

Abstract
Recent genome-wide association studies (GWASs) have identified common variants at 16 autosomal regions influencing the risk of developing colorectal cancer (CRC). To decipher the genetic basis of the association signals at these loci, we performed a meta-analysis of data from five GWASs, totalling 5626 cases and 7817 controls, using imputation to recover un-typed genotypes. To enhance our ability to discover low-frequency risk variants, in addition to using 1000 Genomes Project data as a reference panel, we made use of high-coverage sequencing data on 253 individuals, 199 with early-onset familial CRC. For 13 of the regions, it was possible to refine the association signal identifying a smaller region of interest likely to harbour the functional variant. Our analysis did not provide evidence that any of the associations at the 16 loci being a consequence of synthetic associations rather than linkage disequilibrium with a common risk variant.
INTRODUCTION
Many colorectal cancers (CRC) develop in genetically susceptible individuals, most of whom are not carriers of germline mismatch-repair or APC mutations (1–3). Recent genome-wide association studies (GWASs) have validated the hypothesis that part of the heritable risk of CRC is attributable to common variation identifying susceptibility loci at 1q41, 3q26,2, 6p21.2, 8q23.3, 8q24.21, 10p14, 11q13.4, 11q23.1, 12q13, 14q22.2, 15q13.3, 16q22.1, 18q21.1, 19q13.11, 20p12.3, 20q13.33 and Xp22.2 (4–11).
While the associations identified by GWAS provide novel insights, for example, into the development of CRC highlighting the role of TGF-β signalling in disease aetiology, since the tag single nucleotide polymorphisms (tagSNPs) genotyped are generally not strong candidates for causality elucidating the functional basis of associations is challenging.
Fine-mapping of disease loci has traditionally been undertaken using a combination of re-sequencing and direct typing of SNPs within regions of association. This strategy is, however, costly and time consuming and the ability to impute unobserved genotypes in GWAS data sets using a reference panel provides an attractive and practical alternative. The fidelity of imputation is dependent in part on the extent to which all variants are catalogued within reference panels and the quality of these data.
It has recently been proposed that many GWAS signals are a consequence of ‘synthetic associations’ resulting from the combined effect of one or more rare causal variants rather than simply linkage disequilibrium (LD) with a common risk variant (12). Under such a scenario many risk variants will have carrier frequencies below the threshold of representation in sequencing of population-based reference panels. To maximize the utility of imputation as a means of fine-mapping CRC loci, it is therefore highly desirable to also use high-coverage sequencing data on CRC cases to ensure adequate representation of risk variants.
To decipher the allelic structure underscoring the CRC associations at the 16 autosomal GWAS loci, we performed a meta-analysis of data from five GWASs. We excluded the Xp22.2 locus from the analysis due to the low density of GWAS SNPs on the X chromosome. To ensure recovery of all variants contributing to CRC risk at these loci through imputation in addition to utilizing 1000 Genomes Project data (13) as a reference panel, we made use high-coverage sequencing data on 253 individuals, 199 of whom had familial CRC.
RESULTS
Subjects and definition of genomic regions
We studied five non-overlapping case–control series of Northern European ancestry, which post-QC provided GWAS data on 5626 CRC cases and 7817 controls (Supplementary Material, Table S1). We used Haploview to define the haplotype blocks and recombination hotspots containing the tagSNPs previously found to be associated with CRC risk at 1q41, 3q26,2, 6p21.2, 8q23.3, 8q24.21, 10p14, 11q13.4, 11q23.1, 12q13, 14q22.2, 15q13.3, 16q22.1, 18q21.1, 19q13.11, 20p12.3 and 20q13.33. We did not include the Xp22.2 locus in these analyses due to the low density of GWAS SNPs on the X chromosome and hence the difficulty involved in imputation. To include the possibility of long-range synthetic associations, we imputed the regions defined by at least 1 Mb region surrounding the tagSNP associated with CRC risk at each of the 16 loci. Collectively, the 16 CRC risk loci were captured by 16.2 Mb region of the genome.
Imputation panels
The 1000 genomes Phase I Interim reference panel based on low-coverage (4–6x) sequencing of 1094 individuals from Africa (AFR; n = 246), Asia (ASN; n = 286), Europe (EUR; n = 381) and the Americas (AMR; n = 181) catalogued 203 047 SNPs mapping to the 16.2 Mb region. A total of 92 095 SNPs were monomorphic in all five GWASs. In total, 46 829 of all variants mapping to the 16 regions had frequencies ≥1%, 4658 (10%) of which were not referenced in dbSNP132.
In addition to using 1000 genomes data, we made use of deep sequencing (>30×) data generated on 253 individuals, 199 of whom had been diagnosed with early-onset CRC (henceforth referred to as the CG panel). Depth of sequencing coverage in the CG panel was high across each of the target regions, 48x–58x (Fig. 1). Concordance between Illumina OmniExpress genotype and sequencing data in 84 samples was >99%; 139 668 SNPs and 16 173 indels and substitutions were catalogued within the 16.2 Mb region. Of these, 96 195 were also present in the 1000 genomes panel, and a further 11 653 were monomorphic in the five GWASs. In total, 44 478 of all variants mapping to the 16 regions of association had frequencies ≥1%, 4859 (11%) of which were had not been catalogued by dbSNP132.
Figure 1.

Coverage distribution within each target region. The depth of coverage is calculated based on the number of reads that mapped to that position and is averaged over 100 kb windows. The x-axis denotes the relative position from the start of the target region.
Comparison of the 1000 genomes and CG panel
Figure 2 shows the number and minor allele frequency (MAF) distribution of variants in the 1000 genomes and CG panels. Perhaps not surprisingly a disproportionate number of the variants specific to each panel had MAFs <1% representing private and/or population-specific variants with low frequency in the Northern and Western European (CEU) population. Of the variants specific to the CG panel having MAFs >1, 83.2% were indels. Filtering, requiring an information (INFO) score of ≥0.4, Phet ≥ 0.01 and MAF ≥ 1% excluded 77.9 and 72.9% of variants from the 1000 genomes and CG panel, respectively, resulting in an average of 2814 and 2730 variants successfully imputed per Mb. Thus, through imputation we facilitated a >20-fold increase in the number of variants that could be evaluated for association. Approximately 86% of these variants were shared by both reference panels. The average MAF of variants in the unfiltered 1000 genomes and CG panel were 0.040 and 0.049, respectively. After filtering, the average MAFs of these successfully imputed variants increased to 0.175 and 0.170, respectively. The accuracy of the two reference panels was compared using whole-genome sequencing data on six samples. The genotypes at GWAS tagSNP positions were used to impute these samples and the resulting genotypes compared with the sequenced base calls. This analysis revealed very similar results with 94.9 and 88.6% of heterozygotes being correctly imputed in the imputations with and without the CG panel, respectively.
Figure 2.
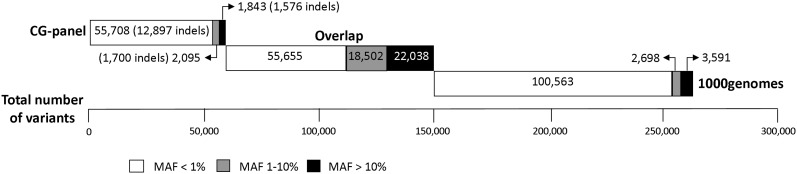
The number of variants specific to 1000 genomes and to the CG-panel reference panels as well as those that are in common between the two panels according to MAF.
Analysis of individual CRC loci
Table 1 shows the number of SNPs directly typed and successfully imputed (INFO score ≥0.4, Phet ≥ 0.01 and MAF ≥ 1%) at each of the 16 loci. Table 2 shows for each region the tagSNP and the most associated SNP along with respective pair wise LD metrics. Details of the 10 most highly associated variants identified in imputation with and without the CG panel are detailed in Supplementary Material, Table S2 (additional data is available at http://tinyurl.com/whiffinetal2013). Regional plots of association results and recombination rates for all 16 regions imputed with the CG panel can be found in Supplementary Material, Figure S1.
Table 1.
The 16 autosomal regions associated with CRC
| GWAS tagSNP | Region | Position | Start | End | Typed SNPs | Successfully imputed variants |
||
|---|---|---|---|---|---|---|---|---|
| 1000 genomes | CG panel | Overlap | ||||||
| rs6691170 | 1q41 | 222 045 446 | 221 300 000 | 222 300 000 | 98 | 3857 | 3803 | 3499 |
| rs10936599 | 3q26.2 | 169 492 101 | 169 000 000 | 170 000 000 | 106 | 2509 | 2439 | 2228 |
| rs1321311 | 6p21.2 | 36 622 900 | 36 100 000 | 37 100 000 | 270 | 2931 | 2993 | 2646 |
| rs16892766 | 8q23.3 | 117 630 683 | 117 100 000 | 118 100 000 | 98 | 2013 | 2002 | 1784 |
| rs6983267 | 8q24.21 | 128 413 305 | 127 900 000 | 128 900 000 | 162 | 3262 | 3136 | 2876 |
| rs10795668 | 10p14 | 8 701 219 | 8 200 000 | 9 200 000 | 189 | 3708 | 3560 | 3285 |
| rs3824999 | 11q13.4 | 74 345 550 | 73 850 000 | 74 850 000 | 94 | 2449 | 2452 | 2233 |
| rs3802842 | 11q23.1 | 111 171 709 | 110 700 000 | 111 700 000 | 76 | 2443 | 2368 | 2130 |
| rs11169552 | 12q13 | 51 155 663 | 50 200 000 | 51 400 000 | 73 | 2199 | 2148 | 1905 |
| rs4444235 | 14q22.2 | 54 410 919 | 54 000 000 | 55 000 000 | 109 | 2282 | 2213 | 2003 |
| rs4779584 | 15q13.3 | 32 994 756 | 32 500 000 | 33 500 000 | 145 | 2411 | 2446 | 2173 |
| rs9929218 | 16q22.1 | 68 820 946 | 68 300 000 | 69 300 000 | 60 | 2325 | 2239 | 2072 |
| rs4939827 | 18q21.1 | 46 453 463 | 46 000 000 | 47 000 000 | 141 | 2882 | 2741 | 2455 |
| rs10411210 | 19q13.11 | 33 532 300 | 33 000 000 | 34 000 000 | 126 | 3730 | 3358 | 3045 |
| rs961253 | 20p12.3 | 6 404 281 | 6 050 000 | 7 050 000 | 160 | 2168 | 2816 | 1950 |
| rs4813802 | 6 699 595 | |||||||
| rs4925386 | 20q13.33 | 60 921 044 | 59 400 000 | 61 400 000 | 123 | 3245 | 2416 | 2224 |
Displayed is the GWAS tagSNP, the imputed region and the numbers of SNPs directly typed and successfully imputed (MAF ≥ 0.01, Phet ≥ 0.01, INFO ≥ 0.4) in each individual panel and those shared between panels.
Table 2.
The original GWAS tagSNP and the most associated variant after imputation with 1000 genomes and CG panel together with pair wise LD metrics (r2, D′)
| tagSNP |
top SNP |
R2 | D′ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Position | OR | P-value | MAF | SNP | Position | OR | P-value | MAF | |||
| 1q41 | rs6691170 | 222 045 446 | 1.08 | 2.39E−03 | 0.397 | rs11118883 | 222 061 022 | 1.12 | 1.18E−04 | 0.348 | 0.393 | 0.719 |
| 3q26 | rs10936599 | 169 492 101 | 1.12 | 2.20E−04 | 0.253 | Indel | 169 583 245 | 1.10 | 9.79E−05 | 0.250 | 0.906a | 1.000a |
| 6p21 | rs1321311 | 36 622 900 | 1.08 | 4.88E−03 | 0.205 | rs9918353 | 36 622 677 | 1.08 | 3.13E−03 | 0.205 | 0.985 | 0.995 |
| 8q23 | rs16892766 | 117 630 683 | 1.24 | 7.84E−07 | 0.092 | rs140355816 | 117 574 515 | 1.61 | 2.58E−09 | 0.017 | 0.092 | 0.724 |
| 8q24 | rs6983267 | 128 413 305 | 1.20 | 1.64E−12 | 0.480 | rs7013278 | 128 414 892 | 1.20 | 1.63E−12 | 0.343 | 0.444 | 1.000 |
| 10p14 | rs10795668 | 8 701 219 | 1.17 | 3.97E−09 | 0.327 | rs11255841 | 8 739 580 | 1.19 | 7.73E−11 | 0.321 | 0.850 | 0.931 |
| 11q13 | rs3824999 | 74 345 550 | 1.16 | 2.57E−09 | 0.471 | rs11604752 | 74 277 144 | 1.16 | 7.29E−10 | 0.476 | 0.919 | 0.973 |
| 11q23 | rs3802842 | 111 171 709 | 1.15 | 4.48E−06 | 0.269 | rs11213801 | 111 119 694 | 1.15 | 1.88E−07 | 0.276 | 0.317 | 0.794 |
| 12q13 | rs11169552 | 51 155 663 | 1.12 | 9.05E−05 | 0.247 | rs61928263 | 50 588 659 | 1.17 | 4.42E−08 | 0.223 | 0.022 | 0.775 |
| 14q22 | rs4444235 | 54 410 919 | 1.10 | 3.34E−04 | 0.483 | — | — | — | — | — | 1.000 | 1.000 |
| 15q13 | rs4779584 | 32 994 756 | 1.21 | 7.28E−09 | 0.191 | rs1406389 | 33 009 478 | 1.24 | 7.57E−12 | 0.222 | 0.269 | 0.652 |
| 16q22 | rs9929218 | 68 820 946 | 1.09 | 5.75E−04 | 0.286 | rs146639854 | 68 429 091 | 1.36 | 8.30E−05 | 0.020 | 0.009 | 1.000 |
| 18q21 | rs4939827 | 46 453 463 | 1.24 | 7.29E−16 | 0.471 | Indel | 46 451 805 | 1.25 | 2.08E−17 | 0.421 | 0.568a | 0.963a |
| 19q13 | rs10411210 | 33 532 300 | 1.25 | 2.11E−06 | 0.096 | rs79812655 | 33 517 923 | 1.23 | 1.27E−07 | 0.131 | 0.419 | 0.717 |
| 20p12 | rs961253 | 6 404 281 | 1.10 | 2.66E−04 | 0.367 | rs990999 | 6 692 529 | 1.13 | 6.49E−06 | 0.377 | 0.001 | 0.028 |
| 20q13 | rs4925386 | 60 921 044 | 1.16 | 8.31E−07 | 0.318 | rs2236202 | 60 985 164 | 1.22 | 2.02E−11 | 0.249 | 0.214 | 0.591 |
aFor indels, the next most associated SNP was used to derive LD statistics.
In 13 of the 16 regions, imputation provided refinement of the association signal identifying a region of interest narrower that the original LD block likely to harbour the functional variant. However, for three loci, 6p21, 12q13 and 16q22.1, the LD structure is large and complex and prohibited a smaller region of association being delineated. In addition, in 10 of the 16 regions, the most associated SNP in the imputation was greater than an order of magnitude more strongly associated with CRC.
In 4 of the 16 regions, 1q41, 15q13.3, 18q21.1 and 20q13.33 imputation results were consistent with and without the CG panel and a variant significantly more associated (P-value over an order of magnitude lower) with CRC than the original tagSNP was uncovered (Supplementary Material, Table S2). In all regions, in silico functional annotation of the most associated variant, using publically available data from ENCODE, revealed that they reside within potential regulatory regions of DNA.
At 1q41, the common imputed SNP rs11118883 was an order of magnitude more strongly associated with CRC compared with the original tagSNP (rs6691170). rs11118883 which is in LD with rs6691170 (r2 = 0.40, D′ = 0.75) localizes 106 bps telomeric to large predicted transcription factor binding site in a region of DNA marked by H3K4Me and H3K27Ac epigenetic markers of enhancer regions.
At 15q13.3, the common imputed SNP rs1406389 is over two orders of magnitude more strongly associated with CRC than rs4779584, the original tagSNP. rs1406389 is 726 bps upstream of GREM1 within a DNaseI hypersensitive site and a binding site for SUZ12 predicted by ChIP-seq, a protein implicated in DNA methylation. It is also within a putative transcript for AX747968 and is in LD with rs4779584 (r2 = 0.67, D′ = 0.82).
At 18q21.1, the common indel, rs4939825, is over an order of magnitude more strongly associated with CRC than rs4929827 when using imputation with the CG panel. The most associated variant in the 1000 genomes imputation, rs4939567 is only 67 bps away within the same predicted MafK-binding site intronic in SMAD7.
At 20q13.33, the common imputed SNPs rs1741640 and rs2236202 provided for four orders of magnitude stronger association than that provided by the original tagSNP, rs4925386. rs1741640 is the most highly associated variant in the imputation with just the 1000 genomes panel and rs2236202 is most highly associated when the CG panel is also used. Both SNPs, which are highly correlated (r2 = 0.56; D′ = 0.76), reside within large predicted ChIP-seq sites in regions of DNA that are hypersensitive to DNaseI and have epigenetic markers of regulatory regions.
In all of these four regions, the most associated variants are common with MAFs similar to that of the original tagSNP. In three regions, 6p21, 8q23 and 16q22, a rare SNP was most associated with CRC in one or both of the imputations. However, in each case, the results were inconsistent across the imputations and/or there was evidence of heterogeneity between studies. Moreover, in the 6p21 and 16q22 regions the strength of association was substantially lower than genome-wide significance.
Conditioning on the best SNP in each region revealed potential second independent hits at 1q41, 14q22.2 and 20p12.13 marked by rs74144285 (Pmeta = 2.91 × 10−4; Pcond = 7.27 × 10−4), rs743275 (Pmeta = 3.66 × 10−4; Pcond = 1.23 × 10−3) and rs6117251 (Pmeta = 1.29 × 10−5; Pcond = 1.25 × 10−5), respectively; consistent with earlier observations for 14q22.2 and 20p12.13 (5). Haplotype analysis did not provide any evidence that associations were not fully captured by individual SNP associations (Supplementary Material, Table S3).
DISCUSSION
Characterizing all genetic variation within each region of association, as we have performed here, is a critical first step in deciphering the allelic architecture underscoring GWAS risk loci. The advantages of characterizing all variants prior to large-scale fine-mapping studies are that the correlations among all genetic variants will be known, which will allow for rapid nomination of specific variants or smaller regions for functional studies.
Imputation of candidate regions has several advantages namely, the increase in power afforded through combination of multiple GWAS which have been carried out on different arrays and the increased density of markers that can be tested for an association. The imputation increased the number of markers by >20-fold leading to an average marker density of ∼3 SNPs/kb. This is in contrast to the 1 SNP/kb density achieved when imputing with the Pilot 1000 Genomes (Jun 2010) and HapMap3 (Feb 2009) combined imputation panels (14) highlighting the value of updated reference panels.
While the 1000 genomes reference panel is based on a large number of samples, only one-third of these are European and the sequence data are low coverage leading to potential under-calling of rare variants and higher sequencing errors in addition to a high proportion of non-CEU variants. In contrast, our new CG panel is based on high-coverage sequence data of 253 samples, 208 of which are of European ancestry. Moreover, 199 are genetically enriched for CRC susceptibility by the virtue of having early-onset disease and a family history of CRC thereby enhancing the ability to recover rare disease causing variants. Furthermore, the high coverage of sequencing minimizes inaccuracy in calling low-frequency variants.
In 13 of the 16 regions, the imputation successfully refined the association signal identifying a smaller region of interest which is the most likely location of a causal variant and/or identifying a good functional candidate. In the remaining three regions, 6p21, 12q13 and 16q22.1, the LD is large and complex leading to many highly correlated variants across a large region making the signal hard to refine. In this study, while we have identified possible candidate variants, further work is required to determine the functional basis of associations.
In our study, we found no evidence to support the existence of ‘synthetic associations’ underscoring the currently identified autosomal GWAS signals for CRC. At 13 of the 16 loci, the variants identified as most associated with CRC in both imputations had MAFs > 10%. At 6p21.2 and 16q22.1, the variant identified in the 1000 genomes and CG-panel imputations, respectively, had MAF < 0.05; however, the P-values of association across the region are low increasing the likelihood of a spurious association. At 8q23.3, the variants identified in both imputations are novel and rare (MAF < 0.05); although these SNPs passed our Phet threshold, there is some evidence that these ‘top’ SNPs are pulled by one study. Although GWAS tagSNPs are unlikely themselves to be functional, they appear much more likely to tag a functional variants of a similar frequency than single or multiple rare causal variants. Here, we have relied on imputation to recover untyped genotypes. As rare variants can be poorly imputed in GWAS, there remains the possibility that low-frequency variants conferring moderate risks might have been missed because of this strategy. To address this possibility, we performed an analysis of haplotypes finding no evidence that any of the association signals at these 16 loci were the result of cryptic rare variants. While inflammatory bowel disease provides support for the existence of ‘synthetic associations’ (12), most of the ‘evidence’ for such a model of disease association comes from simulation studies (12,15–18). Indeed, if such a genetic model was present, such associations would be highly tractable by linkage analysis. No putative linkage signals have, however, been identified in these regions. Moreover, the reproducibility of many GWAS associations across different populations argues against rare variants as a common cause of GWAS signals (19).
Our analysis suggests that rare variants conferring larger risks do not underlie GWAS CRC signals and therefore leaves open the question of where the missing heritability lies. One possibility is that the contribution of common variants has been underestimated and studies with increased power will discover more associated common variants with gradually decreasing effect sizes. This hypothesis is supported by the success of large-scale meta-analysis studies. Although we believe that rare variation does not underlie regions of CRC association identified through GWASs, this is not to say that such variation does not contribute to CRC risk. Current exome and whole-genome sequencing projects will be better powered to detect such variation.
In summary, we have extensively characterized all genetic variation across 16 regions that have been reported to be associated with CRC. In addition to our data providing insight into the allelic architecture of these association signals our study findings provide a resource informing functional analyses aimed at defining the biological basis of risk loci.
MATERIALS AND METHODS
Subjects and data sets
We used GWAS data from five non-overlapping case–control series of Northern European ancestry, which have been previously reported (Supplementary Material, Table S1) (8,20,21). Briefly: UK1: 890 familial colorectal tumour cases and 900 cancer-free controls with self-reported European ancestry from the COloRectal Gene Identification (CORGI) consortium (8); Scotland1: 972 early-onset CRC cases (aged <55 years at diagnosis) and 998 population controls (8); VQ58: 1794 UK cases with stage B/C CRC from the VICTOR (http://www.octo-oxford.org.uk/alltrials/infollowup/vic.html) and QUASAR2 (http://www.octo-oxford.org.uk/alltrials/trials/q2.html) trials, together with publicly available data from 2686 population controls from the UK 1958 Birth Cohort (VQ58) (20); CCFR1: 1175 familial CRC cases and 999 controls from the Colon Cancer Family Registry (CCFR) (http://epi.grants.cancer.gov/CFR/about_colon.html); CCFR2: 795 CRC cases from CCFR and 2234 controls from the Cancer Genetic Markers of Susceptibility studies of breast and prostate cancer (21).
Genotyping
The GWAS samples were genotyped using proprietary Illumina SNP arrays: UK1 on Hap550; Scotland1 on Hap300 + Hap240S; 1958 Birth Cohort on Hap1M and VQ on Hap300, Hap370 or Hap660; CCFR1 and CCFR2 samples using Hap1M, Hap1M-Duo or Omni-express arrays (Supplementary Material, Table S1); general genotyping quality control assessment was as previously described and all SNPs presented in this study passed the required thresholds (11). Duplicate samples were used to monitor genotyping quality. We excluded SNPs from analysis with GenCall scores <0.25; overall call rates <95%; minor allele frequency <0.005; departure from Hardy–Weinberg equilibrium in controls at P < 10−4 or in cases at P < 10−6. We excluded individuals if they failed one or more of the following thresholds: duplication or cryptic relatedness to estimated identity by descent >6.25%, overall successfully genotyped SNPs < 95%, mismatch between predicted and reported gender, outliers in a plot of heterozygosity versus missingness and evidence of non-white European ancestry by principle components analysis-based analysis in comparison with HapMap samples (http://hapmap.ncbi.nlm.nih.gov).
The adequacy of the case–control matching and possibility of differential genotyping of cases and controls was assessed using Q–Q plots of test statistics. λGC values (22) for UK1, Scotland1, VQ58, CFR1 and CFR2 studies were 1.02, 1.01, 1.01, 1.01 and 1.03, respectively, thereby excluding significant differential genotyping or cryptic population substructure.
Imputation reference panels
To impute un-typed genotypes in cases and controls, we made use of two reference panels. First, 1000 genomes Phase I interim data (https://mathgen.stats.ox.ac.uk/impute/impute_v2.html). Secondly, high-coverage sequencing data generated on 199 CRC cases with European ancestry and publically accessible data on 54 healthy unrelated individuals of mixed ancestry (http://media.completegenomics.com/documents/PublicGenomes.pdf) referred to as the CG panel. The CRC cases had been ascertained through CORGI and had been diagnosed with CRC before age 55 and had at least one first-degree relative affected with CRC. Sequencing of these 253 individuals was carried out using unchained combinatorial probe anchor ligation chemistry on arrays of self-assembling DNA nanoballs (23). To identify sequence variation in each sample, paired end reads were aligned to the Human Genome NCBI (National Centre for Biotechnology Information) Build 37. Variation between reference genome and each sample was called and scored using a local de novo assembly algorithm. Phasing of genotypes was performed using the enhanced hidden Markov model chain program SHAPEIT (24).
Statistical and bioinformatic analysis
Analyses were primarily undertaken using R (v2.14.2), STATA v.10 (College Station, TX 77845, USA) and PLINK (v1.07) software. Association statistics, using an additive model, were obtained with SNPtest (https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html). Prediction of the un-typed SNPs was carried out with IMPUTEv2.1.0 (25). All analyses were run twice, once with the 1000 genomes panel only and secondly using the CG panel along with the 1000 genomes. Meta-analyses only included markers with imputed call rates/SNP > 0.9 in all five studies and control MAFs > 0.01. SNPs that significantly (P < 0.001) failed to meet fitness for Hardy–Weinberg proportion in any of the studies were excluded from subsequent analyses (0.6%). Imputed data were analysed using SNPTESTv2.3.0 to account for uncertainties in SNP prediction, and meta-analysis was performed using METAv1.4 with a threshold of 0.4. To filter poorly imputed SNPs, as previously recommended, we excluded variants having overall INFO scores from SNPTESTv2.3.0 of <0.4 (26). Conditional association tests were carried out using PLINK (v1.07) and Haplotype analysis using Haploview (v4.2).
LD metrics were calculated from 1000 genomes pilot release I data and viewed using SNAP (27). Where SNPs had not been catalogued, LD metrics were calculated using in house Perl scripts using the CG-panel data. Regional association plots of LD metrics were then plotted using SNAP. LD blocks were defined on the basis of HapMap recombination rate and were viewed using the Haploview software (v4.2).
To gain insight into the biological basis of associations at each locus, we used the program Mechanize (http://search.cpan.org/dist/WWW-Mechanize/) to perform a comprehensive examination of genomic features associated with variants showing association signals equal to or stronger than that of the region's original tagSNP. Specifically, Mechanize was used to interrogate known genes, COSMIC sites, H3K27Ac, H3K4Me, H3K4Me3, ENCODE ChIP-seq sites, CpG islands, PhastCons conservation, DNaseI hypersensitivity, HapMap SNPs, Vista Enhancers and Affy Exon probe data. We made use of ENCODE data on all available cell types.
WEB ADDRESSES
The R suite can be found at http://www.r-project.org/.
Detailed information on the tagSNP panels can be found at http://www.illumina.com/.
HapMap: http://www.hapmap.org/.
1000 Genomes: http://www.1000genomes.org/.
IMPUTE: https://mathgen.stats.ox.ac.uk/impute/impute.html.
SNPTEST: https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html.
Mechanize: http://search.cpan.org/dist/WWW-Mechanize/.
SNAP: http://www.broadinstitute.org/mpg/snap/.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
In Oxford, additional funding was provided by the Oxford Comprehensive Biomedical Research Centre (C.P. and I.P.T.) and the European Union Framework Programme 7 (FP7) CHIBCHA grant (A.M.J. and I.P.T.). Core infrastructure support to the Wellcome Trust Centre for Human Genetics, Oxford, was provided by grant (090532/Z/09/Z).
In Edinburgh, funding was provided by a Cancer Research UK Programme Grant (C348/A12076) and a Centre Grant from the CORE Charity. Lothian Birth Cohort Illumina genotyping was supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC). Phenotype collection in the Lothian Birth Cohort 1921 was supported by the BBSRC, The Royal Society and The Chief Scientist Office of the Scottish Government. Phenotype collection in the Lothian Birth Cohort 1936 was supported by Research into Ageing (continues as part of Age UK's The Disconnected Mind project). The work on the Lothian Birth Cohorts was undertaken at the University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology, part of the cross-council Lifelong Health and Wellbeing Initiative (G0700704/84698). Funding from the BBSRC, EPSRC, ESRC and MRC is gratefully acknowledged.
Research was also funded by the European Union FP7 (FP7/207-2013) under grant 258236, FP7 collaborative project SYSCOL.
This work of the Colon Cancer Family Registry (CFR) was supported by the US National Cancer Institute, National Institutes of Health (CA-95-011), and through cooperative agreements with members of the Colon CFR and Principal Investigators. Collaborating centres include the Australasian Colorectal Cancer Family Registry (U01 CA097735), the Familial Colorectal Neoplasia Collaborative Group (U01 CA074799), the Ontario Registry for Studies of Familial Colorectal Cancer (U01 CA074783) and the Seattle Colorectal Cancer Family Registry (U01 CA074794). The Colon CFR GWAS was supported by funding from the US National Cancer Institute, National Institutes of Health (U01CA122839 to G.C.).
Supplementary Material
ACKNOWLEDGEMENTS
Cancer Research UK provided principal funding for this study individually to R.S.H. (C1298/A8362–Bobby Moore Fund for Cancer Research UK), I.P.T. and M.G.D. At the Institute of Cancer Research, additional funding was provided by a Centre Grant from Core as part of the Digestive Cancer Campaign, the National Cancer Research Network and the NHS via the Biological Research Centre of the NIHR at the Royal Marsden Hospital NHS Trust. N.W. received a PhD studentship from the Institute of Cancer Research. We are grateful to many colleagues within the UK Clinical Genetics Departments (for CORGI) and to many collaborators who participated in the VICTOR and QUASAR2 trials. We also thank colleagues from the UK National Cancer Research Network (for NSCCG).
This study made use of genotyping data from the 1958 Birth Cohort and NBS samples, kindly made available by the Wellcome Trust Case Control Consortium 2. A full list of the investigators who contributed to the generation of the data is available (see WEB ADDRESSES). Finally, we thank all individuals who participated in the study.
We gratefully acknowledge the work of M. Walker and S. Reid in technical support, R. Wilson (SOCCS3 and COGS study coordinator), G. Barr for data entry in SOCCS studies, the research nurse recruitment teams, the Wellcome Trust Clinical Research Facility for sample preparation and all surgeons, oncologists and pathologists throughout Scotland at contributing centres.
REFERENCES
- 1.Lichtenstein P., Holm N.V., Verkasalo P.K., Iliadou A., Kaprio J., Koskenvuo M., Pukkala E., Skytthe A., Hemminki K. Environmental and heritable factors in the causation of cancer – analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. doi:10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 2.Aaltonen L., Johns L., Jarvinen H., Mecklin J.P., Houlston R. Explaining the familial colorectal cancer risk associated with mismatch repair (MMR)-deficient and MMR-stable tumors. Clin. Cancer Res. 2007;13:356–361. doi: 10.1158/1078-0432.CCR-06-1256. doi:10.1158/1078-0432.CCR-06-1256. [DOI] [PubMed] [Google Scholar]
- 3.Lubbe S.J., Webb E.L., Chandler I.P., Houlston R.S. Implications of familial colorectal cancer risk profiles and microsatellite instability status. J. Clin. Oncol. 2009;27:2238–2244. doi: 10.1200/JCO.2008.20.3364. doi:10.1200/JCO.2008.20.3364. [DOI] [PubMed] [Google Scholar]
- 4.Tomlinson I.P., Webb E., Carvajal-Carmona L., Broderick P., Howarth K., Pittman A.M., Spain S., Lubbe S., Walther A., Sullivan K., et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat. Genet. 2008;40:623–630. doi: 10.1038/ng.111. doi:10.1038/ng.111. [DOI] [PubMed] [Google Scholar]
- 5.Tomlinson I.P., Carvajal-Carmona L.G., Dobbins S.E., Tenesa A., Jones A.M., Howarth K., Palles C., Broderick P., Jaeger E.E., Farrington S., et al. Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet. 2011;7:e1002105. doi: 10.1371/journal.pgen.1002105. doi:10.1371/journal.pgen.1002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tenesa A., Farrington S.M., Prendergast J.G., Porteous M.E., Walker M., Haq N., Barnetson R.A., Theodoratou E., Cetnarskyj R., Cartwright N., et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat. Genet. 2008;40:631–637. doi: 10.1038/ng.133. doi:10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houlston R.S., Webb E., Broderick P., Pittman A.M., Di Bernardo M.C., Lubbe S., Chandler I., Vijayakrishnan J., Sullivan K., Penegar S., et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat. Genet. 2008;40:1426–1435. doi: 10.1038/ng.262. doi:10.1038/ng.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houlston R.S., Cheadle J., Dobbins S.E., Tenesa A., Jones A.M., Howarth K., Spain S.L., Broderick P., Domingo E., Farrington S., et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat. Genet. 2010;42:973–977. doi: 10.1038/ng.670. doi:10.1038/ng.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broderick P., Carvajal-Carmona L., Pittman A.M., Webb E., Howarth K., Rowan A., Lubbe S., Spain S., Sullivan K., Fielding S., et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat. Genet. 2007;39:1315–1317. doi: 10.1038/ng.2007.18. doi:10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- 10.Jaeger E., Webb E., Howarth K., Carvajal-Carmona L., Rowan A., Broderick P., Walther A., Spain S., Pittman A., Kemp Z., et al. Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. Nat. Genet. 2008;40:26–28. doi: 10.1038/ng.2007.41. doi:10.1038/ng.2007.41. [DOI] [PubMed] [Google Scholar]
- 11.Dunlop M.G., Dobbins S.E., Farrington S.M., Jones A.M., Palles C., Whiffin N., Tenesa A., Spain S., Broderick P., Ooi L.Y., et al. Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat. Genet. 2012;44:770–776. doi: 10.1038/ng.2293. doi:10.1038/ng.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickson S.P., Wang K., Krantz I., Hakonarson H., Goldstein D.B. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. doi:10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. doi:10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvajal-Carmona L.G., Cazier J.B., Jones A.M., Howarth K., Broderick P., Pittman A., Dobbins S., Tenesa A., Farrington S., Prendergast J., et al. Fine-mapping of colorectal cancer susceptibility loci at 8q23.3, 16q22.1 and 19q13.11: refinement of association signals and use of in silico analysis to suggest functional variation and unexpected candidate target genes. Hum. Mol. Genet. 2011;20:2879–2888. doi: 10.1093/hmg/ddr190. doi:10.1093/hmg/ddr190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orozco G., Barrett J.C., Zeggini E. Synthetic associations in the context of genome-wide association scan signals. Hum. Mol. Genet. 2010;19:R137–R144. doi: 10.1093/hmg/ddq368. doi:10.1093/hmg/ddq368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang D., Keinan A. Predicting signatures of ‘synthetic associations’ and ‘natural associations’ from empirical patterns of human genetic variation. PLoS Comput. Biol. 2012;8:e1002600. doi: 10.1371/journal.pcbi.1002600. doi:10.1371/journal.pcbi.1002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kent J.W., Jr Rare variants, common markers: synthetic association and beyond. Genet. Epidemiol. 2011;35(Suppl. 1):S80–S84. doi: 10.1002/gepi.20655. doi:10.1002/gepi.20655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kent J.W., Jr, Farook V., Goring H.H., Dyer T.D., Almasy L., Duggirala R., Blangero J. Do rare variant genotypes predict common variant genotypes? BMC Proc. 2011;5(Suppl. 9):S87. doi: 10.1186/1753-6561-5-S9-S87. doi:10.1186/1753-6561-5-S9-S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marigorta U.M., Navarro A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet. 2013;9:e1003566. doi: 10.1371/journal.pgen.1003566. doi:10.1371/journal.pgen.1003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Power C., Elliott J. Cohort profile: 1958 British birth cohort (National Child Development Study) Int. J. Epidemiol. 2006;35:34–41. doi: 10.1093/ije/dyi183. doi:10.1093/ije/dyi183. [DOI] [PubMed] [Google Scholar]
- 21.Newcomb P.A., Baron J., Cotterchio M., Gallinger S., Grove J., Haile R., Hall D., Hopper J.L., Jass J., Le Marchand L., et al. Colon Cancer Family Registry: an international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol. Biomarkers Prev. 2007;16:2331–2343. doi: 10.1158/1055-9965.EPI-07-0648. doi:10.1158/1055-9965.EPI-07-0648. [DOI] [PubMed] [Google Scholar]
- 22.Clayton D.G., Walker N.M., Smyth D.J., Pask R., Cooper J.D., Maier L.M., Smink L.J., Lam A.C., Ovington N.R., Stevens H.E., et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat. Genet. 2005;37:1243–1246. doi: 10.1038/ng1653. doi:10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- 23.Drmanac R., Sparks A.B., Callow M.J., Halpern A.L., Burns N.L., Kermani B.G., Carnevali P., Nazarenko I., Nilsen G.B., Yeung G., et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. doi:10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 24.Delaneau O., Marchini J., Zagury J.F. A linear complexity phasing method for thousands of genomes. Nat. Methods. 2012;9:179–181. doi: 10.1038/nmeth.1785. doi:10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 25.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. doi:10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 26.Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. doi:10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 27.Johnson A.D., Handsaker R.E., Pulit S.L., Nizzari M.M., O'Donnell C.J., de Bakker P.I. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. doi:10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.