

CONSPECTUS
Nitric oxide synthase (NOS) converts L-arginine into L-citrulline and releases the important signaling molecule nitric oxide (NO). In the cardiovascular system NO produced by endothelial NOS (eNOS) relaxes smooth muscle which controls vascular tone and blood pressure.Neuronal NOS (nNOS) produces NO in the brain, where it influences a variety of neural functions such as neural transmitter release. NO can also support immune system, serving as a cytotoxic agent during infections.
Even with all of these important functions, NO is a free radical, and, when overproduced, it can cause tissue damage. This mechanism can operate in many neurodegenerative diseases, and as a result, the development of drugs targeting nNOS is a desirable therapeutic goal. However, the active sites of all 3 human isoforms are very similar, and designing inhibitors specific for nNOS is a challenging problem. It is critically important, for example, not to inhibit eNOS owing to its central role in controlling blood pressure.
In this Account we summarize our efforts in collaboration with Rick Silverman at Northwestern University to develop drug candidates that specifically target NOS using crystallography, computational chemistry, and organic synthesis. As a result we have developed aminopyridine compounds that are 3,800 fold more selective for nNOS than eNOS, some of which show excellent neuro-protective effects in animal models.
Our group has solved approximately 130 NOS-inhibitor crystal structures which have provided the structural basis for our design efforts. Initial crystal structures of nNOS and eNOS bound to selective dipeptide inhibitors showed that a single amino acid difference (Asp in nNOS and Asn in eNOS) results in much tighter binding to nNOS. The NOS active site is open and rigid, which produces few large structural changes when inhibitors bind. However, we have found that relatively small changes in the active site and inhibitor chirality can account for large differences in isoform-selectivity. For example, we expected that the aminopyridine group on our inhibitors would form a hydrogen bond with a conserved Glu inside the NOS active site. Instead, in one group of inhibitors, the aminopyridine group extends outside of the active site where it interacts with a heme propionate. For this orientation to occur, a conserved Tyr side chain must swing out of the way.This unanticipated observation taught us about the importance of inhibitor chirality and active site dynamics.
We also successfully used computational methods to gain insights into the contribution of the state of protonation of the inhibitors to their selectivity. Employing the lessons learned from the aminopyridine inhibitors, the Silverman lab designed and synthesized symmetric double-headed inhibitors with an aminopyridine at each end, taking advantage of their ability to make contacts both inside and outside of the active site.
Crystal structures provided yet another unexpected surprise. Two of the double-headed inhibitor molecules bound to each enzyme subunit, and one molecule participated in the generation of a novel Zn2+ site that required some side chains to adopt alternate conformations. Therefore, in addition to achieving our specific goal, the development of nNOS selective compounds, we have learned how subtle differences in dynamics and structure can control protein-ligand interactions and often in unexpected ways.
Introduction
Structure based approaches to drug design date back to the 1970s with the development of compounds designed to regulate hemoglobin1,2 and the anti-hypertensive drug, captopril.3 However, the wider acceptance of structure based methods coincided with the birth of the biotechnology industry in the early 1980s. With the availability more interesting recombinant proteins, crystallographers had new proteins for structure determination, many of which were important drug targets. The hope was that structure based approaches would streamline drug discovery. In practice, however, the expense of determining crystal structures did not compare favorably with more rapid combinatorial chemistry approaches. To get around this problem was one of the major incentives of the so-called protein structure initiative, generously funded by NIH but met with justifiable skepticism.4 The basic idea is to dramatically lower the cost of structure determination and rapidly provide the structure of drug targets for structure based drug design. This would enable the rational design approach to effectively compete with more “random” synthetic chemistry approaches. It remains to be seen the long-range contributions of the protein structure initiatives but we can ask if the basic approach of structure based drug design works which includes the development of clinically useful molecules. The answer is yes with perhaps the most widely known success story being the HIV protease.5 This also is a spectacular example of what can be achieved by close collaborative efforts to move very quickly in the face of a health emergency.
The focus of this review is our collaborative effort with Prof. Rick Silverman at Northwestern University to develop nitric oxide synthase inhibitors targeting neurodegenerative disorders. A similar review was published in 2009 with a focus on the medicinal chemistry end of this project.6 Here we focus on the protein structural end with an emphasis on new discoveries made since 2009.
NOS Structure
NOS catalyzes the oxidation of L-arginine to L-citrulline and nitric oxide (NO).
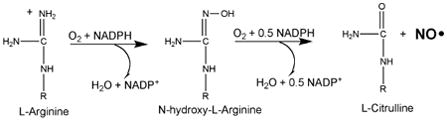
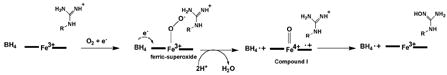
The first step of the reaction is very similar to cytochromes P450 with the exception that the tetrahydrobiopterin (BH4) cofactor serves as a source of an electron.7,8
The mechanism for the second step, N-hydroxy-L-arginine to NO and L-citrulline, remains unsettled and is still under active investigation.
Like cytochrome P450, the source of electrons is an FMN/FAD reductase which shuttles electrons from NADPH via the flavins to the heme. With NOS, however, the FMN/FAD reductase is fused to the C-terminal end of the heme domain which gives a large polypeptide over 1,000 residues which dimerizes9 through the heme domains. Although the crystal structure of holo-NOS has not yet been determined, the structure of the individual heme10-13 and reductase14 domains are known. The structures show that the substrate, L-Arg, is held in place by a series of H-bonds including the conserved active site Glu (Fig. 1). The BH4 is situated at the dimer interface and H-bonds to the same heme propionates accepting H-bonds from L-Arg.
Figure 1.
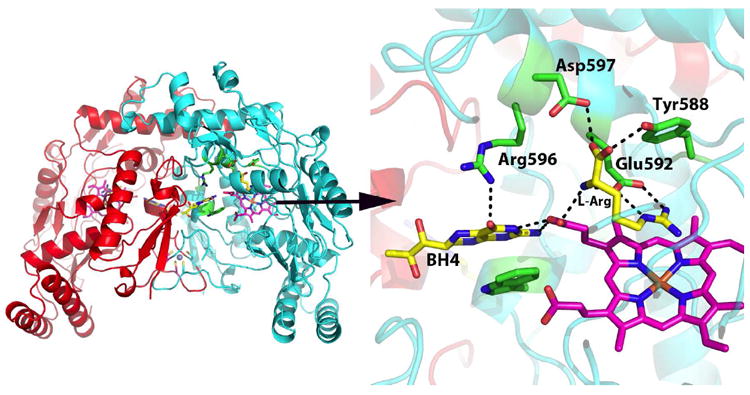
Rat nNOS heme domain dimer strcuture (1OM4). L-Arg is held in place by a series of H-bonds to conserved active site residues as well as one heme propionate. The cofactor, BH4, is positioned at the dimer interface and also H-bonds with the same heme propionate.
NOS as Drug Target
Humans produce three isoforms of NOS: endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS).15 The active site structure is the same for all three with some key differences to be highlighted further on. NO produced by eNOS and nNOS binds to the heme of guanylate cyclase which activates the cyclase to produce cyclic GMP, the final signaling molecule.16 eNOS helps to control proper blood pressure while nNOS-derived NO is involved with neuronal signaling. iNOS is controlled at the level of transcription and is produced by the immune system where NO serves as a cytotoxic agent.
The over production of NO is associated with many neurodegenerative disorders17-22 and thus it is highly desirable to develop nNOS inhibitors. The problem is selectivity. eNOS is critical for maintaining proper blood pressure and thus generic NOS inhibitors might well block the neurotoxic effect of too much NO in the brain but would also result in unwanted hypertension owing to the inhibition of eNOS. The goal of our joint project has been to use crystallography, computational methods, and synthetic chemistry to develop inhibitors that selectively block nNOS but not eNOS.
Initial Discoveries
Although the development of NOS inhibitors began very soon after NOS was discovered, structure based approaches could not be applied until the first crystal structures appeared in 1998.10-13 In our lab studies have focused on rat nNOS and bovine eNOS. From the perspective of isoform-selectivity the NOS crystal structures were disappointing since the active sites for all three isoforms are nearly identical (Fig. 1) which is not too surprising since all three catalyze exactly the same reaction. The earliest NOS inhibitors developed mimicked the substrate, L-Arg. For example, prior to the availability of the crystal structures, L-nitroarginine was reported to be moderately selective for nNOS over iNOS.23
This provided a logical starting point for the development of more selective inhibitors and is where the Silverman lab stepped into the picture. It was reasoned that since the active site where the chemistry of L-Arg oxidation occurs is the same in all NOS isoforms, selectivity is unlikely to be achieved by simple variation on the basic L-Arg skeleton. However, further from the active site toward the surface of the enzyme sequence diversity will be tolerated and thus it might be possible to add a “tail” to L-nitroarginine that extends out of the active site and possibly interact selectively to a unique patch on nNOS and not eNOS. Of course there were no crystal structures although it was known that a conserved Glu is required for activity.24 Therefore, a series of dipeptide inhibitors were developed using a substrate analog, L- or D-nitroarginine, as the NOS active site recognition group linked by a peptide bond with a second amino acid or an other similar chemical group. This worked and resulted in dipeptide inhibitors that were about 1,000-fold more selective for rat nNOS than bovine eNOS.25-27 Our labs then teamed up to try to understand the structural basis for selectivity. The best dipeptide inhibitors that exhibited about 1,500-fold selectivity were chosen for initial crystal structure work. These structures provided the first Eureka moment and the answer to selectivity was shockingly simple.28 As shown in Fig. 2, the dipeptide inhibitor “curls” in nNOS but adopts an extended conformation in eNOS.
Figure 2.
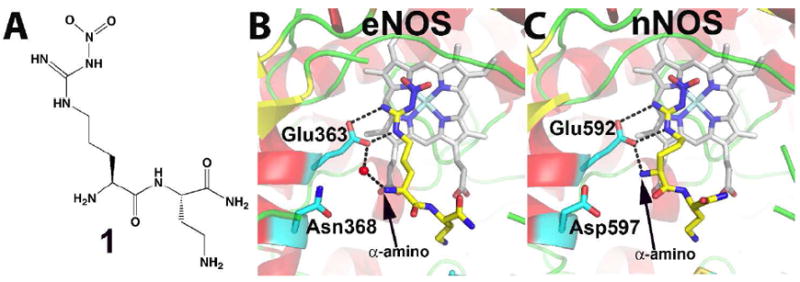
A) Structure of one of the early dipeptide lead compounds, 1, that exhibits excellent isoform selectivity. B) and C) show the crystal structures of the dipeptide inhibitor 1 in the active site of eNOS (1P6L) and nNOS (1P6H). In nNOS the inhibitor “curls” which enables the inhibitor α-amino group to interact with both Glu592 and Asp597. In eNOS Asn368 is the homolog to nNOS Asp597.
This is due to a single amino acid difference in the active site: where nNOS has Asp597, eNOS has Asn368. The α-amino group of the inhibitor maximally interacts with both Glu592 and Asp597 in nNOS if the inhibitor curls thus providing additional electrostatic stabilization not possible in eNOS. This hypothesis was proven by swapping the Asp/Asn side chains in eNOS and nNOS and, indeed, replacing the Asn with Asp in eNOS resulted in tighter binding and a curled conformation and just the opposite happened when the Asp in nNOS was replaced with Asn (Table 1).
Table 1.
Ki values for initial dipeptide inhibitors used for crystal structure determination28
| Ki (μM) of Inhibitors | |||
|---|---|---|---|
| I | II | III | |
| WT eNOS | 107.0 | 80.0 | 110.0 |
| WT nNOS | 0.30 | 0.15 | 0.10 |
| nNOS D597N | 67.0 | 34.0 | 21.0 |
| eNOS N368D | 9.5 | 4.6 | 5.1 |
| |||
The Importance of Chirality
The next generation of inhibitors has an aminopyridine head, a pyrrolidine bridge group, and a tail. A novel fragment hopping computational method was used by the Silverman lab in the design process.29 The basic idea was for the aminopyridine to mimic the guanidinium group of L-Arg to H-bond to Glu592 while the pyrrolidine N atom, mimicking the α-amino N atom in the dipeptide, would be rigidly held by the ring between Glu592 and Asp597 (Asn in eNOS) thereby providing additional electrostatic stabilization in nNOS compared to eNOS (Fig. 3).
Figure 3.
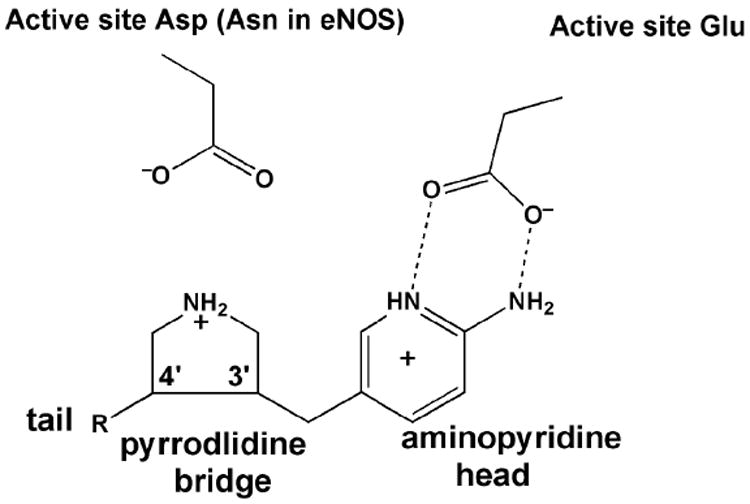
The initial design of aminopyridine-pyrrolidine inhibitors. The aminopyridine mimics the guanidinium group of L-Arg but exhibits a much lower pKa thus increasing bioavailability. In addition the aromatic aminopyrridine should stack over the heme ring. The pyrrolidine NH group extends up toward the active site Asp597 in nNOS.
Subsequent crystal structures showed that the initial design and docking studies closely matched the crystal structure.30 These inhibitors have two chiral centers (Figs. 3 and 4) and up to this point we were working with racemic mixtures.
Figure 4.
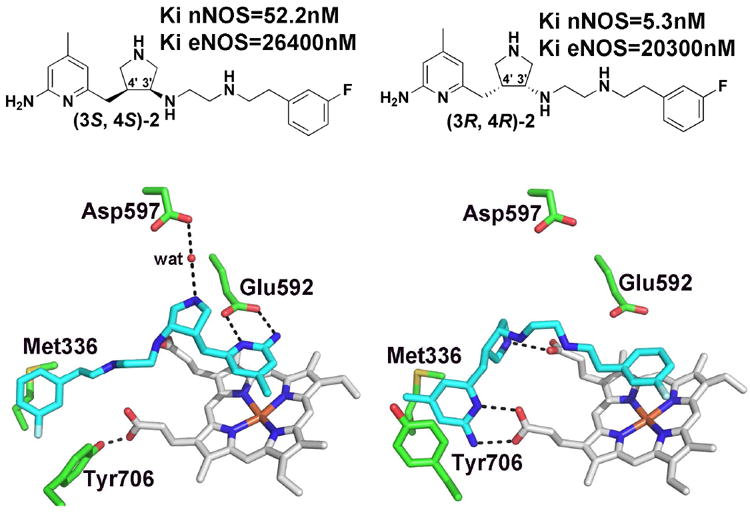
Crystal structures of the two enantiomers of cis-2 bound to nNOS. (3S,4S)-2 (3JWS) binds as expected with the aminopyridine positioned over the heme near Glu592. However, (3R,4R)-2 (3JWT) binds in the flipped mode which places the aminopyridine near a heme propionate. This requires Tyr706 to adopt the “out” rotamer conformation.
Most importantly a racemic mix of compound 2 (Fig. 4) exhibits excellent protection from ischemic brain damage in a cerebral palsy animal model.31 The crystal structure of enantiopure 2 complexed to nNOS provided a surprise.32 (3S,4S)-2 bound as expected with the aminopyridine H-bonded to the active site Glu592. The trans versions of 2 also bound as expected. However, (3R,4R)-2 flipped 180° placing the aminopyridine in position to H-bond with the heme propionate. In order for this to happen Tyr706 must adopt a new rotamer conformation. The reorientation of Tyr706 requires that the Tyr706-heme propionate H-bond be broken, a conserved interaction in all NOS isoforms for which structures are available. The new rotamer position enables the Tyr706 aromatic ring to stack against the inhibitor aminopyridine.
These structures and inhibition data presented two key questions. First, why is (3R,4R)-2 some 3,800-fold more selective for nNOS over eNOS while (3S,4S)-2 is only about 500-fold selective and second, why is the Ki for (3R,4R)-2 10-fold lower than (3S,4S)-2? The structure of eNOS complexed with (3R,4R)-2 exhibited exactly the same flipped binding mode as in nNOS so unlike the dipeptide inhibitors, the isoform-selectivity in this case is not attributed to differences in the inhibitor conformation and binding mode. The first question was addressed by determining the Ki of various nNOS mutants designed to mimic eNOS. The two most important are Asp597 and Met336 in nNOS which are Asn and Val, respectively, in eNOS. Although Asp597 does not directly contact 2 in the flipped binding mode, the negative charge on Asp597 should increase electrostatic stabilization of (3R,4R)-2 especially in the relatively low dielectric environment of the active site. In this mutagenetic analysis we also included the effects of Tyr706 which must move for inhibitors to bind in the flipped mode. We have consistently found that various inhibitors that bind in the flipped mode, Tyr706 forms a better stacking interaction with the aminopyridine in nNOS than eNOS. The only way to test the relative effects of this Tyr side chain was to simply covert it to Ala. The mutant that was most important to compare is the triple D597N/M336V/Y706A nNOS mutant with the Y477A eNOS mutant. For the nNOS triple mutant Ki = 1,290 nM, a 240-fold increase, while Ki = 35,200 nM for the Y477A eNOS mutant, a modest 1.7 fold increase. It thus appears that a combination of additional electrostatic interactions and better non-bonded contacts in nNOS contribute to isoform selectivity.
More recently we found that some aminopyridine-pyrrolidine inhibitors bind in the flipped mode in both eNOS and nNOS but the Tyr moves only in nNOS (unpublished). This must mean Tyr706 is more flexible in nNOS than in eNOS. As shown in Fig. 5 when Tyr706 rotates out to make room for the inhibitor Tyr706 contacts Leu367 and we have found that non-bonded contacts in this region are, indeed, important.33 Probing this region via mutagenesis, however, is problematic since the last well defined residue is Pro338. After Pro338 nNOS electron density maps effectively disappear for about 10 residues clearly showing that this region is highly flexible. eNOS is similar with 10 residues disordered after Pro108 (equivalent to Pro338 in nNOS) in most of structures. However, with some inhibitor complexes the electron density extends a few more residues suggesting that this region of eNOS is slightly more rigid. If this region is more flexible in nNOS then it might be easier for Tyr706 to adopt the rotamer conformation favorable for the flipped binding mode. In this scenario Tyr706 is an equilibrium mix of both the “in” and “out” rotamers and the inhibitor binds best to the “out” rotamer. In nNOS the effective population of the “out” rotamer is higher than in eNOS which accounts in part for isoform selectivity.
Figure 5.
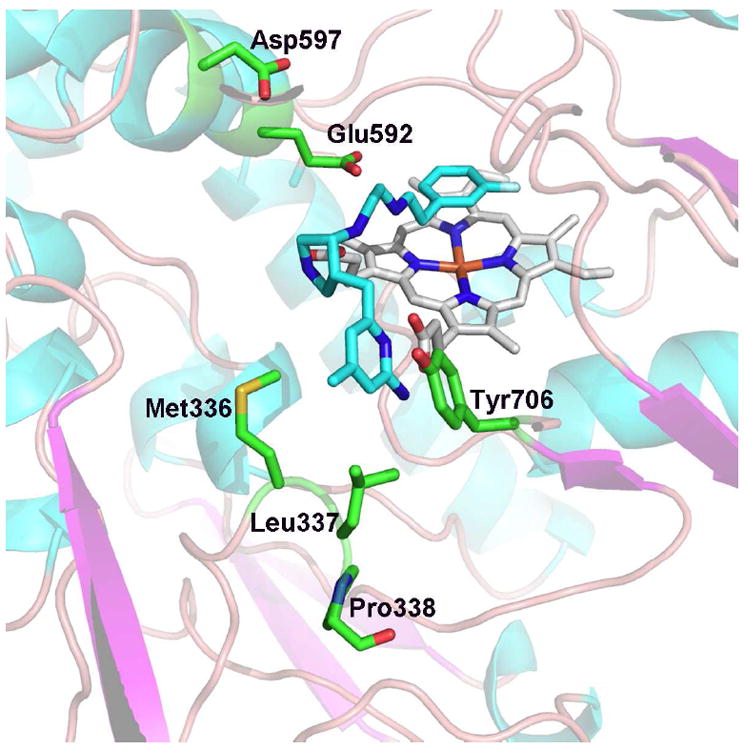
The nNOS active site showing the inhibitor in the flipped mode (3JWT). Note that the aminopyridine and Tyr706 are close to residues Met336 and Leu337. There is a break in the electron density right after Pro338 suggesting that this region may be particularly flexible in nNOS which enables Tyr706 to more readily adopt the out conformation relative to eNOS.
The final question to address is why (3R,4R)-2 binds about 10-fold better to nNOS than (3S,4S)-2. Even though the binding modes are totally different, the structures alone did not provide much insight into why there should be a difference. Here is where computational approaches proved useful. The Molecular Mechanic/Poisson-Boltzman Surface Area (MM-PBSA) procedure is a proven tool for calculating ligand binding free energies.34 The overall molecular mechanical internal energy (electrostatics, non-bonded, bond angles, distance, etc), the electrostatic component of the solvation energy, and the nonpolar component of the solvation energy are calculated and the results from all three are summed to give the total free energy of the complex, Gcomplex. Then the inhibitor is removed from the protein and exactly the same calculations are carried out to give the free energy of the protein (receptor) alone, Greceptor, and of the inhibitor alone, Ginhibitor. The total free energy of binding is then
The most challenging part of these types of calculations is the entropy. As others have done35 we ignored the entropy term which for the aminopyridine inhibitors is justified since the size and number of rotatable bonds is about the same for most of the inhibitors yet the affinities cover a wide range. This precludes the estimate of absolute ΔG but does provide a relative ΔG that can be compared to experimental values and enables the generation of “standard curves” as shown in Fig. 6.
Figure 6.
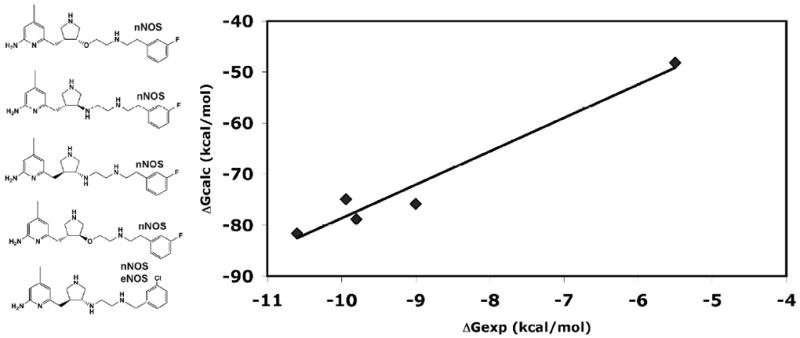
A total of 7 crystal structures of the 5 different aminopyridine inhibitors bound to either nNOS or eNOS were used for the free energy calculations. The correlation between the relative ΔGcalc and experimental ΔGexp (extracted from measured Ki values) is excellent.
For such studies parameters for the ligand are required which includes total charge. Compound 2 and related aminopyridines have 2-3 groups which can carry a full positive charge. For 2 this is the aminopyridine, the pyrrolidine, and the NH group closest to fluoro-phenyl ring so 2 can potentially carry a +3 charge. In the normal binding mode of (3S,4S)-2 the pyrrolidine N atom is about 4.1 Å from the aminopyridine. While both are interacting with Glu592, there is no net charge neutralization which raises the possibility that the aminopyridine, with a pKa near neutrality, is only partially protonated. However, in the flipped binding mode the pyrrolidine N atom and aminopyridine are 4.8 Å apart and both propionates of the heme are close so there is a net charge neutralization if the aminopyridine carries a full +1 charge. We used the MM-PBSA approach to compute the difference between these two models. In model 1 we assume that in both the flipped and normal binding modes the aminopyridine is fully protonated so the entire molecule has a +3 charge. In model 2 we assume that the charge is +3 only in the flipped mode but in the normal binding mode the aminopyridine carries only a +0.5 charge so the net charge is +2.5. The MM-PBSA calculation was carried out for 11 aminopyridine-nNOS crystal structures that provided a mix of flipped and normal binding modes and then the calculated free energies plotted against experimental free energies. In the charge +3 model 1 the correlation coefficient of the plot was 0.52 but for the charge +2.5 model 2 the correlation coefficient increased to 0.81. The calculations and chemical intuition agree: the state of protonation of the inhibitor is highly dependent on inhibitor conformation and interactions with the protein and clearly contributes to binding affinity and isoform-selectivity.
Are Two Heads Better than One?
Since the aminopyridines can bind in two different orientations, the next obvious step was to build a double headed symmetric inhibitor of the form shown in Fig. 7
Figure 7.
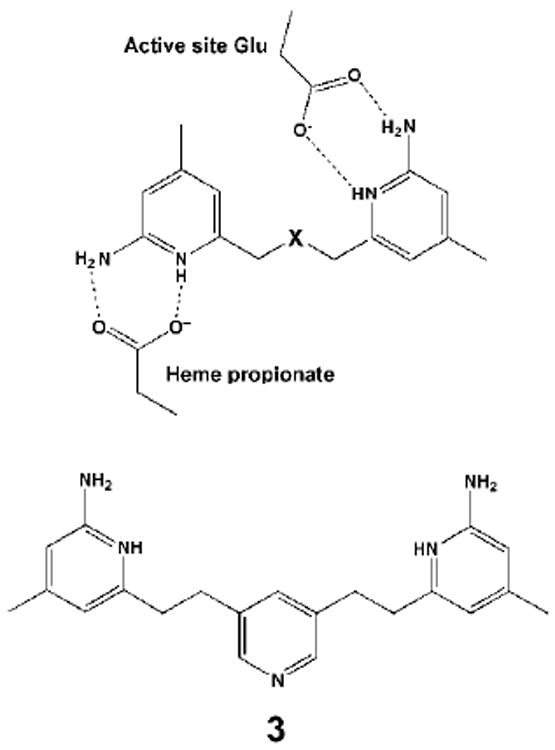
The initial design of double-headed symmetric inhibitors.
Another advantage of these double headed compounds over the original parent inhibitors such as 2 is an increase in lipophilicity which should present fewer problems in crossing the blood-brain barrier.36 We focus on compound 3. The Ki for nNOS is 25 nM and for eNOS 2,680 nM giving a selectivity of about 107. Compound 3 clearly is not as an effective inhibitor as 2 nor is it as selective but the better bioavailability might overcome these disadvantages. Indeed, compound 3 exhibits an IC50 of 5 μM using a cell-based nNOS assay which is several fold better than 2.36
Once again, however, the crystal structures provided an unexpected surprise.37 As shown in Fig. 8, there are two molecules of 3 bound to the nNOS active site.32 One of the molecules binds exactly as predicted. One aminopyridine interacts with the active site Glu while the other displaces Tyr706 and H-bonds with the heme propionate. The second molecule of 3 displaces the BH4 cofactor enabling one of the two aminopyridines to H-bond with the other heme propionate. Normally Arg596 H-bonds with BH4 but when the second molecule of 3 binds, Arg596 must swing out of the way where it now interacts with Asp597. This places the bridging pyridine of the second molecule of 3 in position to complete formation of a Zn2+ binding site. X-ray anomalous dispersion proves that this is a Zn2+ ion.32 In addition to the pyridine of 3, Asp600 and His692 together with a Cl- ion complete the Zn2+ coordination sphere. This new Zn2+ site is close to the dimer interface and one of the ligands, His692, belongs to molecule B of the dimer. Thus there is a slight tightening of the dimer interface in order to form the Zn2+ pocket. The affinity for Zn2+ must be fairly tight since Zn2+ was not included in protein purification or crystallization.
Figure 8.
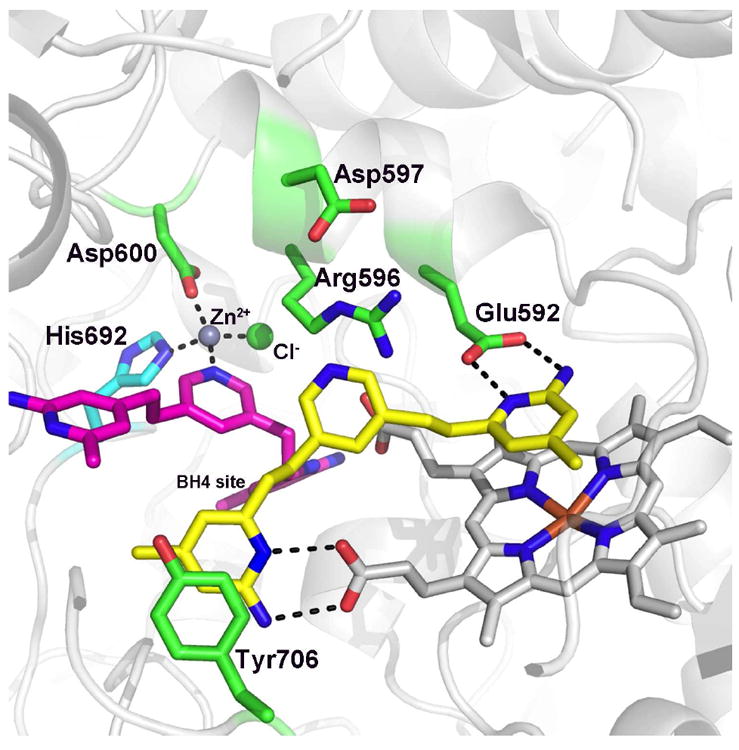
Crystal structure of 3 complexed to nNOS (3N5W). One molecule of 3 binds as predicted with one aminopyridine interacting with Glu592 and other with the heme propionate. The second molecule of 3 displaces BH4 thus enabling an aminopyridine to interact with the second heme propionate. This places the bridging pyridine in position to complete a tetrahedral coordination sphere around a Zn2+ ion. In order for the Zn2+ to bind, Arg596, which normally interacts with BH4, must swing out of the way. In addition, the dimer interface must slightly tighten to enable His692 from molecule B of the dimer to move close enough for Zn2+ coordination.
With eNOS only one molecule of 3 binds even though eNOS has exactly the same potential Zn2+ ligands as nNOS. Many mutants have been generated to try to understand this difference but no consistent picture has emerged. One possible explanation is that the dimer interface in eNOS is tighter than in nNOS. As noted the dimer interface must slightly tighten in order to enable His692 to be in position to coordinate Zn2+. If eNOS has restricted motion in this region, then the Zn2+ affinity will be much lower and the second molecule of 3 will not bind. This explanation is not particularly satisfying since there is no straightforward experimental test. Even so nNOS-3 and additional structures of compounds that are variations of 332 provide the structural underpinning for alternative strategies in the development of isoform selective inhibitors.
Bioavailability
While the structural basis for isoform-selectivity is now understood and some of these compounds show great promise as therapeutic agents for neurodegenerative disorders,31,38 bioavailability remains a problem. The main limitation is too much positive charge. Several well-known approaches have been taken in the Silverman lab to solve this problem. For example, introducing electron withdrawing fluorine atoms will lower the pKa of a neighboring NH group and so compounds 4 and 5 (Fig. 9) were synthesized.39-41 The CF2 group in 5 should lower the pKa of the NH group to about 5.5-6 and turned out to be the most bioavailable inhibitor. The crystal structures of the NOS-5 complex (3NLX and 3NLU) showed that 5 binds in the flipped binding mode as expected. The computed free energy of binding of 5 best matched experimental free energies when the NH group was assigned a +0.5 charge. In fact, the agreement was remarkably good, ΔGcalc = -10.4 kcal/mol and ΔGexp = -10.2 kcal/mol, especially since the flurophenyl group adopted multiple conformations so it was necessary to compute each structure and then average the results. 5 exhibits a Ki = 36 nM for nNOS and 140,000 nM for eNOS so the selectivity is about 3,800-fold. Intravenous dosing of rats showed that 4 (5 without the two gem-fluorines) was 0.33 hours and oral bioavailability essentially zero while 5 exhibited a half-life of 7.5 hours and a 22% oral availability.39
Figure 9.
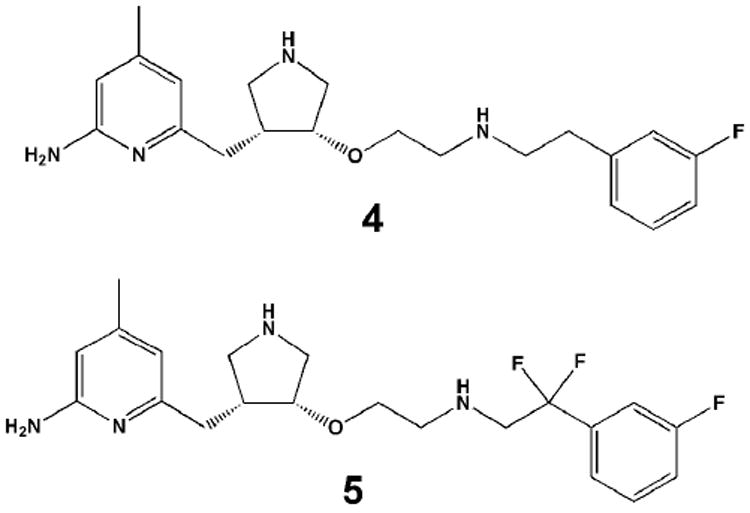
Variations on inhibitor 2 designed for better bioavailability.
Summary
Although challenging, it has been possible to generate inhibitors selective for nNOS. Similar approaches to those we have described here also have been employed to develop iNOS inhibitors that exhibit exceptional selectivity over eNOS.42 The basis for selectivity is quite different than we found with nNOS and is due primarily to inhibitors promoting formation of a novel binding pocket presumably due to greater flexibility near the iNOS active site.43 While the problem of isoform selectivity is basically solved, the next step of moving into human clinical trials faces the bioavialbility hurdle. Even so some of the compounds we have developed exhibit excellent neuro-protective effects in animal models. NuerAxon also has developed nNOS-selective inhibitors that work well in animal migraine models.44 These efforts provide examples of how a combination of computational modeling, crystallography, mutagenesis, organic synthesis, and close collaborative research can solve an important drug design problem. This effort also has provided some interesting insights into how unexpected structural changes contribute to isoform-selectivity. The role of chirality in inhibitor binding modes, the binding of two of the symmetric double-headed inhibitors resulting in the “creation” of a novel Zn2+ site, and the formation of a novel binding pocket in iNOS43 were unexpected. NOS has thus provided a platform for both further sharpening the tools of structure-based drug design and for probing the more basic questions on protein-ligand interactions.
Acknowledgments
We are grateful to those who have contributed to this work including Mack Flinspach, Silvia Delker, Jotaro Igarashi, and Joumana Jamal. We are indebted to Prof. Rick Silverman and the many postdocs and students in his lab who have made this collaborative effort possible. Financial support was provided by NIH grant GM57353 (Poulos) and NIH grant GM49725 (Silverman). We also gratefully acknowledge the beamline staff at the Stanford Synchrotron Radiation Lab and the Advanced Light Source.
Biographies
Huiying Li was born on July 26, 1951 in Beijing, P.R. China. He received a Bachelor degree in Chemistry in 1977 from Shanxi University, Taiyuan, PRC. He next earned a PhD in Physical Chemistry in 1989 from University of South Carolina. In 1990 he joined the Poulos lab as a postdoctoral fellow at the University of Maryland’s Center for Advanced Research in Biotechnology. Here is where he initiated his work on cytochromes P450 and was the first to solve the structure of a P450 in both the “open” and “closed” forms which provided the initial structural insights into the dynamics involved in substrate binding. He moved with Prof. Poulos to the University of California, Irvine (UCI) in 1992 where he played a key role in establishing protein crystallography at UCI. Dr. Li initiated the early work on nitric oxide synthase crystallography which ultimately resulted in the crystal structure of all 3 mammalian isoforms. Dr. Li currently holds the title of Project Scientist where he leads the lab’s effort in nitric oxide synthase.
Thomas L. Poulos was born in Monterey California on Feb. 1, 1947. After graduation from Carmel High School he earned a B.A. degree in Zoology at the University of California, Santa Barbara in 1968 followed by a PhD in Biology in 1972 at the University of California at San Diego (UCSD). He then moved to the Chemistry Department at UCSD for postdoctoral work in the protein crystallography lab of Joe Kraut. While at UCSD he solved the first heme enzyme crystal structure, cytochrome c peroxidase, and initiated work on P450s. In 1983 he was recruited to Genex Corp. in Gaithersburg MD where he held the position of Principal Research Scientist and then Director of Protein Engineering. It was during this time that he solved the first cytochrome P450 structure. In 1987 he moved to the University of Maryland where he was a Professor of Chemistry and Director of the Center for Advanced Research in Biotechnology. In 1992 he moved to the Department of Molecular Biology and Biochemistry at UCI where he now holds the title of Chancellor’s Professor and joint appointments in the Departments of Chemistry and Pharmaceutical Sciences. In 1991 he won the Presidential Meritorious Service Award from the University of Maryland and in 2004 the Brodie Award from the American Society of Experimental Pharmacology and Therapeutics. His primary research interests are in heme enzyme structure and function and structure-based drug design.
References
- 1.Goodford PJ. Drug design by the method of receptor fit. J Med Chem. 1984;27:558–564. doi: 10.1021/jm00371a001. [DOI] [PubMed] [Google Scholar]
- 2.Walkinshaw MD. Protein targets for structure-based drug design. Med Res Rev. 1992;12:317–372. doi: 10.1002/med.2610120403. [DOI] [PubMed] [Google Scholar]
- 3.Cushman DW, Cheung HS, Sabo EF, Ondetti MA. Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry. 1977;16:5484–5491. doi: 10.1021/bi00644a014. [DOI] [PubMed] [Google Scholar]
- 4.Petsko GA. An idea whose time has gone. Genome Biol. 2007;8:107. doi: 10.1186/gb-2007-8-6-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wlodawer A, Erickson JW. Structure-based inhibitors of HIV-1 protease. Annu Rev Biochem. 1993;62:543–585. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]
- 6.Silverman RB. Design of selective neuronal nitric oxide synthase inhibitors for the prevention and treatment of neurodegenerative diseases. Acc Chem Res. 2009;42:439–451. doi: 10.1021/ar800201v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurshman AR, Krebs C, Edmondson DE, Huynh BH, Marletta MA. Formation of a pterin radical in the reaction of the heme domain of inducible nitric oxide synthase with oxygen. Biochemistry. 1999;38:15689–15696. doi: 10.1021/bi992026c. [DOI] [PubMed] [Google Scholar]
- 8.Wei CC, Wang ZQ, Wang Q, Meade AL, Hemann C, Hille R, Stuehr DJ. Rapid kinetic studies link tetrahydrobiopterin radical formation to heme-dioxy reduction and arginine hydroxylation in inducible nitric-oxide synthase. J Biol Chem. 2001;276:315–319. doi: 10.1074/jbc.M008441200. [DOI] [PubMed] [Google Scholar]
- 9.Tzeng E, Billiar TR, Robbins PD, Loftus M, Stuehr DJ. Expression of human inducible nitric oxide synthase in a tetrahydrobiopterin (H4B)-deficient cell line: H4B promotes assembly of enzyme subunits into an active dimer. Proc Natl Acad Sci U S A. 1995;92:11771–11775. doi: 10.1073/pnas.92.25.11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 11.Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Structural characterization of nitric oxide synthase isoforms reveals striking active-site conservation. Nat Struct Biol. 1999;6:233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Raman CS, Glaser CB, Blasko E, Young TA, Parkinson JF, Whitlow M, Poulos TL. Crystal structures of zinc-free and -bound heme domain of human inducible nitric-oxide synthase. Implications for dimer stability and comparison with endothelial nitric-oxide synthase. J Biol Chem. 1999;274:21276–21284. doi: 10.1074/jbc.274.30.21276. [DOI] [PubMed] [Google Scholar]
- 13.Raman CS, Li H, Martasek P, Kral V, Masters BS, Poulos TL. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 14.Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- 15.Bredt DS, Snyder SH. Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- 16.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3’:5’-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A. 1977;74:3203–3207. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 18.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci. 2004;27:283–289. doi: 10.1016/j.tins.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 20.Hantraye P, Brouillet E, Ferrante R, Palfi S, Dolan R, Matthews RT, Beal MF. Inhibition of neuronal nitric oxide synthase prevents MPTP-induced parkinsonism in baboons. Nat Med. 1996;2:1017–1021. doi: 10.1038/nm0996-1017. [DOI] [PubMed] [Google Scholar]
- 21.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 22.Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci U S A. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furfine ES, Harmon MF, Paith JE, Garvey EP. Selective inhibition of constitutive nitric oxide synthase by L-NG-nitroarginine. Biochemistry. 1993;32:8512–8517. doi: 10.1021/bi00084a017. [DOI] [PubMed] [Google Scholar]
- 24.Chen PF, Tsai AL, Berka V, Wu KK. Mutation of Glu-361 in human endothelial nitric-oxide synthase selectively abolishes L-arginine binding without perturbing the behavior of heme and other redox centers. J Biol Chem. 1997;272:6114–6118. doi: 10.1074/jbc.272.10.6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silverman RB, Huang H, Marletta MA, Martasek P. Selective inhibition of neuronal nitric oxide synthase by N omega-nitroarginine-and phenylalanine-containing dipeptides and dipeptide esters. J Med Chem. 1997;40:2813–2817. doi: 10.1021/jm970200u. [DOI] [PubMed] [Google Scholar]
- 26.Gomez-Vidal JA, Martasek P, Roman LJ, Silverman RB. Potent and selective conformationally restricted neuronal nitric oxide synthase inhibitors. J Med Chem. 2004;47:703–710. doi: 10.1021/jm030297m. [DOI] [PubMed] [Google Scholar]
- 27.Hah JM, Roman LJ, Martasek P, Silverman RB. Reduced amide bond peptidomimetics. (4S)-N-(4-amino-5-[aminoakyl]aminopentyl)-N’-nitroguanidines, potent and highly selective inhibitors of neuronal nitric oxide synthase. J Med Chem. 2001;44:2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]
- 28.Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah JM, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Structural basis for dipeptide amide isoform-selective inhibition of neuronal nitric oxide synthase. Nat Struct Mol Biol. 2004;11:54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 29.Ji H, Stanton BZ, Igarashi J, Li H, Martasek P, Roman LJ, Poulos TL, Silverman RB. Minimal pharmacophoric elements and fragment hopping, an approach directed at molecular diversity and isozyme selectivity. Design of selective neuronal nitric oxide synthase inhibitors. J Am Chem Soc. 2008;130:3900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Igarashi J, Li H, Jamal J, Ji H, Fang J, Lawton GR, Silverman RB, Poulos TL. Crystal structures of constitutive nitric oxide synthases in complex with de novo designed inhibitors. J Med Chem. 2009;52:2060–2066. doi: 10.1021/jm900007a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji H, Tan S, Igarashi J, Li H, Derrick M, Martasek P, Roman LJ, Vasquez-Vivar J, Poulos TL, Silverman RB. Selective neuronal nitric oxide synthase inhibitors and the prevention of cerebral palsy. Ann Neurol. 2009;65:209–217. doi: 10.1002/ana.21555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delker SL, Ji H, Li H, Jamal J, Fang J, Xue F, Silverman RB, Poulos TL. Unexpected binding modes of nitric oxide synthase inhibitors effective in the prevention of a cerebral palsy phenotype in an animal model. J Am Chem Soc. 2010;132:5437–5442. doi: 10.1021/ja910228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xue F, Li H, Fang J, Roman LJ, Martasek P, Poulos TL, Silverman RB. Peripheral but crucial: a hydrophobic pocket (Tyr(706), Leu(337), and Met(336)) for potent and selective inhibition of neuronal nitric oxide synthase. Bioorg Med Chem Lett. 2010;20:6258–6261. doi: 10.1016/j.bmcl.2010.08.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Massova I, Kollman PA. Computational alanine scanning to probe protein-protein interactions: A novel approach to evaluate binding free energies. J Amer Chem Soc. 1999;121:8133–8143. [Google Scholar]
- 35.Brown SP, Muchmore SW. High-througput calculation of protein-ligand binding affinities: Modification and adaption of the MM-PBSA protocol to enterprise grdi computing. J Chem Inf Model. 2006;46:999–1005. doi: 10.1021/ci050488t. [DOI] [PubMed] [Google Scholar]
- 36.Xue F, Fang J, Delker SL, Li H, Martasek P, Roman LJ, Poulos TL, Silverman RB. Symmetric double-headed aminopyridines, a novel strategy for potent and membrane-permeable inhibitors of neuronal nitric oxide synthase. J Med Chem. 2011;54:2039–2048. doi: 10.1021/jm101071n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delker SL, Xue F, Li H, Jamal J, Silverman RB, Poulos TL. Role of zinc in isoform-selective inhibitor binding to neuronal nitric oxide synthase. Biochemistry. 2010;49:10803–10810. doi: 10.1021/bi1013479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rao S, Lin Z, Drobyshevsky A, Chen L, Ji X, Ji H, Yang Y, Yu L, Derrick M, Silverman RB, Tan S. Involvement of neuronal nitric oxide synthase in ongoing fetal brain injury following near-term rabbit hypoxia-ischemia. Dev Neurosci. 2011;33:288–298. doi: 10.1159/000327241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xue F, Li H, Delker SL, Fang J, Martasek P, Roman LJ, Poulos TL, Silverman RB. Potent, highly selective, and orally bioavailable gem-difluorinated monocationic inhibitors of neuronal nitric oxide synthase. J Am Chem Soc. 2010;132:14229–14238. doi: 10.1021/ja106175q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawton GR, Ralay Ranaivo H, Chico LK, Ji H, Xue F, Martasek P, Roman LJ, Watterson DM, Silverman RB. Analogues of 2-aminopyridine-based selective inhibitors of neuronal nitric oxide synthase with increased bioavailability. Bioorg Med Chem. 2009;17:2371–2380. doi: 10.1016/j.bmc.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ji H, Delker SL, Li H, Martasek P, Roman LJ, Poulos TL, Silverman RB. Exploration of the active site of neuronal nitric oxide synthase by the design and synthesis of pyrrolidinomethyl 2-aminopyridine derivatives. J Med Chem. 2010;53:7804–7824. doi: 10.1021/jm100947x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheshire DR, Aberg A, Andersson GM, Andrews G, Beaton HG, Birkinshaw TN, Boughton-Smith N, Connolly S, Cook TR, Cooper A, Cooper SL, Cox D, Dixon J, Gensmantel N, Hamley PJ, Harrison R, Hartopp P, Kack H, Leeson PD, Luker T, Mete A, Millichip I, Nicholls DJ, Pimm AD, St-Gallay SA, Wallace AV. The discovery of novel, potent and highly selective inhibitors of inducible nitric oxide synthase (iNOS) Bioorg Med Chem Lett. 2011;21:2468–2471. doi: 10.1016/j.bmcl.2011.02.061. [DOI] [PubMed] [Google Scholar]
- 43.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat Chem Biol. 2008;4:700–707. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Annedi SC, Maddaford SP, Ramnauth J, Renton P, Rybak T, Silverman S, Rakhit S, Mladenova G, Dove P, Andrews JS, Zhang D, Porreca F. Discovery of a potent, orally bioavailable and highly selective human neuronal nitric oxide synthase (nNOS) inhibitor, N-(1-(piperidin-4-yl)indolin-5-yl)thiophene-2-carboximidamide as a pre-clinical development candidate for the treatment of migraine. Eur J Med Chem. 2012:1–14. doi: 10.1016/j.ejmech.2012.07.006. in press. [DOI] [PubMed] [Google Scholar]