


Abstract
Detection of differential expression in RNA-Seq data is currently limited to studies in which two or more sample conditions are known a priori. However, these biological conditions are typically unknown in cohort, cross-sectional and nonrandomized controlled studies such as the HapMap, the ENCODE or the 1000 Genomes project. We present DEXUS for detecting differential expression in RNA-Seq data for which the sample conditions are unknown. DEXUS models read counts as a finite mixture of negative binomial distributions in which each mixture component corresponds to a condition. A transcript is considered differentially expressed if modeling of its read counts requires more than one condition. DEXUS decomposes read count variation into variation due to noise and variation due to differential expression. Evidence of differential expression is measured by the informative/noninformative (I/NI) value, which allows differentially expressed transcripts to be extracted at a desired specificity (significance level) or sensitivity (power). DEXUS performed excellently in identifying differentially expressed transcripts in data with unknown conditions. On 2400 simulated data sets, I/NI value thresholds of 0.025, 0.05 and 0.1 yielded average specificities of 92, 97 and 99% at sensitivities of 76, 61 and 38%, respectively. On real-world data sets, DEXUS was able to detect differentially expressed transcripts related to sex, species, tissue, structural variants or quantitative trait loci. The DEXUS R package is publicly available from Bioconductor and the scripts for all experiments are available at http://www.bioinf.jku.at/software/dexus/.
INTRODUCTION
The advent of next-generation sequencing has greatly expanded our knowledge about transcriptomes. New transcripts and splice variants have been found and break points of known transcripts determined more accurately (1–6). However, in RNA-Seq experiments, quantification of the expression of transcripts can be difficult (7). Without biological variability, transcripts that are differentially expressed between two conditions can be detected reliably (8). In studies with biological variability, however, detection of differential expression between two conditions remains challenging (9). A transcript that is differentially expressed between many conditions is hard to detect because read count variation due to differential expression and due to high overdispersion can only be distinguished with many samples and high coverage. See Supplementary Section S2 for more details. To detect differentially expressed transcripts, we therefore assume that the number of conditions is small compared with the number of samples.
Identifying differential expression is currently limited to particular study designs
Current methods for analyzing RNA-Seq data can identify differential expression between two conditions. For example, in a case-control study, only transcripts that are differentially expressed between cases and controls can be identified. Similarly, in a randomized controlled study, differential expression between treated and untreated subjects can be detected. These study designs can be generalized to more case groups or more treatments, which leads to multiple (more than two) known conditions. For example, multiple conditions may be due to different tissue types, as in the ‘Allen Brain Atlas’ (10), the ‘Gene Expression Nervous System Atlas’ (11), and the ‘BioGPS’ (12).
Identification of differential expression in RNA-Seq data requires a priori known conditions. In cohort, cross-sectional and nonrandomized controlled studies, the biological conditions are unknown or only partially known. Cohort and cross-sectional studies are observational studies in which the conditions of the subjects are unknown. Examples of observational studies include the HapMap (13), ENCODE (6) and the 1000 Genomes (14) project, for which RNA-Seq data are available (15,16). Nonrandomized controlled studies are treatment studies in which conditions such as genetic, environmental or treatment effects are not completely known. In nonrandomized controlled studies, unknown genetic variations such as single-nucleotide polymorphisms (SNPs), copy number variations and unknown environmental factors may result in differential expression between treated subjects. Furthermore, individual unknown treatment effects may cause variation in gene expression, for instance, responses of cell lines to the addition of compounds (17). Other examples are found in oncology, where unknown cancer subtypes or unknown cancer stages are characterized by a particular gene expression profile (18,19).
In nonclinical studies, the conditions are also often unknown. During development, the transcriptome regulates and controls cell growth, differentiation, movement and morphogenesis. Genes are differentially expressed between different time points and between different tissues; even within one tissue, gene expression may vary spatially. For two samples taken at different times or from different locations it is often unknown whether the conditions differ. Another example is in vivo or in vitro gene expression in mice treated with drug candidates (20,21). Unknown factors such as individual responses or side effects lead to differentially expressed transcripts between the samples.
The detection of differential expression in RNA-Seq studies with unknown conditions is important to obtain new biological knowledge. Current RNA-Seq methods, however, require the conditions to be known. For microarray data, a method for identifying unknown conditions in gene expression has been suggested (22). However, this method cannot be applied to RNA-Seq data with unknown conditions because a primary modeled factor is required and the noise is assumed to be Gaussian, which is not appropriate for RNA-Seq count data (23). We therefore present DEXUS, a method capable of detecting differential expression in RNA-Seq studies with unknown conditions.
A summary of study designs and methods that can detect differential expression in them is shown in Table 1.
Table 1.
An overview of study designs and methods that can detect differential expression in them
| Study design | DEXUS | DESeq | edgeR | baySeq | SAMSeq | DEGSeq | PoissonSeq | NOISeq | DSS |
|---|---|---|---|---|---|---|---|---|---|
| Two known conditions | |||||||||
| Case-control study | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Randomized controlled study | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Multiple known conditions | |||||||||
| Multiple case-control study | ✓ | ✓ | ✓ | ✓ | ✓ | ||||
| Multiple treatment RCS | ✓ | ✓ | ✓ | ✓ | ✓ | ||||
| Unknown conditions | |||||||||
| Cross-sectional study | ✓ | ||||||||
| Cohort study | ✓ | ||||||||
| Nonrandomized controlled study | ✓ |
Existing methods for detecting differential expression in RNA-Seq data
Methods that detect differential expression in RNA-Seq data are usually based on read counts, i.e. the number of reads mapping to a DNA region that is transcribed, such as a gene or an exon (32). These methods compare read counts for two conditions. If read counts show a large and consistent difference between the conditions, then the according transcript is differentially expressed. In this subsection, we review methods that detect differential expression in RNA-Seq data. Many methods model read counts by a negative binomial distribution because even after normalization the read counts have high variance. Therefore, we divide methods into two classes: those which do not use negative binomials (class A) and those which do (class B).
The following methods belong to class A.
DEGSeq (28) assumes that the log fold change of mean read counts between the two conditions follows a normal distribution given the log average expression. A differentially expressed gene is identified by a small P-value by means of this distribution.
NOISeq (30) also considers the log fold change of read counts between two given conditions together with their absolute difference. Empirical distributions are calculated using all pairs of replicates from different conditions. NOISeq identifies a gene as differentially expressed if the log fold change of read counts and the absolute difference of read counts between the two conditions have both a small P-value for the empirical distributions.
SAMSeq (27) performs a Wilcoxon test for each transcript testing the counts of one condition against the counts of the other. Because standard normalization techniques are not applicable, subsampling is used to normalize the read counts. SAMSeq requires a relatively high number of samples per condition to obtain significance for differential expression.
PoissonSeq (29) fits a Poisson log-linear model to the read counts after transforming them. A score statistic on the model parameters determines the significance for differential expression.
The following class B methods use negative binomial distributions to model the read counts.
edgeR (25) uses a quantile-adjusted conditional maximum likelihood estimator for the overdispersion parameter of the negative binomial distribution. This estimator is more accurate than the standard maximum likelihood estimator when only few replicates per condition are available (33). Borrowing information across transcripts allows the dispersion parameter to be adjusted toward a consensus value using an empirical Bayes procedure (34). Finally, edgeR uses an exact test to determine whether the counts of the two conditions come from the same negative binomial distribution.
DESeq (24) pools together transcripts with similar expressions values to improve the estimate of the overdispersion parameter. The overdispersion is assumed to be a function of the mean read count and is therefore estimated per condition. To determine whether a transcript is differentially expressed, the distribution parameters of the two conditions are tested by an exact test for equality of means.
baySeq (26) determines the distribution of the overdispersion parameter by applying a quasi-likelihood method to the read counts of one condition. The resulting distribution is used as prior for estimating the overdispersion parameter when fitting the model to the read count data.
DSS (31) is similar to baySeq. A negative binomial distribution is fitted to the read count data using a prior on the overdispersion parameter. This prior is a log-normal distribution, whose parameters are optimized using the dispersion parameters of each condition. Finally, a Wald test is used to determine differential expression.
In summary, the class B methods, which use negative binomial distributions, i.e. DESeq, baySeq, DSS and edgeR, mainly differ in the way they estimate the overdispersion parameter. Estimating the overdispersion parameter is crucial for the performance and not trivial because the maximum likelihood estimator is biased and has high variance if the sample size is small (33). The subsequent statistical test has a smaller effect on the results than the parameter estimates (23,31).
Extensions to multiple known conditions
McCarthy et al. (32) extended the R package edgeR to more than two conditions. A generalized linear model is fitted to the data, and then coefficients are tested for being different from zero, which leads to the final P-values. Again, the estimation of the overdispersion parameter for a transcript borrows information from other transcripts. DESeq, baySeq and SAMSeq have also been extended to more than two conditions.
MATERIALS AND METHODS
Method overview
Our goal is to identify differentially expressed transcripts in studies with unknown conditions. A transcript is differentially expressed if the mean expression levels for different conditions are different and read counts are observed under more than one condition. Therefore we assume a small number of conditions because, as mentioned above, the detection of differential expression for many conditions is difficult. RNA-Seq expression data are usually represented as read counts per transcript, or alternatively by exon or gene. It was observed that read counts from a single condition follow a negative binomial distribution (24–26,31). DEXUS therefore models read counts as a finite mixture of negative binomial distributions.
The model that best explains the observed read counts is selected from a set of models. In a Bayesian framework, model selection is based on finding the parameter which maximizes the posterior, the maximum a posteriori (MAP) parameter. The MAP model is found by an expectation maximization (EM) algorithm, where E-step and M-step are alternated repeatedly. The E-step estimates the unknown conditions based on actual model parameters, and the M-step optimizes the model parameters based on the E-step estimates. Models that use only one condition to explain the read counts are preferred by means of a prior distribution. One condition is the null hypothesis, which is rejected only if the data show strong evidence for more than one condition. Therefore, the parameters of the prior distribution determine how much DEXUS prefers to select models that explain the data without differential expression. Consequently, via the prior parameters, DEXUS can be adjusted to have a low false discovery rate at the detection of differential expression.
In the following subsections, we first describe the model in more detail and then explain the EM algorithm for model selection. Model selection includes prior assumptions that lower the false discovery rate and lead to more accurate estimates. Finally, we show how to call differentially expressed transcripts on the basis of an informative/noninformative (I/NI) value.
The model
Read count x per transcript is explained by a mixture of n negative binomial distributions:
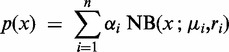 |
(1) |
where 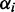 is the probability of being in condition i out of n possible conditions. In condition i, read counts are drawn from a negative binomial distribution with mean
is the probability of being in condition i out of n possible conditions. In condition i, read counts are drawn from a negative binomial distribution with mean 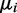 and size ri, where the size parameter ri is the inverse of the overdispersion
and size ri, where the size parameter ri is the inverse of the overdispersion 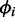 . Note that we use the
. Note that we use the 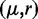 instead of the usual
instead of the usual 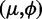 parametrization to locally accumulate parameters that are associated with large overdispersions. This accumulation is essential to define a prior within a Bayesian framework.
parametrization to locally accumulate parameters that are associated with large overdispersions. This accumulation is essential to define a prior within a Bayesian framework.
A nondegenerate DEXUS model is identifiable (see Supplementary Section S3.1.3), as required for the maximum likelihood and the maximum a posterior estimator to be consistent. Consistency means that the estimator converges to the true parameter values with more data points, which is important for identifying differential expression. If the mean read count exceeds the variance, the maximum likelihood estimate of r tends to 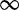 and the negative binomial converges to a Poisson distribution (see Supplementary Section S3.2.2).
and the negative binomial converges to a Poisson distribution (see Supplementary Section S3.2.2).
Model selection
We perform model selection in a Bayesian framework by maximizing the posterior, i.e. by a MAP approach (35–37). Therefore, the parameters  ,
, 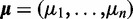 and
and 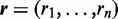 are considered to be random variables, and the likelihood p(x) in Equation (1) becomes the conditional probability
are considered to be random variables, and the likelihood p(x) in Equation (1) becomes the conditional probability 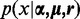 . The objective of the model selection is to maximize the posterior of the parameters:
. The objective of the model selection is to maximize the posterior of the parameters:
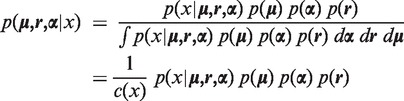 |
(2) |
where the priors on 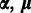 and
and  are assumed to be independent of each other, and are defined in the following.
are assumed to be independent of each other, and are defined in the following.
Dirichlet prior for probabilities of conditions
First we choose the prior 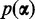 on the probabilities of the conditions. Since the majority of transcripts in a data set are usually not differentially expressed, the model should favor explaining the read counts for a transcript with a single condition. The null hypothesis of one condition should only be rejected if the data contain strong evidence for more than one condition. The prior reduces the number of falsely discovered differentially expressed transcripts and therefore keeps the false discovery rate low. DEXUS uses a Dirichlet prior
on the probabilities of the conditions. Since the majority of transcripts in a data set are usually not differentially expressed, the model should favor explaining the read counts for a transcript with a single condition. The null hypothesis of one condition should only be rejected if the data contain strong evidence for more than one condition. The prior reduces the number of falsely discovered differentially expressed transcripts and therefore keeps the false discovery rate low. DEXUS uses a Dirichlet prior 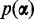 on
on  with parameters
with parameters 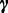 to incorporate the preference for only one condition:
to incorporate the preference for only one condition:
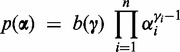 |
(3) |
where  is an n-dimensional probability vector. Each component
is an n-dimensional probability vector. Each component 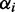 is distributed according to a beta distribution with
is distributed according to a beta distribution with 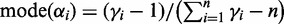 .
.
To express the prior knowledge that most transcripts are not differentially expressed and are generated under only one condition, we set 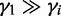 (for
(for 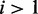 ). This setting assumes that most read counts are generated under condition i = 1, which we call the major condition, while conditions
). This setting assumes that most read counts are generated under condition i = 1, which we call the major condition, while conditions 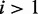 are called minor conditions. The vector of hyperparameters
are called minor conditions. The vector of hyperparameters 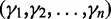 can be simplified to one hyperparameter G (Supplementary Section S3.2.1). In Supplementary Section S3.4 we show that DEXUS is not sensitive to the choice of the hyperparameter G. Therefore DEXUS is easy to use as good results are obtained with the default setting of G = 1 (see Supplementary Section S3.4). Without having seen the data, we assume that only the major condition is present, which means that the transcript is not differentially expressed. Only when the data show strong evidence also for minor conditions, does the posterior assign nonzero probabilities to minor conditions and the transcript is called differentially expressed.
can be simplified to one hyperparameter G (Supplementary Section S3.2.1). In Supplementary Section S3.4 we show that DEXUS is not sensitive to the choice of the hyperparameter G. Therefore DEXUS is easy to use as good results are obtained with the default setting of G = 1 (see Supplementary Section S3.4). Without having seen the data, we assume that only the major condition is present, which means that the transcript is not differentially expressed. Only when the data show strong evidence also for minor conditions, does the posterior assign nonzero probabilities to minor conditions and the transcript is called differentially expressed.
Truncated exponential priors for overdispersions
In DEXUS model selection, the second prior is on the size parameter r of the negative binomial distribution, which determines the overdispersion. A prior on r improves the estimation of r if the number of samples is small. The maximum likelihood estimator of r is biased for few samples and overestimates the true size parameter (38,39), as confirmed in Supplementary Section S3.2.5. In a Bayesian approach, the influence of the prior decreases with an increasing number of samples, and therefore the MAP estimator is asymptotically (number of samples tending to infinity) unbiased.
To keep the estimate of r small, the prior pushes r toward zero. We choose an exponential distribution as prior:
| (4) |
where η is a hyperparameter.
Like DESeq (24), we truncate the size parameter at the right-hand tail by using the constraint 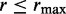 . The upper bound
. The upper bound 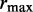 on the size parameter is equivalent to a lower bound on the overdispersion and ensures minimal overdispersion for the read counts of each transcript. Further, this bound makes the parameter space compact, which is required for a consistent estimator. The same exponential prior is used for each component of
on the size parameter is equivalent to a lower bound on the overdispersion and ensures minimal overdispersion for the read counts of each transcript. Further, this bound makes the parameter space compact, which is required for a consistent estimator. The same exponential prior is used for each component of  . The hyperparameter η for the exponential prior on r is transformed to a hyperparameter θ (see Supplementary Section S3.2.5). Like the hyperparameter G, also θ is robust and supplies good results with
. The hyperparameter η for the exponential prior on r is transformed to a hyperparameter θ (see Supplementary Section S3.2.5). Like the hyperparameter G, also θ is robust and supplies good results with 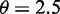 .
.
Uniform priors for means
Finally, DEXUS model selection uses a prior on the mean μ of the negative binomial distribution. If in one condition all read counts were close to zero (transcripts are not present), the estimate of the mean of the negative binomial would not converge. Therefore, 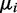 is lower bounded by
is lower bounded by 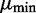 . To ensure a compact parameter space as required for a consistent estimator, we use a uniform prior on
. To ensure a compact parameter space as required for a consistent estimator, we use a uniform prior on 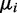 on the compact interval
on the compact interval 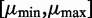 , where
, where 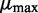 can be set to the largest observed read count.
can be set to the largest observed read count.
In summary, DEXUS has only few parameters which in most applications need not be adjusted by the user, as their default values give good results.
EM algorithm
With the priors defined, the model with maximum parameter posterior can be selected. The EM algorithm (40) is used to minimize an upper bound on the negative log-posterior of the parameters. The E-step of the EM algorithm estimates the probability that a read count is generated under a particular condition. The M-step optimizes the model parameters based on the E-step estimates.
In the DEXUS model, 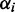 is the probability of condition i without observing any data. The model posterior
is the probability of condition i without observing any data. The model posterior 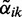 estimates the probability that read count xk is generated under condition i (the probability of condition i after observing data xk):
estimates the probability that read count xk is generated under condition i (the probability of condition i after observing data xk):
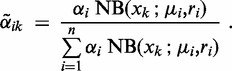 |
(5) |
This equation is the E-step (expectation step) of the EM algorithm. Using the posterior estimates 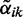 , we obtain following update rules for the M-step (maximization step):
, we obtain following update rules for the M-step (maximization step):
-
∙estimate for
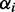 :
:
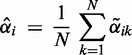
(6) -
∙
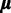 update:
update:
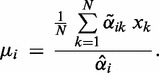
(7) -
∙
 update:
update: - The new ri is obtained by solving the following equation for ri:
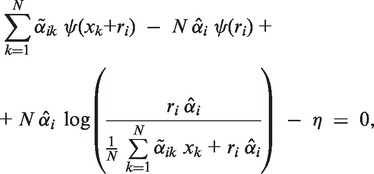
(8) where ψ is the digamma function. The equation is solved numerically for ri by means of a ‘bisection’ procedure.
-
∙
 update:
update:
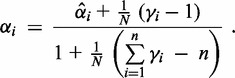
(9)
The complete derivation of the EM algorithm can be found in the Supplementary Section S3.2.1.
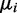 and ri are initialized by using the results of k-means clustering (see Supplementary Section S3.2.4). The values
and ri are initialized by using the results of k-means clustering (see Supplementary Section S3.2.4). The values 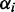 are simply initialized with the maximum entropy setting
are simply initialized with the maximum entropy setting 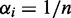 .
.
I/NI value: evidence of differential expression
The Bayesian framework allows definition of an I/NI call (36,37,41,42). The I/NI call serves to extract differentially expressed transcripts with a desired specificity (1 − significance level or 1 − type I error rate) or sensitivity (power or 1 – type II error rate). DEXUS first computes the I/NI value, which quantifies the contribution of differential expression to the read count variation. Transcripts are then called informative if the I/NI value exceeds a threshold.
Unlike 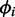 or ri, which capture noise variation,
or ri, which capture noise variation,  captures variation arising from differentially expressed transcripts. The posterior
captures variation arising from differentially expressed transcripts. The posterior 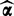 of
of  indicates differential expression in the data in the form of minor conditions with probabilities larger than zero. The larger the posterior value
indicates differential expression in the data in the form of minor conditions with probabilities larger than zero. The larger the posterior value 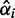 of a minor condition
of a minor condition 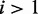 , the stronger the evidence for its presence. Further, evidence is also required that the minor condition is different from the major condition in terms of mean read counts. Although identifiability of the DEXUS model ensures that the negative binomials of different conditions are different, they may still be close to one another. The more the mean
, the stronger the evidence for its presence. Further, evidence is also required that the minor condition is different from the major condition in terms of mean read counts. Although identifiability of the DEXUS model ensures that the negative binomials of different conditions are different, they may still be close to one another. The more the mean 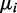 of the minor condition
of the minor condition 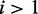 differs from the mean of the major condition, the stronger is the evidence that the minor condition is different from the major condition. In conclusion, evaluating the evidence of differential expression (the I/NI value) should consider two factors: (i)
differs from the mean of the major condition, the stronger is the evidence that the minor condition is different from the major condition. In conclusion, evaluating the evidence of differential expression (the I/NI value) should consider two factors: (i) 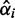 as the evidence for the presence of the minor condition
as the evidence for the presence of the minor condition 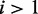 ; (ii) the difference between the means of the major and minor conditions as evidence that they are indeed different.
; (ii) the difference between the means of the major and minor conditions as evidence that they are indeed different.
The difference between the means is expressed by the log difference 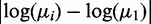 . Factor (I) is incorporated into the I/NI value by weighting these differences by
. Factor (I) is incorporated into the I/NI value by weighting these differences by 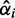 , which yields
, which yields
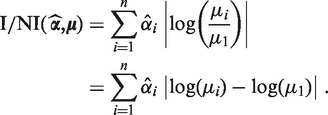 |
(10) |
The I/NI value is the expected log fold change of read counts with respect to the mean read count of the major condition given a noise-free model. ‘Noise-free’ refers to the assumption that under each condition, only the mean read count is observed. For a mathematical interpretation of the I/NI value see Supplementary Section S3.3.2.
Experiments
We evaluated DEXUS on simulated and real-world data sets. The simulated data sets had various library sizes, different numbers of replicates and different ratios between mean read counts under the different conditions. DEXUS was tested on the following real-world RNA-Seq data sets: (i) ‘Nigerian HapMap’, based on 69 Nigerian HapMap individuals, (ii) ‘European HapMap’, based on 60 European HapMap individuals, (iii) ‘Primate Liver’, based on liver tissue samples from humans, chimpanzees and rhesus macaques, (iv) ‘Maize Leaves’, using samples from different locations of maize plant leaves, and (v) ‘Mice Strains’, based on different strains of mice (Supplementary Section S4.2.4).
First we report the performance of DEXUS on 2400 simulated data sets for which the conditions were known but withheld from DEXUS. We then present tests on real-world data sets with either unknown conditions (‘Nigerian HapMap’, ‘European HapMap’) or partially known conditions (‘Primate Liver’, ‘Maize Leaves’). In the latter case the conditions were withheld from DEXUS to ascertain whether it can identify them.
DEXUS for known conditions
Before we tested DEXUS on data with unknown conditions, we assessed how well it performs if the conditions of interest are known. For known conditions, DEXUS estimates only the parameters of a negative binomial for each condition. Therefore, we compared the parameter estimates of DEXUS to previously suggested methods in terms of detecting differentially expressed transcripts, namely the following eight state-of-the-art methods: DSS (31), DESeq (24), baySeq (26), edgeR (25), DEGseq (28), NOISeq (30), PoissonSeq (29) and SAMseq (27).
If only few samples per condition are available, the performance of DEXUS is below the best performing other methods. For medium and large sample numbers and small library size (1e6) DEXUS is second and third best method. For medium and large sample numbers and large library sizes (1e7 and 1e8) DEXUS outperforms all other methods. The experiments and the respective results are described in detail in Supplementary Section S4.2.
Simulated RNA-Seq data
Generating simulated RNA-Seq data
We simulated data sets from different experimental settings following the suggestions of Robinson et al. (34), Hardcaste and Kelly (26) and Wu et al. (31). For all samples of a data set, the library size was 1e6, 1e7 or 1e8 to cover a wide range of applications. Keeping the library size and the read quality constant for each sample in a data set avoids the need for normalization of the read counts, i.e. it avoids normalization biases. For each experiment, we choose a particular number of replicates per condition to evaluate DEXUS for different sample sizes and for unbalanced data. In case of two conditions, the numbers of replicates were 6/6, 9/3, 10/2, 11/1, 12/12, 18/6, 20/4, 22/2 (condition1/condition2). Each experiment consisted of 100 data sets with 10 000 transcripts each. The conditions were known and used for evaluation but withheld from DEXUS.
For the simulation we assumed that under condition i the reads x for a transcript are distributed according to a negative binomial 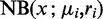 . After Wu et al. (31), we took the mean
. After Wu et al. (31), we took the mean 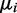 and the size ri from the ‘Mice Strains’ benchmark RNA-Seq data set (43) using only data from one particular biological condition. For a randomly selected transcript, the value
and the size ri from the ‘Mice Strains’ benchmark RNA-Seq data set (43) using only data from one particular biological condition. For a randomly selected transcript, the value 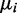 was obtained as the median read count of the condition.
was obtained as the median read count of the condition.
The overdispersion 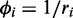 tends to decrease with increasing mean read counts (see Supplementary Figure S15). Therefore, we fitted a regression line to overdispersion values by least squares. After sampling
tends to decrease with increasing mean read counts (see Supplementary Figure S15). Therefore, we fitted a regression line to overdispersion values by least squares. After sampling 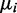 values, the corresponding
values, the corresponding 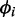 values were obtained by means of the regression line to which zero-one normally distributed noise was added. Thirty percent of the genes were chosen to be differentially expressed. Differential expression was modeled by adjusting the means of the negative binomials to obtain log fold changes of 0.5, 1 and 1.5 between the mean of the major and the minor condition. The fold change values are randomly chosen with equal probability, such that all 3-fold change categories have about the same number of genes in each data set.
values were obtained by means of the regression line to which zero-one normally distributed noise was added. Thirty percent of the genes were chosen to be differentially expressed. Differential expression was modeled by adjusting the means of the negative binomials to obtain log fold changes of 0.5, 1 and 1.5 between the mean of the major and the minor condition. The fold change values are randomly chosen with equal probability, such that all 3-fold change categories have about the same number of genes in each data set.
Evaluation criteria for simulated RNA-Seq data
We formulate the detection of differential expression as a classification task: DEXUS must decide whether a transcript is differentially expressed (positive prediction) or not (negative prediction). For the simulated data, we knew which transcripts were differentially expressed (the positives) and which were not (the negatives). DEXUS ranks the transcripts by the I/NI value from Equation (10). For a given I/NI threshold (the I/NI call), we can determine true positives, false positives, true negatives and false negatives. Using these numbers, we computed the specificity and the sensitivity of DEXUS. The specificity corresponds to ‘1 − significance level’ or ‘1 − type I error rate’. The type I error rate is the ratio between false detections and all negatives. The sensitivity corresponds to the ‘power’ or ‘1 − type II error rate’. The type II error rate is the ratio between missed positives and all positives.
Results on simulated RNA-Seq data
We tested DEXUS on the simulated RNA-Seq data using its default parameters. Table 2 shows the results in terms of sensitivity and specificity for library size 1e8 at different thresholds for the I/NI value. Transcripts with an I/NI value above the threshold are called informative or (equivalently) differentially expressed. Results for other library sizes are presented in Supplementary Tables S12 and S13. The specificity of DEXUS is high across various numbers of replicates, whereas the sensitivity varies considerably. High specificity means that few transcripts are falsely identified as being differentially expressed. In highly unbalanced experiments, i.e. 11/1 and 22/2 replicates, differentially expressed transcripts are detected only at low I/NI thresholds of 0.025 and 0.05. Note that the minor condition i = 2 (smaller subgroup) leads to a small 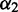 and therefore to a small I/NI value. For unbalanced data, the few minor condition samples must be distinguished from random outliers of the major condition.
and therefore to a small I/NI value. For unbalanced data, the few minor condition samples must be distinguished from random outliers of the major condition.
Table 2.
The performance of DEXUS in terms of sensitivity and specificity in detecting differential expression with unknown conditions
| I/NI threshold | 0.025 |
0.05 |
0.1 |
|||
|---|---|---|---|---|---|---|
| C1/C2 | Specificity | Sensitivity | Specificity | Sensitivity | Specificity | Sensitivity |
| 6/6 | 0.893 ± 0.003 | 0.775 ± 0.009 | 0.951 ± 0.002 | 0.720 ± 0.009 | 0.985 ± 0.002 | 0.646 ± 0.009 |
| 9/3 | 0.893 ± 0.004 | 0.827 ± 0.006 | 0.951 ± 0.002 | 0.766 ± 0.007 | 0.985 ± 0.001 | 0.580 ± 0.008 |
| 10/2 | 0.893 ± 0.003 | 0.819 ± 0.008 | 0.950 ± 0.002 | 0.656 ± 0.009 | 0.985 ± 0.001 | 0.325 ± 0.009 |
| 11/1 | 0.893 ± 0.003 | 0.677 ± 0.009 | 0.951 ± 0.002 | 0.351 ± 0.008 | 0.985 ± 0.001 | 0.020 ± 0.003 |
| 12/12 | 0.945 ± 0.002 | 0.735 ± 0.008 | 0.982 ± 0.001 | 0.665 ± 0.008 | 0.996 ± 0.001 | 0.610 ± 0.009 |
| 18/6 | 0.945 ± 0.003 | 0.816 ± 0.008 | 0.982 ± 0.002 | 0.743 ± 0.009 | 0.996 ± 0.001 | 0.570 ± 0.011 |
| 20/4 | 0.945 ± 0.003 | 0.810 ± 0.008 | 0.982 ± 0.002 | 0.625 ± 0.009 | 0.996 ± 0.001 | 0.308 ± 0.009 |
| 22/2 | 0.946 ± 0.002 | 0.650 ± 0.009 | 0.982 ± 0.001 | 0.325 ± 0.008 | 0.996 ± 0.001 | 0.006 ± 0.002 |
| Mean | 0.919 ± 0.028 | 0.764 ± 0.069 | 0.966 ± 0.017 | 0.606 ± 0.172 | 0.991 ± 0.006 | 0.383 ± 0.261 |
The first column ‘C1/C2’ contains the numbers of replicates for the first and second condition. The other columns list sensitivity and specificity (with standard deviations) of DEXUS at different I/NI thresholds as the average across 100 data sets. The last row (‘Mean’) gives the average of the results in the columns. The library size was 1e8 for all experiments.
Real-world RNA-Seq data
‘Nigerian HapMap’
Pickrell et al. (16) sequenced RNA from 69 Nigerian HapMap individuals to study expression quantitative trait loci (eQTLs). The read count data were provided by the ReCount repository (44). As in previous experiments, DEXUS was applied to these data with its default parameters and ranked genes according to the I/NI value. The read counts of top-ranked genes and the conditions identified by DEXUS are visualized as a heatmap in Figure 1.
Figure 1.

Heatmap of the normalized read counts of the 12 genes with the largest I/NI values for the ‘Nigerian HapMap’ data set. Colors range from white for low expression to blue for high expression. Different individuals are denoted along the x-axis, while the top-ranked genes are denoted by their gene symbols along the y-axis. Red crosses indicate samples that belong to the minor condition. At the right side of the heatmap, each gene is annotated by the minimum (‘>’), the median of two conditions (‘m1’ and ‘m2’) and the maximum (‘<’) read count.
Five out of the 12 top-ranked genes are located on the Y chromosome (RPS4Y1, CYorf15A, EIF1AY, TMSB4Y, RPS4Y2). For these genes, the identified conditions are related to the sex. For four of the 12 top-ranked genes, at least one eQTL is known. For ZFP57, the associated eQTL is the SNP rs1736924 with a minor allele frequency (MAF) of 0.14 (16). CDH1 has 6 eQTLs, one of which is SNP rs7196495 with a MAF of 0.22 (45). CLLU1OS possesses the eQTL SNP rs12580153 with a MAF of 0.19 (46). L1TD1 has 2 eQTLs, one of which is SNP rs12137088 with a MAF 0.30 (47). Because the MAFs are high, it is plausible that the minor alleles are observed in the HapMap data set and that they lead to differential expressions of the associated genes. The conditions that were found by DEXUS are related to the alleles of corresponding SNPs.
Because the HapMap samples are lymphoblastoid cells, we confirmed that the genes detected by DEXUS are indeed expressed in lymphoblastoid cell lines. The gene NLRP2, ranked 11th by DEXUS, is expressed in lymphoblastoid cells but with large variability (48), as shown in Supplementary Figure S17. NLRP2 is expressed in the HapMap individuals, but in some at a low level. Schlattl et al. (49) identified a copy number variable region that partially covers NLRP2 and causes its differential expression. Therefore, the conditions that DEXUS identified for NLRP2 may be related to copy number states of the samples. Copy number states may also be responsible for differential expression of MKRN3, which was ranked 12th by DEXUS. Pinto et al. (50) and Redon et al. (51) identified a copy number variable region covering MKRN3. However, interpreting the MKRN3 conditions is difficult because only the paternal copy of MKRN3 is expressed.
We analyzed the I/NI value ranking of transcripts: genes on the X chromosome were ranked significantly higher than other genes (P = 3.0e−12), which can be explained by sex-related transcripts. An analogous test for the Y chromosome was not significant because too few genes were expressed. However, as already mentioned, five out of the 12 top-ranked genes are located on the Y chromosome. At an I/NI threshold of 0.1, DEXUS called 366 differentially expressed genes. Gene set enrichment analysis showed that the called genes are associated with the extracellular region and the plasma membrane. In total, 20 significant GO terms were found, including ‘extracellular space’, ‘extracellular region part’ and ‘plasma membrane part’ with P = 2.5e−5, P = 8.8e−5 and P = 0.01, respectively. ‘Cell–cell signaling’, ‘chemokine receptor binding’ and ‘chemokine activity’ were also significant at P = 4.0e−3, P = 8.0e−4 and P = 9.8e−4 (P-values were corrected for multiple testing by means of the Benjamini–Hochberg procedure). These GO terms are in agreement with characteristics of lymphoblastoid cells. Supplementary Table S18 provides a complete list of all significant GO terms.
‘European HapMap’
We analyzed the RNA-Seq data of 60 individuals from the HapMap cohort from Montogmery et al. (15), which were provided by the ReCount repository (44). Again, DEXUS was applied to these data with its default parameters and ranked genes according to the I/NI value. The read counts of top-ranked genes and the identified conditions are visualized as a heatmap in Figure 2.
Figure 2.

Heatmap of the normalized read counts of the 12 genes with the largest I/NI values for the ‘European HapMap’ data set. Colors range from white for low expression to blue for high expression. Different individuals are denoted along the x-axis, while the top-ranked genes are denoted by their gene symbols along the y-axis. Red crosses indicate samples that belong to the minor condition. At the right hand side of the heatmap, each gene is annotated by the minimum (‘>’), the median of two conditions (‘m1’ and ‘m2’) and the maximum (‘<’) read count.
RPS4Y1 is the gene with the largest I/NI value, differentially expressed between males and females, and located on the Y chromosome. The genes CYorf15A and TMSB4Y, ranked fourth and fifth according to the I/NI value, are also located on the Y chromosome. As in the ‘Nigerian HapMap’ data set, ZFP57 was detected as being differentially expressed. In addition to ZFP57, two other of the 12 top-ranked genes have eQTLs. CLLU1OS has as eQTL the SNP rs12580153 with a MAF of 0.19 (46). POU2F3 has as eQTL the SNP rs2847497 with a MAF of 0.14 (52). As in the ‘Nigerian HapMap’ data set, some top-ranked genes, such as NLRP2 (again rank 11), were differentially expressed owing to variable copy numbers (49). For the genes T, PRSS21 and RASSF10, DEXUS identified two conditions for which an explanation remains to be found and which may indicate a hitherto unknown source of variability in gene expression. The second-ranked gene T, the third-ranked gene PRSS21 and the 12th-ranked gene RASSF10 are known to be expressed in B-lymphoblastoid cells (6,12), the cell type of the HapMap samples. The high expression variability of T and PRSS21 in the B-lymphoblastoid cell line has already been reported by the ENCODE Project (6). The ENCODE Project expression values for the genes T, PRSS21 and RASSF10 are visualized in Supplementary Figures S19, S20 and S21.
Analyzing the I/NI value ranking, we found that genes on the X chromosome are ranked significantly higher (P = 8.0e−6, Wilcoxon test). The analogous test for the Y chromosome yielded no significant results, as too few genes were expressed. However, three out of the 12 top-ranked genes with the largest I/NI value are located on the Y chromosome.
At an I/NI threshold of 0.1, DEXUS called 680 differentially expressed genes. Gene set enrichment analysis showed that the called genes are associated with ion transport. Significant Gene Ontology (GO) terms were ‘ion transport’, ‘potassium ion transport’ with P = 0.04 and P = 4.3e−03, respectively. Again ‘plasma membrane part’ was significant at P = 0.027. Although 36 of the 680 genes were related to ‘cell–cell signaling’ and 6 to ‘chemokine activity’, these GO terms were not significant in this data set after correction for multiple testing by means of the Benjamini–Hochberg procedure. A table of all significant GO terms can be found in Supplementary Table S19.
‘Primate Liver’
Blekhman et al. (53) investigated the differences in alternative splicing in liver tissue between humans, chimpanzees and rhesus macaques. For this purpose, they performed RNA-Seq on three male and three female liver samples from each species. They focused on the expression values of exons that had reliably determined orthologs in all species. Read counts for exons were provided by Blekhman et al. (53), who used gene models from Ensemble (Release 50). After pooling technical replicates, DEXUS ranked genes according to the I/NI value using its default parameters. The 10 top-ranked genes are visualized in Figure 3, which shows clear differential expression between the species. For these genes, and without having been provided with this information, DEXUS determined one of the three species as minor condition. Interestingly, out of the 10 top-ranked genes, six are human pseudogenes, AC010591.10, AC105383.3, AC093874.3-1, AC105383.3, AL132855.4 and UOX, which are inactive in humans because of recent structural rearrangements (54). Because the rearrangements are recent, their orthologs can be identified reliably in other primates. Differential expression is detected because these orthologs are still transcribed in Pan troglodytes and in Macaca mulatta.
Figure 3.
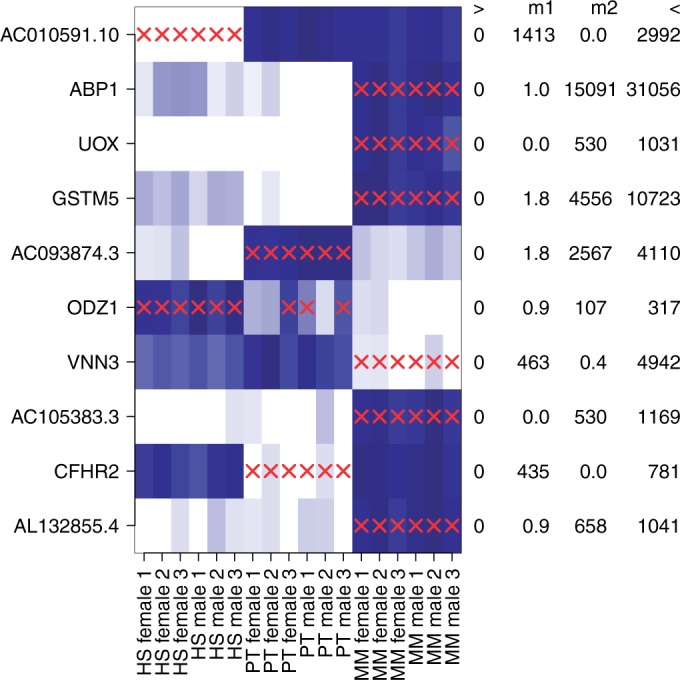
Heatmap of the normalized read counts of the 10 genes with the largest I/NI values for the ‘Primate Liver’ data set. Colors range from white for low expression to blue for high expression. The x-axis shows female and male individuals from the three species human Homo sapiens (HS), chimpanzee P. troglodytes (PT) and rhesus macaques M. mulatta (MM). The y-axis displays top-ranked genes indicated by their gene symbols. Red crosses mark samples that were assigned to the minor condition. At the right side of the heatmap, each gene is annotated by the minimum (‘>’), the median of two conditions (‘m1’ and ‘m2’) and the maximum (‘<’) read count.
Several of the 10 top-ranked genes are associated with liver pathways and are therefore expressed in liver tissues. Differential expression of these genes between species may have arisen from different diets. Examples of such genes are the human pseudogene UOX, which catalyze the oxidation of uric acid to allantoin in M. mulatta, ABP1 and GSTM5, which participate in degradation and detoxification pathways, VNN3, which helps to recycle vitamin B5, and CHFR2, which is associated with lipoproteins.
Thresholding the I/NI call at 0.1, DEXUS called 3384 genes (16% of all genes) as differentially expressed. A gene set enrichment analysis found the GO terms ‘intrinsic to plasma membrane’ (P = 7.9e−7) and ‘integral to plasma membrane’ (P = 4.0e−6) to be significant. Thus, genes that encode membrane proteins seem to be differentially expressed between species more often than other genes. Interestingly, also ‘response to extracellular stimulus’, ‘response to nutrient’ and ‘response to nutrient levels’ were significant (all P-values <7.6e−5), which supports the hypothesis that some genes are differentially expressed owing to the different diets of the species. All P-values were corrected by means of the Benjamini–Hochberg procedure.
‘Maize Leaves’
Using RNA-Seq data from various locations on maize plant leaves, Li et al. (55) studied the developmental dynamics of the maize transcriptome. For each location, two biological replicates were sequenced with Illumina’s Genome Analyzer II. The reads were mapped to the TE-masked Zea maize ZmB73 reference genome version 2 (AGPv2), release 5a, using the GSNAP splicing short read mapper (56). We counted the overlaps between mapped reads and the Z. maize gene definitions from the Ensemble Plants database (Release 14). Reads that have multiple possible alignments or that overlap with more than one gene were discarded. DEXUS was applied to this data with its default parameters.
Figure 4 shows the genes with the largest I/NI value and the conditions that were identified by DEXUS. DEXUS found differences in gene expressions between different locations on the leaf despite this information being withheld. Further, it almost always assigned the two replicates to the same condition without knowledge of replicates or leaf locations. Thus, DEXUS assigns conditions reliably.
Figure 4.
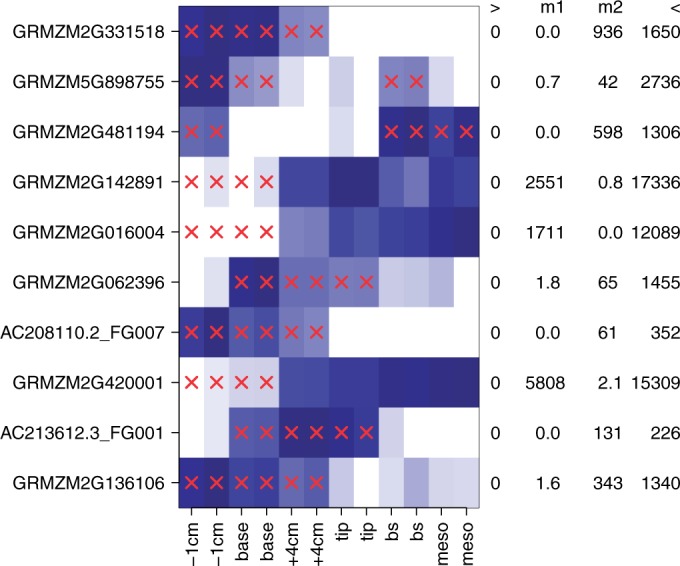
Heatmap of the normalized read counts of the 10 genes with the largest DEXUS I/NI values for the ‘Maize Leaves’ data set. Colors range from white for low expression to blue for high expression. The x-axis shows samples from different locations on the maize plant leaf. The y-axis displays different genes denoted by their gene symbols. Red crosses indicate that the according samples belong to the minor condition. At the right hand side of the heatmap, each gene is annotated by the minimum (‘>’), the median of two conditions (‘m1’ and ‘m2’) and the maximum (‘<’) read count.
Eight of the 10 top-ranked genes were also measured by means of microarrays across different leaf locations of Z. mays (57). In this microarray experiment, all eight genes show an absolute log fold change of at least 1 between base and tip. Six of these eight genes show an absolute log fold change greater than four.
The two remaining genes, GRMZM2G331518 and AC213612.3_FG001, were not annotated on the microarray. The function of the top-ranked gene GRMZM2G331518 is not known. However, the associated peptide is similar to the defensin-like protein 91 of Arabidopsis thaliana, which plays a role in immune response. The gene ranked ninth, AC213612.3_FG001, is a glycine-rich cell wall structural protein, which is compatible with cell walls at different locations having different structure.
At a threshold of 0.1 for the I/NI call, DEXUS called 15 756 differentially expressed genes. Gene set enrichment analysis using the R package goseq (58) yielded to the significant GO terms ‘chloroplast’ (P = 1.8e−92) and ‘plasma membrane’ (P = 1.3e−34). Further, the GO terms ‘cytosolic ribosome’ (P = 9.8e−32), ‘chloroplast thylakoid membrane’ (P = 5.4e−31) and ‘chloroplast stroma’ (P = 1.8e−30) were significant. All P-values were corrected by means of the Benjamini–Hochberg procedure. It is plausible that that chloroplast also differs at different locations on the maize plant leaf. The GO term ‘cell wall’ was highly significant (P = 3.9e−18), which supports the above-mentioned hypothesis that the cell walls differ at different locations on the plant leaf.
CONCLUSION
We have introduced DEXUS, an algorithm that identifies differentially expressed transcripts in RNA-Seq data with unknown conditions. DEXUS is appropriate for use with data from cohort, cross-sectional and nonrandomized controlled studies, where conditions are often unknown. In experiments with simulated and real-world data with known conditions, DEXUS successfully found differential expressed transcripts and conditions, although the conditions were withheld from DEXUS. For HapMap individuals, DEXUS detected differentially expressed transcripts, the vast majority of which are related to sex, eQTLs or structural variants. We envisage that DEXUS will evolve into an important tool for analyzing RNA-Seq data in studies with unknown conditions and thus for obtaining new biological and medical knowledge.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Funds from the Institute of Bioinformatics, Johannes Kepler University Linz.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z, Gerstein M, Snyder M. RNA-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 6.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labaj PP, Leparc GG, Linggi BE, Markillie LM, Wiley SH, Kreil DP. Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics. 2011;27:i383–i391. doi: 10.1093/bioinformatics/btr247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansen KD, Wu Z, Irizarry RA, Leek JT. Sequencing technology does not eliminate biological variability. Nat. Biotechnol. 2011;29:572–573. doi: 10.1038/nbt.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones AR, Overly CC, Sunkin SM. The Allen Brain Atlas: 5 years and beyond. Nat. Rev. Neurosci. 2009;10:821–828. doi: 10.1038/nrn2722. [DOI] [PubMed] [Google Scholar]
- 11.Heintz N. Gene expression nervous system atlas (GENSAT) Nat. Neurosci. 2004;7:483. doi: 10.1038/nn0504-483. [DOI] [PubMed] [Google Scholar]
- 12.Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. International HapMap 3 Consortium, Altshuler,D.M., Gibbs,R.A., Peltonen,L., Altshuler,D.M., Gibbs,R.A., Peltonen,L., Dermitzakis,E., Schaffner,S.F., Yu,F. et al. (2010) Integrating common and rare genetic variation in diverse human populations. Nature, 467, 52–58. [DOI] [PMC free article] [PubMed]
- 14.The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, Guigo R, Dermitzakis ET. Transcriptome genetics using second generation sequencing in a caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras J-B, Stephens M, Gilad Y, Pritchard JK. Understanding mechanisms underlying human gene expression variation with rna sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J-P, Subramanian A, Ross KN, et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 18.The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lal A, Lash AE, Altschul SF, Velculescu V, Zhang L, McLendon RE, Marra MA, Prange C, Morin PJ, Polyak K, et al. A public database for gene expression in human cancers. Cancer Res. 1999;59:5403–5407. [PubMed] [Google Scholar]
- 20.Uehara T, Ono A, Maruyama T, Kato I, Yamada H, Ohno Y, Urushidani T. The Japanese toxicogenomics project: application of toxicogenomics. Mol. Nutr. Food Res. 2010;54:218–227. doi: 10.1002/mnfr.200900169. [DOI] [PubMed] [Google Scholar]
- 21.Chen M, Vijay V, Shi Q, Liu Z, Fang H, Tong W. FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov. Today. 2011;16:697–703. doi: 10.1016/j.drudis.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 22.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:e161. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardcastle TJ, Kelly KA. baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics. 2010;11:422. doi: 10.1186/1471-2105-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Tibshirani R. Finding consistent patterns: a nonparametric approach for identifying differential expression in RNA-seq data. Stat. Methods Med. Res. 2011;22:519–536. doi: 10.1177/0962280211428386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Witten DM, Johnstone IM, Tibshirani R. Normalization, testing, and false discovery rate estimation for RNA-sequencing data. Biostatistics. 2012;13:523–538. doi: 10.1093/biostatistics/kxr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tarazona S, Garca-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21:2213–2223. doi: 10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H, Wang C, Wu Z. A new shrinkage estimator for dispersion improves differential expression detection in rna-seq data. Biostatistics. 2012;14:232–243. doi: 10.1093/biostatistics/kxs033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9:321–332. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 34.Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–2887. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- 35.Hochreiter S, Clevert D-A, Obermayer KA. A new summarization method for Affymetrix probe level data. Bioinformatics. 2006;22:943–949. doi: 10.1093/bioinformatics/btl033. [DOI] [PubMed] [Google Scholar]
- 36.Clevert D-A, Mitterecker A, Mayr A, Klambauer G, Tuefferd M, Bondt AD, Talloen W, Göhlmann H, Hochreiter S. cn.FARMS: a latent variable model to detect copy number variations in microarray data with a low false discovery rate. Nucleic Acids Res. 2011;39:e79. doi: 10.1093/nar/gkr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klambauer G, Schwarzbauer K, Mayr A, Clevert D-A, Mitterecker A, Bodenhofer U, Hochreiter S. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012;40:e69. doi: 10.1093/nar/gks003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lloyd-Smith JO. Maximum likelihood estimation of the negative binomial dispersion parameter for highly overdispersed data, with applications to infectious diseases. PLoS One. 2007;2:e180. doi: 10.1371/journal.pone.0000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piegorsch WW. Maximum likelihood estimation for the negative binomial dispersion parameter. Biometrics. 1990;46:863–867. [PubMed] [Google Scholar]
- 40.Dempster AP, Laird NM, Rubin DB. Maximum Likelihood from Incomplete Data via the EM Algorithm. J. R. Stat. Soc. B Met. 1977;39:1–38. [Google Scholar]
- 41.Talloen W, Clevert D-A, Hochreiter S, Amaratunga D, Bijnens L, Kass S, Göhlmann H. I/NI-calls for the exclusion of non-informative genes: a highly effective filtering tool for microarray data. Bioinformatics. 2007;23:2897–2902. doi: 10.1093/bioinformatics/btm478. [DOI] [PubMed] [Google Scholar]
- 42.Talloen W, Hochreiter S, Bijnens L, Kasim A, Shkedy Z, Amaratunga D. Filtering data from high-throughput experiments based on measurement reliability. Proc. Natl Acad. Sci. USA. 2010;107:173–174. doi: 10.1073/pnas.1010604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bottomly D, Walter NAR, Hunter JE, Darakjian P, Kawane S, Buck KJ, Searles RP, Mooney M, McWeeney SK, Hitzemann R. Evaluating gene expression in C57BL/6J and DBA/2J mouse striatum using RNA-Seq and microarrays. PLoS One. 2011;6:e17820. doi: 10.1371/journal.pone.0017820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frazee AC, Langmead B, Leek JT. ReCount: a multi-experiment resource of analysis-ready RNA-seq gene count datasets. BMC Bioinformatics. 2011;12:449. doi: 10.1186/1471-2105-12-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, Maouche S, Germain M, Lackner K, Rossmann H, et al. Genetics and beyond–the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Arcelus MG, Sekowska M, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veyrieras J-B, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M, Pritchard JK. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halbritter F, Vaidya HJ, Tomlinson SR. GeneProf: analysis of high-throughput sequencing experiments. Nat. Methods. 2011;9:7–8. doi: 10.1038/nmeth.1809. [DOI] [PubMed] [Google Scholar]
- 49.Schlattl A, Anders S, Waszak SM, Huber W, Korbel JO. Relating CNVs to transcriptome data at fine resolution: assessment of the effect of variant size, type, and overlap with functional regions. Genome Res. 2011;21:2004–2013. doi: 10.1101/gr.122614.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pinto D, Marshall C, Feuk L, Scherer SW. Copy-number variation in control population cohorts. Hum. Mol. Genet. 2007;16:R168–R173. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- 51.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blekhman R, Marioni JC, Zumbo P, Stephens M, Gilad Y. Sex-specific and lineage-specific alternative splicing in primates. Genome Res. 2010;20:180–189. doi: 10.1101/gr.099226.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balasubramanian S, Zheng D, Liu Y-J, Gang Fang AF, Carriero N, Robilotto R, Philip Cayting MG. Comparative analysis of processed ribosomal protein pseudogenes in four mammalian genomes. Genome Biol. 2009;10:R2. doi: 10.1186/gb-2009-10-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR, et al. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 2010;42:1060–1067. doi: 10.1038/ng.703. [DOI] [PubMed] [Google Scholar]
- 56.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sekhon RS, Lin H, Childs KL, Hansey CN, Buell CR, deLeon N, Kaeppler SM. Genome-wide atlas of transcription during maize development. Plant J. 2011;66:553–563. doi: 10.1111/j.1365-313X.2011.04527.x. [DOI] [PubMed] [Google Scholar]
- 58.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.