

Abstract
In bilaterian animals, such as humans, flies and worms, hundreds of microRNAs (miRNAs), some conserved throughout bilaterian evolution, collectively regulate a substantial fraction of the transcriptome. In addition to miRNAs, other bilaterian small RNAs, known as Piwi-interacting RNAs (piRNAs), protect the genome from transposons. Here we identified small RNAs from animal phyla that diverged before the emergence of the Bilateria. The cnidarian Nematostella vectensis (starlet sea anemone), a close relative to the Bilateria, possesses an extensive repertoire of miRNA genes, two classes of piRNAs and a complement of proteins specific to small-RNA biology comparable to that of humans. The poriferan Amphimedon queenslandica (sponge), one of the simplest animals and a distant relative of the Bilateria, also possesses miRNAs, both classes of piRNAs and a full complement of the small-RNA machinery. Animal miRNA evolution seems to have been relatively dynamic, with precursor sizes and mature miRNA sequences differing greatly between poriferans, cnidarians and bilaterians. Nonetheless, miRNAs and piRNAs have been available as classes of riboregulators to shape gene expression throughout the evolution and radiation of animal phyla.
The RNA interference (RNAi) pathway, which processes long double-stranded RNA into small interfering RNAs (siRNAs) and uses them to mediate gene silencing, is present in diverse eukaryotes, presumably with a role in transposon silencing or viral defence since early in eukaryotic evolution1. Building on this basal pathway, which includes the Dicer endonuclease and the argonaute (Ago) effector protein, some eukaryotic lineages have acquired additional pathways, each using unique classes of small RNAs to guide silencing. MicroRNAs, ∼21–24-nucleotide RNAs that derive from distinctive hairpin precursors, pair to messenger RNAs to direct their post-transcriptional repression2. More than one-third of human genes are under selective pressure to maintain pairing to miRNAs, implying that these riboregulators influence the expression of much of the transcriptome3. Piwi-interacting RNAs are longer, ∼27–30 nucleotides, with incompletely characterized biogenic pathways. In mammals and flies, piRNA expression is restricted to the germ line, where they have crucial roles in transposon defence, although one class of mammalian piRNAs, highly expressed at the pachytene stage of sperm development, has unknown function4, 5.
The plant and algal miRNAs have gene structure, biogenesis and targeting properties distinct from those of animals6–8. These differences, considered together with the absence of miRNAs in fungi and all other intervening lineages examined, have led to the conclusion that miRNAs of animals and plants had independent origins6. Of the many miRNAs reported in Bilateria (Fig. 1), ∼30 appear to have been present in ancestral bilaterians9–12; however, none have been reported in the earliest branching animal lineages, leading to the hypothesis that bilaterian complexity might, in part, be due to miRNA-mediated regulation11. Likewise, piRNAs have not been reported outside of Bilateria, raising the question of whether a rich small-RNA biology is characteristic of more complex animals, or whether these small RNAs might have emerged earlier in metazoan evolution.
Figure 1. Phylogenetic distribution of annotated miRNAs.

Cladogram of selected eukaryotes, with organisms investigated in this study indicated in red. Branching order of Bilateria is according to ref. 28 and the references therein, and that of basal Metazoa is according to ref. 17 (Supplementary Discussion). Annotated miRNA tallies are from miRBase (v10.1)29.
Diverse microRNAs of the starlet sea anemone
Eumetazoa includes the Bilateria as well as the Cnidaria, which among sequenced genomes is represented by the starlet sea anemone, Nematostella vectensis13. To explore whether cnidarians have miRNAs, we sequenced complementary DNA libraries generated from 15–30-nucleotide RNAs isolated from Nematostella. High-throughput sequencing yielded 2.9 million reads perfectly matching the Nematostella genome (Fig. 2a). To identify miRNAs, we considered properties that have proved useful for distinguishing bilaterian miRNAs from other types of small RNAs represented in sequencing data14,15. The first criterion was the presence of reads mapping to an inferred RNA hairpin with pairing characteristics of known miRNA hairpins. The second was the presence of reads from both arms of the hairpin that, when paired to each other, formed a duplex with 2-nucleotide 3′ overhangs. This duplex corresponds to an intermediate of miRNA biogenesis in which the miRNA and opposing segment of the hairpin, called the miRNA*, are excised from the hairpin through successive action of Drosha and Dicer RNase III endonucleases2. The third criterion was homogeneity of the miRNA 5′ terminus. Because pairing to miRNA nucleotides 2–8 is crucial for target recognition3, reads matching bilaterian miRNAs display less length variability at their 5′ termini than at their 3′ termini14,15.
Figure 2. The miRNAs of N. vectensis.
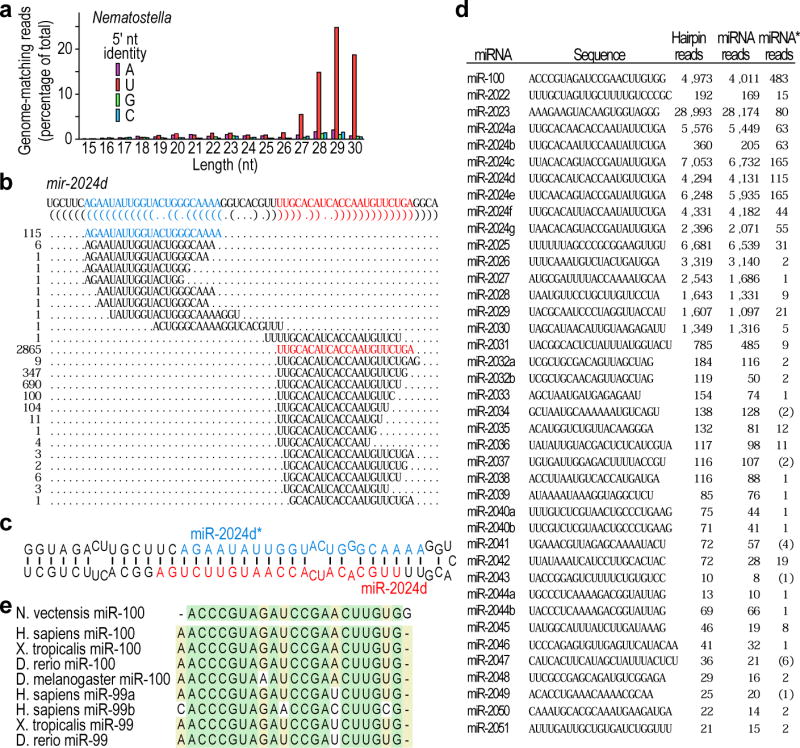
a, Length distribution of genome-matching sequencing reads representing small RNAs, plotted by 5′-nucleotide (nt) identity. Matches to ribosomal DNA were omitted. b, Sequencing reads matching the mir-2024d hairpin. The sequence of the mir-2024d hairpin is depicted above the bracket-notation of its predicted secondary structure. The sequenced small RNAs mapping to the hairpin are aligned below, with the number of reads shown on the left, and the designated miRNA and miRNA* species coloured red and blue, respectively. Analogous information is provided for the other newly identified miRNAs (Supplementary Data 1). c, Predicted secondary structure of the mir-2024d hairpin, indicating the miRNA and miRNA* species. d, The 40 Nematostella miRNAs. MicroRNA read counts include those sharing the dominant 5′ terminus but possessing variable 3′ termini. Occasionally the only sequenced miRNA* species corresponded to a variant miRNA species rather than the major species (counts in brackets). e, Alignment of miR-100 homologues (Danio rerio, D. rerio; Xenopus tropicalis, X. tropicalis).
As exemplified by mir-2024d (Fig. 2b, c), 40 distinct Nematostella loci met these criteria (Fig. 2d and Supplementary Data 1; identical hairpins were not counted because they might have arisen from genome-assembly artefacts). Additional features, not used as selection criteria, resembled those of bilaterian miRNAs2, thereby increasing confidence in our annotations. For example, the loci usually mapped between annotated protein-coding genes (31 loci) or within introns in an orientation suitable for processing from the pre-mRNA (8 loci). The Nematostella miRNAs also had a tight length distribution (centring on 22 nucleotides, Fig. 2d), and 5 groups of miRNAs (corresponding to 13 miRNAs) mapped near to each other in an orientation suitable for production from the same primary transcript (Supplementary Data 1), as occurs in bilaterians2. With the exception of two miRNA pairs (miR-2024a,b and miR-2024f,d), the Nematostella miRNAs had unique sequences at nucleotides 2–8, suggesting a notable diversity of miRNA targeting in this simple animal.
Previous studies that explored the possibility that cnidarians might have miRNAs searched for Nematostella homologs of the ∼30 miRNA families broadly conserved within the Bilateria by probing RNA blots and examining candidate hairpin sequences11,12. These studies reported the possible presence of miR-10, miR-33 and miR-100 family members in Nematostella. None of our reads matched the proposed miR-10, miR-33 or miR-100 homologues, and none matched the proposed hairpin precursors of miR-10 or miR-33. Such discrepancies were not unexpected because detection of distantly related miRNAs by hybridization is prone to false-positives, and many genomic sequences can fold into hairpins. However, one of the newly identified miRNAs arose from the hairpin of the reported miR-100 homologue. The actual miRNA was truncated by one nucleotide at its 5′ terminus compared to bilaterian miR-100 family members (Fig. 2e). Because miRNA-targeting is defined primarily by nucleotides 2–8, this truncation is expected to alter target recognition substantially, with the Nematostella version primarily recognizing mRNAs containing CUACGGG and UACGGGA heptanucleotide sites and the bilaterian versions recognizing mRNAs with two different sites, UACGGGU and ACGGGUA3.
Despite this wholesale shift in their predicted targeting, the Nematostella and bilaterian versions of miR-100 had similarity throughout the RNA, suggesting common origins (Fig. 2e). This result confidently extends the inferred origin of metazoan miRNAs back to at least the last common ancestor of these eumetazoans. Systematic comparison to annotated miRNAs did not reveal any additional Nematostella miRNAs with similarity exceeding that of shuffled control sequences (Supplementary Fig. 1). Although the short length of miRNAs may cause sequence divergence to obscure common ancestry, it is noteworthy that only one of the 40 Nematostella miRNAs appeared homologous to extant bilaterian miRNAs, and even this one seemed to have profoundly different targeting properties.
MicroRNAs near the base of the metazoan tree
To determine whether miRNAs might be present in more deeply branching lineages, we generated 2.5 million genome-matching reads from the small RNAs of the demosponge A. queenslandica, a poriferan thought to represent the earliest diverging extant animal lineage16,17 (Figs 1 and 3a). Eight miRNA genes were identified in Amphimedon adult and embryo samples (Fig. 3b and Supplementary Data 2), exemplified by mir-2018 (Fig. 3c). Six mapped between annotated protein-coding genes; two fell within introns. As is typical for bilaterian miRNAs2 and is also found in Nematostella (Fig. 2d), reads from one arm of the hairpin usually greatly exceeded those from the other arm, enabling unambiguous annotation of the miRNA and mRNA* (Fig. 3b). However, the number of reads from the two arms of the mir-2015 hairpin did not differ substantially, suggesting that each might have similar propensities to enter the silencing complex and target mRNAs. Moreover, the species from the 3′ arm (miR-2015-3p) dominated in adult tissue, whereas the one from the 5′ arm (miR-2015-5p) dominated in embryonic tissue (Fig. 3d), supporting the notion that this single hairpin produces two distinct miRNAs, and implying an intriguing, developmentally controlled differential loading into the silencing complex.
Figure 3. The miRNAs of Amphimedon queenslandica.
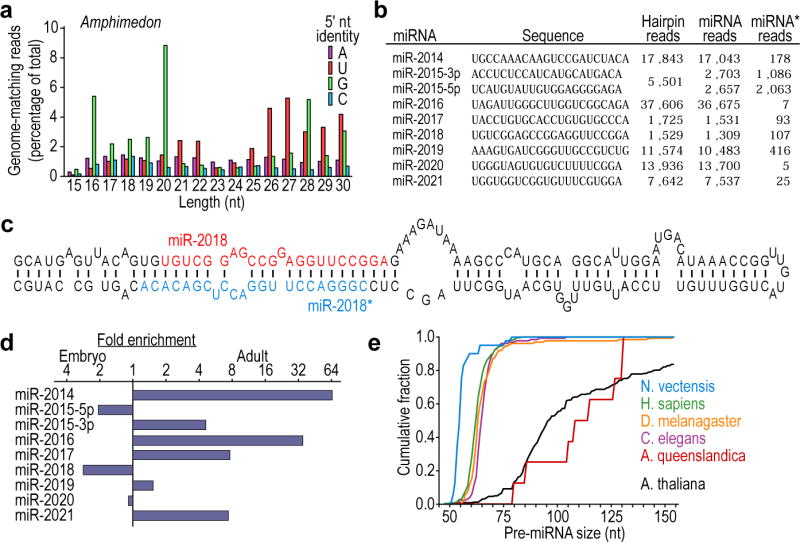
a, Length distribution of genome-matching sequencing reads representing small RNAs, plotted by 5′-nucleotide identity. Matches to ribosomal DNA were omitted. b, The Amphimedon miRNAs, shown as in Fig. 2d. Information analogous to that of Fig. 2b is provided for these miRNAs (Supplementary Data 2). c, Predicted secondary structure of the mir-2018 hairpin. d, Relative expression of Amphimedon miRNAs, as indicated by sequencing frequency from adult and embryo samples. e, Cumulative distributions of pre-miRNA lengths from miRNA transcripts of the species indicated. Amphimedon pre-miRNAs were significantly larger than those from any other animal species examined (P < 10−5, Wilcoxon rank-sum test), whereas those from Nematostella were significantly smaller (P < 10−5).
In Amphimedon, pre-miRNA hairpins were larger than most of those of other metazoans (Fig. 3e). The Nematostella pre-miRNAs (including mir-100) fell at the other end of the spectrum, with a median length less than that of bilaterian pre-miRNAs (Fig. 3e). None of the Amphimedon miRNAs shared significant similarity with any previously described miRNAs (Supplementary Fig. 1), or with the miRNAs found in Nematostella. This observation, combined with their unusually large pre-miRNA hairpins, raised the possibility of an origin independent from that of eumetazoan miRNAs. Arguing against this possibility, we found Amphimedon homologues of Drosha and Pasha proteins (Table 1), which recognize the miRNA primary transcript and cleave it to liberate the pre-miRNA hairpin18. Homologues of these proteins appeared to be absent in all lineages outside the Metazoa, indicating a single origin for these processing factors early in metazoan evolution and implying a single origin for their miRNA substrates.
Table 1. The small-RNA machinery of representative eukaryotes.
| Species | Ago | Piwi | Dicer | Drosha | Pasha | Hen1 |
|---|---|---|---|---|---|---|
| Homo sapiens | 4 | 4 | 1 | 1 | 1 | 1 |
| Drosophila melanogaster | 2 | 3 | 2 | 1 | 1 | 1 |
| Caenorhabditis elegans* | 5 | 3 | 1 | 1 | 1 | 1 |
| Nematostella vectensis† | 3 | 3 | 2 | 1 | 1 | 1 |
| Trichoplax adhaerens† | 1 | 0‡ | 5 | 1 | 0§ | 0‡ |
| Amphimedon queenslandica† | 2 | 3 | 4 | 1 | 1 | 2 |
| Monosiga brevicollis | 0‡ | 0‡ | 0‡ | 0 | 0 | 0‡ |
| Saccharomyces cerevisiae | 0‡ | 0‡ | 0‡ | 0 | 0 | 0‡ |
| Schizosaccharomyces pombe║ | 1 | 0‡ | 1 | 0 | 0 | 0‡ |
| Arabidopsis thaliana | 10 | 0‡ | 4 | 0 | 0 | 2 |
| Physcomitrella patens | 6 | 0‡ | 5 | 0 | 0 | 1 |
| Chlamydomonas reinhardtii | 2 | 0‡ | 3 | 0 | 0 | 1 |
Omitted is a nematode-specific clade of proteins related to the Ago and Piwi protein families but distinct from both27.
Protein sequences are listed in Supplementary Data 3.
Inferred loss based on presence in earlier-diverging lineages.
Inferred loss based on presence in earlier-diverging lineages when assuming that Amphimedon diverged before Trichoplax (Supplementary Discussion).
Ago and Dicer, but not Piwi, Drosha, Pasha or Hen1, were also identified in each of the additional fungal species examined (Aspergillus nidulans, Neurospora crassa and Sclerotinia sclerotiorum).
A third animal lineage branching basal to the Bilateria is Placozoa, represented by the sequenced species Trichoplax adhaerens17. Although earlier analyses of mitochondrial genes suggested that Trichoplax diverged before Amphimedon, genomic data indicate that Trichoplax had a common ancestor with cnidarians and bilaterians more recently than with Amphimedon17 (Fig. 1 and Supplementary Discussion). Our study of Trichoplax small RNAs failed to find miRNAs, despite acquiring many more reads than required to identify miRNAs in all other animals and plants examined (Supplementary Figs 2 and 3). Thus, despite the formal possibility that Trichoplax miRNAs are expressed at levels so low that we failed to detect them, we favour the hypothesis that all miRNA genes have been lost in this lineage. Trichoplax is thought to have derived from a more complex ancestor, having lost, for example, the hedgehog and Notch signalling pathways17. Supporting our hypothesis, no Pasha homologue was found in the Trichoplax genome, although we did find the core RNAi proteins—argonaute and Dicer—suggesting the production and use of siRNAs (Table 1). Drosha, which partners with Pasha during miRNA biogenesis18, was found also but might be required in the absence of miRNAs for ribosomal RNA maturation19. Of the proteins involved in canonical miRNA biogenesis, Pasha is the one without known functions outside the miRNA pathway, and it was the one that appeared to have been discarded, together with all miRNAs, from the Trichoplax genome (Table 1).
We also sequenced small RNAs from the single-celled organism Monosiga brevicollis (Supplementary Fig. 1), which represents the closest known outgroup to the Metazoa20. We failed to detect any plausible miRNAs, a result consistent with our subsequent finding that Monosiga seems to lack all genes specific to small-RNA biology (Table 1). The absence of Dicer and argonaute seemed to be derived rather than ancestral, as the common ancestor of Monosiga and metazoans possessed these core RNAi proteins1 (Table 1). The possibility that the absence of miRNAs in Monosiga might likewise be derived prevented us from setting an early bound on the origin of metazoan miRNAs.
In summary, miRNAs appear to have been available to shape gene expression since at least very early in animal evolution. Nonetheless, the numbers identified in simpler animals (8 unique miRNAs in Amphimedon and 40 in Nematostella) were lower than those reported in more complex animals (Fig. 1). Although miRNAs expressed only under specific conditions or at restricted developmental stages were possibly missed in these and other animals, our results are consistent with the idea that increased organismal complexity in Metazoa correlates with the number of miRNAs and presumably with the number of miRNA-mediated regulatory interactions.
Piwi-interacting RNAs in deeply branching animals
We next turned to the possibility that piRNAs also might have early origins. Piwi proteins, the effectors of bilaterian piRNA pathways, are found in diverse eukaryotic lineages (although not in plants or fungi, Table 1), implying their presence in early eukaryotes1. In cases characterized, however, the small RNAs associated with non-metazoan Piwi proteins resemble siRNAs more than bilaterian piRNAs (deriving, for example, from Dicer-catalyzed cleavage of long double-stranded RNA21), raising the question of when piRNAs of the types found in Bilateria might have emerged. The genomes of both Amphimedon and Nematostella, but not that of Trichoplax, encode Piwi proteins (Table 1) and express many ∼27-nucleotide RNAs with a 5′-terminal uridine (5′-U) (Figs 2a and 3a)—features reminiscent of piRNAs in vertebrates and flies5. Moreover, 45% of Nematostella 5′-U 27–30-nucleotide RNAs originated from only 89 genomic loci (together comprising 0.4% of the genome), the largest of which was 62 kilobases, and essentially all of these small RNAs derived from one strand of each locus (Fig. 4a and Supplementary Table 3). In these respects the genomic loci producing a large fraction of the Nematostella reads closely resembled the loci producing bilaterian piRNAs, particularly the pachytene piRNAs5. We observed a similar clustering of genomic matches of Amphimedon 5′-U 24–30-nucleotide RNAs, although the loci were smaller and accounted for fewer reads (10% of the reads originating from 73 loci comprising 0.2% of the genome, Supplementary Table 4).
Figure 4. The piRNAs of basal metazoans.
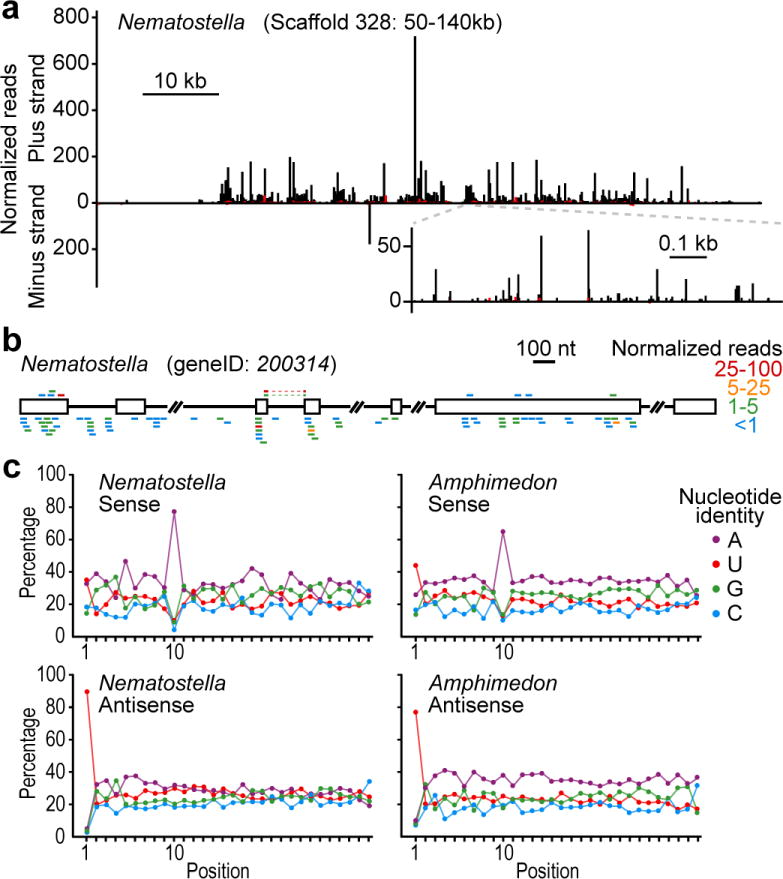
a, Distribution of reads matching a Nematostella piRNA locus. Plotted is the number of matching reads with 5′ nucleotide falling within each 100-nucleotide window (main graph) or at each nucleotide (higher-resolution inset) spanning the genomic region. Bars above and below the x-axis indicate matches to the indicated strand, with black bars indicating reads with a 5′-U and red bars indicating the sum of all other reads. For reads also matching other genomic loci, counts were normalized by total genome matches. Other annotated piRNA loci are presented in Supplementary Tables 3 and 4. kb, kilobases. b, An annotated pre-mRNA corresponding to numerous small RNAs resistant to periodate treatment. Annotated coding segments (open boxes) and intron segments (black line) are indicated. The gene was homologous to endonuclease/reverse transcriptases of other genomes and presumed to be a transposase. Small RNAs with unique 5′ ends are represented by coloured bars above or below the transcript (sense and antisense, respectively), with colours indicating the read numbers (normalized to account for the number of transcriptome matches). Small RNAs matching splice junctions (observed only for sense reads) are represented by discontinuous bars, linked by dashed lines. Other Nematostella and Amphimedon coding regions matching candidate piRNAs are listed in Supplementary Tables 3 and 4. c, Nucleotide composition of periodate-resistant small RNAs matching the indicated strand of Nematostella or Amphimedon annotated coding regions.
Another characteristic of piRNAs is that they undergo Hen1-mediated methylation of their terminal 2′ oxygen22. To test for this modification, we treated RNA from Nematostella and Amphimedon with periodate and then re-sequenced from both treated and untreated samples (Supplementary Fig. 4). Piwi-interacting RNAs and other RNAs modified at their 2′ oxygen remain unchanged with this treatment and are sequenced, whereas those with an unmodified 2′,3′ cis-diol are oxidized, which renders them refractory to sequencing23. In contrast to the Amphimedon miRNAs and many of the Nematostella miRNAs (Supplementary Tables 1 and 2), reads corresponding to the candidate piRNA clusters in both Nematostella and Amphimedon were not reduced after treatment (Supplementary Tables 3 and 4), indicating that their terminal 2′,3′ cis-diol was modified. This modification, considered together with their other features characteristic of vertebrate and fly piRNAs, including the length of 25–30 nucleotides, the 5′-U bias, and the single-stranded, clustered organization of their genomic matches, provided evidence that these small RNAs represented the piRNAs of Nematostella and Amphimedon.
The piRNAs were the type of small RNAs most abundantly sequenced in Nematostella and Amphimedon (Figs 2a and 3a, and Supplementary Discussion). A similar phenomenon is observed in mammalian testes, in which the pachytene piRNAs greatly outnumber the miRNAs and initially obscured detection of a second class of mammalian piRNAs, which resemble the most abundant Drosophila piRNAs with respect to both their biogenesis and their apparent role in suppressing transposon activity24. Most of the Nematostella and Amphimedon genomic loci with clustered piRNA matches resembled the first class of piRNAs, in that they tended to fall outside of annotated genes (P < 10−3, Wilcoxon rank-sum test) and spawned piRNAs predominately from only one DNA strand (>99% and 96% from one strand, Nematostella and Amphimedon, respectively). To determine whether the second class of piRNAs might also exist in deeply branching lineages, we analysed the sequences from periodate-treated samples, focusing on the minority that matched annotated protein-coding genes (Fig. 4b). As expected for class II piRNAs, these piRNAs did not have such a strong tendency to match only one strand of the DNA (62% and 64% antisense for Nematostella and Amphimedon, respectively). Moreover, among the predicted coding regions with the most matches to the piRNAs, a significant fraction (18 of 50 in Nematostella, P < 10−3; 12 of 40 in Amphimedon, P = 0.03, Supplementary Tables 5 and 6) were homologous to transposases.
Having found small RNAs resembling bilaterian class II piRNAs we looked for evidence that they were generated through the same feed-forward biogenic pathway4,25. In this pathway, primary piRNAs from transcripts antisense to transposable elements pair to transposon messages and direct their cleavage. This cleavage defines the 5′ termini of secondary piRNAs generated from the transposon message, and these secondary piRNAs pair to piRNA transcripts, directing cleavage and thereby defining the 5′ termini of additional piRNAs resembling the primary piRNAs. Because the primary piRNAs typically begin with a 5′-U and direct cleavage at the nucleotide that pairs to position 10, the secondary piRNAs typically have an A at position 10. Examination of all 27–30-nucleotide periodate-resistant reads antisense to Nematostella coding regions revealed a propensity for a 5′-U, characteristic of primary piRNAs (Fig. 4c). The sense-strand piRNAs lacked this 5′-U bias and instead displayed a propensity for an A at position 10 (Fig. 4c and Supplementary Fig. 5). Moreover, sense and antisense reads that paired to each other tended to have 10 base pairs formed between their 5′ ends (Supplementary Fig. 6). For the 24–30-nucleotide periodate-resistant reads from Amphimedon, the same hallmark features of the back-and-forth, or ping-pong, amplification cycle for piRNA biogenesis4,25 were observed (Fig. 4c and Supplementary Fig. 6). We conclude that the two classes of piRNAs found previously in mammals and flies have existed since the origin of metazoans: the class I piRNAs, represented by the mammalian pachytene piRNAs, which have unknown function during germline development, and the class II piRNAs, which use the ping-pong cleavage and amplification cascade to quiet expression of certain genes, particularly those of transposons. Indeed, the sequence-based transposon silencing by piRNAs, which by virtue of the feed-forward amplification process focuses on the most active transposon species, might be one of the principle drivers of transposon diversity in animals.
Taken together, our results indicate that miRNAs and piRNAs, as classes of small riboregulators, have been present since the dawn of animal life, and indeed might have helped to usher in the era of multicellular animal life. However, metazoan miRNA evolution seems to have been very dynamic: all miRNAs have been lost in Trichoplax, and the pre-miRNAs of Porifera, Cnidaria and Bilateria have assumed distinct sizes. In addition, no miRNAs have recognizable conservation between poriferans, cnidarians and bilaterians, with only one of the Nematostella miRNAs displaying recognizable homology to bilaterian miRNAs, either because it is the only homologue of extant bilaterian miRNAs or because divergence has obscured common ancestry of other miRNAs. The wholesale shifts in miRNA function implied by this plasticity are congruent with the report that, although thousands of miRNA–target interactions have been maintained within each of the nematode, fly and vertebrate lineages, very few appear to be conserved throughout all three lineages26. The plasticity of miRNA sequences over long timescales helps to explain why the rich small-RNA biology in basal organisms had escaped detection for so long.
Methods Summary
The M. brevicollis library was constructed as described14 and sequenced by 454 Life Sciences. All other libraries (Supplementary Table 7) were constructed using an analogous method and sequenced on the Illumina platform.
Methods
Small RNA sequencing
Samples of N. vectensis (mixed developmental stages, including adult), A. queenslandica (adult tissue, stored in RNAlater, Ambion) and M. brevicollis were ground under liquid nitrogen, and then RNA was extracted with Trizol (Invitrogen). RNA from T. adhaerens (mixed developmental stages, including adult) and A. queenslandica (mixed embryos, from cleavage stage to the larval stage30, stored in RNAlater) was extracted directly with Trizol. The M. brevicollis library was constructed as described14 and sequenced by 454 Life Sciences. All other libraries (Supplementary Table 7) were sequenced on the Illumina platform, and prepared as follows. The 18–30-nt RNAs were purified from total RNA (typically 5 μg) using denaturing polyacrylamide-urea gels. Before purification, trace amounts of 5′-32P-labeled RNA size markers (AGCGUGUAGGGAUCCAAA and GGCAUUAACGCGGCCGCUCUACAAUAGUGA) were mixed with the total RNA and used to monitor this purification and subsequent ligations and purifications. The gel-purified RNA was ligated to pre-adenylated adaptor DNA (AppTCGTATGCCGTCTTCTGCTTG-[3′-3′ linkage]-T) using T4 RNA ligase (10 units ligase, GE healthcare, 10 μl reaction, 50 pmol adaptor ATP-free ligase buffer31, for 2 h at 21–23°). Gel-purified ligation products were ligated to a 5′-adaptor RNA (guucagaguucuacaguccgacgauc), again using T4 RNA ligase (as above, except with 20 units ligase, 15 μl reaction supplemented with 4 nmol ATP, 400 pmol adaptor, for 18 h at room temperature). Gel-purified ligation products were reverse-transcribed (SuperScript II, Invitrogen, 30 μl reaction with the reverse transcription primer CAAGCAGAAGACGGCATA) and then RNA was base-hydrolysed with addition of 5 μl of 1 M NaOH and inclubation at 90 °C for 10 min, followed by neutralization with addition of 25 μl 1 M HEPES, pH 7.0, and desalting (Microspin G-25 column, Amersham). The resulting cDNA library was amplified with the RT primer and PCR primer (AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGA) for a sufficient number of cycles (typically ∼20) to detect (SYBR Gold, Invitrogen) a clear band in a 90% formamide, 8% acrylamide gel, used for purification. Gel-purified amplicon (85–105 nucleotides) from each library was subjected to Illumina sequencing. The adaptor and primer sequences enabled cluster generation on the Illumina machine and placed a binding site for the sequencing primer (CGACAGGTTCAGAGTTCTACAGTCCGACGATC) adjacent to the sequence of the small RNA. Periodate-treated libraries were generated identically, except total RNA was first subjected to β-elimination32. Mock-treated libraries omitting periodate were constructed in parallel.
MicroRNA identification and analysis
The N. vectensis, T. adhaerens and M. brevicollis genomes and predicted gene sets13,17,20 were downloaded from JGI (http://jgi.doe.gov); the A. queenslandica genome was a preliminary assembly16. After removing the adaptor sequences, reads were collapsed to a non-redundant set and matched to the appropriate genome. Genome matches were clustered if neighbouring matches fell within either 50 nucleotides (Amphimedon, Nematostella) or 500 nucleotides (Amphimedon) of each other. The increased size of the clustering window used for the Amphimedon analysis (500 nucleotides) was necessary because the 50-nucleotide window was insufficient to identify all Amphimedon miRNAs, owing to the increased size of their pre-miRNAs (Fig. 3e). No additional miRNAs were identified in Nematostella when using a 500-nucleotide window. Sequences of clusters containing 17–25-nucleotide reads cloned at least twice were folded with RNAfold33. If the most frequently sequenced species was located on one arm of a predicted hairpin and the region of the hairpin corresponding to that sequence contained ≥16 base pairs, the candidate locus was examined manually for characteristics of known miRNAs, using criteria described in the main text. Before comparing between adult and embryonic libraries (Fig. 3d), counts corresponding to each mature miRNA from each library were first normalized by the total number of genome-matching reads in that library.
To detect possible homology between previously known miRNAs and either Nematostella or Amphimedon miRNAs, we searched miRBase (version 10.1) for miRNAs similar to our new miRNAs. Because miRNA conservation is most pronounced within the miRNA 5′ region34, we first identified any known and new miRNAs that shared a hexanucleotide within their first eight nucleotides, allowing two-nucleotide offsets. Because of the limited length of the search sequence, and the large number of miRNAs in miRBase, most Nematostella or Amphimedon miRNAs shared a hexanucleotide with miRBase miRNAs. For all such cases, we then searched for extended similarity between the pairs of miRNAs. With the exception of the miR-100 relationship, no more than chance similarity was observed (Supplementary Fig. 1). However, we cannot rule out the possibility that additional homologous relationships are present but undetectable. Because miRNAs are shorter than most other genetically encoded molecules, sequence divergence can more easily obscure homologous relationships, and although they resist changes in the seed region, which is crucial for target recognition, divergence in this 5′ region can be accelerated with the processes of sub- and neo-functionalization15.
Piwi-interacting RNA identification and analysis
Nematostella 27–30-base polymers and Amphimedon 24–30-base polymers were mapped to their respective genome, and at each matching locus counts were normalized, dividing by the number of genome matches for the sequenced RNA. Windows (1 and 10 kilobases) were slid across the genome to identify regions with both high match-normalized read counts (Nematostella: >1,000; Amphimedon: >100) and high unique sequence counts (Nematostella: >500; Amphimedon: >50). Supplementary Tables 3 (Nematostella) and 4 (Amphimedon) list these regions, reporting the proportion of 5′-U match-normalized reads to each strand and the ratio of match-normalized read counts in periodate-treated compared to mock-treated libraries, after normalization for the number of genome-matching reads in each library. The number of predicted transcripts13,16 overlapping genomic piRNA clusters (Supplementary Tables 3 and 4) was calculated and compared to the number overlapping 1,000 random sets equal in size and number to the piRNA clusters. Inferred protein sequences from predicted transcripts matching the greatest number of periodate-resistant, match-normalized reads were compared to annotated protein sequences using BLAST. Transcripts that were significantly similar to annotated transposons, or protein domains implicated as transposases (for example, reverse transcriptases) were considered to encode transposases. A random selection of 100 predicted transcripts was searched similarly to ascertain significance (Nematostella: 3 out of 100; Amphimedon: 6 out of 100). When mapping to annotated protein-coding regions (Fig. 4b), reads with both sense and antisense matches were distributed to both the sense and antisense tallies after weighting by the proportion of their sense and antisense matches.
Cataloguing of the small RNA machinery
To identify homologues of components of the small RNA machinery, all established family members from H. sapiens, D. melanogaster, C. elegans, S. pombe and A. thaliana were used as BLAST query sequences against all annotated protein sequences of each species in Table 1. The top-ranking hits resulting from these initial searches were used reciprocally as query sequences against all annotated protein sequences of H. sapiens, D. melanogaster, C. elegans, S. pombe and A. thaliana. If the top-ranking hits of such reciprocal queries corresponded to an established family member, the query sequence was considered to be a candidate homologue. The domain structure of each candidate sequence was then evaluated35, and candidates lacking the diagnostic domains were discarded. The diagnostic domains used were a Paz and a Piwi domain (for Ago and Piwi family members), two RNase III domains (Dicer and Drosha), a double-stranded RNA-binding domain (Pasha) and a methylase domain (Hen1).
Supplementary Material
Acknowledgments
We thank M. Abedin and E. Begovic for preparing the Monosiga and Trichoplax samples, respectively, W. Johnston for technical assistance, and J. Grenier, C. Mayr, C. Jan and N. Lau for discussions. This work was supported by an NIH postdoctoral fellowship (A.G.), and from grants from the NIH (D.P.B.), Richard Melmon (M.S., N.K. and D.S.R.), the Center for Intergrative Genomics (M.S. and D.S.R.) and the Australian Research Council (B.F., B.J.W. and B.M.D.). D.P.B. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions A.G. constructed the libraries using procedures developed by H.R.C., and analysed the sequencing reads and protein homology. M.S., B.F., B.J.W., N.K., B.M.D. and D.S.R. provided samples for RNA extraction. A.G. and D.P.B. designed the study and prepared the manuscript, with input from other authors.
Author Information RNA sequencing data were deposited in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE12578. Reprints and permissions information is available at www.nature.com/reprints
References
- 1.Cerutti H, Casas-Mollano JA. On the origin and functions of RNA-mediated silencing: from protists to man. Curr Genet. 2006;50:81–99. doi: 10.1007/s00294-006-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 4.Brennecke J, et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128:1089–1103. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 5.Aravin AA, Hannon GJ, Brennecke J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science. 2007;318:761–764. doi: 10.1126/science.1146484. [DOI] [PubMed] [Google Scholar]
- 6.Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol. 2006;57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218. [DOI] [PubMed] [Google Scholar]
- 7.Molnar A, Schwach F, Studholme DJ, Thuenemann EC, Baulcombe DC. miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature. 2007;447:1126–1129. doi: 10.1038/nature05903. [DOI] [PubMed] [Google Scholar]
- 8.Zhao T, et al. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev. 2007;21:1190–1203. doi: 10.1101/gad.1543507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pasquinelli AE, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- 10.Hertel J, et al. The expansion of the metazoan microRNA repertoire. BMC Genomics. 2006;7:25. doi: 10.1186/1471-2164-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sempere LF, Cole CN, McPeek MA, Peterson KJ. The phylogenetic distribution of metazoan microRNAs: insights into evolutionary complexity and constraint. J Exp Zool. 2006;306:575–588. doi: 10.1002/jez.b.21118. [DOI] [PubMed] [Google Scholar]
- 12.Prochnik SE, Rokhsar DS, Aboobaker AA. Evidence for a microRNA expansion in the bilaterian ancestor. Dev Genes Evol. 2007;217:73–77. doi: 10.1007/s00427-006-0116-1. [DOI] [PubMed] [Google Scholar]
- 13.Putnam NH, et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007;317:86–94. doi: 10.1126/science.1139158. [DOI] [PubMed] [Google Scholar]
- 14.Ruby JG, et al. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 15.Ruby JG, et al. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17:1850–1864. doi: 10.1101/gr.6597907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larroux C, et al. Genesis and expansion of metazoan transcription factor gene classes. Mol Biol Evol. 2008;25:980–996. doi: 10.1093/molbev/msn047. [DOI] [PubMed] [Google Scholar]
- 17.Srivastava M, et al. The Trichoplax genome and the nature of placozoans. Nature. 2008;454:955–960. doi: 10.1038/nature07191. [DOI] [PubMed] [Google Scholar]
- 18.Lee Y, Han J, Yeom KH, Jin H, Kim VN. Drosha in primary microRNA processing. Cold Spring Harb Symp Quant Biol. 2006;71:51–57. doi: 10.1101/sqb.2006.71.041. [DOI] [PubMed] [Google Scholar]
- 19.Fukuda T, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nature Cell Biol. 2007;9:604–611. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- 20.King N, et al. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature. 2008;451:783–788. doi: 10.1038/nature06617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao MC, Chao JL. RNA-guided DNA deletion in Tetrahymena: An RNAi-based mechanism for programmed genome rearrangements. Annu Rev Genet. 2005;39:537–559. doi: 10.1146/annurev.genet.39.073003.095906. [DOI] [PubMed] [Google Scholar]
- 22.Horwich MD, et al. The Drosophila RNA methyltransferase, DmHen1, modifies germline piRNAs and single-stranded siRNAs in RISC. Curr Biol. 2007;17:1265–1272. doi: 10.1016/j.cub.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 23.Seitz H, Ghildiyal M, Zamore PD. Argonaute loading improves the 5′ precision of both MicroRNAs and their miRNA strands in flies. Curr Biol. 2008;18:147–151. doi: 10.1016/j.cub.2007.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 2007;316:744–747. doi: 10.1126/science.1142612. [DOI] [PubMed] [Google Scholar]
- 25.Gunawardane LS, et al. A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science. 2007;315:1587–1590. doi: 10.1126/science.1140494. [DOI] [PubMed] [Google Scholar]
- 26.Chen K, Rajewsky N. Deep conservation of microRNA–target relationships and 3′ UTR motifs in vertebrates, flies, and nematodes. Cold Spring Harb Symp Quant Biol. 2006;71:149–156. doi: 10.1101/sqb.2006.71.039. [DOI] [PubMed] [Google Scholar]
- 27.Yigit E, et al. Analysis of the C. elegans Argonaute family reveals that distinct Argonautes act sequentially during RNAi. Cell. 2006;127:747–757. doi: 10.1016/j.cell.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 28.Bourlat SJ, Nielsen C, Economou AD, Telford MJ. Testing the new animal phylogeny: a phylum level molecular analysis of the animal kingdom. Mol Phylogenet Evol. 2008 doi: 10.1016/j.ympev.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 29.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adamska M, et al. Wnt and TGF-β expression in the sponge Amphimedon queenslandica and the origin of metazoan embryonic patterning. PLoS ONE. 2007;2:e1031. doi: 10.1371/journal.pone.0001031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.England TE, Gumport RI, Uhlenbeck OC. Dinucleoside pyrophosphate are substrates for T4-induced RNA ligase. Proc Natl Acad Sci USA. 1977;74:4839–4842. doi: 10.1073/pnas.74.11.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kemper B. Inactivation of parathyroid hormone mRNA by treatment with periodate and aniline. Nature. 1976;262:321–323. doi: 10.1038/262321a0. [DOI] [PubMed] [Google Scholar]
- 33.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim LP, et al. The microRNAs of Caenorhabditis elegans. Genes Dev. 2003;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchler-Bauer A, et al. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007;35:D237–D240. doi: 10.1093/nar/gkl951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.