

Abstract
Agrobacterium-mediated plant transformation is an extremely complex and evolved process involving genetic determinants of both the bacteria and the host plant cells. However, the mechanism of the determinants remains obscure, especially in some cereal crops such as wheat, which is recalcitrant for Agrobacterium-mediated transformation. In this study, differentially expressed genes (DEGs) and differentially expressed proteins (DEPs) were analyzed in wheat callus cells co-cultured with Agrobacterium by using RNA sequencing (RNA-seq) and two-dimensional electrophoresis (2-DE) in conjunction with mass spectrometry (MS). A set of 4,889 DEGs and 90 DEPs were identified, respectively. Most of them are related to metabolism, chromatin assembly or disassembly and immune defense. After comparative analysis, 24 of the 90 DEPs were detected in RNA-seq and proteomics datasets simultaneously. In addition, real-time RT-PCR experiments were performed to check the differential expression of the 24 genes, and the results were consistent with the RNA-seq data. According to gene ontology (GO) analysis, we found that a big part of these differentially expressed genes were related to the process of stress or immunity response. Several putative determinants and candidate effectors responsive to Agrobacterium mediated transformation of wheat cells were discussed. We speculate that some of these genes are possibly related to Agrobacterium infection. Our results will help to understand the interaction between Agrobacterium and host cells, and may facilitate developing efficient transformation strategies in cereal crops.
Introduction
Genetic transformation, as a reverse genetics tool, has been widely used in modification of some economically important plant species. Great successes have been achieved in enhancing the production of major crops such as soybean, maize and cotton, which have contributed a lot to the global agricultural economy and helped to meet the food demand for human and animal worldwide [1]. However, almost no promising progress has occurred on genetically modified wheat [2]. Presently, the most economic strategy of plant transformation is still Agrobacterium- mediated method, which is progressed slowly in wheat even though it was initiated in 1980s when it was successfully applied to obtain transgenic tobacco plants [3].
The mechanism of Agrobaterium-mediated transformation has been explored in both pathogens and plants, and some pathogen or host proteins/genes have been identified to participate in the Agrobacterium infection and T-DNA delivery process [4]–[11]. A few of these genes were proved to result in improved transformation efficiency in some dicot plants such as Arabidopsis and tobacco, and also in several cereal plants such as rice and maize [12], [13]. Taking rice as an example, even though its transformation process is not difficult, Agrobacterium-mediated transformation efficiency for indica rice variety is much lower than that for japonica cultivars. Tie et al. identified the differentially expressed genes by microarray, and the results were very useful to identify genes involved in the process of Agrobacterium-mediated transformation [14].
Agrobacterium infection of plant cells consists of a series of events, including attachment of Agrobacterium on plant tissues, recognition between Agrobacterium and host, production of transferred substrates, transferring of the components into host cell, movement of the substrates into host nucleus, integration of T-DNA into host genome, and expression of the integrated T-DNA, among which the most vital step is the integration of T-DNA into plant genome. During the whole process, several vir genes and chv genes were proved to contribute to the cellular transportation or transformation of the target DNA fragments [15]. However, only a few literatures reported the response of host response to the infection of Agrobacterium by cDNA-AFLP [16] and genome microarray [17]. Tzfra et al. screened an Arabidopsis cDNA library by the yeast two-hybrid method with the Agrobacterium VirE2 protein as a bait and found that the identified plant protein, designated VIP1, was specifically bound with VirE2, and allowed its nuclear import to participate in the early stages of T-DNA expression [18]. Subsequent research indicated that VIP1 is imported into the nucleus of plants via the karyopherin-α dependent pathway, and its over-expression significantly rendered plants more susceptible to genetic transformation mediated by Agrobacterium [18], [19]. Moreover, the ability of VIP1 interacting with VirE2 protein and localizing in nucleus helped the transportation of the foreign DNA transiently into plant cells and nucleus, and its interaction with a host histone protein of H2A is required for the upcoming stable genetic transformation of the alien DNA strands [20]. VIP2 is another Arabidopsis protein which interacts with VIP1, and also plays an important role in the Agrobacterium-mediated transformation in plants [21]. Because of the complexity of the whole transformation process, a lot of host genes are postulated to participate in the delivery process. Identifying more host genes involved in the response to infection and transformation will help us to further understand the process, and improve the efficiency of Agrobacterium-mediated wheat transformation eventually.
However, Agrobacterium-mediated wheat genetic transformation has remained very low efficiency and strong genotype-dependent [22]. Therefore, particle bombardment method is still the major approach for wheat transformation [22]. Up to now, some improved transformation protocols mediated by Agrobacterium have been reported in wheat since 1997 [23], [24]. For example, Hu et al. reported that they obtained more than 3,000 independent transgenic events with average transformation efficiency of 4.4% [25]. However, these results were limited mainly to few wheat varieties, and the methods they used have been proved difficult to follow up [24]–[27] even if the advances and progress on wheat Agrobacterium-mediated transformation approach were described in freshly published papers [23], [28]. Indeed, no wheat variety has been proved to be competent for the transformation mediated by Agrobacterium. Therefore, more work needs to be conducted to find key host genes involved in the T-DNA delivery process after the wheat cells are infected by Agrobacterium.
In the past few years, development of next-generation sequencing (NGS) technologies has provided a new paradigm for genome and transcriptome characterization [29], [30]. RNA sequencing (RNA-seq) has exhibited some obvious advantages over existing approaches. This technique has been proved to be highly repeatable, and is expected to revolutionize the manner of analyzing eukaryotic transcriptomes [31]. On the other hand, some technologies such as mass spectrometry (MS) and two-dimensional electrophoresis (2-DE) have been widely used in proteomics. Evidences showed that proteomics and transcriptome can mutually promote the detection of expressed genes with complementary advantages at low cost [32]. In this study, the expression activities of associated genes with transformation process were analyzed in the infected wheat callus by Agrobacterium using RNA-seq and 2-DE in conjunction with MS strategy. We identified differentially expressed genes that might be involved in the process of Agrobacterium infection and T-DNA delivery. A set of 4,889 differentially expressed genes (DEGs) and 90 differentially expressed proteins (DEPs) were identified, respectively. Most of them are related to chromatin assembly or disassembly and to immune. After comparative analysis, 24 aligned DEPs were identified to be potentially closely related to Agrobacterium infection response and transformation, and involved in 23 pathways.
Materials and Methods
Plant materials and Agrobacterium strain
A semi-winter wheat (Triticum aestivum L.) variety used throughout this study, Yangmai12, which is a largely commercial wheat variety in southeast China with good agronomic characteristics and high regeneration ability of immature embryos, was kindly provided by Prof. Shunhe Chen at Yangzhou Agricultural Institute, Jiangsu Academy of Agricultural Sciences, China. Wheat immature caryopses were collected from Yangmai12 plants 12–14 days post anthesis. The immature embryos were dissected aseptically and cultured on MSD2 medium (MS inorganic salts, 2 mgl–1 dicamba, 3.0% sucrose, 2.4 gl–1 gelrite, pH 5.8) for 4 days at 25°C under dark conditions before infection by Agrobacterium tumefaciens. The Agrobacterium strain used in this study is C58C1, which harbored a binary vector pZP211 carrying a T-DNA without target gene, and was kindly provided by Dr. Tom Clemente at University of Nebraska-Lincoln, USA.
Infection of pre-cultured immature embryos by Agrobacterium
Agrobacterium tumefaciens strain C58C1 with binary vector pZP211 was incubated overnight in 5 ml YEP medium (10 gl–1 tryptone, 10 gl–1 yeast extract, 5 gl–1 NaCl, pH 7.0) with 50 mgl–1 rifampicin, 50 mgl–1 streptomycin, and 50 mgl–1 spectinomycin inside a shaker with 220 rpm at 28°C. The overnight Agrobacterium culture was put into 45 ml fresh YEP medium, and incubated inside a shake for 6 hours at the same conditions as above mentioned. The Agrobacterium cells was pelleted by centrifugation at 4500 rpm for 10 min at room temperature, and re-suspended by adding 25 ml of liquid inoculation medium WCC (1/10 MS basic medium, 4.0 mM 2-(N-morpholine)-ethane sulphonic acid (MES), 0.75 gl–1 MgCl2, 200 µM acetosyringone, 1.0% glucose, 4.0% maltose, 2.0 mgl–1 dicamba, 2.2 mgl–1 picloram, 100 mgl–1 casein hydrolysate (CH), pH 5.4). The cell density was adjusted to an optical density of 0.5 (OD650) for inoculation [24].
About 50 pre-cultured immature embryos (PCIEs) of wheat were transferred into the prepared Agrobacterium suspension in a petri dish (35 mm×15 mm) containing 3 ml of Agrobacterium culture. In total, 100 PCIEs were infected by Agrobacterium in two plates. Another 100 PCIEs were transferred into 6 ml 1/10 WCC as a control [24]. The inoculation was performed at room temperature for 30 min, then the cell clusters were blotted on sterile filter paper and transferred to larger plates (90 mm×20 mm) containing a piece of sterile filter paper for co-cultivation at 23–24°C in the dark for 36 hours [33]. The infection experiment was designed by three repeats, and RNA isolation was performed from every repeat.
RNA isolation, cDNA library preparation and sequencing
Total RNA was isolated with TRIZOL (Invitrogen, Carlsbad, CA, USA) from the Agrobacterium infected and non-infected PCIEs, which were treated in a solution containing 200 mgl–1 carbenicillin disodium salt (Amresco, USA) for 10 min and then washed with sterile water for 3 times, according to the manufacturer's instructions. Then the RNA-seq were performed in BGI (Beijing Genomics Institute).
Three RNA samples from each treatment were mixed, respectively, and treated with RNase-free DNase I for 30 min at 37°C to remove residual DNA. Beads with oligo (dT) were used to isolate poly (A) mRNA. Next, the mRNA was broken into short fragments (about 200 bp) after adding fragmentation buffer. First strand cDNA was synthesized using random hexamer-primer and reverse transcriptase (Invitrogen, Carlsbad, CA, USA). The second strand cDNA was synthesized using RNase H (Invitrogen, Carlsbad, CA, USA) and DNA polymerase I (Invitrogen, Carlsbad, CA, USA) [34]. The double strand cDNA was purified with QiaQuick PCR extraction kit (Invitrogen, Carlsbad, CA, USA), and washed with EB buffer. A single adenosine was added to the cDNA using Klenowexo–fragment with dATP. Sequencing adaptors were ligated onto the repaired ends of the fragments. The required fragments were purified by agarose gel electrophoresis and enriched by PCR amplification. Finally, the library products were sequenced via Illumina HiSeq™ 2000 (Illumina, San Diego, CA, USA). All the reads sequences have been submitted to the Sequence Read Archive, NCBI. Accession numbers of experiment-SRX273368 run-SRR837407 for treatment group dataset, and experiment-SRX276082 run-SRR847734 for control group dataset have been given.
Raw reads filtering and clean reads aligning with reference sequences
The original image data were transferred into sequence data by base calling, which is defined as raw data or raw reads. Before data analysis, it was prerequisite to remove the dirty raw reads. The filtering steps included (1) removing the reads with adaptors, (2) removing the reads in which unknown bases were more than 10%, and (3) removing low-quality reads (the percentage of the low-quality bases with which value≤5 was more than 50% in a read). Next, the clean reads were aligned to reference sequences using SOAPaligner/soap2 [35], and mismatches less than 2 bases were allowed in the alignment. The reference unigene or EST (Expressed sequence tags) database and annotation data were downloaded from the websites of http://compbio.dfci.harvard.edu/cgi-bin/tgi/tc_ann.pl?gudb=wheat and http://www.ncbi.nlm.nih.gov/nucest/. The ratio we used to assess the percentage of the gene coverage by reads was the quotient of the base numbers in a target gene covered by unique mapping reads divided by the total base numbers of this target gene.
Screening and analysis of differentially expressed genes (DEGs)
The gene expression level was calculated by counting the number of reads which mapped to the reference genes. Gene expression levels were measured as reads per kilo base per million reads (RPKM) method using the formula previously described by Mortazavi et al. [36]. RPKM were calculated from the following formula:
To find genes that have different expression levels between the two samples, we developed a strict algorithm according to the method reported previously [37]. If every gene's expression occupies only a small part of the whole library, p(x) will closely follow the Poisson distribution, in which the amount of unambiguous clean reads from gene A is denoted as x, and the probability of gene A expression is presented by p(x).
If the amount of clean reads for sample 1 and sample 2 is N1 and N2, respectively, gene A holds x reads in sample 1 and y tags in sample 2. The probability of expression quantity of gene A in sample 1 as much as in sample 2 can be calculated by the following formula:
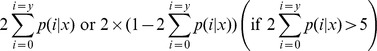 |
P-value corresponds to the test of differential gene expression. We threw in FDR (False discovery rate) to determine the threshold of P-value in multiple tests, and preset the FDR to a number no bigger than 0.01 [38]. The standard (FDR≤0.001 and the absolute value of |log2|ratio≥1) was used as the threshold to judge the significance of gene expression difference. More stringent criteria with smaller FDR and greater fold-change value are used to identify DEGs.
In order to remove the disturbances of the genes from Agrobacterium, we checked out the whole dataset and finally deleted the bacterium genes.
Gene ontology analysis and pathway enrichment analysis of DEGs
DEGs were categorized according to the genome gene ontology (GO) annotations. GO enrichment analysis provides all GO terms which are significantly enriched in DEGs compared with the genome background and filter the DEGs that correspond to biological functions. Using this method all DEGs can be primarily mapped to GO terms in the database (http://www.geneontology.org/), calculating gene numbers for every term, then hyper geometric test was used to find significantly enriched GO terms in DEGs compared with the genome background. This analysis is able to recognize the main biological functions that DEGs play. The calculating formula is as follows:
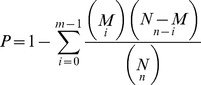 |
In this formula, N stands for the number of all genes with GO annotation, n for the number of DEGs in N, M for the number of all genes that are annotated to the certain GO terms, and m for the number of DEGs in M. The calculated p-value goes through Bonferroni Correction, taking corrected p-value≤0.05 as a threshold. GO terms fulfilling this condition are defined as significantly enriched GO terms in DEGs.
We also analyzed the gene functions employing pathway database, and extracted the metabolic annotation data from KEGG [39]. Significantly enriched metabolic pathways or signal transduction pathways in DEGs can be achieved using the method of enrichment analysis compared with the whole genome background. The calculating formula is the same as that in GO analysis, but here N means the number of all genes with KEGG annotation, n for the number of DEGs in N, M for the number of all genes annotated to specific pathways and m for the number of DEGs in M.
2-DE analysis of total protein from wheat callus infected and non-infected by Agrobacterium
Total protein extraction from the 3 replicated samples, respectively, was carried out following the standard protocol of TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) after extraction of total RNA. Roughly 600 µg total protein from each sample was first separated by isoelectric focusing (IEF) over a pH range of 3–10 using precast first-dimension dry strip (GE Healthcare, Waukesha, WI, USA). The first-dimension strips were equilibrated in equilibration buffer (50 mM Tris-HCl (pH 8.8), 6 M urea, 30% [v/v] glycerol, 2% [w/v] SDS and trace of bromophenol blue) plus 1% DTT for 15 min, and then equilibrated in equilibration buffer plus 4% of iodoacetamide instead of 1% DTT. The equilibrated first-dimension strip was loaded on a 12% SDS-PAGE. The prepared gels were stained with colloidal Coomassie Brilliant blue G-250 (Sigma, St. Louis, MO, USA), and then different stains compared to the control were selected for MALDI-TOF/TOF analysis.
MALDI-TOF/TOF analysis
The MALDI-TOF/TOF analysis was performed in Shanghai Applied Protein Technology Co.Ltd. Quantitative image analysis was performed with ImageMaster 2D Platinum Software Version 5.0 (Amersham Biosciences). and then the interested spots (vol.%≥2 fold and p-value≤0.05) were excised from the Coomassie Blue-stained gels for MALDI-TOF/TOF analyses, which was carried out on an ABI 4800 proteomic analyzer MALDI-TOF/TOF MS (Applied Biosystems/MDS Sciex, USA). The MS together with MS/MS spectra were searched against the NCBI non-redundant green plant database using GPS explorer software (Applied Biosystems, Grand Island, NY, USA) and MASCOT (Matrix Science, Boston, MA, USA) through the following parameters: maximum missed cleavage was 2, peptide mass tolerance was set to ±0.2 Dalton (Da), and fragment tolerance set to ±0.3 Da. The proteins with both protein score confidence interval (CI) and total ion score CI above 95% were identified as credible results for the MS/MS.
Quantitative reverse transcription PCR (qRT-PCR) analysis
qRT-PCR was performed on ABI 7300 (ABI, Foster City, CA, USA) according to the manufacturer's instructions (TaKaRa, Dalian, China) to assess the transcription levels determined by RNA-seq and protein 2-D gel, in which TaActin was used as an internal standard and amplified with its genome-specific primers at the same time. The cDNA derived from the total RNA used in the process of RNA-seq was used as template. The cycle threshold values (CT) were determined through using ADP ribosylation factor (ADP) as the endogenous reference genes [40]. Next, the relative different expression ratios were calculated by the 2−ΔΔCT mathematical model [41]. Each experiment was repeated by three times. Two experiments for the two independent cDNA samples were performed to confirm the reproducibility of the results.
Results
Summary of RNA-seq results
A total of 11,589,085 reads (567,865,165 base pairs) were obtained from the RNA of wheat callus co-cultured with Agrobacterium tumefaciens C58C1 (accession number of Sequence Read Archive, NCBI: experiment-SRX273368 run-SRR837407) and 11,601,434 reads (568,470,266 base pairs) were obtained from control callus which was not infected with C58C1 (accession number of NCBI: experiment-SRX276082 run-SRR847734). Over 95% of the reads from both samples were clean reads (File S1), and over 80% of these reads were mapped to the reference unigenes (File S2). The randomness and sequencing saturation analysis showed that the reads location on the gene was standardized to a relative position, and the number of detected genes reached saturation (File S3). The results of the gene coverage statistics are shown in File S4. In both infected and non-infected samples, more than 11% unigenes demonstrated very high levels of gene coverage (coverage>80%).
Transcription profiles reveal DEGs between infected and non-infected samples
We used the RPKM method to identify the gene expression levels. The gene expression is calculated by the number of reads mapped to the reference sequence; the ratio of RPKM (infected)/RPKM (control) was used to determine the different expression level of each gene. According to the datasets of RPKM of 93,508 unigenes (or ESTs), compared to non-infected samples, the infected samples had 4,889 unigenes (or ESTs) showing different levels of transcription (|log2|ratio (infected/control)≥1 and FDR (false discovery rate) ≤0.001) (File S5). Among them, 2,503 unigenes were up-regulated, and 2,386 were down-regulated. The DEGs that had the mean of |log2|ratio (infected/control)≥5 are listed in Table 1 and Table 2.
Table 1. The differentially expressed genes (|log2|ratio (infected/control)≥5).
| Gene ID | Control -RPKM | Infected -RPKM | |Log2|ratio | P-value | FDR | Description |
| CA594889 | 0.001 | 27.68816 | 14.75698 | 1.72E−06 | 0.000046 | Ta cDNA clone: WT012_D04 |
| EB512672 | 0.001 | 24.2069 | 14.56313 | 1.06E−07 | 3.75E−06 | Blue copper-binding protein homolog |
| TC374940 | 0.001 | 21.75113 | 14.4088 | 0 | 0 | Pathogenesis-related 1a |
| GH722147 | 0.001 | 14.4799 | 13.82176 | 1.97E−10 | 1.19E−08 | Xylanase inhibitor precursor |
| TC420579 | 0.001 | 14.32455 | 13.8062 | 6.01E−12 | 4.65E−10 | Response regulator |
| CA683879 | 0.001 | 14.02145 | 13.77535 | 7.96E−10 | 4.33E−08 | Oxalate oxidase 2 precursor |
| TC440260 | 0.001 | 13.67654 | 13.73942 | 6.47E−09 | 2.94E−07 | Putative disease resistance protein RGA3-like |
| CF133353 | 0.001 | 12.02828 | 13.55414 | 0.000014 | 0.000287 | |
| CJ654139 | 0.001 | 10.98716 | 13.42353 | 2.12E−07 | 7.04E−06 | Os07g0137900 protein |
| CA661174 | 0.001 | 10.48387 | 13.35588 | 1.72E−06 | 4.61E−05 | Prion-like-(Q/n-rich)-domain-bearing protein protein 75 |
| TC397228 | 0.001 | 10.1623 | 13.31094 | 6.47E−09 | 2.94E−07 | |
| TC416495 | 0.001 | 10.04916 | 13.29479 | 3.47E−06 | 8.54E−05 | Mannose phosphate isomerase (MPI) |
| TC451603 | 0.001 | 9.740984 | 13.24985 | 1.6E−09 | 8.17E−08 | LOC496215 protein |
| CA693299 | 0.001 | 9.60027 | 13.22886 | 2.82E−05 | 0.000523 | |
| BE414946 | 0.001 | 9.059047 | 13.14514 | 1.72E−06 | 0.000046 | 4-coumarate-CoA ligase 4CL2 |
| BJ236765 | 0.001 | 8.983681 | 13.13309 | 1.72E−06 | 4.61E−05 | |
| TC455795 | 0.001 | 8.654863 | 13.0793 | 3.47E−06 | 8.53E−05 | |
| TC458503 | 0.001 | 8.165747 | 12.99537 | 2.61E−08 | 1.05E−06 | Early nodulin protein |
| CJ875280 | 0.001 | 7.524698 | 12.87742 | 6.97E−06 | 0.000156 | |
| CA663181 | 0.001 | 7.492188 | 12.87117 | 5.66E−05 | 0.00095 | Os04g0630300 protein |
| TC442697 | 0.001 | 7.417109 | 12.85664 | 0.000014 | 0.000287 | |
| CA638794 | 0.001 | 7.413078 | 12.85586 | 2.82E−05 | 0.000523 | Protein kinase domain containing protein |
| TC426144 | 0.001 | 7.374602 | 12.84835 | 2.82E−05 | 0.000523 | Vitis vinifera Chromosome chr8 scaffold_106 |
| CA499029 | 0.001 | 7.253538 | 12.82447 | 6.97E−06 | 0.000156 | Nitrate-induced NOI protein |
| BM135604 | 0.001 | 6.897282 | 12.75181 | 2.82E−05 | 0.000523 | |
| CJ696409 | 0.001 | 6.893563 | 12.75103 | 3.47E−06 | 8.54E−05 | Transcription initiation factor |
| TC418073 | 0.001 | 5.809657 | 12.50424 | 1.06E−07 | 3.75E−06 | Phenylalanine ammonia-lyase |
| TC415483 | 0.001 | 5.710553 | 12.47941 | 4.27E−07 | 1.32E−05 | Glucosyltransferase |
| TC392329 | 0.001 | 5.470306 | 12.41741 | 1.72E−06 | 4.61E−05 | Reponse regulator 6 |
| TC381923 | 0.001 | 4.514375 | 12.14031 | 1.72E−06 | 4.61E−05 | Vitis vinifera Chromosome chr2 scaffold_105 |
| TC458205 | 0.001 | 3.898996 | 11.92889 | 6.97E−06 | 0.000156 | High molecular weight glutenin subunit |
| CK211359 | 0.001 | 3.700793 | 11.85362 | 5.66E−05 | 0.000951 | NADH dehydrogenase subunit I |
| TC379055 | 0.001 | 2.43027 | 11.2469 | 5.66E−05 | 0.000951 | UDP-glucosyltransferase |
| TC414250 | 5.548758 | 867.0767 | 7.287851 | 1.7E−06 | 4.56E−05 | Wali6 protein |
| TC415670 | 7.672419 | 1009.571 | 7.039846 | 8.1E−08 | 2.95E−06 | Wali3 protein |
| TC416295 | 3.794324 | 390.9363 | 6.686947 | 9.61E−06 | 0.000207 | Wali6 protein |
| TC426400 | 3.150807 | 126.3979 | 5.326108 | 9.61E−06 | 0.000207 | ATP-dependent Clp protease proteolytic subunit |
| TC447088 | 17.75787 | 0.001 | −14.1162 | 2.11E−06 | 5.52E−05 | |
| CA501626 | 13.80722 | 0.001 | −13.7531 | 1.66E−05 | 0.000333 | |
| CA678188 | 13.02965 | 0.001 | −13.6695 | 2.68E−07 | 8.68E−06 | Os10g0329400 protein |
| TC396751 | 12.65317 | 0.001 | −13.6272 | 1.09E−09 | 5.73E−08 | Histone H2B.4 |
| TC374009 | 12.05666 | 0.001 | −13.5575 | 2.82E−13 | 2.89E−11 | Agrostis stolonifera Crs-1 |
| CA606062 | 11.30046 | 0.001 | −13.4641 | 2.11E−06 | 5.52E−05 | Vacuolar-processing enzyme gamma- isozyme precursor |
| CA654969 | 10.32932 | 0.001 | −13.3345 | 8.35E−06 | 0.000183 | |
| CD931119 | 9.342799 | 0.001 | −13.1896 | 4.2E−06 | 0.000101 | Kinesin heavy chain |
| CA642440 | 9.315299 | 0.001 | −13.1854 | 3.31E−05 | 0.000598 | TonB-like protein |
| CJ536742 | 8.682779 | 0.001 | −13.0839 | 3.31E−05 | 0.000598 | |
| TC414261 | 6.403385 | 0.001 | −12.6446 | 3.31E−05 | 0.000598 | ERN2 |
| TC390944 | 6.107886 | 0.001 | −12.5765 | 8.35E−06 | 0.000183 | Pathogenesis related protein-1 |
| TC409167 | 5.647619 | 0.001 | −12.4634 | 1.66E−05 | 0.000334 | |
| TC441032 | 5.39911 | 0.001 | −12.3985 | 2.11E−06 | 5.52E−05 | Mitochondrial ATP synthase |
| CK205455 | 5.16714 | 0.001 | −12.3352 | 4.2E−06 | 0.000101 | |
| TC416474 | 5.10914 | 0.001 | −12.3189 | 1.66E−05 | 0.000334 | TBP-binding protein-like |
| TC432960 | 5.050132 | 0.001 | −12.3021 | 8.35E−06 | 0.000183 | |
| TC392126 | 4.7211 | 0.001 | −12.2049 | 8.35E−06 | 0.000183 | Germin-like protein 6a |
| TC434510 | 28.3137 | 0.366668 | −6.27088 | 1.98E−22 | 5.33E−20 | Peroxidase PXC2 precursor |
Table 2. GO analysis for DEGs (|log2|ratio (infected/control)≥5).
| Gene ID | Cellular component | Molecular function | Biological process |
| TC374940 | 0016023 | ||
| TC420579 | 0005575 0005739 | 0030528 0000156 | 0006355 0009736 0000160 0019827 0009735 |
| TC416495 | 0009536 | ||
| TC458503 | 0009536 | 0003674 | |
| TC418073 | 0005634 0000786 | 0003677 | 0006334 0007283 0007076 |
| TC415483 | 0005739 0016023 | ||
| TC392329 | 0005575 0005634 0005739 | 0003677 0030528 0000156 | 0006355 0006950 0009736 0019827 0000160 0009735 |
| TC381923 | 0005575 0016023 | 0016706 | 0019748 |
| TC458205 | 0009536 | 0003677 | 0006310 |
| TC396751 | 0008021 0042589 0030141 0031201 0030672 0005576 0016021 0000786 0005575 0005634 | 0000149 0003677 0003674 0005516 0017075 0005543 0017022 | 0017157 0008285 0006334 0008150 0050829 0017156 0006944 0009792 0016079 0050830 0051276 0006955 0040007 0002119 |
| TC390944 | 0005576 0016023 | 0003674 | 0008150 |
| TC409167 | 0016023 | ||
| TC392126 | 0016023 | 0008150 |
Furthermore, we classified the differentially expressed unigenes (or ESTs) by transcribed genes into three GO categories: cellular component, molecular function, and biological process. All of the differentially expressed unigenes shared 2,020 GO terms, including 289 cellular component terms (File S6), 382 molecular function terms (File S7), and 1,349 biological processes (File S8). To demonstrate further relationships of the DEGs and the biochemical processes occurring in the infection, the DEGs were categorized into 8, 13, and 15 groups according to cellular component, molecular function, and biological process independently (Figure 1). Based on the results of gene ontology analysis, most of the DEGs were related to various organelles (50.73%). For examples, lots of them were related to mitochondria, and about half of the DEGs had function of enzyme, coenzyme, or cofactor (24.52%) and the rests were related to the function of the unclear binding (20.5%). For the biological process of GO term, about a quarter of DEGs were involved in the metabolism process (22.9%), 15.77% of DEGs were involved in the chromatin assembly or disassembly process, and another 9.68% of DEGs were related to the process of immunity.
Figure 1. Categorization of the GO terms based on the differentially expressed genes.

Categorization of GO terms with a p-value greater than or equal to 1: cellular component terms (A), molecular function terms (B) and biological process terms (C). All of the differentially expressed genes were classified based on GO analysis. By this method all DEGs are firstly mapped to GO terms in the database (http://www.geneontology.org/), calculating gene numbers for every term, then using hypergeometric test to find significantly enriched GO terms in DEGs comparing to the genome background. Each category is labeled with different colors, and the numbers refer to ratio of these categories to the all dataset.
According the analysis of pathway enrichment, 2295 DEGs were involved in 111 pathways (Table 3). Distribution of all DEGs in the pathways was shown in File S9. 507 DEGs were related to metabolic pathways, and account for the largest portion (16.72%). However, these DEGs were not mapped in KEGG database and the metabolic pathways they were involved were quite broad, and almost incorporated all aspects of the metabolic processes, such as starch and sucrose metabolism and fatty acid metabolism. Most of the DEGs that were classed into metabolic pathways were found also to be presented in other pathways. For example, phenylalanine ammonia-lyase (TC418073) was involved in metabolic pathways, but it also participated in phenylalanine metabolism, phenylpropanoid biosynthesis and nitrogen metabolism when the function of this enzyme was concretely implemented. Besides metabolic pathways, the most important bioprocess in Agrobacterium response is biosynthesis of secondary metabolites (9.74%). Furthermore, the rate of the pathways on plant-pathogen interaction, phenylpropanoid biosynthesis and spliceosome were more than 3%. The most weakly tested pathways were about biotin metabolism, arachidonic acid metabolism, photosynthesis and photosynthesis-antenna proteins (0.03%).
Table 3. Pathway analysis of DEGs based on KEGG database.
| Pathway | Annotation (number) | Annotation rate (%) | P-value | Q-value | Pathway ID |
| Metabolic pathways | 507 | 16.72 | 1 | 1.00E+00 | ko01100 |
| Biosynthesis of secondary metabolites | 287 | 9.47 | 0.999988 | 1.00E+00 | ko01110 |
| Plant-pathogen interaction | 111 | 3.66 | 0.940559 | 1.00E+00 | ko04626 |
| Phenylpropanoid biosynthesis | 102 | 3.36 | 0.078706 | 3.49E−01 | ko00940 |
| Spliceosome | 95 | 3.13 | 0.008077 | 5.27E−02 | ko03040 |
| Ribosome | 85 | 2.80 | 1 | 1.00E+00 | ko03010 |
| Starch and sucrose metabolism | 79 | 2.61 | 0.082119 | 3.51E−01 | ko00500 |
| Purine metabolism | 73 | 2.41 | 2.22E−06 | 4.93E−05 | ko00230 |
| Phenylalanine metabolism | 72 | 2.37 | 0.007202 | 5.00E−02 | ko00360 |
| Protein processing in endoplasmic reticulum | 67 | 2.21 | 0.998354 | 1.00E+00 | ko04141 |
| Glutathione metabolism | 65 | 2.14 | 0.002017 | 2.24E−02 | ko00480 |
| Ubiquitin mediated proteolysis | 51 | 1.68 | 0.149354 | 5.42E−01 | ko04120 |
| Endocytosis | 50 | 1.65 | 0.006456 | 4.78E−02 | ko04144 |
| Amino sugar and nucleotide sugar metabolism | 49 | 1.62 | 0.024959 | 1.39E−01 | ko00520 |
| Pyrimidine metabolism | 48 | 1.58 | 0.004626 | 3.67E−02 | ko00240 |
| DNA replication | 41 | 1.35 | 1.32E−11 | 1.47E−09 | ko03030 |
| Alpha-Linolenic acid metabolism | 39 | 1.29 | 0.000108 | 1.50E−03 | ko00592 |
| RNA degradation | 38 | 1.25 | 0.003272 | 2.79E−02 | ko03018 |
| Nucleotide excision repair | 36 | 1.19 | 3.35E−06 | 6.20E−05 | ko03420 |
| Peroxisome | 35 | 1.15 | 0.774472 | 1.00E+00 | ko04146 |
| Flavonoid biosynthesis | 34 | 1.12 | 0.539443 | 1.00E+00 | ko00941 |
| Cysteine and methionine metabolism | 33 | 1.09 | 0.971505 | 1.00E+00 | ko00270 |
| Glycolysis/Gluconeogenesis | 33 | 1.09 | 1 | 1.00E+00 | ko00010 |
| Nitrogen metabolism | 32 | 1.06 | 0.220608 | 7.65E−01 | ko00910 |
| Galactose metabolism | 31 | 1.02 | 0.021792 | 1.27E−01 | ko00052 |
| Phagosome | 31 | 1.02 | 0.999595 | 1.00E+00 | ko04145 |
| ABC transporters | 30 | 0.99 | 3.41E−10 | 1.89E−08 | ko02010 |
| Base excision repair | 29 | 0.96 | 6.56E−07 | 2.19E−05 | ko03410 |
| Cyanoamino acid metabolism | 29 | 0.96 | 0.110194 | 4.37E−01 | ko00460 |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | 29 | 0.96 | 0.693333 | 1.00E+00 | ko00945 |
| Tryptophan metabolism | 26 | 0.86 | 0.4863 | 1.00E+00 | ko00380 |
| Oxidative phosphorylation | 26 | 0.86 | 1 | 1.00E+00 | ko00190 |
| Fructose and mannose metabolism | 25 | 0.82 | 0.903134 | 1.00E+00 | ko00051 |
| Zeatin biosynthesis | 23 | 0.76 | 0.002555 | 2.58E−02 | ko00908 |
| Carbon fixation in photosynthetic organisms | 23 | 0.76 | 1 | 1.00E+00 | ko00710 |
| Mismatch repair | 22 | 0.73 | 8.79E−06 | 1.39E−04 | ko03430 |
| Phosphatidylinositol signaling system | 22 | 0.73 | 0.133323 | 5.10E−01 | ko04070 |
| Limonene and pinene degradation | 21 | 0.69 | 0.899491 | 1.00E+00 | ko00903 |
| RNA polymerase | 20 | 0.66 | 0.001317 | 1.62E−02 | ko03020 |
| Selenoamino acid metabolism | 20 | 0.66 | 0.353576 | 1.00E+00 | ko00450 |
| Pyruvate metabolism | 20 | 0.66 | 0.99903 | 1.00E+00 | ko00620 |
| Fatty acid metabolism | 19 | 0.63 | 0.784428 | 1.00E+00 | ko00071 |
| Sulfur metabolism | 17 | 0.56 | 0.003233 | 2.79E−02 | ko00920 |
| Inositol phosphate metabolism | 17 | 0.56 | 0.362768 | 1.00E+00 | ko00562 |
| Aminoacyl-tRNA biosynthesis | 17 | 0.56 | 0.420217 | 1.00E+00 | ko00970 |
| Alanine, aspartate and glutamate metabolism | 17 | 0.56 | 0.941328 | 1.00E+00 | ko00250 |
| Citrate cycle (TCA cycle) | 17 | 0.56 | 0.989953 | 1.00E+00 | ko00020 |
| Circadian rhythm - plant | 16 | 0.53 | 0.68574 | 1.00E+00 | ko04712 |
| Arginine and proline metabolism | 15 | 0.49 | 0.998069 | 1.00E+00 | ko00330 |
| Linoleic acid metabolism | 14 | 0.46 | 0.468498 | 1.00E+00 | ko00591 |
| Proteasome | 14 | 0.46 | 0.999554 | 1.00E+00 | ko03050 |
| Homologous recombination | 13 | 0.43 | 0.054938 | 2.54E−01 | ko03440 |
| Glycerolipid metabolism | 13 | 0.43 | 0.888763 | 1.00E+00 | ko00561 |
| Caffeine metabolism | 12 | 0.40 | 7.91E−07 | 2.19E−05 | ko00232 |
| Valine, leucine and isoleucine biosynthesis | 12 | 0.40 | 0.430602 | 1.00E+00 | ko00290 |
| Biosynthesis of unsaturated fatty acids | 12 | 0.40 | 0.82953 | 1.00E+00 | ko01040 |
| Benzoxazinoid biosynthesis | 12 | 0.40 | 0.921449 | 1.00E+00 | ko00402 |
| Tyrosine metabolism | 12 | 0.40 | 0.927114 | 1.00E+00 | ko00350 |
| Pentose phosphate pathway | 12 | 0.40 | 0.999553 | 1.00E+00 | ko00030 |
| Glyoxylate and dicarboxylate metabolism | 12 | 0.40 | 1 | 1.00E+00 | ko00630 |
| Sphingolipid metabolism | 11 | 0.36 | 0.050595 | 2.50E−01 | ko00600 |
| N-Glycan biosynthesis | 10 | 0.33 | 0.38074 | 1.00E+00 | ko00510 |
| Glycerophospholipid metabolism | 10 | 0.33 | 0.985818 | 1.00E+00 | ko00564 |
| Ascorbate and aldarate metabolism | 10 | 0.33 | 0.997898 | 1.00E+00 | ko00053 |
| Other glycan degradation | 9 | 0.30 | 0.01401 | 8.64E−02 | ko00511 |
| SNARE interactions in vesicular transport | 9 | 0.30 | 0.604893 | 1.00E+00 | ko04130 |
| Fatty acid biosynthesis | 9 | 0.30 | 0.756422 | 1.00E+00 | ko00061 |
| Terpenoid backbone biosynthesis | 9 | 0.30 | 0.791906 | 1.00E+00 | ko00900 |
| Lysine degradation | 9 | 0.30 | 0.87531 | 1.00E+00 | ko00310 |
| Butanoate metabolism | 9 | 0.30 | 0.974329 | 1.00E+00 | ko00650 |
| Glycine, serine and threonine metabolism | 9 | 0.30 | 0.976647 | 1.00E+00 | ko00260 |
| Pantothenate and CoA biosynthesis | 8 | 0.26 | 0.381927 | 1.00E+00 | ko00770 |
| Valine, leucine and isoleucine degradation | 8 | 0.26 | 0.996475 | 1.00E+00 | ko00280 |
| Ether lipid metabolism | 7 | 0.23 | 0.386485 | 1.00E+00 | ko00565 |
| Tropane, piperidine and pyridine alkaloid biosynthesis | 7 | 0.23 | 0.474718 | 1.00E+00 | ko00960 |
| Diterpenoid biosynthesis | 7 | 0.23 | 0.515453 | 1.00E+00 | ko00904 |
| Regulation of autophagy | 7 | 0.23 | 0.696261 | 1.00E+00 | ko04140 |
| Natural killer cell mediated cytotoxicity | 7 | 0.23 | 0.788938 | 1.00E+00 | ko04650 |
| Propanoate metabolism | 7 | 0.23 | 0.99625 | 1.00E+00 | ko00640 |
| Indole alkaloid biosynthesis | 6 | 0.20 | 0.099823 | 4.10E−01 | ko00901 |
| Basal transcription factors | 6 | 0.20 | 0.892823 | 1.00E+00 | ko03022 |
| Pentose and glucuronate interconversions | 6 | 0.20 | 0.975639 | 1.00E+00 | ko00040 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 6 | 0.20 | 0.980414 | 1.00E+00 | ko00400 |
| Flavone and flavonol biosynthesis | 6 | 0.20 | 0.982781 | 1.00E+00 | ko00944 |
| Protein export | 6 | 0.20 | 0.993342 | 1.00E+00 | ko03060 |
| Isoquinoline alkaloid biosynthesis | 5 | 0.16 | 0.70927 | 1.00E+00 | ko00950 |
| Glucosinolate biosynthesis | 5 | 0.16 | 0.811041 | 1.00E+00 | ko00966 |
| Porphyrin and chlorophyll metabolism | 5 | 0.16 | 0.999916 | 1.00E+00 | ko00860 |
| Non-homologous end-joining | 4 | 0.13 | 0.03947 | 2.09E−01 | ko03450 |
| Ubiquinone and other terpenoid- quinone biosynthesis | 4 | 0.13 | 0.966216 | 1.00E+00 | ko00130 |
| Histidine metabolism | 4 | 0.13 | 0.982783 | 1.00E+00 | ko00340 |
| Steroid biosynthesis | 4 | 0.13 | 0.987955 | 1.00E+00 | ko00100 |
| beta-Alanine metabolism | 4 | 0.13 | 0.998679 | 1.00E+00 | ko00410 |
| Glycosphingolipid biosynthesis - ganglio series | 3 | 0.10 | 0.151237 | 5.42E−01 | ko00604 |
| Glycosaminoglycan degradation | 3 | 0.10 | 0.340238 | 1.00E+00 | ko00531 |
| Lysine biosynthesis | 3 | 0.10 | 0.625386 | 1.00E+00 | ko00300 |
| One carbon pool by folate | 3 | 0.10 | 0.899855 | 1.00E+00 | ko00670 |
| Carotenoid biosynthesis | 3 | 0.10 | 0.960924 | 1.00E+00 | ko00906 |
| Anthocyanin biosynthesis | 2 | 0.07 | 0.051858 | 2.50E−01 | ko00942 |
| Vitamin B6 metabolism | 2 | 0.07 | 0.431126 | 1.00E+00 | ko00750 |
| C5-Branched dibasic acid metabolism | 2 | 0.07 | 0.497579 | 1.00E+00 | ko00660 |
| Glycosphingolipid biosynthesis - globo series | 2 | 0.07 | 0.622102 | 1.00E+00 | ko00603 |
| Folate biosynthesis | 2 | 0.07 | 0.684606 | 1.00E+00 | ko00790 |
| Synthesis and degradation of ketone bodies | 2 | 0.07 | 0.684606 | 1.00E+00 | ko00072 |
| Thiamine metabolism | 2 | 0.07 | 0.697287 | 1.00E+00 | ko00730 |
| Nicotinate and nicotinamide metabolism | 2 | 0.07 | 0.793251 | 1.00E+00 | ko00760 |
| Polyketide sugar unit biosynthesis | 2 | 0.07 | 0.95041 | 1.00E+00 | ko00523 |
| Biotin metabolism | 1 | 0.03 | 0.658762 | 1.00E+00 | ko00780 |
| Arachidonic acid metabolism | 1 | 0.03 | 0.982434 | 1.00E+00 | ko00590 |
| Photosynthesis | 1 | 0.03 | 1 | 1.00E+00 | ko00195 |
| Photosynthesis - antenna proteins | 1 | 0.03 | 1 | 1.00E+00 | ko00196 |
Identification and classification of differentially expressed proteins
Soluble proteins were extracted from infected and non-infected samples, and proteomic dynamics was investigated by high-resolution 2-DE. Protein spots displaying reproducible patterns were identified, and their expression patterns were analyzed. Among the Agrobacterium-infected and the control samples, a total of 867 reproducible protein spots were detected. The expression abundances (vol.%) of 132 protein spots changed by more than two folds, and thus were treated as DEPs. Due to the limited number of protein entries in the database, only 90 proteins spots were identified eventually through MALDI-TOF/TOF (Table 4). The maps are shown in Figure 2. Among these proteins, nine potential isoforms were targeted, and each of them had two or three spots located at different positions in the same gel. For example, in the infected tissues spots I216, I217 and I219 were identified as methionine synthase 1, and spots I320 and I343 were fructose-bisphosphate aldolase. In non-infected tissues, the isoforms include fructose-bisphosphate aldolase GTPase-activating protein-binding protein 1-like (spots N214 and N216), phosphoglucomutase (spots N219 and N229), predicted: pyruvate dehydrogenase E1 component subunit beta (spots N307 and N306), elongation factor 1-beta (spots N356 and N355), and glutathione transferase (spots I385 and I386). Furthermore, there were two groups of unnamed protein (spots N322 and N331/N338 and N342). These isoforms might represent post-translational modification forms of the same protein (Table 4).
Table 4. Differentially expressed proteins under control and infected conditions.
| Group ID | I/C (rate of % vol.) | Accession No. | Protein score | Protein score C. I. % | Total ion score C. I. % | Protein name |
| I198 | 0.111434/0 | gi|42391858 | 200 | 100 | 100 | cold shock domain protein 3 |
| I206 | 0.094232/0 | gi|222834414 | 172 | 100 | 100 | predicted protein |
| I223 | 0.045147/0 | gi|158513193 | 468 | 100 | 100 | pyruvate decarboxylase isozyme 2 |
| I216(I217 I219) | 0.210277/0 | gi|68655495 | 159 | 100 | 100 | methionine synthase 1 enzyme |
| I209 | 0.135266/0 | gi|313661595 | 503 | 100 | 100 | lipoxygenase-1 |
| I181 | 0.079847/0 | gi|326510251 | 394 | 100 | 100 | predicted protein |
| I190 | 0.191266/0 | gi|326514130 | 324 | 100 | 99.998 | predicted protein |
| I226 | 0.118491/0 | gi|357113565 | 483 | 100 | 100 | predicted: succinate dehydrogenase, ubiquinone, flavoprotein subunit, mitochondrial |
| I267 | 0.481638/0 | gi|14018051 | 466 | 100 | 100 | putative alanine aminotransferase |
| I239 | 0.166667/0 | gi|119388723 | 486 | 100 | 100 | alcohol dehydrogenase ADH1A |
| I276 | 0.23262/0 | gi|6561606 | 149 | 100 | 100 | ATPase alpha subunit |
| I288 | 0.937654/0 | gi|57635161 | 235 | 100 | 100 | peroxidase 8 |
| I299 | 0.189651/0 | gi|129916 | 138 | 100 | 85.61 | phosphoglycerate kinase |
| I302 | 0.466078/0 | gi|326527793 | 893 | 100 | 100 | predicted protein |
| I313 | 0.129406/0 | gi|4158232 | 322 | 100 | 100 | glycosylated polypeptide |
| I317 | 0.488077/0 | gi|229358240 | 731 | 100 | 100 | cytosolic malate dehydrogenase |
| I326 | 0.217391/0 | gi|226316439 | 420 | 100 | 100 | fructose-bisphosphate aldolase |
| I309 | 0.473017/0 | gi|326492375 | 150 | 100 | 99.962 | predicted protein |
| I320 (I343) | 0.812175/0 | gi|300681519 | 317 | 100 | 100 | fructose-bisphosphate aldolase, chloroplast precursor |
| I339 | 0.634438/0 | gi|326497973 | 746 | 100 | 100 | predicted protein |
| I347 | 0.301374/0 | gi|159895412 | 329 | 100 | 100 | NADPH-dependent thioredoxin reductase isoform 2 |
| I346 | 0.236009/0 | gi|242058197 | 104 | 99.996 | 100 | hypothetical protein |
| I242 | 0.288839/0 | gi|585032 | 399 | 100 | 100 | cysteine synthase |
| I364 | 0.409977/0 | gi|326492319 | 278 | 100 | 100 | predicted protein |
| I360 | 0.122988/0 | gi|357133190 | 137 | 100 | 99.907 | predicted protein |
| I390 | 0.47657/0 | gi|20067415 | 384 | 100 | 100 | glutathione transferase |
| I429 | 0.479179/0 | gi|728594 | 218 | 100 | 100 | glycine rich protein, RNA binding protein |
| I310 | 6.23836 | gi|146216737 | 367 | 100 | 100 | SGT1 |
| I254 | 5.987146 | gi|222872490 | 136 | 100 | 100 | predicted protein |
| I350 | 5.771895 | gi|326505660 | 177 | 100 | 100 | predicted protein |
| I395 | 3.330519 | gi|27544804 | 405 | 100 | 100 | phospholipid hydroperoxide glutathione peroxidase |
| I232 | 3.201333 | gi|133872360 | 551 | 100 | 100 | Bp2A protein, partial |
| I409 | 3.036805 | gi|9230743 | 64 | 58.026 | 100 | sucrose synthase-2 |
| I391 | 2.829641 | gi|259017810 | 274 | 100 | 100 | dehydroascorbate reductase |
| I199 | 2.5657 | gi|108862362 | 146 | 100 | 100 | oxidoreductase, zinc-binding dehydrogenase family |
| I200 | 2.513553 | gi|49425361 | 382 | 100 | 100 | spermidine synthase |
| I246 | 2.456281 | gi|11124572 | 399 | 100 | 100 | triosephosphat-isomerase |
| I380 | 2.320409 | gi|3688398 | 483 | 100 | 100 | ascorbate peroxidase |
| I353 | 2.202462 | gi|357158835 | 501 | 100 | 100 | predicted: glucose-6-phosphate isomerase-like |
| I422 | 2.117428 | gi|326494858 | 187 | 100 | 100 | predicted protein |
| N193 | 0/0.080562 | gi|326495130 | 503 | 100 | 100 | predicted protein |
| N214 (N216) | 0/0.835538 | gi|357167359 | 261 | 100 | 100 | predicted: ras GTPase-activating protein-binding |
| N206 | 0/0.384636 | gi|326533372 | 365 | 100 | 100 | predicted protein |
| N219(N229) | 0/0.303445 | gi|18076790 | 612 | 100 | 100 | phosphoglucomutase |
| N259 | 0/0.195904 | gi|164565159 | 418 | 100 | 100 | ribulose-1,5-bisphosphate carboxylase/oxygenase |
| N242 | 0/0.121703 | gi|212275097 | 224 | 100 | 100 | uncharacterized protein |
| N269 | 0/0.190315 | gi|357110692 | 510 | 100 | 100 | 6-phosphogluconate dehydrogenase,decarboxylating |
| N289 | 0/0.278852 | gi|28172907 | 510 | 100 | 100 | cytosolic 3-phosphoglycerate kinase |
| N293 | 0/0.557188 | gi|326500176 | 428 | 100 | 100 | predicted protein |
| N302 | 0/0.055462 | gi|326528557 | 521 | 100 | 100 | predicted protein |
| N307 (N306) | 0/0.242551 | gi|357148637 | 458 | 100 | 100 | pyruvate dehydrogenase E1 component subunit beta, mitochondrial |
| N335 | 0/0.869609 | gi|326512374 | 585 | 100 | 100 | predicted protein |
| N347 | 0/0.470007 | gi|326506676 | 275 | 100 | 100 | predicted protein |
| N322 (N331) | 0/0.090235 | gi|326499686 | 327 | 100 | 100 | predicted protein |
| N338 (N342) | 0/0.120011 | gi|326518738 | 542 | 100 | 100 | predicted protein |
| N356 (N355) | 0/0.645651 | gi|232033 | 219 | 100 | 100 | elongation factor 1-beta |
| N352 | 0/0.393733 | gi|326507956 | 314 | 100 | 100 | predicted protein |
| N361 | 0/0.381971 | gi|15808779 | 273 | 100 | 100 | ascorbate peroxidase |
| N350 | 0/0.479167 | gi|18146827 | 527 | 100 | 100 | chitinase 2 |
| N353 | 0/0.119597 | gi|326489985 | 227 | 100 | 100 | predicted protein |
| N346 | 0/0.221933 | gi|357130336 | 190 | 100 | 100 | 26S proteasome non-ATPase regulatory subunit 14 |
| N385 | 0/0.214757 | gi|2499932 | 741 | 100 | 100 | adenine phosphoribosyl transferase 1 |
| N416 | 0/0.113163 | gi|125548641 | 115 | 100 | 96.242 | hypothetical protein OsI_16233 |
| N420 | 0/0.261293 | gi|326534206 | 224 | 100 | 100 | predicted protein |
| N409 | 0/0.977368 | gi|326497111 | 567 | 100 | 100 | predicted protein |
| N406 | 0/0.44138 | gi|22535646 | 81 | 99.143 | 99.653 | hypothetical protein |
| N411 | 0/0.264197 | gi|48475065 | 80 | 99.016 | 75.904 | contains ubiquitin carboxyl-terminal hydrolase |
| N404 | 0/0.154189 | gi|326496833 | 161 | 100 | 100 | predicted protein |
| N408 | 0/0.278555 | gi|326517577 | 81 | 99.143 | 81.951 | predicted protein |
| N403 | 0/1.02402 | gi|40363759 | 561 | 100 | 100 | putative glycine-rich protein |
| N441 | 0/0.226741 | gi|8980491 | 116 | 100 | 100 | thioredoxin h |
| I321 | 0.492256 | gi|40317418 | 158 | 100 | 99.997 | glutamine synthetase isoform GSr2 |
| I356 | 0.398343 | gi|300807845 | 210 | 100 | 100 | profilin |
| I282 | 0.340188 | gi|326499079 | 236 | 100 | 100 | predicted protein |
| I385 (I386) | 0.329162 | gi|20067423 | 293 | 100 | 100 | glutathione transferase |
| I268 | 0.296689 | gi|525291 | 1,030 | 100 | 100 | ATP synthase beta subunit |
| I258 | 0.282746 | gi|9408184 | 147 | 100 | 100 | F0-F1 ATPase alpha subunit |
| I401 | 0.181083 | gi|112821176 | 217 | 100 | 100 | hypothetical protein |
| I273 | 0.169152 | gi|164422240 | 545 | 100 | 100 | ATP1 |
| I363 | 0.099712 | gi|40781605 | 530 | 100 | 100 | 14-3-3 protein |
I/C: infected/control. C.I.: chemical ionization; I: infected; N: non-infected.
Figure 2. 2-DE patterns of proteins extracted from control callus (A) and infected callus (B).

2-DE patterns of proteins extracted from CK (uninfected) PCIEs (A) and infected PCIEs (B). A protein sample of 1200 µg was loaded on each IPG strip (pH 3–10) and protein spots were visualized using coomassie brilliant blue staining. The experiment was repeated three times, 132 differentially expressed protein spots showing significant volume change under Agrobacterium infection. 90 protein spots which were changed by more than two folds were identified by MALDI-TOF/TOF analysis, and labeled on the figure.
Comparative analysis between the results of RNA-seq and proteomics
To describe the differently expressed genes in the process of transcription more accurately, we compared the results of RNA-seq with proteomics. 24 DEPs (26 spots) from the proteomics dataset were in consistent with the RNA-seq dataset (Table 5). On the basis of the pathway analysis of DEGs, the aligned 24 DEPs were involved in 23 pathways (Table 5), which are shown in Figure 3.
Table 5. The compared proteins between proteinomics and RNA-seq datasets.
| Protein spot ID (rate of % vol.) | Gene ID (|log2|ratio) | Pathways |
| gi|42391858 (0.111434/0) | BE431040 (2.13714081) | |
| gi|158513193 (0.045147/0) | TC379241 (1.108033409), TC385701 (1.068969307), TC419215 (1.014521523) | Tryptophan metabolism, Glycolysis/Gluconeogenesis, Metabolic pathways (no map in kegg database) |
| gi|313661595 (0.135266/0) | CA733413 (2.196724855), TC388136 (2.156815406) | Alpha-Linolenic acid metabolism, Linoleic acid metabolism, Metabolic pathways (no map in kegg database) |
| gi|357113565 (0.118491/0) | TC384162 (−1.03191405) | |
| gi|14018051 (0.481638/0) | TC419278 (1.587471506), TC451694 (1.49691429) | Alanine, aspartate and glutamate metabolism, Metabolic pathways (no map in kegg database), Carbon fixation in photosynthetic organisms |
| gi|119388723 (0.166667/0) | TC434396 (1.831048091), TC383270 (1.529094696) | Phenylpropanoid biosynthesis, Biosynthesis of secondary metabolites (no map in kegg database), Metabolic pathways (no map in kegg database) |
| gi|9408184 (0.282746) | TC460547 (3.29707538) | |
| gi|57635161 (0.937654/0) | TC389044 (1.315388003) | Phenylalanine metabolism, Phenylpropanoid biosynthesis, Metabolic pathways (no map in kegg database), Biosynthesis of secondary metabolites (no map in kegg database) |
| gi|4158232 (0.129406/0) | TC369736 (−1.380338094) | |
| gi|229358240 (0.488077/0) | TC457520 (3.014521523), TC372580 (−1.122982) | Citrate cycle (TCA cycle), Pyruvate metabolism, Biosynthesis of secondary metabolites (no map in kegg database), Glyoxylate and dicarboxylate metabolism, Metabolic pathways (no map in kegg database), Carbon fixation in photosynthetic organisms |
| gi|585032 (0.288839/0) | TC419796 (2.599484024), TC373702 (1.751487117), TC376351 (1.501967518) | Sulfur metabolism, Cyanoamino acid metabolism, Selenoamino acid metabolism, Cysteine and methionine metabolism, Metabolic pathways (no map in kegg database) |
| gi|357133190 (0.122988/0) | TC420420 (−1.340011237) TC397562 (−1.160565183) | Proteasome |
| gi|40317418 (0.492256) | TC419727 (3.101984365), TC369687 (2.059845514) | Glutathione metabolism |
| gi|728594 (0.479179/0) | TC400906 (1.599484024) | |
| gi|18076790 (0/0.303445) | TC406505 (−1.090831477) | Purine metabolism, Galactose metabolism, Amino sugar and nucleotide sugar metabolism, Starch and sucrose metabolism, Pentose phosphate pathway, Biosynthesis of secondary metabolites (no map in kegg database), Glycolysis/Gluconeogenesis, Metabolic pathways (no map in kegg database) |
| gi|232033 (0/0.645651) | TC376420 (−1.000125253) | |
| gi|15808779 (0/0.381971) | TC389590 (−1.052592673) | Glutathione metabolism, Ascorbate and aldarate metabolism |
| gi|18146827 (0/0.479167) | CK201148 (1.325723212) | Amino sugar and nucleotide sugar metabolism |
| gi|2499932 (0/0.214757) | CK155765 (−1.3639901) | Purine metabolism, Metabolic pathways (no map in kegg database) |
| gi|9230743 (3.036805) | TC370347 (1.175549678) | Starch and sucrose metabolism, Metabolic pathways (no map in kegg database) |
| gi|8980491 (0/0.226741) | TC396636 (−1.064190453) | |
| gi|525291 (0.296689) | TC411471 (−1.32428039) | Oxidative phosphorylation, Metabolic pathways (no map in kegg database) |
| gi|11124572 (2.456281) | CV767688 (−1.491369406) | Inositol phosphate metabolism, Fructose and mannose metabolism, Biosynthesis of secondary metabolites (no map in kegg database), Glycolysis/Gluconeogenesis, Metabolic pathways (no map in kegg database), Carbon fixation in photosynthetic organisms |
| gi|27544804 (3.330519) | CD908771 (1.284973401), CA729147 (2.336449618) |
Figure 3. Pathway network consist by the 18 DEPs out of the aligned ones.

Pathway network constructed based on the pathways of the 18 from the 24 aligned DEPs in both RNA-seq and proteomics datasets. Up-regulated genes are colored as black box, down-regulated genes as gray. Dashed arrow means that some steps are omitted, and solid arrows means the direct processes.
According to their functions, the aligned proteins were categorized into 5 groups (Figure 4). Half of these proteins (12 proteins) were involved in stress or immune responses, including cold shock domain protein 3, pyruvate decarboxylase isozyme 2, alanine aminotransferase 2, alcohol dehydrogenase ADH1A, peroxidase 8, cysteine synthase, glutamine synthetase isoform GSr2, elongation factor 1-beta, ascorbate peroxidase, thioredoxin h, hospholipid hydroperoxide glutathione peroxidase, and chitinase 2. Eight proteins (33.3%) are related to the process of metabolism, including lipoxygenase-1, cytosolic malate dehydrogenase, proteasome subunit alpha type-3-like, phosphoglucomutase, adenine phosphoribosyltransferase 1, reversibly glycosylated polypeptide and sucrose synthase-2, and triosephosphat-isomerase. In addition, succinate dehydrogenase is a key enzyme of the respiratory chain [42]. F0-F1 ATPase alpha subunit and ATP synthase beta subunit are involved in the energy metabolism, and glycine-rich protein or RNA binding protein is a kind of nucleic acid binding protein.
Figure 4. Categorization of the 24 aligned DEPs in proteomics dataset with the RNA-seq dataset.

Categorizing of the 24 aligned DEPs based on their functions. Each category is labeled with the different colors, and the numbers means the percentage of each DEPs to total aligned DEPs.
For these 24 proteins, their variations on up/down-regulation in proteomics and RNA-seq datasets are not completely consistent (Table 5). Succinate dehydrogenase and triosephosphat-isomerase displayed up-regulation in the proteomics dataset but down-regulation in the RNA-seq dataset. On the contrary, glutamine synthetase isoform GSr2, F0-F1 ATPase alpha subunit and chitinase 2 showed down-regulation in the proteomics dataset but up-regulation in the RNA-seq dataset. This inconsistent phenomenon might be caused by post-translational modification of the target gene and different metabolism process of the corresponding protein.
Expression analysis using quantitative reverse transcription PCR (qRT-PCR)
To verify the DEG and DEP data, we used qRT-PCR to analyze the expression levels of 21 genes including 14 found in both DEGs and DEPs, and 7 other DEGs (|log2|ratio (infected/control)>10) (File S10). The results showed that 11 genes were up-regulated, and 3 genes were down-regulated in the infected callus compared with the non-infected callus, while 7 genes had no changes in expression levels (Figure 5). Although the qRT-PCR data did not match the RNA-seq data perfectly, some genes did show consistent expression patterns in both datasets. For examples, CF133353 (expressed protein), CJ654139 (Os07g0137900 protein), TC416495 (MPI), TC451603 (LOC496215 protein), BJ236765 (unknown), TC458205 (high molecular weight glutenin subunit) and TC379055 (unknown) got vastly different expression both from qRT-PCR and RNA-seq. And the difference of expression from the two analysis both significantly reduced in TC419727 (glutathione transferase), TC388136 (lipoxygenase 1), TC434396 (cinnamyl alcohol dehydrogenase), TC419278 (alanine aminotransferase 2) and CK201148 (chitinase 2). At last, TC406505 (phosphoglucomutase). TC411471 (ATP synthase subunit a), TC420420 (proteasome subunit alpha type-5), CK155765 (adenine phosphoribosyltransferase 1) and CV767688 (triosephosphate isomerase) were down regulated.
Figure 5. Expression level and trend line of certain DEGs according to RNA-seq and qRT-PCR.

Assessment of the expression level of 21 DEGs, and contrasting of the results with RNA-seq. Ranging the DEGs in size of |log2|ratio plotted horizontal axis (from largest to smallest). The black bars mean the |log2|ratio, and the gray bars mean the 2−ΔΔCT of the DEGs. And the gene annotations are as bellow. CF133353, expressed protein; CJ654139, Os07g0137900 protein; TC416495, MPI; TC451603, LOC496215 protein; BJ236765, unknown; TC458205, high molecular weight glutenin subunit; TC379055, unknown; TC419727, glutathione transferase; TC388136, lipoxygenase 1; TC434396, cinnamyl alcohol dehydrogenase; TC419278, alanine aminotransferase 2, CK201148, chitinase 2; TC389044, peroxidase 8; TC370347, sucrose synthase type I; TC379241, pyruvate decarboxylase isozyme 2; TC389590, thylakoid-bound ascorbate peroxidase; TC406505, phosphoglucomutase; TC411471, ATP synthase subunit a; TC420420, proteasome subunit alpha type-5; CK155765, adenine phosphoribosyltransferase 1; CV767688, triosephosphate isomerase.
Discussion
Transferring process of T-DNA from Agrobacterium cells into wheat genome
Agrobacterium tumefaciens is a kind of pathogenic bacteria that causes crown gall disease (the formation of tumours) by the insertion of a T-DNA from a plasmid into plant cells in over 140 species of dicots under natural conditions. Unlike some tumor-inducing viruses, Agrobacterium T-DNA insertion into a host genome is a semi-random process [43] that causes an antibacterial response in the host. Up to date, even though many strains of Agrobacterium can be used in plant genetic transformation for T-DNA delivery, each strain has its suitable species to infect on. In this study, C58C1 strain was chosen because it was successfully used in many reports on wheat transformation [24]. According to some published papers [24], [26], [27], wheat transformation process mediated by Agrobacterium was finished within 48 hours. Especially, the growth peak of Agrobacterium on the surface of the host cells was observed when the co-culture period of Agrobacterium and wheat cells was proceeded for 36 h (File S11), and the expression of T-DNA was very intense after co-culture for 36 h [44]. Therefore, we expect that all of the transformation steps (attraction of Agrobacterium, T-DNA transportation and interaction) were lancing within this time since the infection. Therefore, in this investigation we chose the wheat immature embryos infected with Agrobacterium for 36 hours as materials for RNA-seq and proteomics analysis.
Investigating the host response to Agrobacterium infection process will contribute to understanding the interaction process and find some valuable clues on developing or optimizing of Agrobacterium- mediated transformation process. In our present study, 4,889 DEGs and 90 DEPs were identified to be closely related to Agrobacterium infection and all the DEGs involved in 111 pathways. Actually, RNA-seq is much more sensitive than 2D proteomics analysis, but the number of DEPs is much fewer than the DEGs. This kind of inconsistence is partly due to translational/post-translational regulation, but the most significant reason is that a DEP does not correspond to only one DEG (Table 5). In the response process of wheat cells to Agrobacterium, the genes related to secondary metabolites metabolic played the most important roles according to the results from pathway analysis and gene ontology analysis for biological process (Table 3, File S8). On the contrary, minimum of genes relate to photosynthesis pathway were detected. It indicated that the photosynthesis related genes avoided participating in the process of the transformation.
Potential roles of related metabolism process proteins or secondary metabolites in Agrobacterium mediated approach
A large portion of the DEGs and DEPs in our datasets were found to be involved in the metabolism process. Some of them may play important roles in the interaction between wheat cells and Agrobacterium. Among them, sucrose synthase is an attractive functional protein. This carbohydrate was proved to participate both in sucrose synthesis and cleavage in plants, and catalyzes the chemical reaction of UDP-glucose+D-fructose←→UDP+sucrose [45].In this study, we found that sucrose synthase-2 was up-regulated according to RNA-seq and qRT-PCR, but was down-regulated according to proteomics dataset. The reason might be that the protein is degraded dramatically or transformed into other homologous type very soon although the transcription is activated. The up-regulation of this synthase at the level of transcription was also found in Arabidopsis thaliana under the same situation [46]. As sucrose synthase is beneficial to root nodule organogenesis in legumes [47], this corresponding gene might be related to the process of Agrobacterium-mediated genetic transformation. Furthermore, UDP-glucose is the substrate of UDP-glycosyltransferase, which was confirmed to participate in the response to pathogens [48]. In addition, UDP-glycosyltransferase has also been found to detoxify deoxynivalenol in Fusarium [49]. Recently, an Arabidopsis hat mutant over-expressing a UDP- glucosyltransferase gene was found to be resistant to Agrobacterium-mediated transformation, in which many defense genes were down-regulated [10]. And in our results, we also found that both sucrose synthase and UDP-glucosyltransferase were up-regulated at the level of transcription after infection by Agrobacterium. It is implied that saccharo metabolism might affect the infection process.
Some DEPs were involved in proteasome, such as proteasome subunit alpha type-3-like. proteasomes, played a straightforward and critical role in the process of plant immune system [50]. 26S proteasome and ubiquitin emerge as a key regulatory mechanism in selective protein degradation [51]. This pathway was involved in a wide variety of cellular processes in plants, such as hormone signaling, photomorphogenesis, flower development, embryo development, and defense response [52]. Meanwhile, ubiquitin-protein ligase (CA714086) was identified in our RNA-seq datasets and remarkably up-regulated (|log2|ratio = 4.080610714). The activation of ubiquitin is a typical reaction during the process of pathogen infection [53]. In addition, E3 ubiquitin ligase is required for cell death and defense response in plants [54]. Therefore, ubiquitin-protein ligase might be also related to the Agrobacterium mediated DNA delivery. On the other hand, 26S proteasome subunit was proved to be involved in innate immunity in Arabidopsis [55], In plant, selective removal of short-lived regulatory proteins is a very important controlling strategy for physiology, growth, and development [56]. However, in our study, proteasome maturation factor (TC379459) is down-regulated according to RNA-seq (|log2|ratio = −1.44004434). In wheat cells, some regulatory proteins might produce a favorable environment for Agrobacterium infection. Thus, to defend the resistance from plant cells, Agrobacterium might suppress the proteasomes from plant. Function of above candidate genes screened from this study in wheat Agrobacterium-mediated transformation needs to be further investigated.
Relationship of plant phenylpropanoid biosynthesis and Agrobacterium infection
To our knowledge, UDP-glycosyltransferase mediates the transfer of glycosyl residues from nucleotide sugars to acceptor molecules (aglycones), such as plant secondary metabolites [57]. Most plant secondary metabolites might play important roles during Agrobacterium infection process. For examples, plant phenolics such as acetosyringone is an essential inducer for Agrobacterium infection. Acetosyringone is widely used in the protocol of Agrobacterium-mediated plant transformation. Other phenolics such as protocatechuic acid, β-resorcylic acid and protocatechuate also launch into the Agrobacterium-mediated transformation [58]. In our database, phenylalanine ammonia-lyase (PAL) was found up-regulated dramatically (|log2|ratio = 12.50423728). PAL catalyzes the first step in the biosynthesis of phenylpropanoids, which are further modified into a wide variety of phenolic compounds [59]. Another important secondary metabolites is flavonoid which involved in several biological process for plant development and defense [60]. In our research, two unknown proteins (TC413199, TC430821) were found to be involved in flavonoid biosynthetic process according to the GO analysis. Flavonoid is also a class of plant antibiotics. Sakuranetin, as a member of flavonoid, is recently demonstrated to have anti-inflammatory, anti-mutagenic, and anti-pathogenic activities.
Expression of stress response related proteins during the interaction between plant and Agrobacterium
Based on the gene ontology analysis of the RNA-seq dataset, we found 9.68% DEGs were involved in the immunity process. According to the pathway analysis, 111 DEGs were found to be related to the plant-pathogen interaction pathway. In consideration of Agrobacterium being a kind of plant-pathogen in nature, the stress and pathogen response genes should be the focuses of the transformation process.
Most of the DEGs are involved in responses to reactive oxygen species (ROS) stresses. As we know, oxidative burst is the first defense of plants against pathogen attacks [61]. The ROS, stimulated by stress from pathogen attack and generated from both plant and pathogen [62], plays a key role in the crosstalk between biotic and abiotic stress signaling [63]. Plants generated ROS by activating various oxidases and peroxidases [64]. In the meanwhile, it was found in wheat that Agrobacterium infection induces plant cell to produce hydrogen peroxide (H2O2) rapidly and leads wheat cell death severally [65]. In plant, a series of peroxidases can eliminate ROS, such as catalase, which activity was confirmed to be closely related to efficient regeneration potential of wheat immature embryos during the somatic embryogenesis [66]. There are 3 kinds of peroxidases (peroxidase 8 (TC389044), phospholipid hydroperoxide glutathione peroxidase (CD908771 and CA729147) and ascorbate peroxidase (TC389590)) found in our datasets. In the datasets of RNA-seq and proteomics, peroxidase 8 was up-regulated (|log2|ratio = 1.31539) during the infection process while ascorbate peroxidase was down-regulated (|log2|ratio = −1.05259). Furthermore, according to the results of qRT-PCR, the expression level of peroxidase 8 was up-regulated 1.7 times but the expression level of ascorbate peroxidase almost had no change (File S10). By coincidence, peroxidase was also identified in Ageratum conyzoides and Arabidopsis thaliana responding to Agrobacterium tumefaciens infection [16], [67],. Peroxidase has the function of interrupting the cascades of uncontrolled oxidation [68]. Peroxidase 8 is a kind of peroxidase belonging to Class-III in Triticum monococcum, which was a component of defense system responding to powdery mildew attack [46]. Ascorbate peroxidase scavenges hydrogen peroxide in plants, and is essential to protect cell constituents from lesion by hydrogen peroxide and other hydroxyl radicals produced from the interaction process of plant cells and pathogens [67], [69], Hydrogen peroxide is a kind of inhibitor for the invading pathogens, but it also contributes some virulence to pathogens [61]. Thereby, hydrogen peroxide might be an advantageous compound for both host cells and Agrobacterium, and it might be necessary to repress the accumulation of hydrogen peroxide-scavenging enzyme such as ascorbate peroxidase in the infection process.
Phospholipid hydroperoxide glutathione peroxidase was determined to be related to the metabolism of glutathione, which is an effective antioxidant preventing damage of important cellular components caused by ROS [70]. Moreover, phospholipid hydroperoxide glutathione peroxidase is a monomer, and the donor substrate of this peroxidase is not only restricted to glutathione (GSH) but also binds to specific mitochondrial proteins. In present research, phospholipid hydroperoxide glutathione peroxidase was detected to be up-regulated according to RNA-seq but down-regulated according to proteomics in the infection process. It is possible that phospholipid hydroperoxide glutathione peroxidase binds on mitochondrial proteins dramatically when the activity of mitochondrial is elevated. It makes protein of phospholipid hydroperoxide glutathione peroxidase decrease although the gene's expression was activated.
The rest of DEPs identified are related to be biotic stress response, such as chitinase (CK201148) and thioredoxin h (TC396636). As chitin is an important component of the cell wall of fungi and chitinases are generally found in organisms that dissolve and digest the chitin of fungi [71] plant chitinases are thought to be related to pathogen resistance [72]. Chitinase was up-regulated in our RNA-seq datasets and there was almost no diversity according to qRT-PCR result (2−ΔΔCT = 0.982055), but was down-regulated in proteomics datasets. Moreover, by reducing the defense of host cells, chitinases enable symbiotic interaction with nitrogen-fixing bacteria or mycorrhizal fungi [73]. To remove the barriers of infection, Agrobacterium suppressed the accumulation of chitinase from plant cells although the gene's expression was already activated. hioredoxin h also has potential capability against pathogens attaching, and evidence showed thioedoxin h gene was strongly induced within 4 hours in Arabidopsis cell suspensions treated with fungal elicitors, which contained wide range stress inducing agent [74]. In our study, we found that thioredoxin h was down-regulated both in RNA-seq and proteomics datasets which means the immune response of the host was impaired during the infection process.
Relationship of T-DNA integration and host proteins related to nucleic acid binding and nucleotide excision repair
The ultimate aim of Agrobacterium transformation is to import the T-DNA into plant genome. So, several nucleic acid binding proteins should take part in the two steps: T-DNA nuclear import and integration. And the typically nucleic acid binding proteins are thought to be the T-complex ones from Agrobacterium [75]. Now some host proteins have shown to be important in the last two steps for T-DNA delivery [7]. In this study the nucleic acid binding proteins take a very large part of the DEGs based on the GO analysis. The pathway analysis indicated that the spliceosome proteins should be paid more attention. T-DNA integration into plant chromosome actually belongs to the way of non-homologous (illegitimate) recombination (NHR), even when the T-DNA shares high homology with the host genome. As for the pattern of the T-DNA integration, the double-strand-break repair (DSBR) model and single-strand-gap repair (SSGR) model were originally proposed [76]. Above findings suggest that nucleotide excision repair proteins are the key players in the process of T-DNA integration.
Particularly, histone was demonstrated to play an important role in the transformation process mediated by Agrobacterium [7]. Especially, histone H2A, histone H4, and histone H3-11 in Arabidopsis can increase transformation susceptibility. Other plant proteins related to the transformation process include BTIs (VirB2-interacting proteins), AtRAB8, and DIG3. Hwang et al. used the C-terminal-processed portion of VirB2 as the bait to search the interaction protein by yeast two-hybrid in Arabidopsis, and found that BTI1, BTI2, BTI3, and a membrane-associated GTPase, AtRAB8 interact with VirB2. Their further study showed the positive meaning of these proteins in the infection process of Agrobacterium [8]. DIG3, found in tomato, encodes an enzymatically active type 2C serine/threonine protein phosphatase, which interacts with VirD2. Over-expression of DIG3 in tobacco protoplasts inhibited nuclear import of VirD2 nuclear localization [9]. In our study, Histone and GTPase related protein genes were also identified, such as TC396751 encoding Histone H2B. In the process of Agrobacterium infection, H2B was down-regulated dramatically (|log2|ratio = −13.62721073). In the previous research [13], H2B did not lead to increased transformation susceptibility. However, according to our results, we assumed that H2B might have a negative effect during the transformation process. Therefore, the expression level of H2B might be depressed by Agrobacterium. Besides, we also obtained several serine/threonine-protein kinases such as TC440175 (Serine/threonine-protein kinase Nek5, (|log2|ratio = 2.297455487)). Some of them probably are similar to DIG3, and interact with VirD2. Beyond that, a big part of the DEGs have the function of nucleic acid binding and protein-protein interaction based on the categorization of the GO terms (Figure 1B). It is suggested that some genes among the DEGs should play roles in the nucleic importing and integration into genome of T-DNA.
Conclusions
In this study, we identified a set of 4988 DEGs and 90 DEPs in Agrobacterium-infected wheat tissues. After comparative analysis, 24 of the 90 DEPs were detected in RNA-seq and proteomics datasets simultaneously. The expressions of the most DEGs were found to be uniformly up/down-regulated between RNA-seq and qRT-PCR datasets, which proved the authenticity of the results from RNA-seq. According to GO analysis, we found that a big part of these differentially expressed genes were related to the process of stress or immunity response, and other major part of DEGs involved in the process of molecular modification. We believe that some of these genes are closely related to the transformation process mediated by Agrobacterium. The findings achieved in this study will help to further exploit the interaction between Agrobacterium and host cells, and may facilitate the development of efficient plant transformation strategies.
Supporting Information
Categorization of row reads of the control material A and the infected material B.
(DOC)
Alignment statistics of total reads.
(DOC)
Randomness assessment A and sequencing saturation analysis B.
(DOC)
Gene coverage statistics.
(DOC)
The differentially expressed genes (|log2|ratio (infected/control)≥1).
(XLS)
Gene Ontology analysis information for cellular component.
(DOC)
Gene Ontology analysis information for molecular function.
(DOC)
Gene Ontology analysis information for biological process.
(DOC)
Distribution of all GEGs on the pathways.
(DOC)
Comparison of expression level between the data of real-time PCR and RNA-seq.
(DOC)
Detection of the Agrobacterium attachment on wheat callus by scanning electron microscope (SEM). The adsorption of Agrobacterium to wheat callus after co-culture for 30 minutes, 12 hours, 24 hours, 36 hours, and 48 hours.
(TIF)
Acknowledgments
The authors are grateful to Ms. Sarah Hancock for language editing (13-119-J) from the Kansas Agricultural Experiment Station.
Funding Statement
This research was financially supported by grants from the Ministry of Agriculture of China (2011ZX08010-004), and the National Natural Science Foundation of China (30971776). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sanvido O, Romeis J, Bigler F (2011) Environmental change challenges decision-making during post-market environmental monitoring of transgenic crops. Transgenic Res 20: 1191–1201. [DOI] [PubMed] [Google Scholar]
- 2. Shrawat AK, Lorz H (2006) Agrobacterium-mediated transformation of cereals: a promising approach crossing barriers. Plant Biotechnol J 4: 575–603. [DOI] [PubMed] [Google Scholar]
- 3. De Block M, Herrera-Estrella L, Van Montagu M, Schell J, Zambryski P (1984) Expression of foreign genes in regenerated plants and in their progeny. EMBO J 3: 1681–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dafny-Yelin M, Levy A, Tzfira T (2008) The ongoing saga of Agrobacterium-host interactions. Trends Plant Sci 13: 102–105. [DOI] [PubMed] [Google Scholar]
- 5. Gelvin SB (2009) Agrobacterium in the genomics age. Plant Physiol 150: 1665–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lacroix B, Zaltsman A, Citovsky V (2010) Agrobacterium-mediated transformation. In: Stewart CN, Touraev A, Citovsky V, Tzfira T, editors. Plant Transformation Technologies. Oxford: Blackwell Publishing Ltd. pp. 1–29. [Google Scholar]
- 7. Tzfira T, Citovsky V (2002) Partners-in-infection: host proteins involved in the transformation of plant cells by Agrobacterium . Trends Cell Biol 12: 121–129. [DOI] [PubMed] [Google Scholar]
- 8. Hwang HH, Gelvin SB (2004) Plant proteins that interact with VirB2, the Agrobacterium tumefaciens pilin protein, mediate plant transformation. Plant Cell 16: 3148–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tao Y, Rao PK, Bhattacharjee S, Gelvin SB (2004) Expression of plant protein phosphatase 2C interferes with nuclear import of the Agrobacterium T-complex protein VirD2. Proc Natl Acad Sci USA 101: 5164–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gelvin SB (2010) Plant proteins involved in Agrobacterium-mediated genetic transformation. Annu Rev Phytopathol 48: 45–68. [DOI] [PubMed] [Google Scholar]
- 11.Cheng M, Chang YF, Olhoft PM, Cardoza V, Lai FM (2011) Cells/tissues conditioning for facilitating T-DNA delivery. In: Dan.Y, Ow.D.W., editors. Historical technology developments in plant transformation. Sharjah: Bentham Science Publishers Ltd. pp. 77–107. [Google Scholar]
- 12. Frame BR, Shou H, Chikwamba RK, Zhang Z, Xiang C, et al. (2002) Agrobacterium tumefaciens-mediated transformation of maize embryos using a standard binary vector system. Plant Physiol 129: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tenea GN, Spantzel J, Lee LY, Zhu Y, Lin K, et al. (2009) Overexpression of several Arabidopsis histone genes increases Agrobacterium-mediated transformation and transgene expression in plants. Plant Cell 21: 3350–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tie W, Zhou F, Wang L, Xie W, Chen H, et al. (2012) Reasons for lower transformation efficiency in indica rice using Agrobacterium tumefaciens-mediated transformation: lessons from transformation assays and genome-wide expression profiling. Plant Mol Biol 78: 1–18. [DOI] [PubMed] [Google Scholar]
- 15. McCullen CA, Binns AN (2006) Agrobacterium tumefaciens and plant cell interactions and activities required for interkingdom macromolecular transfer. Annu Rev Cell Dev Biol 22: 101–127. [DOI] [PubMed] [Google Scholar]
- 16. Ditt RF, Nester EW, Comai L (2001) Plant gene expression response to Agrobacterium tumefaciens . Proc Natl Acad Sci USA 98: 10954–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deeken R, Engelmann JC, Efetova M, Czirjak T, Müller T, et al. (2006) An integrated view of gene expression and solute profiles of Arabidopsis tumors: a genome-wide approach. Plant Cell 18: 3617–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tzfira T, Vaidya M, Citovsky V (2001) VIP1, an Arabidopsis protein that interacts with Agrobacterium VirE2, is involved in VirE2 nuclear import and Agrobacterium infectivity. EMBO J 20: 3596–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tzfira T, Vaidya M, Citovsky V (2002) Increasing plant susceptibility to Agrobacterium infection by overexpression of the Arabidopsis nuclear protein VIP1. Proc Natl Acad Sci USA 99: 10435–10440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li J, Krichevsky A, Vaidya M, Tzfira T, Citovsky V (2005) Uncoupling of the functions of the Arabidopsis VIP1 protein in transient and stable plant genetic transformation by Agrobacterium . Proc Natl Acad Sci USA 102: 5733–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anand A, Krichevsky A, Schornack S, Lahaye T, Tzfira T, et al. (2007) Arabidopsis VIRE2 interacting protein2 is required for Agrobacterium T-DNA integration in plants. Plant Cell 19: 1695–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J, Ye X, An B, Du L, Xu H (2012) Genetic transformation of wheat: current status and future prospects. Plant Biotechnol Rep 6: 183–193. [Google Scholar]
- 23. Harwood WA (2012) Advances and remaining challenges in the transformation of barley and wheat. J Exp Bot 63: 1791–1798. [DOI] [PubMed] [Google Scholar]
- 24. Cheng M, Fry JE, Pang S, Zhou H, Hironaka CM, et al. (1997) Genetic transformation of wheat mediated by Agrobacterium tumefaciens . Plant Physiol 115: 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu T, Metz S, Chay C, Zhou H, Biest N, et al. (2003) Agrobacterium-mediated large-scale transformation of wheat (Triticum aestivum L.) using glyphosate selection. Plant Cell Rep 21: 1010–1019. [DOI] [PubMed] [Google Scholar]
- 26. Jones HD, Doherty A, Wu H (2005) Review of methodologies and a protocol for the Agrobacterium-mediated transformation of wheat. Plant methods 1: 5 doi:10.1186/1746-4811-1181-1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tom C, Amitava M (2004) Genetic engineering of wheat: ptotocols and use to enhance stress tolerance. In: Liang GH, Skinner DZ, editors. Genetically modified crops: their development, uses, and risks: Food products press. pp. 131–163.
- 28. Bińka A, Orczyk W, Nadolska-Orczyk A (2012) The Agrobacterium-mediated transformation of common wheat (Triticum aestivum L.) and triticale (x Triticosecale Wittmack): role of the binary vector system and selection cassettes. J Appl Genetics 53: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Metzker ML (2010) Sequencing technologies-the next generation. Nat Rev Gen 11: 31–46. [DOI] [PubMed] [Google Scholar]
- 30. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, et al. (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lopez-Casado G, Covey PA, Bedinger PA, Mueller LA, Thannhauser TW, et al. (2012) Enabling proteomic studies with RNA-Seq: The proteome of tomato pollen as a test case. Proteomics 12: 761–774. [DOI] [PubMed] [Google Scholar]
- 33. Cheng M, Hu T, Layton J, Liu CN, Fry JE (2003) Desiccation of plant tissues post-Agrobacterium infection enhances T-DNA delivery and increases stable transformation efficiency in wheat. In Vitro CellDev-Pl 39: 595–604. [Google Scholar]
- 34. D'Alessio JM, Gerard GF (1988) Second-strand cDNA synthesis with E. coli DNA polymerase I and RNase H: the fate of information at the mRNA 5′ terminus and the effect of E. coli DNA ligase. Nucleic Acids Res 16: 1999–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. [DOI] [PubMed] [Google Scholar]
- 36. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- 37. Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7: 986–995. [DOI] [PubMed] [Google Scholar]
- 38. Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann stat 29: 1165–1188. [Google Scholar]
- 39. Hooper SD, Bork P (2005) Medusa: a simple tool for interaction graph analysis. Bioinformatics 21: 4432–4433. [DOI] [PubMed] [Google Scholar]
- 40. Carvalho K, de Campos M, Pereira L, Vieira L (2010) Reference gene selection for real-time quantitative polymerase chain reaction normalization in “Swingle” citrumelo under drought stress. Anal Biochem 402: 197–199. [DOI] [PubMed] [Google Scholar]
- 41. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 42. Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, et al. (1998) A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394: 694–697. [DOI] [PubMed] [Google Scholar]
- 43. van Attikum H, Bundock P, Hooykaas PJ (2001) Non-homologous end-joining proteins are required for Agrobacterium T-DNA integration. EMBO J 20: 6550–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Finer K, Finer J (2000) Use of Agrobacterium expressing green fluorescent protein to evaluate colonization of sonication-assisted Agrobacterium-mediated transformation-treated soybean cotyledons. Lett Appl Microbiol 30: 406–410. [DOI] [PubMed] [Google Scholar]
- 45. Elling L (1995) Effect of metal ions on sucrose synthase from rice grains-a study on enzyme inhibition and enzyme topography. Glycobiology 5: 201–206. [DOI] [PubMed] [Google Scholar]
- 46. Liu G, Sheng X, Greenshields DL, Ogieglo A, Kaminskyj S, et al. (2005) Profiling of wheat class III peroxidase genes derived from powdery mildew-attacked epidermis reveals distinct sequence-associated expression patterns. Mol Plant-Micr Inter 18: 730–741. [DOI] [PubMed] [Google Scholar]
- 47. Röhrig H, Schmidt J, Miklashevichs E, Schell J, John M (2002) Soybean ENOD40 encodes two peptides that bind to sucrose synthase. Proc Natl Acad Sci USA 99: 1915–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. von Saint Paul V, Zhang W, Kanawati B, Geist B, Faus-Keßler T, et al. (2011) The Arabidopsis glucosyltransferase UGT76B1 conjugates isoleucic acid and modulates plant defense and senescence. Plant Cell 23: 4124–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Poppenberger B, Berthiller F, Lucyshyn D, Sieberer T, Schuhmacher R, et al. (2003) Detoxification of the Fusarium mycotoxin deoxynivalenol by a UDP-glucosyltransferase from Arabidopsis thaliana . J Biol Chem 278: 47905–47914. [DOI] [PubMed] [Google Scholar]
- 50. Spoel SH, Mou Z, Tada Y, Spivey NW, Genschik P, et al. (2009) Proteasome-mediated turnover of the transcription coactivator NPR1 plays dual roles in regulating plant immunity. Cell 137: 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hochstrasser M (1995) Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr Opin Cell Biol 7: 215–223. [DOI] [PubMed] [Google Scholar]
- 52. Vierstra RD (2003) The ubiquitin/26S proteasome pathway, the complex last chapter in the life of many plant proteins. Trends Plant Sci 8: 135–142. [DOI] [PubMed] [Google Scholar]
- 53. Edelmann MJ, Kessler BM (2008) Ubiquitin and ubiquitin-like specific proteases targeted by infectious pathogens: Emerging patterns and molecular principles. BBA-Mol Basis Dis 1782: 809–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang CW, González-Lamothe R, Ewan RA, Rowland O, Yoshioka H, et al. (2006) The E3 ubiquitin ligase activity of Arabidopsis PLANT U-BOX17 and its functional tobacco homolog ACRE276 are required for cell death and defense. Plant Cell 18: 1084–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yao C, Wu Y, Nie H, Tang D (2012) RPN1a, a 26S proteasome subunit, is required for innate immunity in Arabidopsis . Plant J 71: 1015–1028. [DOI] [PubMed] [Google Scholar]
- 56. Smalle J, Vierstra RD (2004) The ubiquitin 26S proteasome proteolytic pathway. Annu Rev Plant Biol 55: 555–590. [DOI] [PubMed] [Google Scholar]
- 57. Gachon CM, Langlois-Meurinne M, Saindrenan P (2005) Plant secondary metabolism glycosyltransferases: the emerging functional analysis. Trends Plant Sci 10: 542–549. [DOI] [PubMed] [Google Scholar]
- 58. Bhattacharya A, Sood P, Citovsky V (2010) The roles of plant phenolics in defence and communication during Agrobacterium and Rhizobium infection . Mol Plant Pathol 11: 705–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cheng GW, Breen PJ (1991) Activity of phenylalanine ammonia-lyase (PAL) and concentrations of anthocyanins and phenolics in developing strawberry fruit. J Am Soc Hortic Sci 116: 865–869. [Google Scholar]
- 60. Hichri I, Barrieu F, Bogs J, Kappel C, Delrot S, et al. (2011) Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J Exp Bot 62: 2465–2483. [DOI] [PubMed] [Google Scholar]
- 61. Bolwell GP, Bindschedler LV, Blee KA, Butt VS, Davies DR, et al. (2002) The apoplastic oxidative burst in response to biotic stress in plants: a three-component system. J Exp Bot 53: 1367–1376. [PubMed] [Google Scholar]
- 62. Rolke Y, Liu S, Quidde T, Williamson B, Schouten A, et al. (2004) Functional analysis of H2O2-generating systems in Botrytis cinerea: the major Cu-Zn-superoxide dismutase (BCSOD1) contributes to virulence on French bean, whereas a glucose oxidase (BCGOD1) is dispensable. Mol Plant Pathol 5: 17–27. [DOI] [PubMed] [Google Scholar]
- 63. Fujita M, Fujita Y, Noutoshi Y, Takahashi F, Narusaka Y, et al. (2006) Crosstalk between abiotic and biotic stress responses: a current view from the points of convergence in the stress signaling networks. Curr Opin Plant Biol 9: 436–442. [DOI] [PubMed] [Google Scholar]
- 64. Li JL, Sulaiman M, Beckett RP, Minibayeva FV (2010) Cell wall peroxidases in the liverwort Dumortiera hirsuta are responsible for extracellular superoxide production, and can display tyrosinase activity. Physiol Plantarum 138: 474–484. [DOI] [PubMed] [Google Scholar]
- 65. Parrott DL, Anderson AJ, Carman JG (2002) Agrobacterium induces plant cell death in wheat (Triticum aestivum L.). Physiol Mol Plant Pathol 60: 59–69. [Google Scholar]
- 66. She M, Yin G, Li J, Li X, Du L, et al. (2012) Efficient regeneration potential is closely related to auxin exposure time and catalase metabolism during the somatic embryogenesis of immature embryos in Triticum aestivum L. Mol Biotechnol 54: 451–460. [DOI] [PubMed] [Google Scholar]
- 67. Ditt RF, Kerr KF, de Figueiredo P, Delrow J, Comai L, et al. (2006) The Arabidopsis thaliana transcriptome in response to Agrobacterium tumefaciens . Mol Plant-Micr Inter 19: 665–681. [DOI] [PubMed] [Google Scholar]
- 68. Hong CY, Hsu YT, Tsai YC, Kao CH (2007) Expression of ascorbate peroxidase 8 in roots of rice (Oryza sativa L.) seedlings in response to NaCl. J Exp Bot 58: 3273–3283. [DOI] [PubMed] [Google Scholar]
- 69. Asada K (1992) Ascorbate peroxidase-a hydrogen peroxide-scavenging enzyme in plants. Physiol Plantarum 85: 235–241. [Google Scholar]
- 70. Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF (2003) The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol 66: 1499–1503. [DOI] [PubMed] [Google Scholar]
- 71. Collinge DB, Kragh KM, Mikkelsen JD, Nielsen KK, Rasmussen U, et al. (1993) Plant chitinases. Plant J 3: 31–40. [DOI] [PubMed] [Google Scholar]
- 72. Schlumbaum A, Mauch F, Vögeli U, Boller T (1986) Plant chitinases are potent inhibitors of fungal growth. Nature 324: 365–367. [Google Scholar]
- 73. Kasprzewska A (2003) Plant chitinases-regulation and function. Cell MolBiolLett 8: 809–824. [PubMed] [Google Scholar]
- 74. Reichheld JP, Mestres-Ortega D, Laloi C, Meyer Y (2002) The multigenic family of thioredoxin h in Arabidopsis thaliana: specific expression and stress response. Plant Physiol Biochem 40: 685–690. [Google Scholar]
- 75. Zupan JR, Zambryski P (1995) Transfer of T-DNA from Agrobacterium to the plant cell. Plant Physiol 107: 1041–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tzfira T, Li J, Lacroix B, Citovsky V (2004) Agrobacterium T-DNA integration: molecules and models. TRENDS Genet 20: 375–383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Categorization of row reads of the control material A and the infected material B.
(DOC)
Alignment statistics of total reads.
(DOC)
Randomness assessment A and sequencing saturation analysis B.
(DOC)
Gene coverage statistics.
(DOC)
The differentially expressed genes (|log2|ratio (infected/control)≥1).
(XLS)
Gene Ontology analysis information for cellular component.
(DOC)
Gene Ontology analysis information for molecular function.
(DOC)
Gene Ontology analysis information for biological process.
(DOC)
Distribution of all GEGs on the pathways.
(DOC)
Comparison of expression level between the data of real-time PCR and RNA-seq.
(DOC)
Detection of the Agrobacterium attachment on wheat callus by scanning electron microscope (SEM). The adsorption of Agrobacterium to wheat callus after co-culture for 30 minutes, 12 hours, 24 hours, 36 hours, and 48 hours.
(TIF)