

Abstract
IL-4 is an important regulator of the activation, proliferation, and differentiation of many hematopoetic cells. Many of these biological effects result from the activation of Janus kinases (JAK)1 and JAK3 and the transcription factor Stat6. Recent data suggest that members of the SOCS (suppressor of cytokine signaling) family of proteins can inhibit JAK-STAT signaling. We have examined the ability of SOCS family members to suppress IL-4 signaling, and we have found that SOCS-1 potently inhibits the activation of JAK1 kinase and Stat6 in response to IL-4. Furthermore, SOCS-1 can inhibit the induction of CD23 expression by IL-4. SOCS-2 does not inhibit induction of signaling by IL-4, while inhibition of IL-4 signaling by SOCS-3 can be detected in transient transfection systems, but not in stable cell lines. These studies implicate SOCS-1 in modulation of IL-4 signaling and suggest that SOCS-1 may play a role in regulating the immune response.
Interleukin-4 is a cytokine involved in the activation, proliferation, and differentiation of a variety of hematopoetic cells, including B cells, T cells, and mast cells (1). Central to the response of cells to IL-4 is the activation of the nonreceptor tyrosine kinases Janus kinase (JAK)3 1 and JAK3, and the subsequent phosphorylation of a number of signaling molecules, including a member of the STAT family of transcription factors, Stat6. Phosphorylated Stat6 activates transcription of genes involved in B cell differentiation, including the Ig germline ε and γ1 genes and the low-affinity FcεII receptor (CD23). The observations that cell lines deficient in JAK1 are unable to phosphorylate Stat6 (2, 3), and that B cells from Stat6-deficient mice are deficient in class-switching to IgE (4–7), underscore the essential role of JAK1 and Stat6 in IL-4 function.
Although much is known about IL-4-mediated activation of the JAK-STAT signaling pathway, much less is known about how the pathway is inactivated. Recently, a novel suppressor of cytokine signaling, SOCS-1/SSI-1/JAB (8–10), was identified, both as a JAK2 binding protein and as an inhibitor of IL-6 signal transduction. SOCS-1 inhibits IL-6-induced phosphorylation of gp130, JAK2, and Stat3, and subsequent differentiation of monocytes into macrophages (8–10). Recent data suggest that SOCS-1 can bind and inhibit the kinase activity of all four JAK family members (8, 10). Interestingly, expression of SOCS-1 mRNA in mouse bone marrow is induced by several cytokines, suggesting that the induction of SOCS-1 may represent a more general negative feedback loop that modulates the responsiveness of cells to cytokine (9).
SOCS-1 is a member of a larger gene family. Database searches have identified seven other genes that share with SOCS-1 a small C-terminal region of homology, termed the “SOCS box,” and a central Src homology 2 domain (9, 11–13). Outside of these two regions of homology, the SOCS family members have very divergent amino acid sequences, and it is unclear, given this limited homology, whether the other family members also play a role in inhibition of cytokine signaling.
In bone marrow cells, IL-4 has been shown to induce the expression of SOCS-1, -2, and -3. In this study, we examined whether these SOCS family members may play a role in the suppression of IL-4 signaling. We have demonstrated biochemically and functionally that constitutive expression of SOCS-1, but not SOCS-2, can inhibit IL-4-induced activation of JAK1 and Stat6. SOCS-1 can also inhibit IL-4-induced gene transcription mediated by Stat6. Furthermore, we have demonstrated that, like SOCS-1, SOCS-3 can suppress IL-4-induced gene transcription in transient transfection assays, but that in stable cell lines, SOCS-3 does not suppress IL-4 signaling.
Materials and Methods
Cell lines, cytokines and Abs
The M12.4.1 cell line has been previously described (3). The 293T human embryonic kidney cell line is a gift from Dr. Chris Schindler of Columbia University (New York, NY). Murine rIL-4 is a gift from Dr. Robert Coffman of DNAX Research Institute (Palo Alto, CA), and human rIL-4 is a gift from Dr. Satwant Narula of Schering-Plough (Kenilworth, NJ). Phycoerythrin-conjugated rat anti-mouse CD23 Ab was purchased from PharMingen (San Diego, CA). Rabbit anti-human/mouse JAK1 and anti-phosphotyrosine Abs were purchased from Upstate Biotechnology (Lake Placid, NY). Rabbit anti-mouse JAK1 and M-20 anti-Stat6 Abs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Plasmids
Plasmid p(Iε-IL4RE)4-Luc was previously described (14). pSV40-βgal is a gift from Dr. Schindler. The SOCS expression vectors were previously described (9).
Transfections
M12 cells were transfected as previously described (15) with the following modifications. Cells (4 × 106) were electroporated at 300 V, 1250 μF in complete RPMI (cRPMI) containing 10 μg/ml DEAE-dextran. For stable transfections, clones were selected for 2 wk in cRPMI containing G418 (Life Technologies, Grand Island, NY) at 2 mg/ml, and clones were screened for SOCS gene expression by Northern blot analysis. 293T cells were transiently transfected as previously described (14). Luciferase and β-galactosidase measurements were performed as previously described (14).
Electrophoretic mobility shift assay (EMSA), immunoprecipitation (IP), and in vitro kinase assay
Whole cell extracts were prepared and EMSA, IP, and in vitro kinase assays were performed as previously described (16).
FACS analysis
Cells were treated with IL-4 for 48 h, washed, and resuspended in 3% BSA/0.02% sodium azide in PBS. A total of 2 × 105 cells/100 μl were incubated at 4°C for 30 min with Ab at 0.2 μg/100 μl. Cells were washed, resuspended in PBS/BSA, and analyzed on a flow cytometer (FACScan; Becton Dickinson, Mountain View, CA).
Results
Inhibition of IL-4-induced transcription by SOCS-1 and SOCS-3, but not SOCS-2, in a transient transfection assay
The system we chose to study the effect of the SOCS proteins on IL-4 signaling was the M12.4.1 B cell line. M12 cells do not express, either constitutively or IL-4 inducibly, detectable SOCS-1, -2 or -3 RNA (data not shown). This cell line is therefore an ideal system with which to examine the effect of each of these SOCS genes on IL-4 signaling independently. To determine whether SOCS-1, -2, or -3 can inhibit IL-4-induced transcription, we transiently transfected M12 cells with a Stat6-responsive luciferase reporter construct (p(Iε-IL4RE)4-Luc) and expression vectors containing each of the SOCS genes. Cotransfection of SOCS-1 or SOCS-3 resulted in a dose-dependent decrease in induction of pIε-Luc, while cotransfection of SOCS-2 had little effect on reporter activity (Fig. 1A). We repeated the transient transfections in 293T cells with similar results (Fig. 1B). These transient transfection assays indicate that expression of SOCS-1 and SOCS-3, but not SOCS-2, can inhibit IL-4-induced activation of a Stat6-driven reporter, and that the level of inhibition is proportional to the amount of transfected SOCS DNA.
Figure 1.
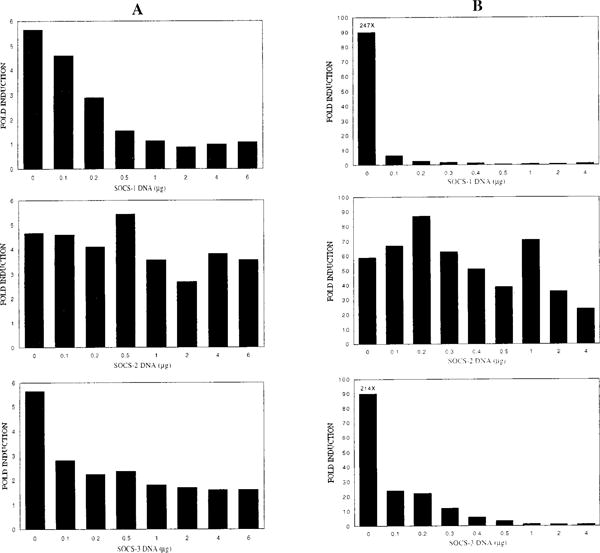
Suppression of IL-4-induced activation of a Stat6-responsive reporter gene by SOCS-1 and SOCS-3, but not SOCS-2, in transient transfection. A, M12 cells were transfected with increasing amounts of SOCS expression vector, 5 μg of p(Iε-IL4RE)4-Luc reporter and 1 μg of pSV40-βgal by DEAE-dextran and electroporation. Total amount of transfected DNA was kept constant at 12 μg by addition of salmon testes DNA. One-half of each transfection was treated with IL-4 and, 24 h after transfection, cells were harvested and luciferase activity was determined. B, 293T cells were transfected with increasing amounts of SOCS expression vector, 5 μg of p(Iε-IL4RE)4-Luc reporter, 1 μg of pSV40-βgal, and 0.5 μg HuStat6 expression vector by calcium phosphate precipitation. Total amount of DNA was kept constant at 10.5 μg by addition of salmon testes DNA. IL-4 was added to the media 24 h after transfection, and, 48 h after transfection, cells were harvested and luciferase activity was determined. Luciferase values are adjusted for transfection efficiency by standardizing with the β-galactosidase activity of each sample. The results shown are averages of three independent transfections: SOCS-1 (top panels), SOCS-2 (middle panels), and SOCS-3 (bottom panels).
Inhibition of IL-4-induced Stat6-DNA binding activity by SOCS-1, but not by SOCS-2 or SOCS-3, in SOCS stable transfectants
To further characterize the effect of SOCS gene expression on IL-4 signaling, we generated M12 stable transfectants expressing SOCS-1, -2 or -3, and compared their IL-4 inducibility to that of control transfectants expressing the selectable marker alone. The effect of SOCS expression on IL-4-induced Stat6-DNA complex formation was determined by EMSA using an oligonucleotide containing a consensus γ-IFN-activated sequence from the IFN regulatory factor-1 promoter known to bind Stat6 in vitro (17). As expected, expression of SOCS-1 resulted in decreased formation of the Stat6-DNA complex in response to IL-4 (Fig. 2A), and expression of SOCS-2 had no effect on complex formation (Fig. 2B). To our surprise, however, expression of SOCS-3 did not inhibit formation of the Stat6-DNA complex, which was equivalent in all three of the SOCS-3 clones analyzed and the control (Fig. 2C). Thus, stable expression in M12 cells of SOCS-1, but not SOCS-2 or SOCS-3, suppresses the IL-4-induced DNA binding activity of Stat6.
Figure 2.
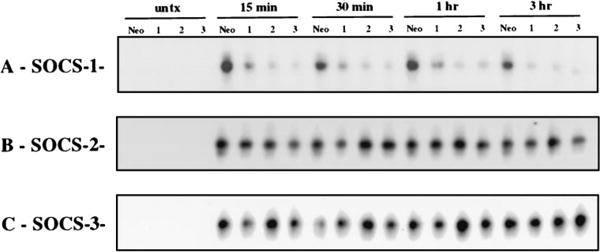
Suppression of IL-4-induced Stat6-DNA binding activity by SOCS-1, but not SOCS-2 or SOCS-3, in M12 stable cell lines. EMSA was done with whole cell extracts from cells untreated (lanes 1–4) and treated with a timecourse of IL-4 (lanes 5–20): control cells (A-C; Neo), SOCS-1 clones (A: 1, 2, 3), SOCS-2 clones (B: 1, 2, 3), and SOCS-3 clones (C: 1, 2, 3). The probe used was from the IFN regulatory factor-1 γ-IFN-activated sequence.
Inhibition of IL-4-induced activation of JAK1 and Stat6 by SOCS-1, but not by SOCS-2 or SOCS-3
Phosphorylation and activation of JAK1 and Stat6 are essential for induction of Stat6 DNA binding activity (2). To ascertain whether the decrease in Stat6 DNA binding activity in the SOCS-1 stable transfectants was due to inhibition of JAK1 kinase activity, we immunoprecipitated lysates from cells untreated or treated with IL-4 with Abs to JAK1 or Stat6 and probed with Ab to phosphotyrosine. Induction of JAK1 and Stat6 phosphorylation in the SOCS-1 stable clones was reduced when compared with control (Fig. 3A), while induction in the SOCS-2 stable clones (Fig. 3B)and in the SOCS-3 stable clones (Fig. 3C)was similar to that of controls. To further confirm that SOCS-1 suppresses JAK1 activation, we measured the IL-4-induced kinase activity of JAK1 in the SOCS-1 stable clones by in vitro kinase assay. The kinase activity of JAK1 was suppressed in the SOCS-1 clones when compared with control cells (Fig. 3D). Thus, it appears that the IL-4-induced tyrosine phosphorylation of JAK1 and Stat6, and the activation of JAK1 kinase activity, are suppressed by SOCS-1, but not by SOCS-2 or SOCS-3, in stable transfectants.
Figure 3.
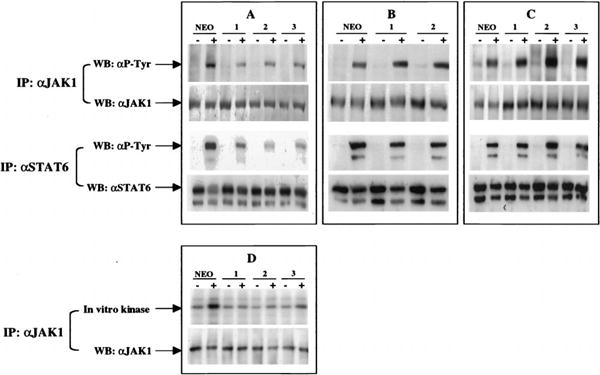
Suppression of activation of JAK1 and Stat6 by SOCS-1, but not SOCS-2 or SOCS-3. M12 stable transfectants were starved for 4 h in serum-free medium and then cultured with medium alone or medium plus IL-4, for 10 min for JAK1 IPs and in vitro kinase assays, and for 30 min for Stat6 IPs. Control (A-D: Neo), SOCS-1 (A and D: 1–1, 1–2, 1–3), SOCS-2 (B: 2–1, 2–2), and SOCS-3 (C: 3–1, 3–2, 3–3) extracts were precleared with normal rabbit serum and JAK1 or Stat6 was IPed with Abs to JAK1 or Stat6 as indicated (IP). Immune complexes in A-C were fractionated by SDS-PAGE (7% gel) and blotted with Abs to phosphotyrosine, JAK1, or Stat6 as indicated (WB). Immune complexes in D were incubated in in vitro kinase reactions with γ-ATP, and the reactions were split and fractionated on two SDS-PAGE gels, one analyzed by autoradiography and the other blotted with JAK1 Ab.
Inhibition of IL-4-induced gene transcription by SOCS-1, but not SOCS-2 or SOCS-3
Recent studies have suggested that control of STAT function may occur at other levels than simply regulation of DNA binding (18–20). To determine whether stable expression of SOCS-2 or SOCS-3 can suppress IL-4-induced gene transcription by another mechanism than inhibition of Stat6 tyrosine phosphorylation and DNA binding, we examined the responsiveness of an IL-4 inducible gene, CD23, in the SOCS stable clones. Up-regulation of CD23 was blocked in the SOCS-1 transfectants (Fig. 4b), which is consistent with the observation that IL-4 induction of CD23 expression is dependent on activation of Stat6. Up-regulation of CD23 surface expression in the SOCS-2 and SOCS-3 clones (Fig. 4, c and d), however, was comparable to that of control (Fig. 4a), indicating that stable expression of SOCS-2 and SOCS-3 do not block IL-4 induction of CD23 expression.
Figure 4.

Suppression of IL-4-induced gene expression by SOCS-1, but not SOCS-2 or SOCS-3. Cell surface expression of CD23 is suppressed in SOCS-1-expressing cells, but not in SOCS-2- or SOCS-3-expressing cells. Control (a), SOCS-1 (b), SOCS-2 (c), and SOCS-3 (d) stable transfectants were cultured for 48 h in medium alone (closed histograms) or medium plus IL-4 (open histograms), and cells were immunostained with phycoerythrin-conjugated anti-murine CD23. The results shown are representative of those obtained from all analyzed clones.
Discussion
In this study, we examined the effect of SOCS-1, -2, and -3 expression on IL-4 signaling. We found that expression of SOCS-1, but not SOCS-2, resulted in inhibition of signaling. SOCS-1 was able to inhibit IL-4-induced activation of a Stat6 reporter in transient transfection, and IL-4-induced activation of Stat6-DNA binding activity, phosphorylation of JAK1 and Stat6, activation of JAK1 kinase activity, and activation of gene transcription, in stable cell lines. Furthermore, we found that transient overexpression of SOCS-3 was capable of inhibiting IL-4-induced gene transcription, but that stable expression of SOCS-3 had no detectable effect on IL-4 signaling.
The role of SOCS-1 in regulating IL-4 function in vivo remains to be determined. A number of cytokines that signal through JAKs, including IL-4, have been shown to induce bone marrow expression of SOCS-1 RNA within 1 h of cytokine treatment (9). However, the activation of JAK kinases in response to IL-4 occurs within minutes of exposure to cytokine. As SOCS-1 expression is undetectable in unstimulated bone marrow, it is unlikely that SOCS-1 regulates the initiation of IL-4 signaling in these cells. It is more likely that, after SOCS-1 expression is induced by IL-4, it can serve to regulate the amplitude or the duration of signaling in a cell. Recently, mice deficient in SOCS-1 have been generated (21, 22), and thymocytes from these mice exhibit enhanced proliferation and prolonged duration of Stat6 phosphorylation in response to IL-4 (21). It is possible that induction of SOCS-1 gene expression functions to alter the future responsiveness of a cell to cytokine after it has received an initial signal, in essence establishing a memory of past signaling events in the cell. Alternately, SOCS-1 may regulate the activity of kinases or other signaling molecules further downstream in IL-4 signaling than JAKs. The nature of the in vivo role of SOCS-1 in regulation of cytokine signaling must await a more detailed analysis of the mechanism of SOCS-1 action and the regulation of its expression and activity.
Several observations have suggested that SOCS-3, like SOCS-1, may be a JAK kinase inhibitor and, moreover, that SOCS-3 may preferentially inhibit JAK2 function. SOCS-3 has been shown to interact with and inhibit the activity of JAK2 (12), and to inhibit signaling by erythropoetin and growth hormone (12, 23), which utilize JAK2, but not signaling by IFN-β (24), which utilizes JAK1 and Tyk2. In addition, growth hormone preferentially induces SOCS-3 expression in mouse liver (23). We have demonstrated that overexpression of SOCS-3 can inhibit the IL-4-induced activation of a Stat6 reporter in transient transfection. As IL-4 signals through JAK1 and JAK3, and does not activate JAK2, this demonstrates that the suppressive effect of SOCS-3 seen in transient transfections is not a JAK2-specific phenomenon. However, we did not see any effect of SOCS-3 expression on IL-4 responsiveness in stable transfectants. This was not due to lack of expression of SOCS-3. In fact, levels of SOCS-3 protein were considerably greater than levels of SOCS-1 protein in the stable clones, as measured by Western blotting (data not shown). SOCS-3 has a greater degree of homology to SOCS-1 than to SOCS-2 (13). It is possible that the suppressive effect of SOCS-3 detected in the transient assays may be a consequence of high levels of protein expression obtained in transient transfections and that, although well-expressed in the stable clones, SOCS-3 levels were not sufficient in the clones to inhibit JAK activation.
The JAK kinases, and the molecules that regulate their activity, are central to normal development and immune function, and aberrant control of JAK-STAT signaling has been implicated in a number of pathological states. Defining the mechanisms by which cells regulate JAK kinase activity is therefore of paramount importance in understanding how and why regulation fails and in understanding resulting pathology.
Acknowledgments
We thank Dr. Robert Coffman for recombinant murine IL-4, Dr. Satwant Narula for recombinant human IL-4, and Nika Danial for critical reading of the manuscript.
Footnotes
This work was supported by The Leukemia Society of America and funded by National Institutes of Health Grant R01-AI-33450.
Abbreviations used in this paper: JAK, Janus kinase; SOCS, suppressor of cytokine signaling; SSI, STAT-induced STAT inhibitor; JAB, JAK-binding protein; EMSA, electrophoretic mobility shift assay; IP, immunoprecipitation.
References
- 1.Nelms K, Huang H, Ryan J, Keegan A, Paul WE. Interleukin-4 receptor signaling mechanisms and their biological significance. Adv Exp Med Biol. 1998;452:37. doi: 10.1007/978-1-4615-5355-7_5. [DOI] [PubMed] [Google Scholar]
- 2.Chen XH, Patel BK, Wang LM, Frankel M, Ellmore N, Flavell RA, LaRochelle WJ, Pierce JH. Jak1 expression is required for mediating interleukin-4-induced tyrosine phosphorylation of insulin receptor substrate and Stat6 signaling molecules. J Biol Chem. 1997;272:6556. doi: 10.1074/jbc.272.10.6556. [DOI] [PubMed] [Google Scholar]
- 3.Reichel M, Nelson BH, Greenberg PD, Rothman PB. The IL-4 receptor α-chain cytoplasmic domain is sufficient for activation of JAK-1 and STAT6 and the induction of IL-4-specific gene expression. J Immunol. 1997;158:5860. [PubMed] [Google Scholar]
- 4.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 6.Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C, Quelle FW, Nosaka T, Vignali DA, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 7.Linehan LA, Warren WD, Thompson PA, Grusby MJ, Berton MT. STAT6 is required for IL-4-induced germline Ig gene transcription and switch recombination. J Immunol. 1998;161:302. [PubMed] [Google Scholar]
- 8.Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 9.Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 10.Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S, Kishimoto T. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- 11.Minamoto S, Ikegame K, Ueno K, Narazaki M, Naka T, Yamamoto H, Matsumoto T, Saito H, Hosoe S, Kishimoto T. Cloning and functional analysis of new members of STAT induced STAT inhibitor (SSI) family: SSI-2 and SSI-3. Biochem Biophys Res Commun. 1997;237:79. doi: 10.1006/bbrc.1997.7080. [DOI] [PubMed] [Google Scholar]
- 12.Masuhara M, Sakamoto H, Matsumoto A, Suzuki R, Yasukawa H, Mitsui K, Wakioka T, Tanimura S, Sasaki A, Misawa H, et al. Cloning and characterization of novel CIS family genes. Biochem Biophys Res Commun. 1997;239:439. doi: 10.1006/bbrc.1997.7484. [DOI] [PubMed] [Google Scholar]
- 13.Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, Nicola NA. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci USA. 1998;95:114. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu B, Reichel M, Fisher DA, Smith JF, Rothman P. Identification of a STAT6 domain required for IL-4-induced activation of transcription. J Immunol. 1997;159:1255. [PubMed] [Google Scholar]
- 15.Gauss G, Lieber M. DEAE-dextran enhances electroporation of mammalian cells. Nucleic Acids Res. 1992;20:6739. doi: 10.1093/nar/20.24.6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danial NN, Pernis A, Rothman PB. Jak-STAT signaling induced by the v-abl oncogene. Science. 1995;269:1875. doi: 10.1126/science.7569929. [DOI] [PubMed] [Google Scholar]
- 17.Berton MT, Linehan LA. IL-4 activates a latent DNA-binding factor that binds a shared IFN-γ and IL-4 response element present in the germ-line γ 1 Ig promoter. J Immunol. 1995;154:4513. [PubMed] [Google Scholar]
- 18.Wen Z, Darnell JE., Jr Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997;25:2062. doi: 10.1093/nar/25.11.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508. doi: 10.1128/mcb.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Blenis J, Li HC, Schindler C, Chen-Kiang S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990. doi: 10.1126/science.7701321. [DOI] [PubMed] [Google Scholar]
- 21.Naka T, Matsumoto T, Narazaki M, Fujimoto M, Morita Y, Ohsawa Y, Saito H, Nagasawa T, Uchiyama Y, Kishimoto T. Accelerated apoptosis of lymphocytes by augmented induction of bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proc Natl Acad Sci USA. 1998;95:15577. doi: 10.1073/pnas.95.26.15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, Hilton DJ, Alexander WS. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci USA. 1998;95:14395. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams TE, Hansen JA, Starr R, Nicola NA, Hilton DJ, Billestrup N. Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J Biol Chem. 1998;273:1285. doi: 10.1074/jbc.273.3.1285. [DOI] [PubMed] [Google Scholar]
- 24.Sakamoto H, Yasukawa H, Masuhara M, Tanimura S, Sasaki A, Yuge K, Ohtsubo M, Ohtsuka A, Fujita T, Ohta T, et al. A Janus kinase inhibitor, JAB, is an interferon-γ-inducible gene and confers resistance to interferons. Blood. 1998;92:1668. [PubMed] [Google Scholar]