

Abstract
Inosine 5′-monophosphate dehydrogenase (IMPDH) catalyzes the first unique step of the GMP branch of the purine nucleotide biosynthetic pathway. This enzyme is found in organisms of all three kingdoms. IMPDH inhibitors have broad clinical applications in cancer treatment, as antiviral drugs and as immunosuppressants, and have also displayed antibiotic activity. We have determined three crystal structures of Bacillus anthracis IMPDH, in a phosphate ion-bound (termed “apo”) form and in complex with its substrate, inosine 5′-monophosphate (IMP), and product, xanthosine 5′-monophosphate (XMP). This is the first example of a bacterial IMPDH in more than one state from the same organism. Furthermore, for the first time for a prokaryotic enzyme, the entire active site flap, containing the conserved Arg-Tyr dyad, is clearly visible in the structure of the apoenzyme. Kinetic parameters for the enzymatic reaction were also determined, and the inhibitory effect of XMP and mycophenolic acid (MPA) has been studied. In addition, the inhibitory potential of two known Cryptosporidium parvum IMPDH inhibitors was examined for the B. anthracis enzyme and compared with those of three bacterial IMPDHs from Campylobacter jejuni, Clostridium perfringens, and Vibrio cholerae. The structures contribute to the characterization of the active site and design of inhibitors that specifically target B. anthracis and other microbial IMPDH enzymes.
Inosine 5′-monophosphate dehydrogenase (IMPDH, EC 1.1.1.205) catalyzes the oxidation of inosine 5′-mono-phosphate (IMP) to xanthosine 5′-monophosphate (XMP) with the concomitant reduction of NAD+ to NADH. The reaction is a branch point between the adenine and guanine nucleotide biosynthesis, and a rate-limiting step of GMP biosynthesis. As a regulator of the intracellular guanine nucleotide pool, IMPDH is crucial for DNA and RNA synthesis, signal transduction, and other processes involved in cell proliferation. Inhibition of IMPDH causes an overall reduction in the size of guanine nucleotide pools, and as GTP is a cofactor in the conversion of IMP to AMP, adenylate pools are also affected.1,2 Many inhibitors of human IMPDHs are used clinically as anticancer and immunosuppressive agents.1,3–6 IMPDH is also a potential antibacterial target.7
Discovery of new antibiotic drugs against Bacillus anthracis, the causative agent of anthrax, is an important goal of the biodefense program.8,9 Mycophenolic acid (MPA), a potent inhibitor of eukaryotic IMPDHs, has antibacterial activity against B. anthracis10 and Staphylococcus aureus.11,12 However, new antibiotics are needed to combat the emergence of multidrug resistant bacteria.
Crystal structures of IMPDH enzymes from several organisms in complex with substrates, cofactors, and inhibitors have been determined.13–22 IMPDH is a homotetramer with 4-fold symmetry. Most IMPDH monomers contain two domains: the catalytic domain, which is a classic eight-fold β/α-barrel (TIM barrel),23 and the CBS subdomain (homologous to a domain in cystathionine β-synthase).24,25 The active site is composed of residues located in loops on the proximal face of the TIM barrel near the C-terminal end with the adjacent subunits also contributing residues. The active site loops are disordered in most IMPDH structures, indicating conformational flexibility required by the IMPDH-catalyzed reaction. Binding of the substrate and the cofactor results in conformational changes involving two mobile elements of the IMPDH active site, with one being the active site loop containing catalytic Cys308 (residues 302–317 in B. anthracis numbering) and the second, another loop, commonly called the active site “flap” (residues 380–430). Interactions mediated by these loops in the substrate-cofactor complexes vary between IMPDH enzymes from different sources (bacterial vs human), and they are partly responsible for a different sensitivity of these enzymes to inhibitors.26
The mechanism of IMPDH has been studied extensively (reviewed in ref 3). It involves a two-step reaction that consists of a fast redox step and a rate-limiting hydrolysis step. Cys308 from the active site loop attacks C2 of IMP and forms a covalent adduct. The NAD+ cofactor is bound; a hydride ion is transferred from C2 to NAD+, and NADH is released (Figure 1). The catalytic flap moves into the site previously occupied by NADH, positioning a conserved Arg-Tyr dyad (B. anthracis residues 404 and 405, respectively) in the active site for hydrolysis of the covalently bound thioimidate intermediate, E-XMP* (Figure 1).3,27 An active site water molecule activated through proton abstraction by the catalytic dyad hydrolyzes E-XMP*,20,28,29 and XMP is released. IMPDH catalyzes two very different chemical transformations, and the enzyme has two mutually exclusive conformations, an open conformation for ligand binding, redox reaction, and product release and a closed conformation for hydrolysis.3 Both of these conformations can be targeted for inhibitors.
Figure 1.

(A) Mechanism of the IMPDH reaction. The covalent enzyme–thioimidate intermediate is shown as E-XMP*. B. anthracis numbering is used. (B) Inhibitors targeting human IMPDH.
All IMPDH enzymes require monovalent cations such as K+ for activity. The ion is proposed to play a dynamic role, possibly binding transiently near the active site cysteine residue, stabilizing the E-XMP* complex, and influencing NAD+ binding.30,31
Although IMPDHs from all organisms are similar in sequence and structure, the eukaryotic and prokaryotic enzymes differ significantly in their kinetic properties and sensitivity to inhibitors.3,7,32 For example, mycophenolic acid (MPA) is a significantly more potent inhibitor of the human enzyme than the bacterial IMPDHs.7,15 MPA binds in the nicotinamide portion of the NAD+ binding pocket and traps the E-XMP* intermediate.33,34 The selectivity of MPA stems from its competition with the flap for the NAD+ site. Because the closed conformation is required for the hydrolysis of E-XMP*, hydrolysis will occur faster in the enzymes in which the closed conformation is preferred. Human IMPDHs favor the open conformation, while bacterial enzymes prefer the closed one.7,35 The closed conformation makes the prokaryotic IMPDHs resistant to MPA.3
Thus far, parasite-specific inhibitors have only been reported for Cryptosporidium parvum. The X-ray structure of C. parvum IMPDH in complex with IMP and one such inhibitor, compound C64, revealed a new binding mode in which the inhibitor interacts with the purine ring of IMP as expected but bends across Ala165 toward the dimer interface to interact with Tyr358 in the adjacent subunit.22 Mammalian IMPDHs contain substitutions at positions 165 and 358 (Ala253 and Tyr445, respectively, in B. anthracis numbering), which explain their resistance to these inhibitors. C. parvum IMPDH is closely related to prokaryotic IMPDHs,36,37 and the C. parvum-specific inhibitors were shown to also inhibit bacterial proteins, such as Helicobacter pylori, Borrelia burgdorferi, and Streptococcus pyogenes.38 Thus, Ala165 and Tyr358 comprise a structural motif that defines the susceptibility of the enzyme to C. parvum inhibitors. Ala165 and Tyr358 are present at corresponding positions in IMPDHs from a wide variety of pathogenic bacteria, including B. anthracis, Campylobacter jejuni, and Clostridium perfringens (Figure 2).
Figure 2.

Multiple-sequence alignment of IMPDHs from selected bacterial pathogens. The following IMPDH sequences were used in the alignment: B. anthracis strain Ames, S. pyogenes, Streptococcus pneumoniae, Cl. perf ringens, Ca. jejuni, Vibrio cholerae, Haemophilus inf luenzae, Francisella tularensis, and Bo. burgdorferi. Identical residues are highlighted in red, and similar residues are shown as red letters. Secondary structure elements derived from B. anthracis IMPDH are depicted as arrows (representing β-strands) and cylinders (representing α-helices and 310-helices) and are numbered consecutively. The positions for the active site cysteine, hydrolysis dyad, and residues from the C. parvum signature motif are marked as black rectangles and labeled.
Currently, there are no examples of an IMPDH structure in more than one state from the same bacterial species. Additionally, there has been no comparison of the active site in noncovalently bound substrate and product complexes for a bacterial IMPDH. Here, we report three crystal structures of IMPDH from B. anthracis, a phosphate ion-bound (Pi-bound) form (2.60 Å), which will be interchangeably termed the “apo” state, and in complex with its substrate (IMP) and product (XMP) (2.38 and 2.65 Å, respectively). We characterize kinetic parameters of the B. anthracis enzyme with respect to the substrates IMP and NAD+, MPA, and two inhibitors of C. parvum IMPDH,22,39,40 a quinolinium-substituted triazole derivative, A110,41 and a 2-pyridinyl derivative of benzimida-zole, C9138 (Figure 7). We compare the inhibition properties of B. anthracis IMPDH to those of three bacterial enzymes from Ca. jejuni, Cl. perfringens, and Vibrio cholerae, and we show that these enzymes bind inhibitors with very different affinities, indicating that small changes in the active site can influence inhibitor binding.
Figure 7.

Biolayer interferometry analysis using Octet RED of binding of A110 and C91 to B. anthracis IMPDH. The analysis was performed in triplicate. For each triplicate, the responses were fit to a 1:1 protein-ligand interaction model. The obtained values were used to generate steady state plots.
MATERIALS AND METHODS
Materials
IMP was purchased from Acros Organics. XMP was purchased from Fluka. NAD+, NADH, and MPA were purchased from Sigma. Two C. parvum-selective inhibitors, compounds A110 (4-{(1R)-[1-(4-chlorophenyl)-1H-1,2,3-tria- zol-4yl]ethoxy}quinoline-1-oxide, C19H15ClN4O2, CAS Regis- try Number 1185388–35–5) and C91 [α-methyl-N-2-naphtha-lenyl-2-(2-pyridinyl)-1H-benzimidazole-1-acetamide, C24H18N4O, CAS Registry Number 125749–66–9], were synthesized as described in refs 41 and 38, respectively. Crystallization reagents were purchased from Hampton Research (Aliso Viejo, CA) and Microlytic (Woburn, MA).
Gene Cloning, Protein Expression, Purification, and Crystallization
The coding sequences of IMPDH enzymes were amplified by polymerase chain reaction (PCR) from chromosomal DNA of B. anthracis (strain Sterne), Ca. jejuni, Cl. perfringens, and V. cholerae using primers compatible with the ligation-independent cloning vector pMCSG7.42 The genes were cloned into pMCSG7 using a modified ligation-independent cloning protocol.42 The recombinant constructs produced fusion proteins with an N-terminal His6 tag and a TEV protease recognition site (ENLYFQ↓S). The fusion proteins were expressed in Escherichia coli strain BL21(DE3) harboring the pMAGIC plasmid encoding one rare E. coli Arg tRNA (covering codons AGG/AGA) in the presence of 100 μg/mL ampicillin and 30 μg/mL kanamycin. The cells were grown in LB medium at 37 °C to an OD600 of 0.6–0.8. This was followed by overnight induction with 0.5 mM isopropyl β-D-thiogalactoside (IPTG) at 20 °C. Cells were harvested, resuspended in lysis buffer [50 mM Hepes (pH 8.0), 500 mM NaCl, 10 mM imidazole, 10 mM 2-mercaptoethanol, and 5% glycerol], and stored at −80 °C.
The selenomethionine (SeMet) derivative of B. anthracis IMPDH was prepared as described previously.43 The BL21-(DE3)/pMAGIC cells were grown at 37 °C in M9 medium supplemented with 0.4% glucose, 8.5 mM NaCl, 0.1 mM CaCl2, 2 mM MgSO4, and 1% thiamine. After the OD600 reached 0.8, Leu, Ile, Lys, Phe, Thr, and Val each at 0.01% (w/v) were added to inhibit the methionine metabolic pathway and increase the level of SeMet incorporation. SeMet was added to the culture (60 mg of SeMet/L of culture), and 15 min later, protein expression was induced with 0.5 mM IPTG. The culture was then incubated at 18 °C overnight. Cells were harvested, resuspended in lysis buffer, and stored at −80 °C.
For inhibitor binding studies using ForteBio Octet, all IMPDH proteins presented here were expressed from the vector pMCSG50. This vector was constructed by modification of the dual-LIC vector pMCSG60. Vector pMCSG60 contains the SmaI LIC site from pMCSG2644 downstream from the Ssp I LIC site in pMCSG7,42 allowing for expression of two proteins from a single vector. A pair of synthetic oligos encoding a 15-amino acid biotinylation signal45 and including the His6 tag was ligated into pMCSG60 between the NdeI and Acc65I restriction sites of pMCSG60. This regenerated the pMCSG7 LIC site with the biotinylation signal inserted between the His6 tag and the TEV protease recognition site. The biotin-protein ligase gene (birA) was synthesized from E. coli BL21 by PCR. The two-step PCR introduced a silent single-base mutation that eliminated the SspI site from the native gene. The birA PCR product was cloned into the SmaI LIC site of the new vector using the standard LIC protocol. The resulting plasmid (pMCSG50) retains the standard pMCSG7 LIC site for protein expression and coexpresses the biotin-protein ligase gene. Target protein overexpressed from this plasmid contains a biotinylated tag (GLNDIFEAQ-KIEWR) that is modified at the lysine residue (underlined). The enzymatic activities of the native enzyme and the biotin-labeled derivative were comparable within 10%.
All IMPDH proteins were purified according to a standard protocol.46 Lysozyme (final concentration of 1 mg/mL) and protease inhibitor cocktail (Roche, Indianapolis, IN; 50 mL/g of wet cells) were added to the thawed cell suspension. The solution was incubated on ice for 20 min and lysed by sonication. The lysate was clarified by centrifugation at 36000g for 1 h and filtered through a 0.44 μm membrane. Clarified lysate was applied to a 5 mL HiTrap Ni-NTA column (GE Healthcare Life Sciences, Piscataway, NJ) on an ÄKTAxpress system (GE Healthcare Life Sciences). The column was washed with lysis buffer containing 20 mM imidazole, and the protein was eluted with the same buffer containing 250 mM imidazole. IMPDHs were dialyzed against crystallization buffer containing 20 mM Hepes (pH 8.0), 150 mM KCl, and 2 mM DTT, concentrated, flash-frozen, and stored in liquid nitrogen. The His6 tag was not cleaved from the protein for either the crystallography or enzyme assays.
Native (i.e., not labeled with SeMet) IMPDH enzyme preparations were used for all enzyme assays, and these were subjected to an additional purification step on a Superdex 300 16/60 size exclusion chromatography column equilibrated with buffer containing 50 mM Tris-HCl (pH 8.0), 100 mM KCl, 1 mM DTT, 3 mM EDTA, and 10% glycerol or containing 20 mM Hepes (pH 8.0), 150 mM KCl, and 2 mM DTT. Fractions containing IMPDH were identified by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, concentrated, and flash-cooled.
Crystallization screens were initially performed by the sitting-drop, vapor-diffusion method at 16 and 18 °C. Crystals containing IMP were optimized using the hanging-drop, vapor-diffusion method. Hanging drops consisted of IMPDH at a concentration of 15 mg/mL and 4 mM IMP mixed with an equal volume of reservoir solution [20% PEG 3350, 0.2 M MgSO4, and 0.1 M Tris-HCl (pH 7.6)]. Crystals with XMP were obtained at 16 °C from a sitting-drop solution containing 8 mg/mL protein and 1.6 mM XMP mixed with an equal volume of reservoir solution [0.5% PEG MME 5000, 0.8 M K/Na tartrate, and 0.1 M Tris-HCl (pH 8.5)]. Phosphate ion-bound crystals were obtained via the sitting-drop method from a solution containing 15 mg/mL protein mixed with an equal volume of reservoir solution [1.0 M Na2HPO4/K2HPO4 (pH 8.2)]. Diffraction quality crystals typically appeared within 3 days. The crystals were mounted on CryoLoops (Hampton Research) and flash-cooled in liquid nitrogen. The cryopro-tectant consisted of 15% glycerol for the IMP-containing crystals and a saturated glucose solution in the crystal mother liquor for the Pi- and XMP-containing crystals. Native and SeMet-labeled IMPDHs crystallized under similar conditions and diffracted X-rays from 2.38 to 2.65 Å.
Data Collection, Structure Determination, and Refine-ment
Diffraction data were collected at 100 K using the Q315r CCD detector (ADSC) at the 19-ID beamline of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory.47 For the crystal of IMPDH complexed with IMP, the three-wavelength inverse-beam multiwavelength anomalous dispersion (MAD) data sets from peak (0.97939 Å), inflection point (0.97930 Å), and high-energy remote (0.95385 Å) were collected from a single SeMet-labeled protein crystal (0.5 mm × 0.3 mm × 0.3 mm) to 2.38 Å resolution, with a 5 s exposure per 1° rotation frame and a 200 mm crystal-detector distance. The total rotation range was 180° on ω as predicted using the strategy module in the HKL3000 suite. The inflection data and the high-energy remote data were not included for the phasing because there was no contribution of data due to crystal decay. Thus, only peak 0.97939 Å was utilized. Similarly, the crystal data for the Pi-bound complex and the IMPDH- XMP complex were collected at the same beamline. The single-wavelength data for the Pi-bound form were collected to 2.6 Å at the 0.97940 Å value covering 130° (on ω), with a 7 s exposure and 1°/frame. For the IMPDH-XMP complex, 2.65 Å single-wavelength data at 0.97918 Å were obtained by collecting 220° (on ω) of diffraction images with a 5 s exposure and 1°/frame. All data were processed and scaled with HKL3000,48 and the detailed crystal and data collection statistics are listed in Table 1.
Table 1.
Crystal and Data Collection Statisticsa
| with Pi | with IMP | with XMP | |
|---|---|---|---|
| space group | I4 | I4 | I4 |
| unit cell parameters (Å) | a = 123.03, c = 140.72 | a = 122.52, c = 140.90 | a = 123.25, c = 141.64 |
| no. of residues/protein | 511 | 511 | 511 |
| no. of molecules/asymmetric unit | 2 | 2 | 2 |
| no. of SeMet molecules/asymmetric unit | 17 | 17 | 17 |
| wavelength (Å) | 0.97940 | 0.97939 | 0.97918 |
| resolution limit (Å) | 2.60 (2.60–2.64) | 2.38 (2.38–2.42) | 2.65 (2.65–2.70) |
| no. of observed unique reflections | 31165 (1554) | 41552 (2105) | 30553 (1502) |
| data completeness (%) | 97.0 (98.4) | 99.9 (100) | 99.9 (100) |
| 〈I/σ(I)〉 | 10.6 (2.6) | 12.9 (3.3) | 10.4 (3.2) |
| redundancy | 5.6 (5.6) | 10.0 (9.6) | 9.1 (9.2) |
| Rmerge b | 0.095 (0.725) | 0.070 (0.776) | 0.094 (0.735) |
Numbers in parentheses are values for the highest-resolution bin.
Rmerge = Σhkl Σi|Ii(hkl) − 〈I(hkl)〉|/Σhkl Σi|〈Ii(hkl)〉|, where Ii(hkl) is the intensity for the ith measurement of an equivalent reflection with indices h, k, and l.
The structure of the IMPDH-IMP complex was determined by SAD phasing with the peak data only up to 2.5 Å resolution using the HKL3000 software package:48 selenium sites were determined by SHELXC49 and SHELXD, the handedness was checked by SHELXE,49 phasing was done by MLPHARE,50 density modification was done by DM,51 and the initial model was built by ARP/wARP,52 which contained 90% of the total of 1022 expected protein residues of the dimer. The initial model was further extended manually using COOT.53 Cycles of manual adjustment of the model using COOT and REFMAC (CCP4 suite)54 and PHENIX55 refinement against the averaged peak data converged the model to the final Rwork of 0.170 and the Rfree of 0.201 with zero σ cutoff (Table 2). The structures of the phosphate ion-bound form of IMPDH and the IMPDH–XMP complex were determined by molecular replacement using MOLREP56 on HKL3000. After the molecular replacement with chain A of the structure of IMPDH from S. pyogenes [Protein Data Bank (PDB) entry 1ZFJ, 69% identical sequence] as a search model to find a dimer, multiple cycles of both rigid-body refinement and restrained refinement were conducted with REFMAC.56 This molecular replacement-produced model with the S. pyogenes protein (PDB entry 1ZFJ) sequence was then applied to AutoBuild routine on PHENIX55 to rebuild in an effort to reduce a model bias and to correct the sequence (from S. pyogenes to B. anthracis IMPDH). Several alternating runs of PHENIX and manual adjustment using COOT further refined the resulting models, both for the phosphate ion-bound form and for the XMP complex. The final Pi- bound structure contains 490 residues (from 511 residues, including uncleaved His6 tag residues) for chain A and 434 residues for chain B with Rwork and Rfree values of 0.181 and 0.242, respectively (Table 2). The final IMPDH–XMP structures with 442 residues for chain A and 426 residues for chain B were converged to Rwork and Rfree values of 0.195 and 0.245, respectively. Both structures, except for approximately half of the CBS domain (residues 94–202), which is partly disordered, overall were well-defined. Native and SeMet-labeled IMPDHs have virtually identical structures.
Table 2.
Phasing and Structure Refinement Statisticsa
| with Pi | with IMP | with XMP | |
|---|---|---|---|
| Phasing | |||
| resolution range (Å) | 37.0–2.65 | 38.7–2.50 | 39.0–2.65 |
| phasing method | MR | SAD | MR |
| search model | chains A and B of 1ZFJ | – | chains A and B of 1ZFJ |
| figure of merit | – | 0.301 | – |
| phasing power | – | 1.465 | – |
| Refinement | |||
| resolution range (Å) | 37.00–2.65 | 38.70–2.38 | 39.00–2.65 |
| no. of reflections | 31065 | 40903 | 29052 |
| σ cutoff | 0.0 | 0.0 | 0.0 |
| Rwork | 0.184 | 0.172 | 0.195 |
| Rfree | 0.242 | 0.201 | 0.245 |
| rmsd | |||
| bond lengths (Å) | 0.010 | 0.011 | 0.009 |
| bond angles (deg) | 1.32 | 1.41 | 1.28 |
| dihedral angles (deg) | 15.5 | 15.3 | 16.0 |
| no. of protein residues | 924 | 843 | 868 |
| other molecules | 124 H2O, 3 PO43− | 416 H2O, 2 IMP, 2 SO42−, 4 glycerol, 1 Cl− | 95 H2O, 2 XMP, 1 tartrate, 1 SO42− |
| mean B factor (Å2) | 61.4 | 41.6 | 82.7 |
| Wilson B factor (Å2) | 49.54 | 27.59 | 56.56 |
| Ramachandran plot [most favored/outliers (%)] | 94.9/0.1 | 98.3/0.0 | 95.7/0.0 |
| PDB entry | 3TSB | 3USB | 3TSD |
Because the structure of the protein–IMP complex was not available at the time when the structures of the phosphate ion-bound form and the complex with XMP were determined, the S. pyogenes structure (PDB entry 1ZFJ) was used as an MR model for these two B. anthracis structures.
The stereochemistry of all three structures was checked with PROCHECK57 and MolProbity.58 Final refinement statistics are listed in Table 2. All figures were prepared using PyMOL (W. L. DeLano, The PyMOL Molecular Graphics System, http://www.pymol.org).
Enzyme Assays
In all kinetic studies, the His6-tagged fusion or biotin-His6-tagged fusion constructs of native recombinant IMPDHs were used. When compared with that of the native enzyme, the activity of the SeMet-labeled B. anthracis enzyme was reduced 2-fold (data not shown). The concentration of the IMPDH fusion constructs was determined by absorbance at 280 nm using an extinction coeficient calculated by the ExPASy ProtParam tool. Except where noted, kinetic studies were performed in an assay buffer [50 mM Tris-HCl (pH 8.0), 100 mM KCl, 3 mM EDTA, and 1 mM DTT] containing varied concentrations of IMP and NAD+ at 25 °C. Reactions were initiated by the addition of IMPDH to a final concentration of 50 nM. The production of NADH was measured by monitoring the increase in absorbance at 340 nm (ε = 6.22 mM−1 cm−1) using a Shimadzu BioSpec-1601, Hitachi U-2000, or Cary 100 Bio spectrophotometer. All IMPDH preparations were shown to be free of contaminating NADH oxidase activity; this was verified with an assay buffer containing 50 μM NADH. No oxidation of NADH was observed during the 40 min incubation period (data not shown). The pH dependence of the B. anthracis enzyme was determined in an assay buffer prepared with MES (pH 5.2–7.2), Tris-HCl (pH 7.2–9.0), or glycine (pH 8.6–10). Monovalent cation dependence was determined in an assay buffer in which KCl was substituted with 100 mM NH4Cl, LiCl, CsCl, or NaCl. Kinetic parameters were determined by collecting initial velocity data at varying concentrations of IMP (5–1000 μM) and NAD+ (100–8000 μM). For analysis of the steady state kinetic data for IMPDH enzymes displaying strong substrate inhibition with respect to NAD+, the method described by Kerr et al. was used.3,59 First, kinetic parameters with respect to NAD+ were obtained by plotting the initial velocity versus IMP at fixed NAD+ concentrations and fitting the data to the Michaelis–Menten equation (eq 1). Apparent Vmax values were then plotted versus NAD+ concentration and fit to the uncompetitive substrate inhibition equation (eq 2). This yielded kcat, Km, and Kii values for NAD+. The Km and kcat for IMP were obtained by plotting the initial velocity versus NAD+ at fixed IMP concentrations, fitting to eq 2, and then plotting the apparent Vmax versus IMP concentration and fitting to eq 1. The kcat values, identical within error, were averaged. Prism 4 (GraphPad Software) or SigmaPlot (SPSS, Inc.) was used for data analysis.
| (1) |
| (2) |
where v is the initial velocity, V is the maximal velocity, A and B are substrate concentrations, KA and KB are the respective Michaelis constants, and Kii is the substrate inhibition constant for NAD+. Inclusion of a biotin tag at the N-terminus of the examined proteins had no significant effect on enzymatic activity.
Inhibitor Kinetics
Inhibition constants for XMP, MPA, A110, and C91 were determined with varying concentrations of IMP, XMP, and NAD+ as indicated. Stock solutions of MPA, A110, and C91 were prepared in DMSO, and the final reaction concentration of DMSO was 5%. Because small amounts of DMSO (up to 15–20%) increased IMP dehydrogenase activity, control reactions for MPA experiments included 5% DMSO. Initial velocity data were fit to the equations for competitive inhibition
| (3) |
noncompetitive inhibition
| (4) |
and uncompetitive inhibition
| (5) |
where S is the concentration of the varied substrate, I is the inhibitor concentration, Km is the Michaelis constant, and Kis and Kii are the slope and intercept inhibition constants, respectively. Global curve fits of the data were performed with Prism 4, and the F test was used to compare models.
Determination of IC50
Enzyme inhibition was assessed by monitoring the production of NADH by absorbance at varying inhibitor concentrations (50 pM to 10 μM). Each enzyme was incubated with an inhibitor for 10 min at room temperature prior to the addition of substrates. The following buffer conditions were used: 50 mM Tris-HCl (pH 8), 100 mM KCl, 1 mM DTT, 3 mM EDTA, 10 nM enzyme, and 1 mM IMP. Each enzyme required a different concentration of NAD+; generally ~2.5Km (NAD+) was used as the NAD+ concentration. IC50 values were calculated for each inhibitor according to eq 6 using SigmaPlot (SPSS, Inc.)
| (6) |
where vi is the initial velocity in the presence of inhibitor I and v0 is the initial velocity in the absence of inhibitor.
Inhibitor Binding Studies
Analysis of the binding of inhibitors to IMPDH proteins was performed using biolayer interferometry (BLI)60 on the Octet RED (ForteBio, Menlo Park, CA). Assays were performed in 96-well black microplates (Fisher Scientific, Pittsburgh, PA) at 25 °C. All volumes were 300 μL. Biotinylated IMPDH was loaded onto Super Streptavidin (SSA) Biosensors (ForteBio) at a concentration of 50 μg/mL in phosphate-buffered saline (PBS). Reference SSA Biosensors were blocked with biocytin at 10 μg/mL in PBS buffer. Each tested inhibitor was titrated from 0.2 nM to 10 μM (a concentration of up to 30 μM was tested for the V. cholerae enzyme) in a buffer containing 50 mM Tris-HCl (pH 8), 100 mM KCl, 1 mM DTT, 3 mM EDTA, 0.1 mg/mL BSA, 5% DMSO, 1 mM IMP, and NAD+ in specific amounts for a given enzyme. Assays were run first using the protein biosensors, followed by the reference biosensors using the same protocol. The reference data were then subtracted from the protein data. The association and dissociation curves were fit using a single-exponential fitting model using analysis software provided by the manufacturer.
Coordinates
The atomic coordinates for the Pi-, IMP-, and XMP-bound structures of B. anthracis IMPDH have been deposited in the PDB as entries 3TSB, 3USB, and 3TSD, respectively.
RESULTS
Overall Structure of IMPDH from B. anthracis
We have determined three crystal structures of B. anthracis IMPDH, a phosphate ion-bound form (an apo form), in complex with the substrate, IMP, and in complex with the product, XMP. Both IMP- and XMP-containing structures were obtained by cocrystallization. The phosphate ion present in the active site of the apo form most likely originated from the crystallization buffer. The IMP-bound structure was determined by single-wavelength anomalous diffraction (SAD) phasing using the HKL3000 software package.48 The structures of the phosphate ion- and XMP-containing IMPDH were determined by molecular replacement.
Size exclusion chromatography shows that B. anthracis IMPDH exists as a homotetramer in solution (Figure S1 of the Supporting Information), and it crystallizes as such. All three structures contain two monomers in the asymmetric unit. The B. anthracis IMPDH monomer has a typical two-domain structure, the catalytic domain, which is a TIM barrel, and the CBS domain (Figure 3). In all structures reported here, each monomer contains a ligand bound in the active site (phosphate anion in the apo structure and IMP and XMP in the substrate-and product-bound structures, respectively). In all the structures, the CBS domains are partially disordered.
Figure 3.

Structure of chain A of the B. anthracis IMPDH complex with IMP (gray space-filling model) showing the TIM barrel and CBS domains (left). Structure of the tetramer in a view parallel to the 4-fold axis (right). The tetramer was generated by performing the appropriate symmetry operation on the more complete monomer (chain A). Each subunit is shown in a different color with a space-filling model of IMP in the active site of each subunit. The inset above the tetramer shows the specific regions of the protein participating in the formation of the tetramer.
The tetrameric structure of IMPDH is stabilized by intermonomer contacts with each of the adjacent subunits. The C-terminus is close (~15 Å) to the catalytic site and on the opposite face of the tetramer from the N-terminus. In the tetramer, all C-termini are close to each other (~12 Å). The subunit interactions can be categorized into three groups depending on their proximity to the catalytic site and level of amino acid sequence conservation. In one group, the first 14 residues of the N-terminus (including 310-helix, η1) project approximately 20 Å from the TIM barrel (Figure 3) and interact with surface residues of an adjacent IMPDH monomer. This regional contact is distal from the catalytic site and involves residues 3–12 of the N-terminus that interact with β-sheet (β19) residues 460–463 of an adjacent subunit. The interaction involves hydrogen bonds and salt bridges between residues that display little sequence conservation. Another loop (residues 18–27) and 310-helix η2 are involved in subunit contacts with the adjacent IMPDH molecule and also form part of the active site pocket of the adjacent subunit. Additional subunit contacts originate from helix α11 (residues 310–315) and a loop (residues 316–319), interacting with β-sheet β1 (residues 16–19) and residues 479–484 in an adjacent IMPDH monomer. These regions are approximately 20 Å from the substrate-binding site.
The catalytic domain (residues 1–90 and 222–487) forms the core of the tetramer and is approximately 40 Å × 40 Å × 50 Å. This domain is a classic eight-fold β/α TIM barrel with the active site loop (residues 302–329) containing catalytic Cys308 positioned on the proximal face of the β/α-barrel near the subunit interface (Figure 3). The CBS domain is composed of tandem motifs (residues 91–221, approximately 20 Å × 20 Å × 40 Å) and projects outward from the tetramer. Of the two monomers in the asymmetric unit, molecule A in the phosphate ion-bound structure has the most complete electron density for the CBS domain. In the IMP-containing structure determined at higher resolution (2.38 Å), four residues are missing in the loop connecting the tandem CBS motifs and 12 residues are missing in the structure with XMP. The structure of B. anthracis IMPDH is most similar to that of IMPDH from S. pyogenes (PDB entry 1ZFJ).15 Alignment of these structures using the SSM server of the European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm)61 yielded a backbone rmsd of 1.34 Å and 69% sequence identity. In addition to the S. pyogenes enzyme, there are four other structures of microbial IMPDHs available in the PDB. These include IMPDHs from Thermotoga maritima [PDB entry 1VRD, 60% sequence identity, 1.03 Å rmsd (DOI 10.2210/pdb1vrd/pdb)], C. parvum (PDB entry 3FFS, 57% sequence identity, 1.03 Å rmsd22), Bo. burgdorferi (PDB entry 1EEP, 55% sequence identity, 0.96 Å rmsd17), and Pyrococcus horikoshii [PDB entry 2CU0, 50% sequence identity, 1.04 Å rmsd (DOI 10.2210/pdb2cu0/pdb)]. All of these enzymes show similar structural features and active sites.
Substrate and Product Binding
IMP dehydrogenase catalyzes the oxidation of IMP to XMP with the concomitant reduction of NAD+ to NADH. During the reaction, IMP forms a covalent adduct with the active site Cys residue (Cys308 in B. anthracis), the hydride ion is transferred from the C2 atom of the hypoxanthine ring to NAD+, and the oxygen atom is placed in this position resulting in the formation of xanthosine. In the structures of B. anthracis IMPDH with IMP and XMP, the substrate or product is bound in a pocket located inside the β-barrel structure. Binding of these ligands is characterized by subtle conformational changes. In both structures, the ribose and phosphate moieties assume virtually identical conformations and are highly coordinated by the protein. The sugar moiety is in the C2′-endo conformation, and its 2′-and 3′-hydroxyl groups are hydrogen-bonded with Asp341. The phosphate group is anchored by a number of amino acid side chains (Ser306, Ser365, and Tyr388; surprisingly, Tyr388 is ordered in the XMP-bound structure but is disordered when IMP is bound) and three main chain nitrogen atoms (Gly343, Gly364, and Ser365) (Figure 4A). The remaining hydrogen bonding potential of the phosphate oxygen atoms is fulfilled by water molecules. The conformation of the purine ring is somewhat different in the two liganded structures. In the structure with IMP, the Cys308 sulfur atom is located 3.5 Å (chain A) and 3.9 Å (chain B) from the C2 atom of the inosine ring and is not covalently linked to the ring. This conformation of inosine seems to be stabilized by the side chains of Ile307, Cys308, and Met51. In the XMP-bound structure, the xanthosine ring is rotated (rotation defined on the basis of the O4-C1′-N9-C4 torsion angle within the IMP and XMP molecule) by ~10° (the difference between the chain A/B-averaged torsion angle for each ligand) with respect to the position of the inosine ring and its C2 atom is closer (3.4 and 3.7 Å for chains A and B, respectively) to the sulfur of Cys308 (Figure 4A). This conformation is typically observed in other XMP-IMPDH complexes21 and causes the Cys308 side chain to rotate away from the XMP molecule. The hydroxyl group of Thr310 forms a hydrogen bonding interaction with the O2 atom of XMP. In the structure of the apoenzyme, the phosphate anion occupies the same position as the IMP or XMP phosphate group and makes the same contacts with the protein that are observed for the phosphate moiety in the structures with IMP and XMP. These include interactions with amino acid side chains of Ser306, Ser365, and Tyr388 and three main chain nitrogen atoms of Gly343, Gly364, and Ser365. In contrast to the Tritrichomonas foetus IMPDH,21 in all three structures of the B. anthracis IMPDH the catalytic loop containing Cys308 is ordered and exhibits very little conformational dynamics (Figure 4A, B). Although potassium chloride was included in the crystallization medium, electron density for a potassium ion was not observed in any of our structures (the metal ion position is occupied by water molecules in our structures).
Figure 4.

Structural alignment of IMPDH active sites. (A) Overlap of the B. anthracis IMPDH structures of Pi (green), IMP (magenta), and XMP (yellow) complexes showing the positions of the IMP and XMP (magenta and yellow sticks, respectively). (B) Overlap of the B. anthracis Pi-bound (green) and IMP-bound (magenta) structures with the S. pyogenes enzyme-IMP complex (PDB entry 1ZFJ) (turquoise). IMP molecules (B. anthracis in magenta, S. pyogenes in turquoise) are depicted as sticks. S. pyogenes residues Met393 and Gly394 interacting with IMP are also depicted as sticks. (C) Overlap of the structures of the B. anthracis Pi-bound enzyme (green) with the T. foetus IMPDH complex with MZP (purple) (PDB entry 1PVN20). The conserved Arg-Tyr dyad within helix η 3 of the flap is depicted as sticks. (D) Overlap of the structures of the B. anthracis Pi-bound enzyme (green) with the T. foetus IMPDH (purple) complex with IMP (omitted for the sake of clarity) and TAD (purple) (PDB entry 1LRT19). A distal portion of the B. anthracis catalytic flap with the conserved Arg-Tyr dyad is clashing with the T. foetus β-methylene-thiazole portion of TAD, indicating that these two elements are occupying the same space within the active site.
Active Site Flap
After the hydride transfer, the E-XMP* intermediate is covalently trapped in the active site and the Cys308–XMP* bond must be hydrolyzed for the product to be released. The active site flap is a mobile element containing the catalytic dyad (Arg404 and Tyr405), which positions itself into the active site during the hydrolysis phase of the reaction. This flap shows a varying degree of disorder in reported structures. The structure of the complete flap has been captured only for the T. foetus IMPDH in the presence of the transition state analogue, mizoribine 5′-monophosphate (MZP).20 In the B. anthracis IMPDH structure with IMP, residues 381–421, which comprise most of the flap region, are disordered. A larger portion of the flap is visible in the structure with XMP, with only residues 394–414 missing. Interestingly, the entire flap is visible in the apo (Pi-bound) structure (Figure 5). Figure 4B shows a comparison of the three structures: the B. anthracis phosphate ion-bound enzyme, the B. anthracis enzyme in complex with IMP, and the S. pyogenes IMPDH-IMP complex, with a partially ordered active site flap. In the S. pyogenes structure, the base of the flap consists of a small, three-stranded antiparallel β-sheet (β16–β18, the “finger domain”). A loop that extends from the second strand of the β-sheet (β17) is partially ordered and contains residues (S. pyogenes Met393 and Gly394) that contact the purine residue of IMP (Figure 4B). The distal portion of this loop, containing the catalytic Arg-Tyr dyad, is disordered in the S. pyogenes structure as it is in most other IMPDH structures but is visible in the structure of the apoenzyme. The portion of the loop ordered in the structure of the apoenzyme contains a small α-helix (α15, residues 394–399) followed by a 310-helix (η3, residues 401–405) (Figure 4B) and a longer loop (residues 406–416). The flap is ordered because several residues (side chains of Lys386, Arg389, Asp403, Arg404, Asn410, Glu416, and Lys425, main chain carbonyl, and amide groups of Gly400, Arg404, Glu419, Arg420, and Gly426) of this structural motif interact with the main body of the enzyme (both within the subunit and with the neighboring subunit). For example, the C-terminal sequence of the neighboring subunit forms a β-strand (β20, residues 476–480) that complements the β-finger of the flap with Lys480 providing an additional bridging interaction with Glu419 of the flap. There are also interactions within the flap unit (for example, Lys402 and Gln407). Arg404 and Tyr405, located within the 310-helix, η3, extend toward the active site. The conformation of these residues is maintained through the bridging interaction with Asp251. An overlap of the B. anthracis phosphate ion-bound structure with the T. foetus structure of the MZP–enzyme complex (PDB entry 1PVN, rmsd on Cα atoms of compared 362 residues of 1.23 Å),20 the only other structure with a completely ordered flap, shows that the active site flaps maintain remarkable structural similarity with only ~40% sequence identity (Figure 4C). Several conserved side chains assume virtually identical conformations [numbers in parentheses are in T. foetus notation, Tyr388 (405), Gly390 (407), Gly392 (409), Ser393 (410), Arg404 (418), Tyr405 (419), Glu416 (431), Gly417 (432), Pro423 (438), and Tyr424 (439)]. The only significant differences are observed in the loop regions of the flap. The small α-helix (α15, residues 394–399) in the B. anthracis structure is replaced by a loop in the T. foetus structure (residues 411–414) (Figure 4C). Also, a portion of the loop (residues 406–414) connecting helix 3 and sheet β18 (residues 406–419) is shifted toward the center of the tetramer in the B. anthracis structure, whereas the corresponding portion of the T. foetus loop faces the other monomer (Figure 4C). Interesting information about the flap movement during catalysis can be obtained by comparing the B. anthracis apo structure with the structure of T. foetus IMPDH in complex with IMP and an NAD+ analogue, β-methylene-thiazole-4-carboxamide-adenine dinucleotide (TAD).20 In that structure, the TAD molecule lies in the cleft between two adjacent monomers; the thiazole ring stacks against the inosine ring of IMP, and a portion of the flap is missing (T. foetus residues 417–429). An overlap of the B. anthracis apo (Pi-bound) structure with the T. foetus enzyme–IMP–TAD complex (PDB entry 1LRT, rmsd for Cα atoms of 338 compared residues of 1.23 Å) indicates that the conserved Arg-Tyr dyad (helix η3) occupies the nicotinamide subsite of NAD+ within the active site (Figure 4D). This observation suggests that the portion of the flap swings in and out of the NAD+ site during catalysis. Because the flap is disordered in all structures containing IMP and XMP, it suggests that binding of a ligand causes the flap to become disordered by disrupting the interaction of this structural motif with the main body of the enzyme. The conformation or ordering of the flap does not seem to depend on the crystal packing because the structures of B. anthracis IMPDHs are isomorphic. The flap environment is very similar in all B. anthracis structures, and the contacts made by the flap are mainly intramolecular with the adjacent monomer within the same tetramer. A possible explanation for the flap stability in the phosphate ion-bound enzyme is the presence of a small hydrophobic core near the active site. This core is formed by the flap (residues Tyr388, Ile418, and Val422), the active site loop (residues Ile307 and Thr309), and the loop from the neighboring subunit (residues His471, Ile478, Tyr485, and Tyr346). Disruption of this core by binding of a ligand would clearly have a detrimental effect on the maintenance of the structure of the flap.
Figure 5.
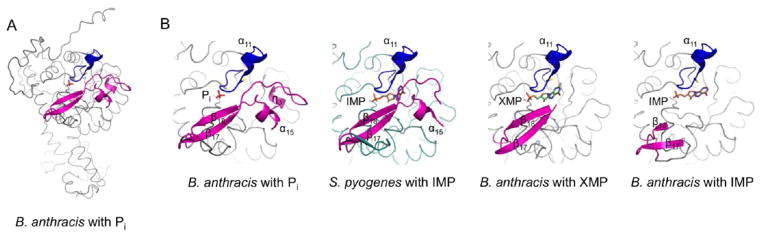
Different degrees of flap disorder in bacterial IMPDHs. (A) B. anthracis monomer (molecule A) of the highly ordered Pi-bound (apo) structure. (B) Detail of the active site, from left to right, of the B. anthracis Pi-bound enzyme (entire flap ordered), the S. pyogenes complex with IMP (missing residues 402–415 that include helix η3), and the B. anthracis complex with XMP (missing residues 394–414, including helices α15 and η3) and with IMP (missing residues 381–421), respectively. The catalytic flap is colored magenta, and the catalytic loop is colored dark blue, with the catalytic Cys represented as sticks. Ligands (Pi, IMP, and XMP) are colored red, orange, and green sticks, respectively.
Kinetic Characterization of B. anthracis IMPDH
The kinetic properties of B. anthracis IMPDH are similar to those of other microbial IMPDH enzymes.15,62–64 The enzyme has a pH optimum of 8.2 and a strict requirement for monovalent cation. Optimal activation occurs with K+ concentrations between 100 and 150 mM; however, inhibition is observed at higher concentrations. The enzyme shows ≤1% activity with 100 mM Na+ or Li+ or in the absence of monovalent cations; 100 mM NH4+ or Cs+ yielded 77 or 36% activity, respectively, relative to the activity in the presence of 100 mM K+, the typical assay concentration. Plots of initial velocity versus IMP concentration at fixed NAD+ concentrations are consistent with the Michaelis–Menten kinetics. Substrate inhibition was observed at high NAD+ concentrations (Figure 6B).3,65 Secondary plots of the initial velocity versus IMP and NAD+ are shown in Figure 6, and the steady state parameters are listed in Table 3. XMP is a competitive inhibitor versus IMP. MPA displays noncompetitive inhibition versus IMP and NAD+ (Table 3). The Michaelis–Menten kinetic characterization was also performed for IMPDHs from three other bacterial species, Ca. jejuni, Cl. perfringens, and V. cholerae. All tested proteins exhibit similar Km values for the substrate, IMP, and have requirements for higher NAD+ concentrations than eukaryotic enzymes (Table 4).
Figure 6.

Steady state kinetics of B. anthracis IMPDH. (A) Secondary plot of kinetic data showing apparent Vmax values (from initial velocity vs NAD+ plots) plotted vs IMP concentration. The solid line represents the best fit to eq 1 for Michaelis–Menten kinetics. (B) Secondary plot of kinetic data showing apparent Vmax values (from initial velocity vs IMP plots) plotted vs NAD+ concentration. The solid line represents the best fit to eq 2 for uncompetitive substrate inhibition.
Table 3.
Kinetic Parameters for Inhibition of B. anthracis IMPDH by XMP and MPA
| inhibitor | inhibition patterna | Kis (μM) | Kii (μM) | |
|---|---|---|---|---|
| XMP | vs IMPb | C | 218 ± 6 | nac |
| MPA | vs IMPb | NC | nac | 4.83 ± 0.04 |
| MPA | vs NAD+ d | NC | nac | 3.9 ± 0.2 |
C, competitive; NC, noncompetitive.
NAD+ concentration fixed at 1.3 mM.
Not applicable.
IMP concentration fixed at 750 μM.
Table 4.
Kinetic Parameters of Bacterial IMPDH Proteins
| B. anthracis | Ca. jejuni | Cl. perfringens | V. cholerae | |
|---|---|---|---|---|
| kcat (s−1) | 1.4 ± 0.2 | 2.2 ± 0.3 | 0.6 ± 0.1 | 2.1 ± 0.2 |
| Km(IMP) (μM) | 18 ± 2 | 25 ±3 | 11 ± 2 | 80 ± 10 |
| Km(NAD) (μM) | 550 ± 50 | 220 ± 20 | 370 ± 4 | 1200 ± 200 |
| Kii(NAD) (mM) | 3.9 ± 0.5 | 20 ± 4 | 13 ± 2 | naa |
Not applicable.
IMPDH Inhibition
C. parvum IMPDH-selective inhibitors have been recently indentified in a high-throughput screen targeting the NAD+-binding site.22,39,40 Because B. anthracis IMPDH contains the Ala253-Tyr445 (C. parvum Ala165-Tyr358) structural motif that defines the susceptibility of the enzyme to C. parvum inhibitors, the B. anthracis enzyme should also be sensitive to these inhibitors. To test this hypothesis, IC50 data for the B. anthracis IMPDH were measured for two of the C. parvum inhibitors, a quinolinium-substituted triazole derivative, A110,41 and a 2-pyridinyl derivative of benzimida-zole, C9138 (Figure 7). To compare the inhibition properties of A110 and C91 for other bacterial enzymes, we also obtained IC50 values for IMPDHs from Ca. jejuni, Cl. perfringens, and V. cholerae. Because V. cholerae IMPDH does not contain the Ala165-Tyr358 structural motif, a much lower affinity for A110 and C91 was expected and the V. cholerae enzyme was used as a negative control. The B. anthracis and Ca. jejuni enzymes exhibited similar sensitivities to A110 and C91 (IC50 values in the range of 51–120 nM), whereas Cl. perfringens IMPDH was generally less sensitive to both inhibitors, with 2- and 10-fold larger IC50 values for A110 (280 ± 30 nM) and C91 (570 ± 20 nM), respectively (Table 5).
Table 5.
Inhibition of IMPDHs by Compounds A110 and C91
|
B. anthracis
|
Ca. jejuni
|
Cl. perfringens
|
V. cholerae
|
|||||
|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | KD (nM) | IC50 (nM) | KD (nM) | IC50 (nM) | KD (nM) | IC50 (nM) | KD (nM) | |
| A110 | 57 ± 7 | 150 ± 20 | 120 ± 20 | 200 ± 18 | 280 ± 30 | 490 ± 42 | >5000a | >5000 |
| C91 | 57 ± 1 | 150 ± 40 | 51 ± 9 | 140 ± 10 | 570 ± 20 | 1100 ± 62 | >5000a | >5000 |
Concentrations of >5 μM were not tested.
In addition, to test an alternative to a typical kinetic assay, biolayer interferometry (BLI)60 was utilized to determine the apparent dissociation constant, KD, for binding of the inhibitors to IMPDHs. The relationship of IC50 to Ki (KD values in BLI) is described by Cheng and Prusoff.66 This relationship depends on substrate concentration [S], the reaction mechanism, and the inhibitor’s mode of inhibition. In the case of competitive inhibition, IC50 values approximate KD when the [S] used in the assay is much lower than Km. For uncompetitive inhibition, IC50 values approximate KD when the [S] in the assay is much higher than Km. For noncompetitive (mixed) inhibition, IC50 does not depend on [S] and will be equal to KD.67 This last mode of inhibition applies to the case of IMPDH inhibition presented here. BLI is a label-free technology for measuring biomolecular interactions. It is an optical technique that analyzes the interference pattern reffected from two surfaces: a layer of the immobilized protein on a biosensor tip and the reference layer. The binding between an immobilized protein and a ligand in solution produces the increase in optical thickness at the biosensor tip, which causes a shift in the interference pattern that can be measured in real time.60,68 Binding affinities of A110 and C91 for B. anthracis, Ca. jejuni, Cl. perfringens, and V. cholerae IMPDHs were measured using BLI (Figure 7). A comparison of IC50 values and KD is presented in Table 5. The values for KD obtained using BLI agree within 3-fold with the IC50 values obtained spectrometrically. Generally, regardless of the mode of inhibition, IC50 values are expected be higher or equal to KD values. All KD values listed in Table 5 are higher than the corresponding IC50 values, but they agree within 3-fold (i.e., 57 ± 7 nM vs 150 ± 20 nM). This correlation is considered to be within the experimental error, given the two vastly different experimental designs.69
DISCUSSION
Overall Structure of Bacterial IMPDHs
All IMPDH enzymes share several common characteristics. The active form of the enzyme is a tetramer with four active sites per oligomer. The reaction proceeds through several distinct steps. IMPDH enzymes have an active site cysteine residue that upon IMP binding forms a covalent intermediate. Here we were able to capture the B. anthracis IMPDH in three different states. A substrate-bound form shows that IMP is anchored in the active site using the phosphosugar moiety. The inosine ring remains quite flexible, and on the basis of our structures and those previously reported, it assumes at least two distinct conformations. The B. anthracis IMPDH product-bound state is very similar to all structures reported to date. In both of these forms, the active site flap is mostly disordered. The third structure is of an apoenzyme, which contains a phosphate ion bound in the active site (corresponds to the position of a phosphate group in IMP and XMP). In this structure, the active site flap is fully ordered and its conformation is consistent with the role of Arg404 and Tyr405 as the catalytic dyad during the hydrolysis step.
A consensus sequence that includes an active site cysteine has been proposed as a signature motif for the IMPDHs as well as GMP reductase enzymes. Alignment of these consensus sequences of bacterial and eukaryotic IMPDH enzymes shows a significant conservation with 30–40% sequence identity among organisms. However, regions that lack homology on the basis of a global comparison show similarity when the comparison is restricted to the bacterial or eukaryotic group. In this restricted comparison, 90% of the residues are conserved in this region of the active site for either the bacterial or eukaryotic group.32 The availability of structural information for several IMPDH enzymes provides a resource for defining the distinct characteristics of bacterial and eukaryotic enzymes. Features such as the catalytic pocket, catalytic loop, and flap are conserved, but some distinct characteristics have been identified for bacterial IMPDHs. These discriminatory signatures of bacterial and mammalian IMPDH enzymes can be exploited in the development of bacterium-specific inhibitors. For example, the presence of the Ala165-Tyr358 structural motif defines the susceptibility of the enzyme to C. parvum inhibitors.
Currently, there are 26 structures of IMPDHs deposited in the PDB, only seven of which (including three B. anthracis structures presented here) are from bacteria and one of which is from archaea. The best structurally characterized is the IMPDH from T. foetus for which several structures with substrate, cofactor, MPA, transition state analogue MZP, and ribavirin 5′-monophosphate (RMP) have been determined. However, there is still the need for more comprehensive structural characterization of bacterial enzymes, which would offer valuable insights into the mechanistic specifics of prokaryotic IMPDHs. Three crystal structures of the B. anthracis IMPDH presented here provide an important contribution in the process of elucidation of the bacterial IMPDH signature.
Catalytic Site and Implications for the Mechanism of Bacterial IMPDH
Covalent tetrahedral intermediate formation has been proposed.70 However, our structures show that IMPDH does not form a covalent bond with the substrate in the absence of the NAD+ cofactor. Therefore, the cofactor not only plays the role of hydride acceptor but also completes the structure of the catalytic pocket. Initiation of the reaction cycle requires alignment of inosine and nicotinamide rings in near parallel fashion and positioning of the C2 atom of the inosine ring close to the C4 atom on the pro-S face of the nicotinamide ring.27,71 It appears that only then can the formation of thioimidate occur. This mechanism is in striking contrast with results obtained with halogenated derivatives of IMP. IMPDH catalyzes the dehalogenation of 2-fluoro- and 2-chloroinosine 5′-monophosphate in the absence of NAD+.72 This suggests that the Cys308 activation system is in place, but the reaction does not proceed with IMP alone because the hydride is a poorer leaving group than chloride and fluoride. However, the inosine ring seems to exist in multiple conformations, some of which may result in a productive covalent adduct, providing a suitable leaving group is present. For IMP, the NAD+ cofactor is required to position the inosine ring for the Cys308 attack and the subsequent transfer of hydride. In the structure of the hamster IMPDH (PDB entry 1JR1), which also contains a molecule of MPA in the active site, the inosine ring is covalently attached to Cys331 (equivalent to Cys308 in our structure). The hamster IMPDH structure represents the covalent thioimidate (E-XMP*) intermediate of the reaction, in which MPA, an uncompetitive inhibitor, prevents the hydrolysis of the thiopurine intermediate by the catalytic dyad (Arg404 and Tyr405) as was originally suggested by Link and Straub.34
Unlike previous structures, with the exception of the T. foetus enzyme complex with MZP,20 the entire flap is well-ordered in the structure of the B. anthracis phosphate ion-bound enzyme. The distal portion of the flap folds into the active site, creating a closed conformation. This closed conformation is stabilized by the interactions of several residues of this structural motif with the main body of the enzyme. Binding of IMP or XMP disrupts these interactions and forces the flap to move away from the active site. This is signified by the disorder of portions of the flap in the structure of substrate and product complexes. This may indicate that for the B. anthracis enzyme, the equilibrium between the open and closed conformation is shifted toward the closed conformation in the absence of IMP and/or NAD+. Binding of the substrate appears to induce the open conformation.
Although potassium chloride was included in the crystallization medium, electron density for a potassium ion was not observed in any of our structures. To date, only five (of 26 deposited) X-ray crystal structures of IMPDHs identified a conserved metal (K+ or Na+) binding site near the loop containing the catalytic Cys residue. K+ ion has been observed in the structures of the Chinese hamster E-XMP*-MPA complex (PDB entry 1JR1)13 and IMP analogue complexes, human type II E-RVP-MAD [PDB entry 1NF7 (DOI 10.2210/pdb1nf7/pdb)] and T. foetus E-MZP (PDB entry 1PVN).20 Na+ ion was located at the same site in the structures of T. foetus E-RVP (PDB entry 1ME8) and E-RVP-MPA (PDB entry 1ME7).18 In the case of K+ ion, its binding site is formed by six main chain carbonyls from two adjacent monomers. For T. foetus, these include three carbonyls from the Cys319 loop (Gly314, Gly316, and Cys319) and three from an α-helix in the C-terminal portion of the adjacent subunit (Glu485, Gly486, and Gly487).18,20 A second potassium site is observed exclusively in T. foetus IMPDH. This site is also located at the interface of two monomers and involves interactions with residues that are not conserved in B. anthracis or any other organism.18,19 The metal binding site associated with the active site Cys-containing loop is thought to be catalytically important. This site is disrupted in many structures that are believed to reffect intermediate stages of the catalytic cycle, suggesting that K+ binds only to the specific state of the enzyme, such as E-XMP*.13,20 However, it was recently shown through biochemical experiments and molecular dynamics simulations that K+ is present throughout the catalytic cycle but plays a major role in influencing the rate of the catalytic flap closure. Specifically, the presence of K+ is proposed to decrease the activation barrier for the flap movement by providing an alternative orientation (or disruption) of the Cys loop and thus facilitating the interaction between the loop and the flap, allowing flap closure.73 Interestingly, T. foetus IMPDH is also activated by Na+ and thus it is not K+-specific. This lack of ion specificity is thought to be associated with a higher flexibility of the catalytic loop that is observed for T. foetus.3 This flexibility is not seen in the B. anthracis enzyme or any other K+-specific IMPDHs such as that of Bo. burgdorferi or S. pyogenes. It was proposed that the presence of a Pro residue within the Cys loop (position 304 in B. anthracis) in the K+-specific IMPDHs prevents the adaptation of the Cys loop to smaller monovalent cations.3 Indeed, the Cys loop seems to be in the same orientation in all three B. anthracis structures presented here (Figure 4A, B). The three structures of the B. anthracis IMPDH represent the specific but static states of the enzyme, with no cofactor present and no reaction occurring. It is possible that because K+ acts as the ball-and-socket joint facilitating the conformational changes, the presence of this ion is necessary during the reaction but not before (structure with IMP) or after (structure with XMP and Pi) the reaction has occurred.
The active site of IMPDH can be subdivided into five subsites. Two of these subsites are occupied by IMP and XMP, with phosphosugar and purine base moieties. The NAD+ binding site has the nicotinamide monophosphate binding subsite, the adenosine monophosphate binding subsite, and the pyrophosphate binding subsite. All C. parvum inhibitors are designed to occupy the NAD+ site.39 Kinetic characterization further localized binding to the nicotinamide monophosphate subsite.39 Because B. anthracis IMPDH possesses the Ala253-Tyr445 minimal structural motif (C. parvum Ala165-Tyr358) that defines the susceptibility of the enzyme to C. parvum inhibitors, the protein is inhibited by inhibitors A110 and C91. Two other bacterial enzymes, those of Ca. jejuni and Cl. perfringens, also exhibit sensitivities to A110 and C91, whereas the V. cholerae protein, which does not contain the Ala165-Tyr358 motif, is not inhibited by these two compounds. The values of IC50 and KD of A110 are within a factor of 2 for B. anthracis, Ca. jejuni, and Cl. perfringens enzymes, indicating that the binding sites of these compounds are conserved. Notably, the Cl. perfringens enzyme displays a different sensitivity to C91, with IC50 and KD values approximately 10-fold higher than the corresponding values for the B. anthracis and Ca. jejuni enzymes. This suggests that C91 binds in a region that is conserved in B. anthracis and Ca. jejuni IMPDHs, but different in Cl. perfringens. It is important to mention that because the KD values obtained for the examined IMPDHs are proportional to the IC50 values calculated in a conventional assay, BLI can be utilized as a convenient method for primary screening of inhibitor candidates.
Bacterial enzymes bind NAD+ poorly (Km > 1 mM)3,74,75 and are inhibited by MPA only at relatively high concentrations (Ki > 3 μM). This suggests that the NAD+ binding pocket is different in bacterial IMPDH. In T. foetus and human IMPDHs, the adenosine portion of NAD+ is stacked with at least one aromatic and/or arginine residue (Arg241-Trp269 in T. foetus, Arg253-Tyr282 in human type I, and His253-Phe282 in human type II enzymes).16,21 This stacking is not present in B. anthracis (Thr230-Gly259) or other bacteria. Other differences include the presence of a serine residue (T. foetus Ser263, human type I Ser275, and human type II Ser276) forming a hydrogen bond with the NAD+ pyrophosphate group.21 In B. anthracis, this residue is replaced with Ala253. All these differences may account for the lower affinity of the B. anthracis and other bacterial enzymes for NAD+. Interestingly, the overall catalytic mechanism seems to remain the same. Bacterial IMPDH enzymes appear to favor a closed conformation (as shown in our phosphate ion-bound structure), in which the catalytic flap occupies the NAD+ site. This closed conformation is needed for the hydrolysis step of the reaction. Therefore, it is possible that the bacterial enzymes, to increase the efficiency of hydrolysis, evolved toward a reduced affinity for the cofactor. This is consistent with the lower NAD+ affinity and higher catalytic rate observed in bacterial IMPDH enzymes. Conversely, the mammalian IMPDH enzymes favor the open conformation. Accordingly, these display higher NAD+ affinities and lower hydrolysis rates.
We have determined three crystal structures of B. anthracis IMPDH, a phosphate ion-bound (apo) form, one with substrate, IMP, and one with product, XMP. The Pi-bound structure is the first bacterial structure with a complete active site flap. In this structure, the distal portion of the flap folds into the active site mimicking a closed conformation, which is required for the hydrolysis stage of the catalysis.
Rising microbial antibiotic resistance creates an urgent need for new drugs for the treatment of bacterial infections such as methicillin resistant St. aureus (MRSA). Because IMPDH is a crucial regulator of the intracellular guanine nucleotide pool, the inhibitors against IMPDH may strongly augment currently applied drugs against many other multidrug resistant strains even in cases where IMPDH is not essential. Some bacteria like Ca. jejuni lack the guanine salvage pathway, making their IMPDH enzymes an excellent targets for drug development. Thus, IMPDH inhibition offers a promising strategy for the development of broad-spectrum antibiotics.
Inhibition studies have, thus far, primarily focused on the active site, with inhibitors occupying the IMP or NAD+ site. NAD+-based inhibitors that target the cofactor binding site can interact with three subsites, the nicotinamide monophosphate binding subsite, the adenosine monophosphate binding subsite, and the pyrophosphate binding subsite. The nicotinamide subsite of NAD+ is highly conserved, and it is likely that more potent and selective inhibitors can be obtained by designing compounds that extend into the more diverged adenosine and pyrophosphate subsites. Because we observe that bacterial enzymes exhibit different sensitivities to the two tested C. parvum inhibitors, this study provides additional support for the strategy that prokaryotic IMPDH-specific inhibitors can be developed by focusing on the divergent portions of the NAD+ site.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract HHSN272200700058C, by National Institutes of Health Grants AI057153 (A.J.) and AI093459-01 (L.H.), and by the U.S. Department of Energy, Office of Biological and Environmental Research, under Contract DE-AC02-06CH11357. This work has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (“Argonne”). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under Contract DE-AC02-06CH11357. The U.S. Government retains for itself, and others acting on its behalf, a paid-up nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government.
We thank all members of the Structural Biology Center at Argonne National Laboratory for their help in conducting these experiments, Lour Lezondra for help with the purification of IMPDH from B. anthracis, William Eschenfeldt for help with the preparation of an expression vector, and Lindy Keller for help in the preparation of the manuscript.
ABBREVIATIONS
- IMPDH
inosine 5′-monophosphate dehydrogenase
- IMP
inosine 5′-monophosphate
- NAD+
nicotinamide adenine dinucleotide
- NADH
reduced nicotinamide adenine dinucleo-tide
- XMP
xanthosine 5′-monophosphate
- CBS
cystathionine β-synthase
- Pi
phosphate ion
- MPA
mycophenolic acid
- MZP
mizoribine 5′-monophosphate
- RMP
ribavirin 5′-monophos-phate
- MAD
mycophenolic adenine nucleotide
- rmsd
root-mean-square deviation
- SAD
single-wavelength anomalous diffraction
- TAD
β-methylene-thiazole-4-carboxamide adenine dinucleotide
- BLI
biolayer interferometry
- MRSA
methicillin resistant St. aureus
Footnotes
The authors declare no competing financial interest.
A size exclusion profile of the B. anthracis IMPDH (Figure S1). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nair V, Shu Q. Inosine Monophosphate Dehydrogenase as a Probe in Antiviral Drug Discovery. Antiviral Chem Chemother. 2007;18:245. doi: 10.1177/095632020701800501. [DOI] [PubMed] [Google Scholar]
- 2.Shu Q, Nair V. Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med Res Rev. 2008;28:219–232. doi: 10.1002/med.20104. [DOI] [PubMed] [Google Scholar]
- 3.Hedstrom L. IMP Dehydrogenase: Structure, Mechanism, and Inhibition. Chem Rev. 2009;109:2903–2928. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weimert NAP, DeRotte M, Alloway RRP, Woodle ESMD, Vinks AAPP. Monitoring of Inosine Monophosphate Dehydrogenase Activity as a Biomarker for Mycophenolic Acid Effect: Potential Clinical Implications. Ther Drug Monit. 2007;29:141–149. doi: 10.1097/FTD.0b013e31803d37b6. [DOI] [PubMed] [Google Scholar]
- 5.Pankiewicz KW, Lesiak-Watanabe KB, Watanabe KA, Patterson SE, Jayaram HN, Yalowitz JA, Miller MD, Seidman M, Majumdar A, Prehna G, Goldstein BM. Novel Mycophenolic Adenine Bis(phosphonate) Analogues As Potential Differentiation Agents against Human Leukemia. J Med Chem. 2002;45:703–712. doi: 10.1021/jm0104116. [DOI] [PubMed] [Google Scholar]
- 6.Dhar TGM, Shen Z, Guo J, Liu C, Watterson SH, Gu HH, Pitts WJ, Fleener CA, Rouleau KA, Sherbina NZ, McIntyre KW, Witmer MR, Tredup JA, Chen BC, Zhao R, Bednarz MS, Cheney DL, MacMaster JF, Miller LM, Berry KK, Harper TW, Barrish JC, Hollenbaugh DL, Iwanowicz EJ. Discovery of N-[2-[2-[[3-Methoxy-4-(5-oxazolyl)phenyl]-amino]-5-oxazolyl]phenyl]-N-methyl-4-morpholineacetamide as a Novel and Potent Inhibitor of Inosine Monophosphate Dehydrogen-ase with Excellent in Vivo Activity. J Med Chem. 2002;45:2127–2130. doi: 10.1021/jm0105777. [DOI] [PubMed] [Google Scholar]
- 7.Hedstrom L, Liechti G, Goldberg JB, Gollapalli DR. The Antibiotic Potential of Prokaryotic IMP Dehydrogenase Inhibitors. Curr Med Chem. 2011;18:1909–1918. doi: 10.2174/092986711795590129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maher C, Lushniak BD. Availability of Medical Countermeasures for Bioterrorism Events: US Legal and Regulatory Options. Clin Pharmacol Ther. 2009;85:669–671. doi: 10.1038/clpt.2009.55. [DOI] [PubMed] [Google Scholar]
- 9.Franco C, Sell TK. Federal Agency Biodefense Funding, FY2011–FY2012. Biosecurity and Bioterrorism: Biodefense Strategy, Practice, and Science. 2011;9:117–137. doi: 10.1089/bsp.2011.0018. [DOI] [PubMed] [Google Scholar]
- 10.Bentley R. Mycophenolic Acid: A One Hundred Year Odyssey from Antibiotic to Immunosuppressant. Chem Rev. 2000;100:3801–3826. doi: 10.1021/cr990097b. [DOI] [PubMed] [Google Scholar]
- 11.Abraham EP. The effect of mycophenolic acid on the growth of Staphylococcus aureus in heart broth. Biochem J. 1945;39:398–408. doi: 10.1042/bj0390398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Florey HW, Jennings MA, Gilliver K, Sanders AG. Mycophenolic Acid, an Antibiotic from Penicillium brevicom-pactum Dierckx. Lancet. 1946;247:46–49. doi: 10.1016/s0140-6736(46)90242-5. [DOI] [PubMed] [Google Scholar]
- 13.Sintchak MD, Fleming MA, Futer O, Raybuck SA, Chambers SP, Caron PR, Murcko MA, Wilson KP. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophe-nolic acid. Cell. 1996;85:921–930. doi: 10.1016/s0092-8674(00)81275-1. [DOI] [PubMed] [Google Scholar]
- 14.Whitby FG, Luecke H, Kuhn P, Somoza JR, Huete-Perez JA, Phillips JD, Hill CP, Fletterick RJ, Wang CC. Crystal structure of Tritrichomonas foetus inosine-5′-monophosphate dehydrogenase and the enzyme-product complex. Biochemistry. 1997;36:10666–10674. doi: 10.1021/bi9708850. [DOI] [PubMed] [Google Scholar]
- 15.Zhang R, Evans G, Rotella FJ, Westbrook EM, Beno D, Huberman E, Joachimiak A, Collart FR. Characteristics and crystal structure of bacterial inosine-5′-monophosphate dehydro-genase. Biochemistry. 1999;38:4691–4700. doi: 10.1021/bi982858v. [DOI] [PubMed] [Google Scholar]
- 16.Colby TD, Vanderveen K, Strickler MD, Markham GD, Goldstein BM. Crystal structure of human type II inosine monophosphate dehydrogenase: Implications for ligand binding and drug design. Proc Natl Acad Sci USA. 1999;96:3531–3536. doi: 10.1073/pnas.96.7.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMillan FM, Cahoon M, White A, Hedstrom L, Petsko GA, Ringe D. Crystal structure at 2.4 Å resolution of Borrelia burgdorferi inosine 5′-monophosphate dehydrogenase: Evidence of a substrate-induced hinged-lid motion by loop 6. Biochemistry. 2000;39:4533–4542. doi: 10.1021/bi992645l. [DOI] [PubMed] [Google Scholar]
- 18.Prosise GL, Wu JZ, Luecke H. Crystal structure of Tritrichomonas foetus inosine monophosphate dehydrogenase in complex with the inhibitor ribavirin monophosphate reveals a catalysis-dependent ion-binding site. J Biol Chem. 2002;277:50654–50659. doi: 10.1074/jbc.M208330200. [DOI] [PubMed] [Google Scholar]
- 19.Gan L, Petsko GA, Hedstrom L. Crystal structure of a ternary complex of Tritrichomonas foetus inosine 5′-monophosphate dehydrogenase: NAD+ orients the active site loop for catalysis. Biochemistry. 2002;41:13309–13317. doi: 10.1021/bi0203785. [DOI] [PubMed] [Google Scholar]
- 20.Gan L, Seyedsayamdost MR, Shuto S, Matsuda A, Petsko GA, Hedstrom L. The immunosuppressive agent mizoribine monophosphate forms a transition state analogue complex with inosine monophosphate dehydrogenase. Biochemistry. 2003;42:857–863. doi: 10.1021/bi0271401. [DOI] [PubMed] [Google Scholar]
- 21.Prosise GL, Luecke H. Crystal structures of Tritrichomonas foetus inosine monophosphate dehydrogenase in complex with substrate, cofactor and analogs: A structural basis for the random-in ordered-out kinetic mechanism. J Mol Biol. 2003;326:517–527. doi: 10.1016/s0022-2836(02)01383-9. [DOI] [PubMed] [Google Scholar]
- 22.MacPherson IS, Kirubakaran S, Gorla SK, Riera TV, D’Aquino JA, Zhang M, Cuny GD, Hedstrom L. The Structural Basis of Cryptosporidium-Specific IMP Dehydrogenase Inhibitor Selectivity. J Am Chem Soc. 2010;132:1230–1231. doi: 10.1021/ja909947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wierenga RK. The TIM-barrel fold: A versatile framework for efficient enzymes. FEBS Lett. 2001;492:193–198. doi: 10.1016/s0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- 24.Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 25.Ignoul S, Eggermont J. CBS domains: Structure, function, and pathology in human proteins. Am J Physiol. 2005;289:C1369–C1378. doi: 10.1152/ajpcell.00282.2005. [DOI] [PubMed] [Google Scholar]
- 26.Hedstrom L, Gan L. IMP dehydrogenase: Structural schizophrenia and an unusual base. Curr Opin Chem Biol. 2006;10:520–525. doi: 10.1016/j.cbpa.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Pankiewicz KW, Goldstein BM. Inosine Monophosphate Dehydrogenase: A Major Therapeutic Target. American Chemical Society; Washington, DC: 2003. Inosine Monophosphate Dehydrogenase and Its Inhibitors: An Overview; pp. 1–17. [Google Scholar]
- 28.Guillen Schlippe YV, Hedstrom L. A twisted base? The role of arginine in enzyme-catalyzed proton abstractions. Arch Biochem Biophys. 2005;433:266–278. doi: 10.1016/j.abb.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 29.Min D, Josephine HR, Li H, Lakner C, MacPherson IS, Naylor GJP, Swofford D, Hedstrom L, Yang W. An Enzymatic Atavist Revealed in Dual Pathways for Water Activation. PLoS Biol. 2008;6:1802–1810. doi: 10.1371/journal.pbio.0060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiang B, Taylor JC, Markham GD. Monovalent Cation Activation and Kinetic Mechanism of Inosine 5′-Mono-phosphate Dehydrogenase. J Biol Chem. 1996;271:1435–1440. doi: 10.1074/jbc.271.3.1435. [DOI] [PubMed] [Google Scholar]
- 31.Markham GD. Monovalent Cation Activation of IMP Dehydrogenase. ACS Symp Ser. 2003;839:169–183. [Google Scholar]
- 32.Zhang R, Evans G, Rotella F, Westbrook E, Huberman E, Joachimiak A, Collart FR. Differential signatures of bacterial and mammalian IMP dehydrogenase enzymes. Curr Med Chem. 1999;6:537–543. [PubMed] [Google Scholar]
- 33.Fleming MA, Chambers SP, Connelly PR, Nimmesgern E, Fox T, Bruzzese FJ, Hoe ST, Fulghum JR, Livingston DJ, Stuver CM, Sintchak MD, Wilson KP, Thomson JA. Inhibition of IMPDH by Mycophenolic Acid: A Dissection of Forward and Reverse Pathways Using Capillary Electrophoresis. Biochemistry. 1996;35:6990–6997. doi: 10.1021/bi9607416. [DOI] [PubMed] [Google Scholar]
- 34.Link JO, Straub K. Trapping of an IMP Dehydrogenase-Substrate Covalent Intermediate by Mycophenolic Acid. J Am Chem Soc. 1996;118:2091–2092. [Google Scholar]
- 35.Digits JA, Hedstrom L. Drug Selectivity Is Determined by Coupling Across the NAD+ Site of IMP Dehydrogen-ase. Biochemistry. 2000;39:1771–1777. doi: 10.1021/bi992288e. [DOI] [PubMed] [Google Scholar]
- 36.Striepen B, Pruijssers AJP, Huang J, Li C, Gubbels MJ, Umejiego NN, Hedstrom L, Kissinger JC. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc Natl Acad Sci USA. 2004;101:3154–3159. doi: 10.1073/pnas.0304686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Striepen B, White MW, Li C, Guerini MN, Malik SB, Logsdon JM, Liu C, Abrahamsen MS. Genetic complementation in apicomplexan parasites. Proc Natl Acad Sci USA. 2002;99:6304–6309. doi: 10.1073/pnas.092525699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gollapalli DR, MacPherson IS, Liechti G, Gorla SK, Goldberg JB, Hedstrom L. Structural Determinants of Inhibitor Selectivity in Prokaryotic IMP Dehydrogenases. Chem Biol. 2010;17:1084–1091. doi: 10.1016/j.chembiol.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Umejiego NN, Gollapalli D, Sharling L, Volftsun A, Lu J, Benjamin NN, Stroupe AH, Riera TV, Striepen B, Hedstrom L. Targeting a Prokaryotic Protein in a Eukaryotic Pathogen: Identification of Lead Compounds against Cryptospor-idiosis. Chem Biol. 2008;15:70–77. doi: 10.1016/j.chembiol.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. Triazole Inhibitors of Cryptosporidium parvum Inosine 5′-Monophosphate Dehydrogenase. J Med Chem. 2009;52:4623–4630. doi: 10.1021/jm900410u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharling L, Liu X, Gollapalli DR, Maurya SK, Hedstrom L, Striepen B. A Screening Pipeline for Antiparasitic Agents Targeting Cryptosporidium Inosine Monophosphate Dehydro-genase. PLoS Neglected Trop Dis. 2010;4:1–12. doi: 10.1371/journal.pntd.0000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expression Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 43.Walsh MA, Dementieva I, Evans G, Sanishvili R, Joachimiak A. Taking MAD to the extreme: Ultrafast protein structure determination. Acta Crystallogr. 1999;D55:1168–1173. doi: 10.1107/s0907444999003698. [DOI] [PubMed] [Google Scholar]
- 44.Eschenfeldt W, Maltseva N, Stols L, Donnelly M, Gu M, Nocek B, Tan K, Kim Y, Joachimiak A. Cleavable C-terminal His-tag vectors for structure determination. J Struct Funct Genomics. 2010;11:31–39. doi: 10.1007/s10969-010-9082-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed bio-tinylation. Protein Sci. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim Y, Babnigg G, Jedrzejczak R, Eschenfeldt WH, Li H, Maltseva N, Hatzos-Skintges C, Gu M, Makowska-Grzyska M, Wu R, An H, Chhor G, Joachimiak A. High-throughput protein purification and quality assessment for crystallization. Methods. 2011;55:12–28. doi: 10.1016/j.ymeth.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, Molitsky MJ, Keefe L, Gonczy J, Rock L, Sanishvili R, Walsh MA, Westbrook E, Joachimiak A. The Structural Biology Center 19ID undulator beamline: Facility specifications and protein crystallographic results. J Synchrotron Radiat. 2006;13:30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: The integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. 2006;D62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 49.Schneider TR, Sheldrick GM. Substructure solution with. SHELXD Acta Crystallogr. 2002;D58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 50.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macro-molecular refinement. Acta Crystallogr. 2001;D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 51.Cowtan K. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. 1994:34–38. [Google Scholar]
- 52.Morris RJ, Perrakis A, Lamzin VS. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 2003;374:229–244. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 54.Collaborative Computational Project Number 4 . The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 55.Adams PD, Afonine RV, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 57.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 58.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, Richardson DC. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kerr KM, Digits JA, Kuperwasser N, Hedstrom L. Asp338 controls hydride transfer in Escherichia coli IMP dehydrogenase. Biochemistry. 2000;39:9804–9810. doi: 10.1021/bi0005409. [DOI] [PubMed] [Google Scholar]
- 60.Schmitt HM, Brecht A, Piehler J, Gauglitz G. An integrated system for optical biomolecular interaction analysis. Biosens Bioelectron. 1997;12:809–816. [Google Scholar]
- 61.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. 2004;D60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 62.Kerr KM, Cahoon M, Bosco DA, Hedstrom L. Monovalent Cation Activation in Escherichial coli Inosine 5′-Mono-phosphate Dehydrogenase. Arch Biochem Biophys. 2000;375:131–137. doi: 10.1006/abbi.1999.1644. [DOI] [PubMed] [Google Scholar]
- 63.Zhou X, Cahoon M, Rosa P, Hedstrom L. Expression, Purification, and Characterization of Inosine 5′-Mono-phosphate Dehydrogenase from Borrelia burgdorferi. J Biol Chem. 1997;272:21977–21981. doi: 10.1074/jbc.272.35.21977. [DOI] [PubMed] [Google Scholar]
- 64.Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. Cryptosporidium parvum IMP Dehydrogenase: Identification of functional, structural, and dynamic properties that can be exploited for drug design. J Biol Chem. 2004;279:40320–40327. doi: 10.1074/jbc.M407121200. [DOI] [PubMed] [Google Scholar]
- 65.Digits JA, Hedstrom L. Species-Specific Inhibition of Inosine 5′-Monophosphate Dehydrogenase by Myco-phenolic Acid. Biochemistry. 1999;38:15388–15397. doi: 10.1021/bi991558q. [DOI] [PubMed] [Google Scholar]
- 66.Cheng YC, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 67.Burlingham BT, Widlanski TS. An Intuitive Look at the Relationship of Ki and IC50: A More General Use for the Dixon Plot. J Chem Educ. 2003;80:214. [Google Scholar]
- 68.Cooper MA. Optical biosensors: Where next and how soon? Drug Discovery Today. 2006;11:1061–1067. doi: 10.1016/j.drudis.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 69.Small Molecule Binding Kinetics. Technical Note 16, ForteBio Home Page.
- 70.Xiang B, Markham GD. Probing the mechanism of inosine monophosphate dehydrogenase with kinetic isotope effects and NMR determination of the hydride transfer stereospecificity. Arch Biochem Biophys. 1997;348:378–382. doi: 10.1006/abbi.1997.0439. [DOI] [PubMed] [Google Scholar]
- 71.Hedstrom L. IMP dehydrogenase: Mechanism of action and inhibition. Curr Med Chem. 1999;6:545–560. [PubMed] [Google Scholar]
- 72.Antonino LC, Wu JC. Human IMP dehydrogenase catalyzes the dehalogenation of 2-fluoro- and 2-chloroinosine 5′-monophosphate in the absence of NAD. Biochemistry. 1994;33:1753–1759. doi: 10.1021/bi00173a018. [DOI] [PubMed] [Google Scholar]
- 73.Riera TV, Zheng L, Josephine HR, Min D, Yang W, Hedstrom L. Allosteric Activation via Kinetic Control: Potassium Accelerates a Conformational Change in IMP Dehydrogen-ase. Biochemistry. 2011;50:8508–8518. doi: 10.1021/bi200785s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Braun-Perez SB, Peetz M. Inosine mono-phosphate dehydrogenase as a target for antiviral, anticancer, antimicrobial, and immunosuppressive therapeutics. Future Med Chem. 2010;2:81–92. doi: 10.4155/fmc.09.147. [DOI] [PubMed] [Google Scholar]
- 75.Pimkin M, Markham GD. Inosine 5′-monophopshate dehydrogenase. Adv Enzymol Relat Areas Mol Biol. 2009;76:1–53. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.