencer. The barcodes were then used to trace the sequence data back to a specific sample. BAM files were generated using LifeTech LifeScope software. Read depth analyses were done using Genomics Suite (Partek). All repeat content analysis was performed using the open web-based platform GALAXY and FASTQ files (Supplementary Figure S1) (32). Briefly, Bowtie (33) was used to map the reads in hg19. We used the commonly used settings except for allowing for a 3 bp mismatch penalty for the 75 bp reads (34). The resulting SAM files were converted to BAM files using SAM Tools (Sequence Read Archive Accession No. SRP028202). Using BED Tools (35), we intersected the BAM alignments with a BED file containing coordinates for Segmental Duplications, Repeat Masked Data, Alpha Satellite DNA or Alu elements. These files were exported from the UCSC Genome Browser. For the analyses of the subsets of Repeat-Masked data, we custom sorted the data to filter out both Alpha Satellite DNA (Alpha/ALR) and Alu elements.
For copy number analyses, 1 µg of genomic DNA was processed by the Genome-Wide Human SNP Nsp/Sty 5.0/6.0 Assay kit (Affymetrix) and hybridized onto Affymetrix SNP6.0 arrays. Comparative data analysis and visualization was done using the Integrated Genome Browser (Affymetrix).
Fluorescence in situ hybridization
Semi-confluent cells in 15 cm dish were collected at 6 h after treatment with 100 ng/ml Colcemid (Sigma). The samples were fixed, denatured and spread as described previously (36). For probes, BAC clones were prepared using BACMAX DNA Purification Kit (Epicentre). The DNA was labeled with Biotin- or DIG-Nick Translation Mix (Roche) and hybridized onto the metaphase samples at 37°C with human CotI DNA (Invitrogen) and salmon sperm DNA. After hybridization, the biotin- or digoxigenin-labeled DNA probe was visualized by the Alexa488-conjugated streptavidin (Invitrogen) and biotinylated anti-streptavidin antibody (Vector) or by Anti-Digoxigenin-Rhodamine (Roche), respectively. The samples were counterstained with ProLong Gold Antifade Reagent with DAPI (Invitrogen).
Breakpoint sequences
The breakpoint sequence for each palindromic junction was obtained using long-PCR and Sanger sequencing (Primer list in Supplementary Table S2). For long-PCR, genomic DNA was amplified using Phusion Hot Start Pol II (Thermo Scientific) and cloned into pSC-B (Stratagene) for sequencing. To reconfirm the breakpoint, genomic DNA was amplified using independent primer sets designed for each breakpoint.
RESULTS
A genomic approach for identifying the locations of sister chromatid fusions
To gain insight into the nucleotide-level mechanism of sister chromatid fusion, we developed an NGS-based approach that pinpoints the location of de novo palindromic junctions in the human genome. The NGS libraries were made through the sequential denaturation and rapid renaturation of genomic DNA (GAPF) (31,37) (Figure 2a). Briefly, intra-strand self complementarities (black and dotted lines) at palindromic junctions facilitate the in vitro folding-back of denatured, ssDNA molecules and produce double-stranded DNA (dsDNA) molecules. In contrast, non-palindromic DNA does not have such self-complementarities and remains as ssDNA molecules. The digestion of resulting DNA with ssDNA-specific nuclease S1 results in the enrichment of DNA from palindromic junctions. The enriched DNA was fragmented, ligated to linkers and PCR-amplified for NGS using a linker-specific primer (GAPF-seq). NGS libraries were obtained for a colorectal cancer cell line Colo320DM (Colo) and normal fibroblast IMR90. We previously showed that a palindromic junction within the CTSK gene sets the telomeric boundary of the 1q21 amplicon in Colo (30). Because this region contains the anti-apoptosis gene MCL1 and is amplified in 10% of all primary tumors (38), understanding the mechanisms of amplification has significance. To confirm that palindromic junctions were enriched in the libraries before NGS, PCR-amplified fragments were hybridized with a probe within the palindromic junction (CTSK cen) and outside of the junction (CTSK tel) (Figure 2b). Fragments with high signal intensity were only seen by the probe within the palindromic junction, thus confirming the enrichment.
Figure 2.
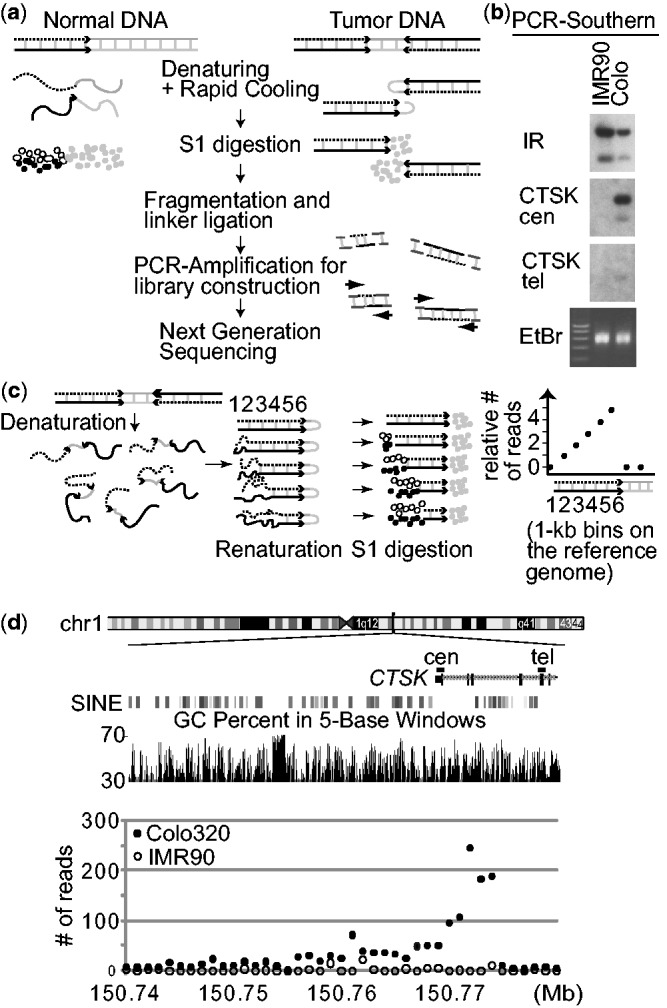
(a) A strategy for enriching DNA from palindromic junctions. DNA is shown by either a black, dotted, or gray line, with the black and dotted lines being complementary to each other. (b) Southern analyses of the PCR products using probes in the human genome. The locations of probes (CTSK cen and tel) are shown in (d). (i) enrichment is only seen in Colo320DM by probe CTSK cen that is within the palindromic junction, but not by probe CTSK tel, and (ii) probes from a naturally existing DNA inverted repeat at chromosome 19p13.2 (chr19:7049026–7058989 in hg19) showed strong enrichment in both IMR90 and Colo320DM. EtBr, ethidium bromide stained gel. (c) Schematic drawings show that in vitro fold-back of palindromic DNA after denaturation/renaturation and subsequent digestion by nuclease S1 (left) can results in the steady increase in read depth (per 1 kb bin) toward a palindromic junction (right). The numbers indicate 1 kb bins on the reference genome. (d) The numbers of GAPF-reads/kb in Colo320DM (closed circles) and IMR90 (open circles) are shown for the 50 kb region of chromosome 1 where the CTSK gene is located. Two genomic features (GC content and SINE elements) within the genomic region are also shown.
Sequencing of the libraries (75-base single end sequencing) yielded 10 579 579 GAPF reads from Colo and 7 448 354 GAPF reads from IMR90 DNA that were mapped to the reference human genome (hg19) (Table 1). Consistent with the fact that our DNA processing steps eliminates non-duplicated DNA, we found a relative depletion of uniquely mapped sequences in the libraries. The intersections between GAPF reads and unique (non-repeat masked) sequences showed that only 26.52% of GAPF reads from Colo and 23.87% of GAPF reads from IMR90 are mapped uniquely to hg19. Considering that nearly 50% of the human genome is unique (49.37% in hg19), the efficiency of depletion is >50%. Instead, repeated sequences are enriched in GAPF reads. For example, we found an enrichment in both alpha satellite DNA (7.33-fold in Colo and 4.39-fold in IMR90) and SINE elements (Alu) (4.08-fold in Colo and 4.74-fold in IMR90). Segmental duplications (low-copy repeats), the recently duplicated genomic segments (39) that account for ∼4.9% of hg19, were moderately enriched (1.4-fold in Colo and 1.5-fold in IMR90).
Table 1.
Enrichment of repeated (duplicated) sequences in the GAPF-seq libraries
| hg19 | GAPF-seq |
||
|---|---|---|---|
| GAPF-seq Reads | COLO320DM | IMR90 | |
| Total reads | 17 498 356 | 15 351 029 | |
| Mapped reads | 10 579 579 | 7 448 354 | |
| Intersected with segmental dup (# of Reads) | 737 758 | 545 540 | |
| % of Mapped | 4.90 | 6.97 | 7.32 |
| Enrichment | 1.42 | 1.50 | |
| Intersected with repeat masker (# of reads) | 7 773 451 | 5 670 447 | |
| % of Mapped | 50.63 | 73.48 | 76.13 |
| Enrichment | 1.45 | 1.50 | |
| Intersected with Alu (# of reads) | 4 196 090 | 3 429 714 | |
| % of Mapped | 9.72 | 39.66 | 46.05 |
| Enrichment | 4.08 | 4.74 | |
| Intersected with alpha satellite (# of reads) | 184 018 | 77 681 | |
| % of Mapped | 0.24 | 1.74 | 1.04 |
| Enrichment | 7.33 | 4.39 | |
We then confirmed the enrichment of reads at the junction at CTSK in Colo and identified a unique pattern of read depth (the number of reads for each 1 kb bin) at and near palindromic junctions. Denatured DNA molecules should fold back more efficiently in the regions close to the middle of palindromic junctions than regions distant from the junctions. This should lead to a steady increase in the number of GAPF reads toward the palindromic junctions and a sharp drop beyond the junction (Figure 2c). To test this idea, read depth analysis was conducted for the entire 1.1 megabase amplicon at 1q21 (Chr 1:149 700 001–150 800 000) (Supplementary Table S1) (Figure 3a). On average, there were 7.98 reads per kb in the library from Colo DNA and 1.64 reads per kb from IMR90 DNA. Indeed, 1 kb bins with markedly increased reads were located within CTSK in Colo, but not in IMR90 (Figure 2d). At a palindromic junction that colocalizes with the boundary of the amplicon (30), Colo had >180 reads per bin for three consecutive bins (150 771 001–150 774 000), whereas IMR90 had only 0–11 reads in the corresponding bins. Furthermore, we found a steady increase of reads toward these three bins from the centromeric side, which was followed by a sharp drop. These results demonstrate the enrichment and unique pattern of read depth at palindromic junctions.
Figure 3.
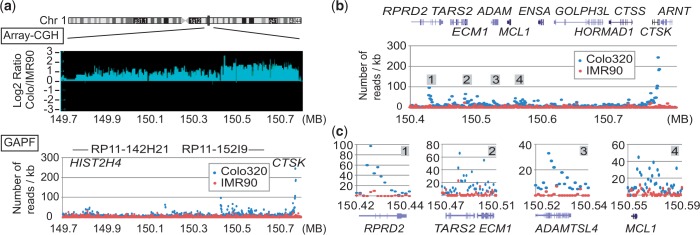
(a) Copy number ratio (Colo/IMR90, Array-CGH, top) and the numbers of GAPF-reads/kb in the 1.1 Mb region (bottom) of chromosome 1. The numbers of GAPF-reads for Colo (blue circles) and IMR90 (red circles) for each 1 kb bin are shown. The locations of BAC clones used for the probes for FISH (Figure 4a) are shown. (b) The number of GAPF-reads/kb over the 400 kb highly amplified region. Four peaks are marked as 1–4. Genes within the region are shown on the top. (c) The detailed views of read depths for GAPF peaks 1–4.
Palindromic junctions cluster within the highly amplified genomic regions
The palindromic junction at the telomeric boundary of the 1q21 amplicon would indicate the inverted duplication of the centromere-proximal part of the long arm of chromosome 1. Such duplication produces a dicentric chromosome that could initiate BFB cycles and gene amplification. Copy number analysis using high-density oligonucleotide microarrays defined the amplicon in Colo (Figure 3a, top). The moderately amplified 700 kb region extended up to a histone H4 gene cluster (HIST2H4) where naturally occurring inverted segmental duplications set the centromeric boundary (30). This moderately amplified region did not have a high peak of GAPF reads (Figure 3a, bottom). In contrast, within the highly amplified 400 kb region on the telomeric side, there was a series of high peaks (Figure 3b). Three peaks (RDRP2, ECM1 and ADAMSL4) had gradual increases of reads toward the centromeric sides (Figure 3c), indicating the palindromic duplication for the genomic region including MCL1. Importantly, the peak at RDRP2 was located at the boundary between the moderately amplified region and the highly amplified region, indicating the role of the palindromic junction in setting the amplicon boundary. The direction of duplication was somewhat ambiguous at the MCL1 locus. There was an increase toward the telomeric end of MCL1 that would indicate the palindromic duplication excluding MCL1. However, there were several bins with increased reads that do not show clear direction, suggesting more than one rearrangement within this region (see later in the text).
Physical locations of amplicons
We sought further evidence for the history of BFB cycles by defining the physical locations of amplicons using fluorescence in situ hybridization (FISH). Publically available Spectoral Karyotyping (SKY) data [SKY/M-FISH and comparative genomic hybridization (CGH) database] show a chromosome with complex fusions; a 5 p chromosome fused to 1q and the 1q fused to 13q (Figure 4a). The fusion breakpoint was at 1q21 in a subset of cells where the CTSK gene is located. Such a fusion suggests a chromosome healing event by a translocation for a broken chromosome undergoing BFB cycles. To further define the physical locations of amplicons, a BAC clone in the highly amplified region (RP11-152I9, red, also in Figure 3a) and a clone from a moderately amplified region (RP11-142H21, green) were labeled and hybridized to metaphase spreads (Figure 4b). Consistent with the SKY data, amplified red signals were seen in the middle of the long chromosome arm in the majority of metaphases. However, in 4 of 22 metaphases, multiple red and green signals are seen at the end of a small chromosome. In metaphases of Colo320HSR, a clone that originated from Colo320DM (40), an amplicon at the end of a small chromosome was common (23/28) (Figure 5c). It is possible that the broken chromosome with the amplicon at the end originally existed in Colo320DM. It could then fuse to chromosome 13 in most of the cells in Colo320DM, but not in Colo320HSR.
Figure 4.
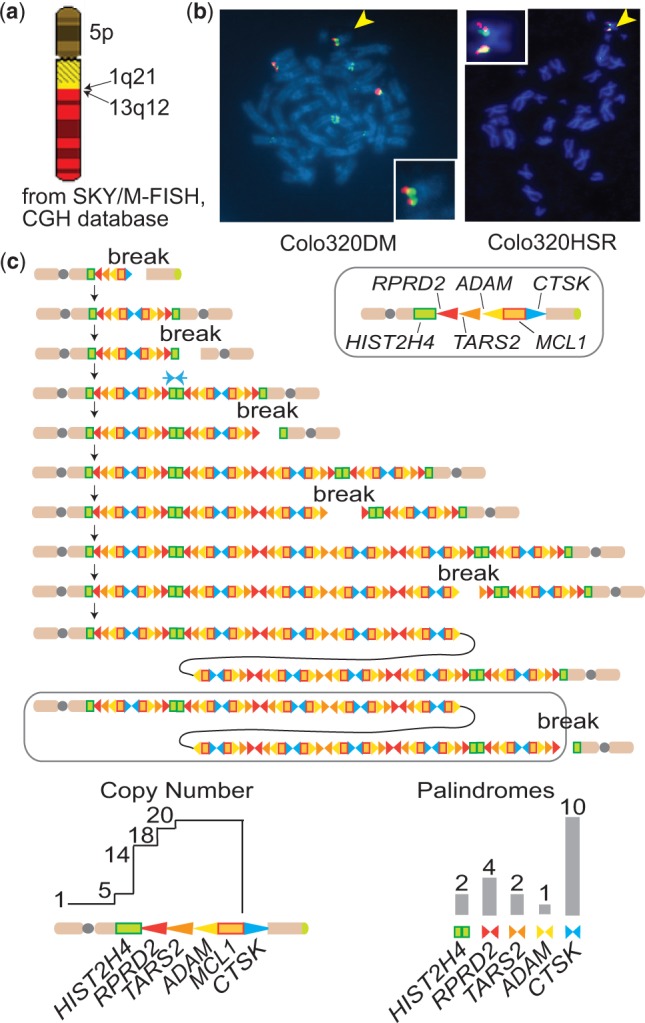
1q21 amplicon at the end of a broken chromosome. (a) The diagram of a chromosome with complex fusions in Colo320DM, obtained from the SKY/M-FISH and CGH database (http://www.ncbi.nlm.nih.gov/sky/skyquery.cgi). Note the fusion breakpoint between 1q21 and 13q12. (b) Pictures from dual color metaphase FISH using the probes for the regions of low (green) and high (red) level amplification are shown for Colo320DM (left) and Colo320HSR (right). The pictures for the broken chromosomes (yellow arrowheads) are magnified and superimposed. The genomic locations of BAC clones are shown in Figure 3a. (c) The BFB cycle-based process for giving rise to the 1q21 amplicon in Colo320 cells. Genomic segments are shown either by rectangles or triangles. Triangles also represent the direction of read-depth increases, and palindromic junctions are shown by pairs of triangles. Both copy number profiles and GAPF read depth predicted by the process are shown at the bottom.
Figure 5.

(a) PCR strategy for the amplification of palindromic fusion breakpoints. Lines represent genomic segments. Two PCR primers are indicated by gray arrows. Inverted Alu repeats (or microhomologies) are indicated by a pair of black arrows. (b) Ethidium bromide-stained agarose gels after PCR amplification for RDRP2 and ADAMSTL4 junctions. Normal fibroblast IMR90 DNA was used as a negative control. (c) DNA sequences of palindromic junctions with Alu inverted repeats. The locations (coordinates of chromosome 1) of inverted Alu repeats and the coordinates (top and bottom) of breakpoints and junction sequences (middle) are shown. Sequence homologies are shown in red. (d) DNA sequences of microhomology-mediated palindromic junctions.
Together with the cluster of palindromic junctions, the physical location of amplicons strongly suggests the history of BFB cycles during the establishment of the 1q21 amplicon (Figure 4c). First, a break at the CTSK gene led to the duplication of the centromere-bearing chromosome and the loss of the distal 1q. The resulting dicentric chromosome broke at HIST2H4 and initiated another duplication, which set the boundary of the entire amplicon. Subsequent breaks and fusions at RDRP2, ADAMSLT4 and MCL1 established the highly amplified region at the end of chromosome (Figure 4c, circled). Such a BFB-based mechanism is also consistent with the copy number and GAPF profiles we observed (Figure 4c, bottom); (i) the palindromic junctions at CTSK and at RDRP2 set the boundaries for the highly amplified region and (ii) palindromic junctions are expected to be the most abundant at CTSK, followed by at RDRP2.
Central asymmetry at the junctions and homology at the breakpoints
Given the evidence of BFB cycles, breakpoint sequences at the palindromic junctions could elucidate the nucleotide-level mechanisms of sister chromatid fusion: how the breaks in mitosis are processed to produce sister chromatid fusion. To sequencse the breakpoints, we designed two PCR primers for the minus-strand in the reference genome near the peaks of sequence reads (Figure 5a). The inverted duplication would bring the minus-strand to the plus-strand in the opposite orientation and thus could enable PCR to amplify breakpoints. Sequencing of the PCR products revealed striking similarities between fusion junctions (Figure 5b). First, there are few kb central asymmetries at the palindromic junctions: 2 1 kb in the RDRP2 junction, 1.7 kb in the ECM1 junction and 3.7 kb in the ADAMSLT4. We recovered two junctions at MCL1, each of which has 2.0 and 3.5 kb asymmetry, respectively. Second, there were sequence homologies at the breakpoints. For RDRP2 and MCL1 junctions, the breakpoints were located within Alu repeats that were a few kb apart in inverted orientations and exhibited high sequence similarities (>80%). In all three cases, there were more than 30 identical nucleotides surrounding the breakpoints (shown in red in Figure 5b). MCL1 junctions were located within a dense cluster of Alu repeats (Supplementary Figure S2), which could promote frequent breakages and fusions and explain the complex pattern of GAPF-read depth (Figure 3c). The ECM1 and ADAMSLT4 junctions were mediated by microhomologies of 3 and 15 bp, respectively. There were no non-template sequence insertions at these breakpoints. The homologies at the breakpoints favor the end processing—capping—replication model in which a broken end undergoes single-strand degradation and the resulting 3′ tail finds the homology in the same strand at few kb internal from the end, followed by DNA synthesis and the formation of a capped end (Figure 1b).
DISCUSSION
A number of studies support a view that the BFB cycle is an important underlying process for genome instability and gene amplification in tumor cells (5,7,14,15). A critical step for BFB cycles leading to gene amplification is the fusion of sister chromatids; however, ambiguity still exists for the nucleotide-level mechanism underlying sister chromatid fusion. In this study, we address this issue using integrated cytogenetic, genomic and nucleotide-level analysis. The results from FISH were consistent with the formation of the 1q21 amplicon through BFB cycles: the duplication of genomic segments at 1q21 at the end of broken chromosome. GAPF-seq showed repeated breakage and sister chromatid fusions within the highly amplified region, as palindromic junctions are seen at 50 kb intervals. Finally, a few kb of central asymmetries and homologies at the breakpoints indicated that the end-capping followed by DNA replication is a primary mechanism of sister chromatid fusion.
Our study also provided a unique opportunity to understand the fate of naturally occurring broken ends in mitosis. The process preceding end-capping is end resection. Whether broken ends undergo end resection depends on the stages of the cell cycle (41,42). End resection initiates homology-dependent repair of DSBs during S and G2 when the sister chromatid is available as repair templates. End resection is not a primary choice for DSB processing in G1, as DSBs are protected and repaired by NHEJ. An important aspect of broken ends in BFB cycles is that broken ends arise in mitosis and are one-ended. Although the fate of broken ends in mitosis has not been studied as extensively as DSBs in other stages of the cell cycles, recent studies indicate that mitotic breaks can undergo end resection. M phase breaks introduced by ionizing radiation (IR) trigger a DNA damage response in human cells. Those breaks are marked by IR-induced foci with γ-H2AX, the MRE11-RAD50-NBS1 (MRN) complex and MDC1 (43). However, 53BP1, a protein that preserves DSB ends for NHEJ (44), is excluded from the M phase IR-induced foci. The lack of 53BP1 hints that the ends are unprotected and are available for end resection. Indeed, Peterson et al. (45) has shown recently that broken DNA treated with the Xenopus M phase extract undergoes CtIP-dependent end resection. CtIP binds to the MRN complex and enhances the endonuclease activity (46). In S phase, the resection by MRN-CtIP removes DNA at short distance from the end and is followed by more extensive resection by EXO1 and DNA2 (47,48). It is currently unknown how extensive the end resection could be in M phase. Given the highly condensed chromatin in M phase, end resection could be limited to a few kb. A few kb of ssDNA tails produced in BFB cycles may have limited access to inter-homologue homology-dependent repair due to the lack of RAD51 loading (45), and instead fold back to find homologies.
We defined a characteristic pattern of breakpoints for sister chromatid fusion: homologies at the breakpoints and a few kb of central asymmetries between duplicated genomic regions. NGS-based paired end sequencing provides a large number of breakpoint sequences in an unbiased fashion. By searching breakpoints with characteristic pattern, we could infer the impact of BFB cycles on genome instability in tumors. A class of breakpoints called fold-back inversion exactly satisfies our criteria. Fold-back inversions are common in pancreatic tumors (16% of 381 breakpoints from 13 pancreatic tumors) and are often associated with amplified genomic regions (25). More than 70% of such fold back inversions have <3 kb of central asymmetries. Microhomologies (>1 bp) are seen in 60% of the breakpoints. Thus, the majority of breakpoints in fold-back inversions could represent sister chromatid fusions. Such inversions are also reported in breast, ovarian and gastric tumors (49,50). The fact that fold-back inversions are common in tumors demonstrates the impact of BFB cycles in genome instability. It is also important to mention that, although less common, there are fold-back inversions with the insertion of non-templated sequences. The insertion of non-templated sequences indicates the NHEJ of two ends as an underlying mechanism; however, it is not known whether this minor class of fold-back inversions derives from M phase DSBs during BFB cycles. It is possible that a chromosome with a spontaneous G1 break replicates in S phase and is repaired by NHEJ with the insertion of non-templated sequences. Such an event could also be important for BFB cycles because it could initiate BFB cycles when a centromere-bearing chromosome is joined and a dicentric chromosome is produced. Furthermore, non-DSB-initiated mechanisms, including a replication fork collapse followed by the strand invasion at an inverted ectopic homology and the subsequent initiation of break-induced replication (27,28), could produce characteristic junctions with central asymmetries and homologies at the breakpoints. Such a mechanism produces complex duplications and triplications of germline origin. However, asymmetries between duplicated segments are much longer, several hundred kb in some cases, suggesting that strand invasion typically occurs at regions of homology that are distant.
We show that BFB cycles use either homologies from divergent Alu repeats or microhomologies (3 and 15 bp) for the capping of broken ends. This is in contrast to the long homology seen at the HIST2H4 locus where a 29 kb segmental inverted duplication (96% sequence identity) could cap the end (30). Long stretches of homology certainly promote fold-back annealing (19,30). Whether divergent Alu repeats and microhomologies are preferred substrates for sister chromatid fusion in cancer cells needs to be addressed in future studies. In this regard, it is noteworthy that, in maize, a chromosome structure called a knob is a preferred site of breaks (13). A knob is a darkly stained, heterochromatic structure with highly repetitive sequences. Thus, breaks could occur non-randomly within or near repeated segments, and such segments could facilitate end-capping. Our targeted genomic approach for sister chromatid fusions will help to elucidate this issue.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Cancer Institute [R01CA149385]; American Cancer Society and Cleveland Clinic (to H.T.). Funding for open access charge: National Cancer Institute [R01CA149385 to H.T.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs M-C Yao, V Boener and A Rattray for comments on the manuscript. The array-CGH experiments were conducted and supported by the Gene Expression and Genotyping Core Facility of the Case Comprehensive Cancer Center (Grant Number P30 CA43703).
REFERENCES
- 1.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 2.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 3.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 4.Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 5.Debatisse M, Malfoy B. Gene amplification mechanisms. Adv. Exp. Med. Biol. 2005;570:343–361. doi: 10.1007/1-4020-3764-3_12. [DOI] [PubMed] [Google Scholar]
- 6.Mondello C, Smirnova A, Giulotto E. Gene amplification, radiation sensitivity and DNA double-strand breaks. Mutat. Res. 2010;704:29–37. doi: 10.1016/j.mrrev.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka H, Yao MC. Palindromic gene amplification—an evolutionarily conserved role for DNA inverted repeats in the genome. Nat. Rev. Cancer. 2009;9:216–224. doi: 10.1038/nrc2591. [DOI] [PubMed] [Google Scholar]
- 8.Kaufman RJ, Brown PC, Schimke RT. Amplified dihydrofolate reductase genes in unstably methotrexate-resistant cells are associated with double minute chromosomes. Proc. Natl Acad. Sci. USA. 1979;76:5669–5673. doi: 10.1073/pnas.76.11.5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nunberg JH, Kaufman RJ, Schimke RT, Urlaub G, Chasin LA. Amplified dihydrofolate reductase genes are localized to a homogeneously staining region of a single chromosome in a methotrexate-resistant Chinese hamster ovary cell line. Proc. Natl Acad. Sci. USA. 1978;75:5553–5556. doi: 10.1073/pnas.75.11.5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marotta M, Chen X, Inoshita A, Stephens R, Thomas Budd G, Crowe JP, Lyons J, Kondratova A, Tubbs R, Tanaka H. A common copy-number breakpoint of ERBB2 amplification in breast cancer colocalizes with a complex block of segmental duplications. Breast Cancer Res. 2012;14:R150. doi: 10.1186/bcr3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith KA, Gorman PA, Stark MB, Groves RP, Stark GR. Distinctive chromosomal structures are formed very early in the amplification of CAD genes in Syrian hamster cells. Cell. 1990;63:1219–1227. doi: 10.1016/0092-8674(90)90417-d. [DOI] [PubMed] [Google Scholar]
- 12.Coquelle A, Pipiras E, Toledo F, Buttin G, Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 1997;89:215–225. doi: 10.1016/s0092-8674(00)80201-9. [DOI] [PubMed] [Google Scholar]
- 13.McClintock B. The stability of broken ends of chromosomes in Zea Mays. Genetics. 1941;26:234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maser RS, DePinho RA. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–569. doi: 10.1126/science.297.5581.565. [DOI] [PubMed] [Google Scholar]
- 15.Murnane JP. Telomere dysfunction and chromosome instability. Mutat. Res. 2012;730:28–36. doi: 10.1016/j.mrfmmm.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tlsty TD. Functions of p53 suppress critical consequences of damage and repair in the initiation of cancer. Cancer Cell. 2002;2:2–4. doi: 10.1016/s1535-6108(02)00088-0. [DOI] [PubMed] [Google Scholar]
- 17.Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC, Alt FW. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–821. doi: 10.1016/s0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 18.Difilippantonio MJ, Petersen S, Chen HT, Johnson R, Jasin M, Kanaar R, Ried T, Nussenzweig A. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J. Exp. Med. 2002;196:469–480. doi: 10.1084/jem.20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka H, Tapscott SJ, Trask BJ, Yao MC. Short inverted repeats initiate gene amplification through the formation of a large DNA palindrome in mammalian cells. Proc. Natl Acad. Sci. USA. 2002;99:8772–8777. doi: 10.1073/pnas.132275999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narayanan V, Mieczkowski PA, Kim HM, Petes TD, Lobachev KS. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell. 2006;125:1283–1296. doi: 10.1016/j.cell.2006.04.042. [DOI] [PubMed] [Google Scholar]
- 21.Shuster MI, Han L, Le Beau MM, Davis E, Sawicki M, Lese CM, Park NH, Colicelli J, Gollin SM. A consistent pattern of RIN1 rearrangements in oral squamous cell carcinoma cell lines supports a breakage-fusion-bridge cycle model for 11q13 amplification. Genes Chromosomes Cancer. 2000;28:153–163. [PubMed] [Google Scholar]
- 22.Ciullo M, Debily MA, Rozier L, Autiero M, Billault A, Mayau V, El Marhomy S, Guardiola J, Bernheim A, Coullin P, et al. Initiation of the breakage-fusion-bridge mechanism through common fragile site activation in human breast cancer cells: the model of PIP gene duplication from a break at FRA7I. Hum. Mol. Genet. 2002;11:2887–2894. doi: 10.1093/hmg/11.23.2887. [DOI] [PubMed] [Google Scholar]
- 23.Hellman A, Zlotorynski E, Scherer SW, Cheung J, Vincent JB, Smith DI, Trakhtenbrot L, Kerem B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. 2002;1:89–97. doi: 10.1016/s1535-6108(02)00017-x. [DOI] [PubMed] [Google Scholar]
- 24.Kitada K, Yamasaki T. The complicated copy number alterations in chromosome 7 of a lung cancer cell line is explained by a model based on repeated breakage-fusion-bridge cycles. Cancer Genet. Cytogenet. 2008;185:11–19. doi: 10.1016/j.cancergencyto.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bignell GR, Santarius T, Pole JC, Butler AP, Perry J, Pleasance E, Greenman C, Menzies A, Taylor S, Edkins S, et al. Architectures of somatic genomic rearrangement in human cancer amplicons at sequence-level resolution. Genome Res. 2007;17:1296–1303. doi: 10.1101/gr.6522707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carvalho CM, Ramocki MB, Pehlivan D, Franco LM, Gonzaga-Jauregui C, Fang P, McCall A, Pivnick EK, Hines-Dowell S, Seaver LH, et al. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat. Genet. 2011;43:1074–1081. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rattray AJ, Shafer BK, Neelam B, Strathern JN. A mechanism of palindromic gene amplification in Saccharomyces cerevisiae. Genes Dev. 2005;19:1390–1399. doi: 10.1101/gad.1315805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanaka H, Cao Y, Bergstrom DA, Kooperberg C, Tapscott SJ, Yao MC. Intrastrand annealing leads to the formation of a large DNA palindrome and determines the boundaries of genomic amplification in human cancer. Mol. Cell. Biol. 2007;27:1993–2002. doi: 10.1128/MCB.01313-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diede SJ, Guenthoer J, Geng LN, Mahoney SE, Marotta M, Olson JM, Tanaka H, Tapscott SJ. DNA methylation of developmental genes in pediatric medulloblastomas identified by denaturation analysis of methylation differences. Proc. Natl Acad. Sci. USA. 2010;107:234–239. doi: 10.1073/pnas.0907606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langmead B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinformatics. 2010 doi: 10.1002/0471250953.bi1107s32. Chapter 11, Unit 11.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe T, Tanabe H, Horiuchi T. Gene amplification system based on double rolling-circle replication as a model for oncogene-type amplification. Nucleic Acids Res. 2011;39:e106. doi: 10.1093/nar/gkr442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka H, Bergstrom DA, Yao MC, Tapscott SJ. Widespread and nonrandom distribution of DNA palindromes in cancer cells provides a structural platform for subsequent gene amplification. Nat. Genet. 2005;37:320–327. doi: 10.1038/ng1515. [DOI] [PubMed] [Google Scholar]
- 38.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 40.Quinn LA, Moore GE, Morgan RT, Woods LK. Cell lines from human colon carcinoma with unusual cell products, double minutes, and homogeneously staining regions. Cancer Res. 1979;39:4914–4924. [PubMed] [Google Scholar]
- 41.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 42.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 43.Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 2010;190:197–207. doi: 10.1083/jcb.200911156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peterson SE, Li Y, Chait BT, Gottesman ME, Baer R, Gautier J. Cdk1 uncouples CtIP-dependent resection and Rad51 filament formation during M-phase double-strand break repair. J. Cell Biol. 2011;194:705–720. doi: 10.1083/jcb.201103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng G, Dai H, Zhang W, Hsieh HJ, Pan MR, Park YY, Tsai RY, Bedrosian I, Lee JS, Ira G, et al. Human nuclease/helicase DNA2 alleviates replication stress by promoting DNA end resection. Cancer Res. 2012;72:2802–2813. doi: 10.1158/0008-5472.CAN-11-3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hillmer AM, Yao F, Inaki K, Lee WH, Ariyaratne PN, Teo AS, Woo XY, Zhang Z, Zhao H, Ukil L, et al. Comprehensive long-span paired-end-tag mapping reveals characteristic patterns of structural variations in epithelial cancer genomes. Genome Res. 2011;21:665–675. doi: 10.1101/gr.113555.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McBride DJ, Etemadmoghadam D, Cooke SL, Alsop K, George J, Butler A, Cho J, Galappaththige D, Greenman C, Howarth KD, et al. Tandem duplication of chromosomal segments is common in ovarian and breast cancer genomes. J. Pathol. 2012;227:446–455. doi: 10.1002/path.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.