

Abstract
Lynch syndrome (LS) is caused by mutations in mismatch repair genes and is characterized by a high cumulative risk for the development of mainly colorectal carcinoma and endometrial carcinoma. Early detection of LS is important since surveillance can reduce morbidity and mortality. However, the diagnosis of LS is complicated by the absence of a pre-morbid phenotype and germline mutation analysis is expensive and time consuming. Therefore it is standard practice to precede germline mutation analysis by a molecular diagnostic work-up of tumours, guided by clinical and pathological criteria, to select patients for germline mutation analysis. In this review we address these molecular analyses, the central role for the pathologist in the selection of patients for germline diagnostics of LS, as well as the molecular basis of LS.
Keywords: Lynch syndrome, mismatch repair genes, MSI analysis, immunohistochemistry, MLH1 promoter methylation assay, BRAF mutation analysis, molecular pathology, review
Lynch syndrome
Colorectal cancer (CRC) is the most common malignancy within the European Union and ranks second to lung cancer as a cause of cancer-related mortality [1]. CRC results from both genetic and environmental factors. The most common genetic susceptibility for CRC is Lynch syndrome (LS), formerly known as hereditary non-polyposis colorectal cancer (HNPCC). LS accounts for approximately 3% of all CRCs [2, 3], and also for 2% of all endometrial cancers [4]. The burden of LS is considerably greater than these percentages imply, as the cancers are diagnosed at a young age and synchronous or metachronous malignancies occur in 30% of the patients [5, 6].
LS is characterized by a high lifetime risk for the development of CRC (20–70%), endometrial cancer (15–70%) and other extra-colonic cancers (<15%) [7–14]. These extra-colonic malignancies include carcinomas of the small intestine, stomach, pancreas and biliary tract, ovarium, brain, upper urinary tract and skin. LS is caused by germline mutations in mismatch repair (MMR) genes [15], and the definitive diagnosis is currently made by identification of an inactivating germline mutation in one of the MMR genes MLH1, MSH2, MSH6 or PMS2[16]. Early detection of LS is of great importance, particularly in pre-symptomatic mutation carriers, since colonoscopic surveillance has proven to reduce CRC morbidity and mortality by 65–70%[17–19] and prophylactic surgery may prevent endometrial and ovarium carcinoma effectively [20]. Individuals with a predisposing mutation are candidates for participation in surveillance programs.
The diagnosis of LS is hampered by the absence of specific diagnostic features and the first manifestation in many patients is the presence of an advanced cancer. Furthermore, DNA mutation analysis is time consuming and expensive. For these reasons, DNA analysis is generally preceded by a molecular diagnostic work-up to select patients as candidates for genetic tests. This molecular diagnostic work-up may be guided by several clinical and pathological criteria such as the presence of LS associated malignancies, number of malignancies and age at cancer diagnosis, family history as well as histological tumour features such as mucinous or signet-ring differentiation. In this review, we address the central role for the pathologist in the selection of patients for germline diagnostics of LS, the molecular analyses to identify LS as well as the molecular basis of LS.
Identification of patients at risk for Lynch syndrome
Different models and strategies have been developed to identify patients with LS. In 1990, the Amsterdam Criteria I were developed to provide a basis for uniformity in collaborative studies to find the disease-causing gene (Table 1) [21]. These criteria were designed to be highly specific at the expense of the sensitivity [3, 22]. They were criticized because extra-colonic tumours were not taken into account, thereby excluding classical LS families. Therefore, the Amsterdam Criteria II were established in 1999 (Table 2) [23]. However, many families with the syndrome (i.e. mutation carriers) do not meet these criteria [24], usually because these families are too small or there is a late onset of the disease. In addition, obtaining a thorough family history is difficult in clinical practice [25] and patients may have limited knowledge of their family history [26, 27].
Table 1.
Amsterdam criteria I [21]
| Families must fulfil all criteria: |
| 1. There should be at least three relatives with a CRC. |
| 2. One should be a first-degree relative of the other two. |
| 3. At least two successive generations should be affected. |
| 4. At least one should be diagnosed before the age of 50 years. |
| 5. Familial adenomatous polyposis (FAP) should be excluded. |
| 6. Tumours should be verified by pathological examination. |
Table 2.
Amsterdam criteria II [23]
| Families must fulfil all criteria: |
| 1. There should be at least three relatives with a LS-associated cancer*. |
| 2. One should be a first-degree relative of the other two. |
| 3. At least two successive generations should be affected. |
| 4. At least one should be diagnosed before the age of 50 years. |
| 5. Familial adenomatous polyposis (FAP) should be excluded in the CRC case(s), if any. |
| 6. Tumours should be verified by pathological examination. |
CRC, cancer of the endometrium, small bowel, ureter or renal pelvis.
In 1997, the Bethesda Guidelines were published to select patients whose tumours should be analysed for molecular features associated with LS, i.e. microsatellite instability (MSI), to identify potential mutation carriers (Table 3) [28]. The Bethesda Guidelines have been revised in 2004 to make them more suitable for use in clinical practice, and are not only based on family history, but also on age at cancer diagnosis, number of LS-associated carcinomas and certain histological tumour features (Table 4) [29]. These histological tumour features, associated with LS, include the presence of tumour-infiltrating lymphocytes, a Crohn’s-like lymphocytic reaction, mucinous or signet-ring cell differentiation and a medullary or undifferentiated and solid growth pattern. The additional value of these pathology characteristics in the selection of tumours for further testing for LS has been described previously [30, 31]. However, these histological features are related to both microsatellite unstable sporadic tumours as well as LS tumours. Therefore the ability to identify LS patients alone on the basis of these tumour features is limited [32]. In addition, the assessment of these histological tumour features indicating MSI is poorly implemented in daily clinical practice [32].
Table 3.
Original Bethesda Guidelines [28]
| Individuals meeting any one of the following should undergo MSI testing: |
| 1. Individuals with cancer in families that meet the Amsterdam criteria. |
| 2. Individuals with two LS-related cancers, including synchronous and metachronous CRCs or associated extracolonic cancers*. |
| 3. Individuals with CRC and a first-degree relative with CRC and/or LS-related extracolonic cancer and/or a colorectal adenoma; one of the cancers diagnosed at age <45 years, and the adenoma diagnosed at age <40 years. |
| 4. Individuals with CRC or endometrial cancer diagnosed at age <45 years. |
| 5. Individuals with right-sided CRC with an undifferentiated pattern (solid/cribriform) on histopathology diagnosed at age <45 years ‡. |
| 6. Individuals with signet-ring-cell-type CRC (more than 50% signet ring cells) diagnosed at age <45 years. |
| 7. Individuals with adenomas diagnosed at age <40 years. |
Endometrial, ovarian, gastric, hepatobiliary or small-bowel cancer or transitional cell carcinoma of the renal pelvis or ureter.
Solid/cribriform defined as poorly differentiated or undifferentiated carcinoma composed of irregular, solid sheets of large eosinophilic cells and containing small gland-like spaces.
Table 4.
Revised Bethesda Guidelines [29]
| Individuals meeting any one of the following should undergo MSI testing: |
| 1. CRC diagnosed in an individual under age 50 years. |
| 2. Presence of synchronous, metachronous colorectal, or other LS-associated tumours*, regardless of age. |
| 3. CRC with the MSI-H histology‡, in a patient <60 years of age. |
| 4. CRC in 1 or more first-degree relatives with a LS-related tumour*, with 1 of the cancers being diagnosed under age 50 years. |
| 5. CRC diagnosed in 2 or more first- or second-degree relatives with LS-related tumours*, regardless of age. |
Endometrial, ovarian, gastric, small bowel, pancreas, hepatobiliary tract, renal pelvis or ureter, and brain tumours, sebaceous gland adenomas and keratoacanthomas.
Presence of tumour-infiltrating lymphocytes, Crohn’s like lymphocytic reaction, mucinous or signet-ring differentiation, or medullary growth pattern.
At present, the most widely accepted recommendation for the identification of patients with LS is based on the combination of these revised Bethesda Guidelines and MSI testing. This combination has proven to be an effective and efficient strategy for LS identification, with a sensitivity for detection of mutation carriers reported from 72%[3] up to 100%[33–36], and a specificity ranging from 77% to 98%[33, 35, 36]. However, these criteria have been criticized because of the use of broad and complex variables, and families with MSH6 and possibly also PMS2 mutations remain undetected [37]. It has also been shown in several studies that these criteria are poorly implemented in clinical practice [32, 38–40].
In 2005, a Dutch group therefore developed a new strategy for the detection of LS [41]. In this strategy the pathologist selects newly diagnosed patients fulfilling one of the following criteria for MSI analysis; (1) CRC before the age of 50 years, (2) Two LS-associated tumours, including synchronous or metachronous CRCs or LS-associated tumours or (3) adenoma before the age of 40 years. These criteria, known as MIPA criteria, simplify the Bethesda guidelines in such a way that pathologists, without knowledge of family history, can easily apply them. These criteria were found to be effective, efficient and feasible in daily practice [41, 42].
In The Netherlands, the diagnosis of LS is currently based on a nationwide guideline for MSI analysis (Table 5), that was introduced in January 2008 (http://www.oncoline.nl). This guideline resembles the MIPA criteria. MSI analysis (and immunohistochemistry of the MMR proteins) is requested by the pathologist in patients newly diagnosed with CRC or endometrial carcinoma before the age of 50 years, or patients with two LS-associated tumours (including synchronous and metachronous CRCs or LS-associated tumours) before the age of 70 years. Presence of multiple LS-associated cancers is registered in PALGA, the nationwide network and registry of histopathology and cytopathology in The Netherlands (http://www.palga.nl). For MSI analysis based on a positive family history, referral to a clinical geneticist is indicated. In those cases MSI analysis will generally be performed when the (revised) Bethesda or Amsterdam Criteria are met and if archival paraffin-embedded tumour tissue can be obtained.
Table 5.
Dutch guideline for MSI testing (http://www.oncoline.nl)
| The pathologist is advised to requests MSI testing (and immunohistochemistry of the MMR proteins) in the following patients: |
| 1. CRC or endometrial carcinoma before the age of 50 years. |
| 2. A second CRC before the age of 70 years. |
| 3. CRC before the age of 70 years AND another synchronous or previous LS-associated tumour*. |
CRC, endometrial, ovarian, gastric, small bowel, pancreas, hepatobiliary tract, renal pelvis or ureter, and brain tumours, sebaceous gland adenomas and keratoacanthomas.
Since clinical criteria do not quantify the likelihood of being a mutation carrier, refined algorithms and multivariable models have been developed to make a quantitative estimation of the risk of carrying a germline MMR-gene mutation, without the requirement of tissue [36]. Several models that combine personal and familial data have been developed, such as the Leiden model, the Edinburgh Model, Premm1,2 and the MMR-pro model [43–46]. One of the advantages of the quantitative models is that the threshold for sensitivity or specificity of the model can be adjusted based upon the clinical situation. However, the role for these models in daily clinical practice remains to be determined.
At present a study (called LIMO and coordinated by the Erasmus MC, Rotterdam, The Netherlands) is performed to determine whether further improvement of LS diagnostics can be obtained by the performance of MSI analysis in CRC patients up to the age of 70 years. MSI analysis is performed in a prospective consecutive series of 1000 newly diagnosed CRC patients ≤70 years, and the results are expected in 2010.
Molecular basis of Lynch syndrome and sporadic MMR-deficient tumours
LS is caused by a germline mutation in one of the MMR genes, most commonly MLH1 and MSH2 (±90%) [47, 48], but also MSH6 and PMS2[37, 49, 50]. LS patients are born with a germline mutation in one of these MMR genes, and acquire inactivation of the second wild-type allele in their tumours, fulfilling Knudson’s two hit hypothesis for inactivation of tumour suppressor genes [51]. Because of the high chance of inactivation of the homologous wild-type allele during life, LS transmits phenotypically in an autosomal dominant fashion. The somatic inactivation of the corresponding wild-type allele occurs almost exclusively by small mutations or (partial) gene loss, and bi-allelic inactivation then leads to complete abolition of the protein function. This results in a defective DNA MMR system, since the protein products of the MMR genes are involved in correction of nucleotide base mismatches and small insertions or deletions that arise during DNA replication [52–54].
The mechanism of MMR has been largely elucidated (Fig. 1). MSH2 (mutS homologue 2) forms a heterodimer with MSH6 (mutS homologue 6), sliding along the DNA as a clamp to identify single nucleotide mispairs and small insertions and deletions [55, 56]. MLH1 (mutL homologue 1) dimerizes with PMS2 (post-meiotic segregation 2) and binds to the MSH2-MSH6 complex. Together this group of four proteins recruits an exonuclease to perform the DNA repair [57, 58]. If any of the four major proteins (MSH2, MLH1, MSH6, or PMS2) is functionally inactive, mismatches are not repaired. A defective DNA MMR system increases the mutation rate and makes the cell vulnerable to mutations in genes controlling cell growth (including tumour suppressor genes and oncogenes), resulting in an elevated cancer risk.
Fig 1.
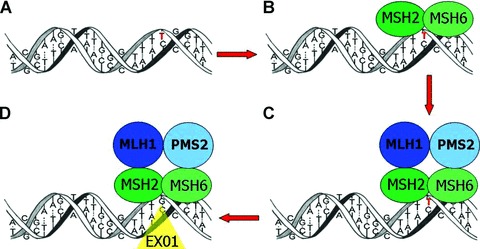
The MMR system. During DNA replication, insertions or deletions of one or more nucleotides and single nucleotide mismatches may occur. For example: (A) A single nucleotide mismatch occurs (G>T in red). (B) MSH2 and MSH6 form a heterodimer and recognize the mismatch. (C) MLH1 and PMS2 dimerize and bind to the MSH2-MSH6 complex. (D) The complex of four proteins activates an exonuclease to perform the DNA repair.
In case of a defective MMR system, mutations occur frequently in small (usually mononucleotide or dinucleotide) repetitive DNA sequences, known as microsatellites [59, 60]. In MMR deficient tumour cells the number of nucleotide repeat units of microsatellites can deviate from the corresponding normal DNA; the number of repeats is usually decreased, but occasionally increased (Fig. 2). This variation in repeat units and thus length or size of microsatellites is called MSI. MSI (formerly referred to as MIN, another abbreviation for MSI, or replication error abbreviated as RER) is the molecular hallmark of LS since approximately 95% of all LS-associated cancers show MSI [61–63]. MSI thereby serves as a reliable phenotypic marker of MMR deficiency which is easy to evaluate in order to pre-select patients for germline mutation analysis of the MMR genes.
Fig 2.
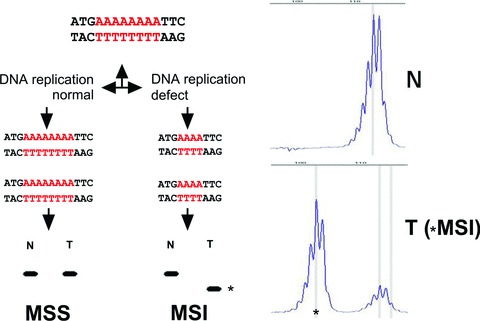
MSI. A schematic microsatellite is indicated (poly A track). When the tumour cells have an intact MMR system the size of the microsatellite will be the same in DNA isolated from normal (N) and from tumour (T) cells: microsatellite stable (MSS) tumour. In case of a defect in MMR the size of the microsatellite (number of repeat units) can change (in most cases becomes shorter) when comparing N with T DNA: microsatellite unstable (MSI) tumour. Asterisks indicate the microsatellite unstable tumour DNA fragment.
Despite the fact that tumour MSI is a reliable marker for MMR deficiency, it is a marker for LS with limited specificity since 15% of sporadic CRCs also demonstrate a MSI phenotype. This is mainly caused by somatic hypermethylation of the MLH1-gene promoter [64, 65]. DNA methylation is an epigenetic DNA modification that specifically targets cytosine residue at CpG dinucleotides. Genomic regions that contain a high frequency of CpG dinucleotides are called CpG islands, present in the promoters of about 40% of all human genes, including the MLH1-gene [66]. Hypermethylation of CpG islands in the MLH1 promoter causes severe inhibition of gene transcription thereby functionally mimicking an inactivating gene mutation. If both copies of the gene are inactivated (mainly by bi-allelic hypermethylation), the DNA MMR function of MLH1 is lost. This leads to microsatellite unstable cancers, especially in older patients [65]. MLH1 deficient microsatellite unstable tumours can be assessed for MLH1 hypermethylation to distinguish sporadic CRCs from LS-related cancers. Theoretically, sporadic hypermethylation of the other MMR genes is possible but has not yet been demonstrated.
Specific activating mutations in the BRAF oncogene, usually V600E missense mutations (formerly reported as V599E), can be detected in 40–87% of all sporadic microsatellite unstable tumours. An oncogenic BRAF mutation has been described only once [67] in numerous investigated LS tumours [68–75, 76]. These results indicate that BRAF mutations are closely correlated with MLH1 methylation in sporadic CRCs [69–72, 76, 77]. Therefore, BRAF mutation status can be used to identify sporadic microsatellite unstable tumours, although it has been demonstrated that determination of hypermethylation of the MLH1 gene promoter is more sensitive to detect sporadic MSI tumours [69].
In addition to sporadic forms of MLH1 promoter hypermethylation, germline epimutations of MLH1 (soma-wide mono-allelic hypermethylation of the gene promoter) have also been reported [78–85]. Germline MLH1 hypermethylation, often showing some degree of mosaicism, is functionally equivalent to an inactivating mutation and produces a clinical phenotype that resembles LS. Inheritance of epimutations is weak as the methylation can be cleared on passage through the germline (germline MLH1 promoter epimutations are reversible during meiosis) and so can display non-Mendelian inheritance. Heritability of epimutations might also be explained by the inheritance of an unknown predisposition to epimutations, rather than the inheritance of the epimutation itself [86]. Although very rare, germline MLH1 promoter methylation should be considered in younger individuals or individuals with multiple LS-associated tumours without a family history who present with an MSI tumour showing loss of MLH1 expression [81, 82].
Besides germline MLH1 hypermethylation, a new mechanism of germline MSH2 hypermethylation has recently been discovered [87]. Ligtenberg et al. showed that a germline deletion of the last two exons of TACSTD1, the gene just upstream of MSH2 encoding epithelial cell adhesion molecule (EpCAM), leads to inactivation of the MSH2 gene by promoter hypermethylation exclusively in tissues expressing EpCAM (mosaic pattern). This mechanism may cause LS in patients with MSH2-deficient microsatellite unstable tumours with an undetectable MSH2 germline mutation. Identification of these cases is possible by the determination of the methylation status of the MSH2 gene promoter in the tumour and in EpCAM expressing normal tissues (e.g. normal colorectal mucosa). In addition, evidence for the presence of MSH2 methylation can be obtained by detection of deletions in the 3′ end of the TACSTD1 gene.
Molecular diagnostics of Lynch syndrome
The molecular diagnostics of LS usually starts with MSI analysis. MSI analysis is traditionally performed with a panel of five microsatellite markers proposed by a NCI (National Cancer Institute) sponsored consensus conference, also known as the Bethesda panel [29]. With these markers, microsatellites in tumour DNA are compared to microsatellites in corresponding DNA from normal tissue. Tumours with more than one unstable marker (or ≥40% of markers) are categorized as having a high degree of MSI (MSI-H), which is suspect for LS or epigenetic MLH1 silencing [88–91]. Those with one unstable marker (20–40% of markers) are categorized as having a low degree of MSI (MSI-L) and tumours with no instability (≤20%) are categorized as being microsatellite stable (MSS), seen in sporadic carcinomas [92]. Although there are no clear differences in clinical or pathological features between MSI-L and MSS tumours, it has been speculated that MSI-L tumours comprise an independent phenotype [93]. However, there is nowadays no role for separating MSI-L from MSS tumours in the diagnostic work-up. Furthermore, MSI testing seems not only important for recognition of LS, but may in the future also improve the clinical management of CRC patients. This is because patients with microsatellite unstable CRCs appear to have a better prognosis than patients with MSS tumours [94–97] and they do not seem to benefit from adjuvant chemotherapy with 5-fluorouracil [98–100].
The Bethesda panel, comprising two mononucleotide repeats (BAT-25 and BAT-26) and three dinucleotide repeats (D2S123, D5S346 and D17S250) [62], does have some limitations, mainly caused by the dinucleotide repeats. These repeats are highly polymorphic and less sensitive and specific in the identification of MSI-H tumours than mononucleotide repeats. Their use in MSI screening requires analysis of corresponding germline DNA [101] and the interpretation of size alterations in dinucleotide repeats is more difficult due to stutter, a PCR artefact. Their use can result in misclassification of MSI-L tumours as MSI-H [102, 103]. Furthermore, MSH6 mutation carriers may develop tumours (predominantly endometrial cancer) without alteration in these dinucleotide repeats leading to false MSI-L or MSS results [104, 105].
The limitations of the Bethesda panel have lead to the development of a pentaplex panel, which comprises five quasi-monomorphic mononucleotide repeats (see below). This panel shows less variation in size among different ethnic populations and has been shown to be superior to the Bethesda panel for the detection of MSI-H tumours [102, 106]. Because the pentaplex analysis is carried out in a single multiplex PCR, this method is simple to use and is free of errors due to mixing samples.
To gain insight into what gene might be affected in patients with MSI-H tumours, MLH1, MSH2, MSH6 and PMS2 protein expression can be assessed by immunohistochemistry. The combination of MSI analysis and MMR protein immunostaining is generally considered as the superior strategy for the identification of suspected LS patients [107]. Absence of MMR protein nuclear staining within the tumour cells can be compared to nuclear staining in the normal cells within the same tumour specimen (and same histological section). The latter then serve as internal positive control.
Due to their heterodimeric nature, different immunohistochemical staining patterns of the MMR proteins can be observed (Table 6). Loss of MLH1 protein due to MLH1 gene mutation or promoter hypermethylation is usually accompanied with absence of PMS2 in the tumour (Fig. 3). Similarly, absence of MSH2 due to MSH2 mutations results in absence of MSH6 (Fig. 3), since MSH6 and PMS2 will disintegrate without their obligatory partners MSH2 and MLH1, respectively. A mutation in either PMS2 or MSH6 does not lead to loss of MLH1 and MSH2 protein, respectively (Fig. 3), because of the formation of other heterodimers than MLH1-PMS2 and MSH2-MSH6. MLH1 can for instance dimerize with either MLH3 or PMS1 [108, 109] and MSH2 can also bind to MSH3 [110]. Due to the binding of MLH1 and MSH2 to other MMR proteins in the absence of PMS2 or MSH6, there is no concurrent loss of MLH1 and MSH2 [111]. To date no bona fide involvement of PMS1, MLH3 or MSH3 (inactivating mutations) has been demonstrated in LS.
Table 6.
Immunohistochemical expression patterns associated with MMR-gene mutations
| MMR-gene mutation | |||||
|---|---|---|---|---|---|
| MLH1 | MSH2 | MSH6 | PMS2 | ||
| Protein expression | MLH1 | −* | + | + | + |
| MSH2 | + | − | + | + | |
| MSH6 | + | − | − | + | |
| PMS2 | − | + | + | − | |
Absent MLH1 protein expression can be associated with either MLH1 germline mutations as well as epigenetic MLH1 silencing by promoter methylation.
+= present nuclear protein expression in tumour cells (as well as in normal cells).
−= absent nuclear protein expression in tumour cells (and present staining in normal cells, thus serving as internal positive control).
Fig 3.
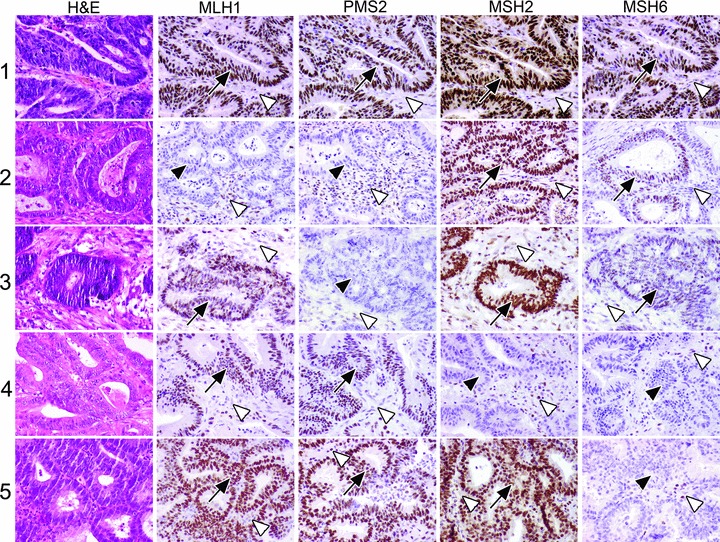
Five CRC cases; haematoxylin and eosin staining and MLH1, PMS2, MSH2 and MSH6 immunohistochemistry (IHC) results. Case 1: normal IHC in the tumour cells. Case 2: absence of MLH1 and PMS2 in the tumour cells. Case 3: absence of PMS2 in the tumour cells. Case 4: absence of MSH2 and MSH6 in the tumour cells. Case 5: absence of MSH6 in the tumour cells. Arrows point to IHC+ tumour cells, filled arrow heads point to IHC– tumour cells and open arrow heads point to IHC+ stromal cells.
In general, absent MSH2, MSH6 or PMS2 expression in tumour cells with present staining in normal cells is suspect for underlying LS and calls for germline testing. Absent MLH1 (and PMS2) expression can indicate either LS or a sporadic tumour with epigenetically silenced MLH1[111]. If epigenetic MLH1 silencing has been excluded by the analysis of MLH1 hypermethylation and/or BRAF mutation analysis, MLH1 germline mutation testing is indicated. Furthermore, it might theoretically be possible that immunohistochemical absence of PMS2 or MSH6 without concomitant absence of MLH1 or MSH2, respectively, is due to mutations in MLH1 or MSH2. These mutations then will not lead to decreased MLH1 and MSH2 immunostaining, while binding to and expression of PMS2 and MSH6, respectively, is abrogated. Therefore, absent PMS2 or MSH6 immunostaining without detectable mutations in the PMS2 or MSH2 gene, asks for mutation analysis of MLH1 or MSH2 respectively.
At our institution, MSI analysis and immunohistochemistry are requested either by the pathologist when patients fulfil the criteria as depicted in Table 5, or by the clinical geneticist (or clinician) when individuals meet the Bethesda Guidelines. The flowchart of the molecular diagnostics of LS in The Netherlands is depicted in Fig. 4. All these different molecular diagnostic procedures will be described in more detail in the next paragraphs.
Fig 4.
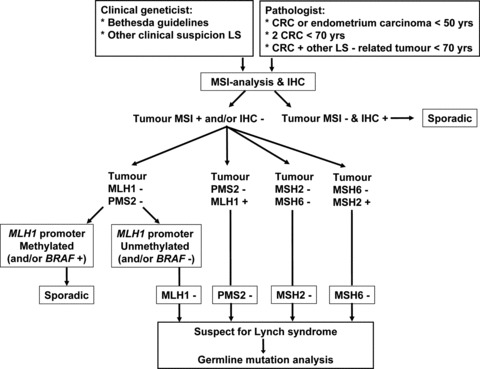
Flowchart for molecular diagnostics of LS in The Netherlands.
Description of molecular analyses
MSI analysis
From routine formalin fixed and paraffin embedded (FFPE) tumour tissue specimens 10 to 20 consecutive sections of 4 μm are cut and routinely glued on microscope glass slides. The number of sections is determined by the size of the tissue fragments that need to be isolated for DNA analysis. All sections are deparaffinized and the first and the last section of the series are routine Mayer haematoxylin and eosin stained. These sections are used as reference for the isolated tissue parts. The intermediate sections are stained in haematoxylin and rinsed in distilled water. The indicated tumour and normal tissue fragments are then manually scraped in distilled water from the glass slide and transferred to Eppendorf vials. From the remaining tissue fragments on the glass slides, routine microscopic preparations are made after additional staining with eosin. With these preparations the isolated tissue fragments can be verified (Fig. 5). Occasionally, when large and easy recognizable tissue fragments can be isolated, scraping is performed from paraffin sections on glass slides without deparaffinization. Furthermore, when the tissue fragments to be isolated are too small for manual isolation, laser microdissection is used on haematoxylin and eosin stained sections glued on membrane containing glass slides (PALM Membrane Slides, P.A.L.M. Microlaser Technologies AG, Bernried, Germany) (Fig. 5).
Fig 5.
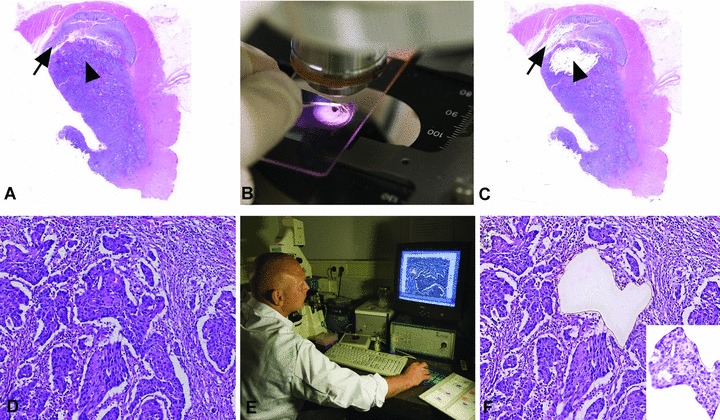
(A, B, C) Manual macrodissection of a normal tissue fragment (arrow) and a region of the tumour composed of a high percentage of tumour cells (arrow head), before, during and after macrodissection, respectively. (D, E, F) Laser capture microdissection of a small tumour tissue fragment surrounded by abundant stromal cells, before, during and after laser capture microdissection, respectively. Insert in (F) shows the microdissected fragment.
Although MSI can be reliably detected even when DNA is isolated from a tissue fragment composed of only 10% neoplastic cells (unpublished data), tumour DNA is isolated preferably from a tissue fragment with a high percentage (>70%) of tumour cells. DNA isolated from tissue with a high percentage of tumour cells can also be used for reliable additional investigations (BRAF mutation and MLH1 hypermethylation). In the case of an adenoma the fragment with the highest grade of dysplasia should be used for DNA isolation. For isolation of normal DNA a tissue fragment composed of normal cells, preferably from the normal epithelial counterpart of the tumour (e.g. normal colorectal or normal endometrial mucosa), is used to circumvent heterogeneity problems that can be caused by mosaicism (e.g. mosaic MLH1 promoter germline hypermethylation or MSH2 promoter hypermethylation only in Epcam expressing cells). However, since these mosaic phenomena are very rare, other normal tissue fragments (e.g. a tumour-negative lymph node) can be used for normal DNA isolation, in cases where there is no, or not easy to isolate, normal mucosa available.
From the microdissected FFPE tissue fragments DNA is extracted by addition of 100 to 200 μl (when very small tissue fragments are used digestion is performed in a volume down to 25 μl) lysis buffer (10 mM Tris/HCL pH 8.0, 1 mM ethylenediaminetetraacetic acid [EDTA] pH 8.0, 0.01% Tween 20) containing 2 mg/ml proteinase K and 5% Chelex 100 resin. Following overnight incubation at 56°C, proteinase K is inactivated at 100°C for 10 min. Next, dissolved DNA is separated from cell debris by centrifugation at maximum speed in a microcentrifuge for 5 min. The DNA-containing supernatant is carefully pipetted from the Chelex resin-containing pellet (Chelex resin inhibits polymerase activity) and transferred to another Eppendorf vial. In case un-deparaffinized sections were used for DNA isolation, the DNA-containing supernatant is collected by carefully poking the pipette tip through the solidified paraffin layer on top of the supernatant.
Different methods for MSI analysis are currently available. In our laboratory we use the MSI analysis system of Promega (Promega, Madison, WI, USA) [103]; a fluorescent multiplex PCR-based assay in which the PCR products are separated by capillary electrophoresis using an ABI PRISM 3130xl genetic analyser (Applied Biosystems, Foster City, CA, USA). PCR is performed according to the kit instructions in a total volume of 10 μl including 2 μl of an about 80-fold dilution of the isolated DNA solution. The output data are analysed with GeneMarker software (SoftGenetics, State College, PA, USA) to determine MSI status of tumour samples. This system includes fluorescently labelled primers for co-amplification of five quasi-monomorphic mononucleotide repeat markers BAT-25, BAT-26, NR-21, NR-24 and MONO-27. In addition, 2 pentanucleotide markers (Penta C and Penta D) characterized by a high level of polymorphism have been added to provide information on possible sample mix-up or contamination. Because of the low size variation in the population of the selected mononucleotide markers this analysis allows, in most cases, that only tumour DNA is investigated for MSI. DNA from a MSS cell line suffices as normal DNA reference. If inconclusive results are obtained, for example due to the infrequent occurrence of bi-allelic variation or borderline shifts of the marker peaks, the assay is repeated with both tumour and patient matched normal DNA. Furthermore, additional mononucleotide MSI markers such as BAT-40 can be used in the case of a MSS tumour with a strong clinical suspicion for underlying LS. Results of MSS and microsatellite unstable tumours are shown in Fig. 6.
Fig 6.
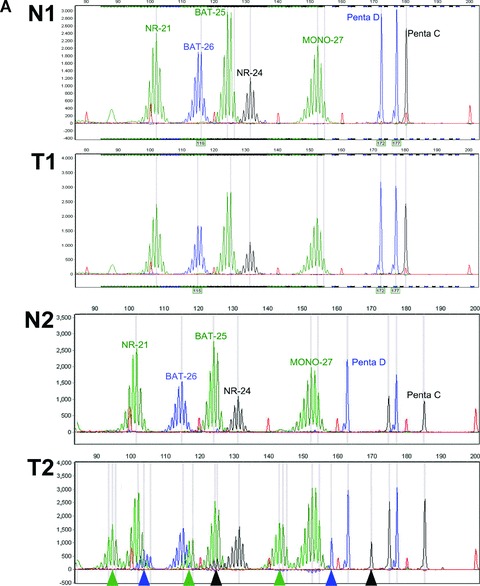
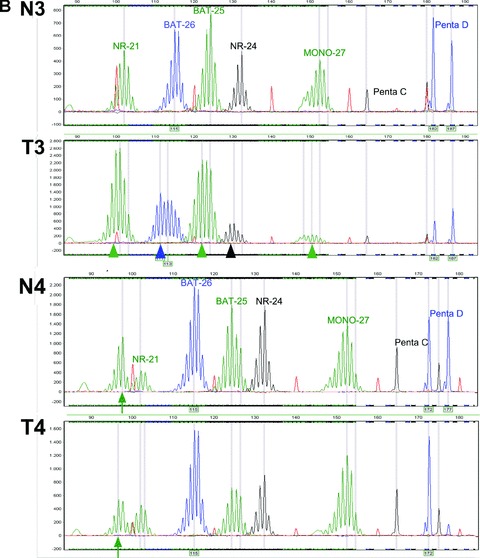
MSI analysis of four cases of paired normal (N) and tumour (T) DNA. (A) Case 1: N1 and T1, CRC no MSI: MSS. Case 2: N2 and T2, CRC demonstrates clear MSI. (B) Case 3: N3 and T3, endometrial carcinoma with MSI (subtle microsatellite shifts). Case 4: N4 and T4, CRC without MSI but with heterozygous NR-21 microsatellite alleles (present in normal and tumour DNA). Arrow heads indicate the MSI shifts, arrows indicate the variant NR-21 allele.
Immunohistochemistry
Our method of immunohistochemistry was described in detail previously [112]. Briefly, FFPE tissue sections (4 μm) are dewaxed, and antigen retrieval is performed in 10 mM Tris-EDTA buffer, (pH 9.0) in a microwave oven for 45 min. at 100°C. Primary antibodies anti-MLH1 (Pharmingen BD, Alphen aan den Rijn, The Netherlands; clone G168–728; dilution, 1:20), anti-MSH2 (Pharmingen BD; clone G219–1129; dilution, 1:300), anti-MSH6 (Pharmingen BD; clone 44; dilution, 1:100) and anti-PMS2 (Pharmingen BD; clone A16–4; dilution, 1:50) are applied for 1 hr at room temperature. After washing, immunoreactivity is visualized with the Envision kit (Dako, Glostrup, Denmark). Subsequently, the sections are counterstained with Mayer haematoxylin and evaluated under a light microscope (Fig. 3).
MLH1 promoter hypermethylation assay
In case of absent MLH1 expression in tumour cells, the methylation status of the MLH1 promoter can be determined by different methods such as methylation-specific PCR [113] and methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) [114]. MS-MLPA is performed with the SALSA MS-MLPA Kit ME011-A1 for MMR genes (MRC-Holland, Amsterdam, The Netherlands). The analysis is performed according to the kit instructions with 3 μl undiluted DNA solution as input. The assay takes advantage of methylation-sensitive endonuclease HhaI, which only cleaves unmethylated DNA fragments. The MS-MLPA kit contains 8 control probe sequences and 21 methylation-sensitive probes of which 5 recognize CpG dinucleotides within the MLH1 promoter. The methylation-sensitive probes contain a restriction site for HhaI. Comparison of a HhaI-digested DNA sample (yielding only signal of methylated DNA) to its undigested counterpart (yielding signal of both methylated and unmethylated DNA) provides insight into the degree of methylation. Details of the MS-MLPA protocol are freely available on the website of the manufacturer (http://www.mrc-holland.com).
Basically, tumour DNA is hybridized to the probe mix. After hybridization, half of the sample is subjected to a ligation step joining both adjacently hybridized fragments of a probe set, whereas the other half of the sample is subjected to both ligation and HhaI digestion, leaving only methylated sequences intact. Subsequent PCR amplification exponentially amplifies all ligated, but undigested, probes. The signal generated with the part of the sample that has undergone both ligation and digestion represents the amount of methylated DNA present in the tumour. For fragment analysis, PCR products are separated by capillary gel electrophoresis using an ABI PRISM 3130xl genetic analyser (Applied Biosystems) and quantified with GeneMarker software version 1.7 (SoftGenetics). The MS-MLPA results are normalized by dividing the peak height of each MLH1 probe signal by the mean peak height of the eight control fragments obtained with the same sample (Fig. 7). The degree of methylation for individual MLH1 probes can be assessed by dividing normalized values of each MLH1 probe within digested DNA samples by normalized values of the probe in corresponding undigested samples. The MS-MLPA assay is performed with both tumour and normal mucosal DNA to detect possible germline MLH1 promoter hypermethylation.
Fig 7.
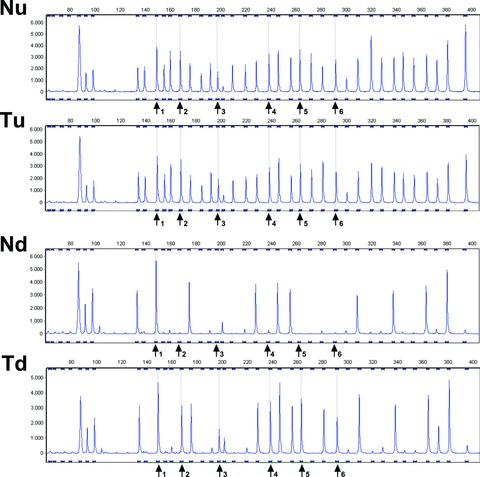
MLH1 promoter hypermethylation assay; MS-MLPA analysis of a MSI CRC with absence of MLH1 and PMS2 expression. Results are shown of paired undigested normal (Nu) and undigested tumour (Tu) DNA and of the methylation-sensitive endonuclease HhaI digested normal (Nd) and tumour (Td) DNA. Arrows indicate six MLH1 promoter probes, nr 1 representative for a fragment without and nrs 2–6 representative for fragments with a HhaI restriction site. In the HhaI digested DNA, probes 2–6 are clearly present in the tumour DNA (Td) and not in the paired normal DNA (Nd) indicating tumour-specific methylation of the MLH1 promoter fragments 2–6.
BRAF mutation analysis
BRAF alterations of mutational hotspot codon V600 are determined by bi-directional cycle sequencing of PCR-amplified fragments. PCR amplification is performed by M13-tailed forward primer 5′-TGT AAA ACG ACG GCC AGT AAA CTC TTC ATA ATG CTT GCT CTG -3′ and M13-tailed reverse primer 5′-CAG GAA ACA GCT ATG ACC GGC CAA AAA TTT AAT CAG TGG AA-3′. PCR products are generated in a 15 μl reaction mixture including 1.0 μl undiluted DNA solution, 10 μmol of each primer, 25 mM MgCl2, 10 mM dNTPs and 1 U Taq polymerase (Promega). The PCR reaction is performed with a thermocycler (Biometra, Göttingen, Germany) with an initial denaturating step (95°C) for 3 min., followed by 35 cycles consisting of denaturation (95°C) for 30 sec., annealing (60°C) for 45 sec. and extension (72°C) for 45 sec. After the final cycle, an extension period of 10 min. at 72°C is performed. The PCR products are sequenced with M13 forward primer 5′-TGT AAA ACG ACG GCC AGT-3′ and M13 reverse primer 5′-CAG GAA ACA GCT ATG ACC-3′ using the ABI PRISM BigDye Terminator v3.1 kit (Applied Biosystems). Sequence analyses are performed on an ABI PRISM 3130xl genetic analyser (Applied Biosystems). Samples are analysed using Mutation Surveyor software (SoftGenetics) and are compared with the public sequence of GenBank (NT_007914). Examples of BRAF mutation analysis results are shown in Fig. 8.
Fig 8.
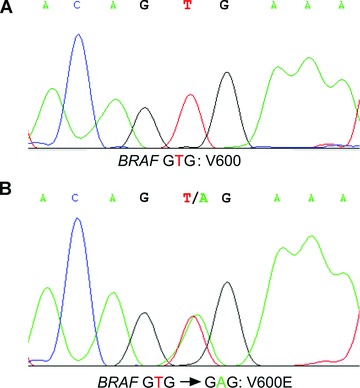
Mutation analysis of codon 600 of the BRAF gene. (A) Tumour DNA without BRAF codon 600 mutation. (B) Tumour DNA with a heterozygous oncogenic mutation GTG-GAG, leading to a V600E amino acid substitution.
Limitations of molecular analyses
Over the last decade, the diagnostics of LS have improved considerably. Nevertheless, there still remain some limitations that need to be addressed. It has to be taken into account that the described procedures provide information on the chance that a certain tumour arose in the context of LS and are not diagnostic for LS in an absolute sense. The false negative rate of MSI analysis is very low (<5%) but cannot be completely ruled out. MSI can be very subtle or escape detection particularly in low grade lesions as adenomas, in endometrial carcinomas (Fig. 6B, panel N3/T3) and in samples with a low percentage of neoplastic cells [115]. These false-negative results may lead to the exclusion of LS patients (and affected family members) from necessary surveillance programs and subsequent failure to detect (secondary) cancers in an early stage. In addition, although rare, sporadic MSS tumours can occur in LS patients and MSI analysis then fails to indicate LS. To exclude false-negative MSI results as much as possible it is necessary to isolate DNA from a tissue fragment with a high percentage of tumour cells. For this, laser microdissection might be preferable instead of manual microdissection. However, laser microdissection is a time consuming and labour-intensive procedure to obtain sufficient tissue fragments for DNA isolation [116]. In general, it is recommended to refer patients with a high clinical or familial suspicion of LS to a clinical genetics department, irrespective of the MSI status. In addition, other hereditary CRC syndromes such as attenuated familial adenomatous polyposis, MYH-associated polyposis, Cowden syndrome or Peutz-Jeghers syndrome might need to be excluded.
Although the assessment of MMR-protein expression by immunohistochemistry is a fast and simple procedure, the interpretation of the results can be difficult. Interpretation may be impeded by absence or low intensity of the nuclear staining in tumour and normal tissue due to fixation artefacts, especially in old archival specimens [117]. In case of missense mutations, the inactive protein may be (partly) expressed and detectable by immunohistochemistry. The interpretation is also hampered by some degree of observer-variation and the value of immunohistochemistry partially depends on the experience of the pathologist [118, 119]. For these reasons immunohistochemistry cannot replace MSI testing to detect LS, and this underlines the importance of the combined application of MSI analysis and MMR protein immunostaining to detect LS.
In the evaluation of MLH1 promoter methylation, it is important to study the correct promoter regions since MLH1 expression only correlates with methylation of the proximal promoter regions (mainly region C, but also region D) [113, 120, 121]. Nevertheless, there are still studies published in which the distal MLH1 promoter regions were analysed, which are not or only poorly associated with gene silencing. Moreover, epigenetic inactivation of the second normal MLH1 allele by promoter methylation (second hit), may also play a role in individuals with LS [122, 123], and it should be realized that the detection of MLH1 promoter methylation can not completely rule out LS. In the case of a strong clinical suspicion, referral to a clinical geneticist is indicated. The exact frequencies of MLH1 promoter methylation in LS patients (either as a second inactivating event, or as a heritable germline epimutation), are unknown. It has been reported that in tumours from MLH1 mutation carriers, the wild-type allele is hypermethylated in 0–46% of the tumours [65, 76, 80, 122, 124–129]. However, only one study evaluated the proximal promoter region (region D) associated with gene silencing in 55 CRCs and endometrial cancers of MLH1 germline mutation carriers [129]. Hypermethylation was seen in 7.3% of all tumours (16% of CRCs). In the other studies, promoter regions not associated with MLH1 silencing were investigated (i.e. the distal promoter regions) [65, 76, 80, 122, 124–128].
There are some other points of concern in the molecular diagnostics of LS. First, the value of MSI testing and immunohistochemistry in other LS-related tumours than CRC is largely unknown [130]. In endometrial tumours, the second most common malignancy in LS, MSI can escape detection by the occurrence of only subtle shifts in the size of the markers [131]. Therefore, MSI analysis in endometrial cancers is performed with patient matched normal DNA as the reference, and molecular pre-screening has been found feasible [4, 132, 133] (Fig. 6B, panel N3/T3). Furthermore, the quality of DNA extracted from FFPE tumours can occasionally be poor and therefore not suitable for MSI analysis [130]. And last but not least, some individuals might have ethical objections against MSI testing or immunohistochemistry, since the diagnosis of LS can be very likely after the described molecular examinations, which might have negative social consequences and raise concerns e.g. about insurance risks. Therefore, we believe that the clinician should inform the patient about the fact that the pathological examinations may not only give information about the nature of the tumour, but may also indicate an elevated risk of an underlying hereditary disorder.
Conclusions
Different diagnostic strategies have been developed for LS as discussed in this review and the optimal method for the identification of LS patients is still debated and in flux. In the previous paragraphs the molecular diagnostic approach of LS in The Netherlands (Erasmus MC, University Medical Center, Rotterdam) has been described (Fig. 4). This approach combines MSI analysis and MMR protein immunostaining and is in our opinion a productive way of pre-selecting patients for germline mutation analysis, with a central role for the pathologist. Nevertheless, if the clinical suspicion for LS is very high, e.g. because of a positive family history for LS-associated cancers or a LS-associated malignancy diagnosed at a very young age, referral to a clinical geneticist is strongly recommended, even in the case of tumours without MMR deficiency (i.e. MSS).
Acknowledgments
The currently ongoing LIMO study is supported by an Erasmus MC-grant. We thank Frank van der Panne for photographic work.
References
- 1.Boyle P, Ferlay J. Cancer incidence and mortality in Europe, 2004. Ann Oncol. 2005;16:481–8. doi: 10.1093/annonc/mdi098. [DOI] [PubMed] [Google Scholar]
- 2.Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998;338:1481–7. doi: 10.1056/NEJM199805213382101. [DOI] [PubMed] [Google Scholar]
- 3.Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26:5783–8. doi: 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66:7810–7. doi: 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- 5.Lynch HT, Smyrk TC, Watson P, et al. Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: an updated review. Gastroenterology. 1993;104:1535–49. doi: 10.1016/0016-5085(93)90368-m. [DOI] [PubMed] [Google Scholar]
- 6.Watson P, Lynch HT. Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer. 1993;71:677–85. doi: 10.1002/1097-0142(19930201)71:3<677::aid-cncr2820710305>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 7.Quehenberger F, Vasen HF, Van Houwelingen HC. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet. 2005;42:491–6. doi: 10.1136/jmg.2004.024299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vasen HF, Wijnen JT, Menko FH, et al. Cancer risk in families with hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis. Gastroenterology. 1996;110:1020–7. doi: 10.1053/gast.1996.v110.pm8612988. [DOI] [PubMed] [Google Scholar]
- 9.Plaschke J, Engel C, Kruger S, et al. Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol. 2004;22:4486–94. doi: 10.1200/JCO.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 10.Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–8. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 11.Vasen HF, Stormorken A, Menko FH, et al. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol. 2001;19:4074–80. doi: 10.1200/JCO.2001.19.20.4074. [DOI] [PubMed] [Google Scholar]
- 12.Hendriks YM, Wagner A, Morreau H, et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology. 2004;127:17–25. doi: 10.1053/j.gastro.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 13.Hampel H, Stephens JA, Pukkala E, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology. 2005;129:415–21. doi: 10.1016/j.gastro.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Senter L, Clendenning M, Sotamaa K, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology. 2008;135:419–28. doi: 10.1053/j.gastro.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lynch HT, De La Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet. 1999;36:801–18. [PMC free article] [PubMed] [Google Scholar]
- 16.Boland CR. Evolution of the nomenclature for the hereditary colorectal cancer syndromes. Fam Cancer. 2005;4:211–8. doi: 10.1007/s10689-004-4489-x. [DOI] [PubMed] [Google Scholar]
- 17.Jarvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–34. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 18.De Jong AE, Hendriks YM, Kleibeuker JH, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology. 2006;130:665–71. doi: 10.1053/j.gastro.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 19.Stupart DA, Goldberg PA, Algar U, et al. Surveillance colonoscopy improves survival in a cohort of subjects with a single mismatch repair gene mutation. Colorectal Dis. 2009;11:126–30. doi: 10.1111/j.1463-1318.2008.01702.x. [DOI] [PubMed] [Google Scholar]
- 20.Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354:261–9. doi: 10.1056/NEJMoa052627. [DOI] [PubMed] [Google Scholar]
- 21.Vasen HF, Mecklin JP, Khan PM, et al. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC) Dis Colon Rectum. 1991;34:424–5. doi: 10.1007/BF02053699. [DOI] [PubMed] [Google Scholar]
- 22.Kievit W, De Bruin JH, Adang EM, et al. Current clinical selection strategies for identification of hereditary non-polyposis colorectal cancer families are inadequate: a meta-analysis. Clin Genet. 2004;65:308–16. doi: 10.1111/j.1399-0004.2004.00220.x. [DOI] [PubMed] [Google Scholar]
- 23.Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–6. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 24.Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA. 2006;296:1507–17. doi: 10.1001/jama.296.12.1507. [DOI] [PubMed] [Google Scholar]
- 25.Church J, McGannon E. Family history of colorectal cancer: how often and how accurately is it recorded. Dis Colon Rectum. 2000;43:1540–4. doi: 10.1007/BF02236735. [DOI] [PubMed] [Google Scholar]
- 26.Katballe N, Juul S, Christensen M, et al. Patient accuracy of reporting on hereditary non-polyposis colorectal cancer-related malignancy in family members. Br J Surg. 2001;88:1228–33. doi: 10.1046/j.0007-1323.2001.01868.x. [DOI] [PubMed] [Google Scholar]
- 27.Sijmons RH, Boonstra AE, Reefhuis J, et al. Accuracy of family history of cancer: clinical genetic implications. Eur J Hum Genet. 2000;8:181–6. doi: 10.1038/sj.ejhg.5200441. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez-Bigas MA, Boland CR, Hamilton SR, et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst. 1997;89:1758–62. doi: 10.1093/jnci/89.23.1758. [DOI] [PubMed] [Google Scholar]
- 29.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–8. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenkins MA, Hayashi S, O’Shea AM, et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: a population-based study. Gastroenterology. 2007;133:48–56. doi: 10.1053/j.gastro.2007.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Truta B, Chen YY, Blanco AM, et al. Tumor histology helps to identify Lynch syndrome among colorectal cancer patients. Fam Cancer. 2008;7:267–74. doi: 10.1007/s10689-008-9186-8. [DOI] [PubMed] [Google Scholar]
- 32.Sanchez JA, Vogel JD, Kalady MF, et al. Identifying Lynch syndrome: we are all responsible. Dis Colon Rectum. 2008;51:1750–6. doi: 10.1007/s10350-008-9414-1. [DOI] [PubMed] [Google Scholar]
- 33.Wolf B, Gruber S, Henglmueller S, et al. Efficiency of the revised Bethesda guidelines (2003) for the detection of mutations in mismatch repair genes in Austrian HNPCC patients. Int J Cancer. 2006;118:1465–70. doi: 10.1002/ijc.21524. [DOI] [PubMed] [Google Scholar]
- 34.Pinol V, Castells A, Andreu M, et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA. 2005;293:1986–94. doi: 10.1001/jama.293.16.1986. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Moranta F, Castells A, Andreu M, et al. Clinical performance of original and revised Bethesda guidelines for the identification of MSH2/MLH1 gene carriers in patients with newly diagnosed colorectal cancer: proposal of a new and simpler set of recommendations. Am J Gastroenterol. 2006;101:1104–11. doi: 10.1111/j.1572-0241.2006.00522.x. [DOI] [PubMed] [Google Scholar]
- 36.Balmana J, Balaguer F, Castellvi-Bel S, et al. Comparison of predictive models, clinical criteria and molecular tumour screening for the identification of patients with Lynch syndrome in a population-based cohort of colorectal cancer patients. J Med Genet. 2008;45:557–63. doi: 10.1136/jmg.2008.059311. [DOI] [PubMed] [Google Scholar]
- 37.Ramsoekh D, Wagner A, Van Leerdam ME, et al. A high incidence of MSH6 mutations in Amsterdam criteria II-negative families tested in a diagnostic setting. Gut. 2008;57:1539–44. doi: 10.1136/gut.2008.156695. [DOI] [PubMed] [Google Scholar]
- 38.Van Dijk DA, Oostindier MJ, Kloosterman-Boele WM, et al. Family history is neglected in the work-up of patients with colorectal cancer: a quality assessment using cancer registry data. Fam Cancer. 2007;6:131–4. doi: 10.1007/s10689-006-9114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Overbeek LI, Hoogerbrugge N, Van Krieken JH, et al. Most patients with colorectal tumors at young age do not visit a cancer genetics clinic. Dis Colon Rectum. 2008;51:1249–54. doi: 10.1007/s10350-008-9345-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Lier MG, De Wilt JH, Wagemakers JJ, et al. Underutilization of microsatellite instability analysis in colorectal cancer patients at high risk for Lynch syndrome. Scand J Gastroenterol. 2009;44:600–4. doi: 10.1080/00365520802706008. [DOI] [PubMed] [Google Scholar]
- 41.Kievit W, De Bruin JH, Adang EM, et al. Cost effectiveness of a new strategy to identify HNPCC patients. Gut. 2005;54:97–102. doi: 10.1136/gut.2004.039123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Bruin JH, Ligtenberg MJ, Nagengast FM, et al. Optimizing the detection of hereditary non-polyposis colorectal cancer: an update. Scand J Gastroenterol Suppl. 2006;243:146–52. doi: 10.1080/00365520600664508. [DOI] [PubMed] [Google Scholar]
- 43.Wijnen JT, Vasen HF, Khan PM, et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med. 1998;339:511–8. doi: 10.1056/NEJM199808203390804. [DOI] [PubMed] [Google Scholar]
- 44.Barnetson RA, Tenesa A, Farrington SM, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354:2751–63. doi: 10.1056/NEJMoa053493. [DOI] [PubMed] [Google Scholar]
- 45.Balmana J, Stockwell DH, Steyerberg EW, et al. Prediction of MLH1 and MSH2 mutations in Lynch syndrome. JAMA. 2006;296:1469–78. doi: 10.1001/jama.296.12.1469. [DOI] [PubMed] [Google Scholar]
- 46.Chen S, Wang W, Lee S, et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA. 2006;296:1479–87. doi: 10.1001/jama.296.12.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition–Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20:269–76. doi: 10.1155/2004/305058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vasen HF, Boland CR. Progress in genetic testing, classification, and identification of Lynch syndrome. JAMA. 2005;293:2028–30. doi: 10.1001/jama.293.16.2028. [DOI] [PubMed] [Google Scholar]
- 49.Miyaki M, Konishi M, Tanaka K, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17:271–2. doi: 10.1038/ng1197-271. [DOI] [PubMed] [Google Scholar]
- 50.Hendriks YM, Jagmohan-Changur S, Van Der Klift HM, et al. Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome) Gastroenterology. 2006;130:312–22. doi: 10.1053/j.gastro.2005.10.052. [DOI] [PubMed] [Google Scholar]
- 51.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–62. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 52.Chung DC, Rustgi AK. DNA mismatch repair and cancer. Gastroenterology. 1995;109:1685–99. doi: 10.1016/0016-5085(95)90660-6. [DOI] [PubMed] [Google Scholar]
- 53.Papadopoulos N, Nicolaides NC, Liu B, et al. Mutations of GTBP in genetically unstable cells. Science. 1995;268:1915–7. doi: 10.1126/science.7604266. [DOI] [PubMed] [Google Scholar]
- 54.Baker SM, Bronner CE, Zhang L, et al. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–19. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- 55.Fishel R, Ewel A, Lescoe MK. Purified human MSH2 protein binds to DNA containing mismatched nucleotides. Cancer Res. 1994;54:5539–42. [PubMed] [Google Scholar]
- 56.Yang Q, Zhang R, Wang XW, et al. The mismatch DNA repair heterodimer, hMSH2/6, regulates BLM helicase. Oncogene. 2004;23:3749–56. doi: 10.1038/sj.onc.1207462. [DOI] [PubMed] [Google Scholar]
- 57.Genschel J, Bazemore LR, Modrich P. Human exonuclease I is required for 5′ and 3′ mismatch repair. J Biol Chem. 2002;277:13302–11. doi: 10.1074/jbc.M111854200. [DOI] [PubMed] [Google Scholar]
- 58.Gruber SB. New developments in Lynch syndrome (hereditary nonpolyposis colorectal cancer) and mismatch repair gene testing. Gastroenterology. 2006;130:577–87. doi: 10.1053/j.gastro.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 59.Ionov Y, Peinado MA, Malkhosyan S, et al. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–61. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 60.Umar A, Boyer JC, Thomas DC, et al. Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J Biol Chem. 1994;269:14367–70. [PubMed] [Google Scholar]
- 61.Aaltonen LA, Peltomaki P, Mecklin JP, et al. Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Res. 1994;54:1645–8. [PubMed] [Google Scholar]
- 62.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57. [PubMed] [Google Scholar]
- 63.Lynch HT, De La Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–32. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 64.Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–11. [PubMed] [Google Scholar]
- 65.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–5. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 67.Wang L, Cunningham JM, Winters JL, et al. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003;63:5209–12. [PubMed] [Google Scholar]
- 68.Miyaki M, Iijima T, Yamaguchi T, et al. Both BRAF and KRAS mutations are rare in colorectal carcinomas from patients with hereditary nonpolyposis colorectal cancer. Cancer Lett. 2004;211:105–9. doi: 10.1016/j.canlet.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 69.McGivern A, Wynter CV, Whitehall VL, et al. Promoter hypermethylation frequency and BRAF mutations distinguish hereditary non-polyposis colon cancer from sporadic MSI-H colon cancer. Fam Cancer. 2004;3:101–7. doi: 10.1023/B:FAME.0000039861.30651.c8. [DOI] [PubMed] [Google Scholar]
- 70.Kambara T, Simms LA, Whitehall VL, et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut. 2004;53:1137–44. doi: 10.1136/gut.2003.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Domingo E, Laiho P, Ollikainen M, et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet. 2004;41:664–8. doi: 10.1136/jmg.2004.020651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Deng G, Bell I, Crawley S, et al. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10:191–5. doi: 10.1158/1078-0432.ccr-1118-3. [DOI] [PubMed] [Google Scholar]
- 73.Young J, Barker MA, Simms LA, et al. Evidence for BRAF mutation and variable levels of microsatellite instability in a syndrome of familial colorectal cancer. Clin Gastroenterol Hepatol. 2005;3:254–63. doi: 10.1016/s1542-3565(04)00673-1. [DOI] [PubMed] [Google Scholar]
- 74.Loughrey MB, Waring PM, Tan A, et al. Incorporation of somatic BRAF mutation testing into an algorithm for the investigation of hereditary non-polyposis colorectal cancer. Fam Cancer. 2007;6:301–10. doi: 10.1007/s10689-007-9124-1. [DOI] [PubMed] [Google Scholar]
- 75.Bessa X, Balleste B, Andreu M, et al. A prospective, multicenter, population-based study of BRAF mutational analysis for Lynch syndrome screening. Clin Gastroenterol Hepatol. 2008;6:206–14. doi: 10.1016/j.cgh.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 76.Nagasaka T, Koi M, Kloor M, et al. Mutations in both KRAS and BRAF may contribute to the methylator phenotype in colon cancer. Gastroenterology. 2008;134:1950–60. doi: 10.1053/j.gastro.2008.02.094. , 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–99. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gazzoli I, Loda M, Garber J, et al. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002;62:3925–8. [PubMed] [Google Scholar]
- 79.Suter CM, Martin DI, Ward RL. Germline epimutation of MLH1 in individuals with multiple cancers. Nat Genet. 2004;36:497–501. doi: 10.1038/ng1342. [DOI] [PubMed] [Google Scholar]
- 80.Miyakura Y, Sugano K, Akasu T, et al. Extensive but hemiallelic methylation of the hMLH1 promoter region in early-onset sporadic colon cancers with microsatellite instability. Clin Gastroenterol Hepatol. 2004;2:147–56. doi: 10.1016/s1542-3565(03)00314-8. [DOI] [PubMed] [Google Scholar]
- 81.Hitchins M, Williams R, Cheong K, et al. MLH1 germline epimutations as a factor in hereditary nonpolyposis colorectal cancer. Gastroenterology. 2005;129:1392–9. doi: 10.1053/j.gastro.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 82.Hitchins MP, Wong JJ, Suthers G, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. 2007;356:697–705. doi: 10.1056/NEJMoa064522. [DOI] [PubMed] [Google Scholar]
- 83.Valle L, Carbonell P, Fernandez V, et al. MLH1 germline epimutations in selected patients with early-onset non-polyposis colorectal cancer. Clin Genet. 2007;71:232–7. doi: 10.1111/j.1399-0004.2007.00751.x. [DOI] [PubMed] [Google Scholar]
- 84.Morak M, Schackert HK, Rahner N, et al. Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur J Hum Genet. 2008;16:804–11. doi: 10.1038/ejhg.2008.25. [DOI] [PubMed] [Google Scholar]
- 85.Hitchins MP, Ward RL. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J Med Genet. 2009;46:793–802. doi: 10.1136/jmg.2009.068122. [DOI] [PubMed] [Google Scholar]
- 86.Dobrovic A, Kristensen LS. DNA methylation, epimutations and cancer predisposition. Int J Biochem Cell Biol. 2009;41:34–9. doi: 10.1016/j.biocel.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 87.Ligtenberg MJ, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat Genet. 2009;41:112–7. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 88.Dietmaier W, Wallinger S, Bocker T, et al. Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res. 1997;57:4749–56. [PubMed] [Google Scholar]
- 89.Thibodeau SN, French AJ, Cunningham JM, et al. Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res. 1998;58:1713–8. [PubMed] [Google Scholar]
- 90.Veigl ML, Kasturi L, Olechnowicz J, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA. 1998;95:8698–702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jass JR, Walsh MD, Barker M, et al. Distinction between familial and sporadic forms of colorectal cancer showing DNA microsatellite instability. Eur J Cancer. 2002;38:858–66. doi: 10.1016/s0959-8049(02)00041-2. [DOI] [PubMed] [Google Scholar]
- 92.Laiho P, Launonen V, Lahermo P, et al. Low-level microsatellite instability in most colorectal carcinomas. Cancer Res. 2002;62:1166–70. [PubMed] [Google Scholar]
- 93.Pawlik TM, Raut CP, Rodriguez-Bigas MA. Colorectal carcinogenesis: MSI-H versus MSI-L. Dis Markers. 2004;20:199–206. doi: 10.1155/2004/368680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000;342:69–77. doi: 10.1056/NEJM200001133420201. [DOI] [PubMed] [Google Scholar]
- 95.Wright CM, Dent OF, Barker M, et al. Prognostic significance of extensive microsatellite instability in sporadic clinicopathological stage C colorectal cancer. Br J Surg. 2000;87:1197–202. doi: 10.1046/j.1365-2168.2000.01508.x. [DOI] [PubMed] [Google Scholar]
- 96.Chang EY, Dorsey PB, Frankhouse J, et al. Combination of microsatellite instability and lymphocytic infiltrate as a prognostic indicator in colon cancer. Arch Surg. 2009;144:511–5. doi: 10.1001/archsurg.2009.40. [DOI] [PubMed] [Google Scholar]
- 97.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–9. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 98.Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–57. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–18. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 100.Jover R, Zapater P, Castells A, et al. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut. 2006;55:848–55. doi: 10.1136/gut.2005.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chialina SG, Fornes C, Landi C, et al. Microsatellite instability analysis in hereditary non-polyposis colon cancer using the Bethesda consensus panel of microsatellite markers in the absence of proband normal tissue. BMC Med Genet. 2006;7 doi: 10.1186/1471-2350-7-5. : 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Suraweera N, Duval A, Reperant M, et al. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology. 2002;123:1804–11. doi: 10.1053/gast.2002.37070. [DOI] [PubMed] [Google Scholar]
- 103.Bacher JW, Flanagan LA, Smalley RL, et al. Development of a fluorescent multiplex assay for detection of MSI-High tumors. Dis Markers. 2004;20:237–50. doi: 10.1155/2004/136734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Akiyama Y, Sato H, Yamada T, et al. Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res. 1997;57:3920–3. [PubMed] [Google Scholar]
- 105.Berends MJ, Wu Y, Sijmons RH, et al. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet. 2002;70:26–37. doi: 10.1086/337944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xicola RM, Llor X, Pons E, et al. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J Natl Cancer Inst. 2007;99:244–52. doi: 10.1093/jnci/djk033. [DOI] [PubMed] [Google Scholar]
- 107.Halvarsson B, Lindblom A, Rambech E, et al. Microsatellite instability analysis and/or immunostaining for the diagnosis of hereditary nonpolyposis colorectal cancer. Virchows Arch. 2004;444:135–41. doi: 10.1007/s00428-003-0922-z. [DOI] [PubMed] [Google Scholar]
- 108.Chen PC, Dudley S, Hagen W, et al. Contributions by MutL homologues Mlh3 and Pms2 to DNA mismatch repair and tumor suppression in the mouse. Cancer Res. 2005;65:8662–70. doi: 10.1158/0008-5472.CAN-05-0742. [DOI] [PubMed] [Google Scholar]
- 109.Cannavo E, Marra G, Sabates-Bellver J, et al. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Res. 2005;65:10759–66. doi: 10.1158/0008-5472.CAN-05-2528. [DOI] [PubMed] [Google Scholar]
- 110.Acharya S, Wilson T, Gradia S, et al. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc Natl Acad Sci USA. 1996;93:13629–34. doi: 10.1073/pnas.93.24.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn. 2008;10:293–300. doi: 10.2353/jmoldx.2008.080031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Poley JW, Wagner A, Hoogmans MM, et al. Biallelic germline mutations of mismatch-repair genes: a possible cause for multiple pediatric malignancies. Cancer. 2007;109:2349–56. doi: 10.1002/cncr.22697. [DOI] [PubMed] [Google Scholar]
- 113.Capel E, Flejou JF, Hamelin R. Assessment of MLH1 promoter methylation in relation to gene expression requires specific analysis. Oncogene. 2007;26:7596–600. doi: 10.1038/sj.onc.1210581. [DOI] [PubMed] [Google Scholar]
- 114.Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30 doi: 10.1093/nar/gnf056. : e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Heinmoller E, Renke B, Beyser K, et al. Pitfalls in diagnostic molecular pathology–significance of sampling error. Virchows Arch. 2001;439:504–11. doi: 10.1007/s004280100450. [DOI] [PubMed] [Google Scholar]
- 116.Muller A, Giuffre G, Edmonston TB, et al. Challenges and pitfalls in HNPCC screening by microsatellite analysis and immunohistochemistry. J Mol Diagn. 2004;6:308–15. doi: 10.1016/S1525-1578(10)60526-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mangold E, Pagenstecher C, Friedl W, et al. Tumours from MSH2 mutation carriers show loss of MSH2 expression but many tumours from MLH1 mutation carriers exhibit weak positive MLH1 staining. J Pathol. 2005;207:385–95. doi: 10.1002/path.1858. [DOI] [PubMed] [Google Scholar]
- 118.Muller W, Burgart LJ, Krause-Paulus R, et al. The reliability of immunohistochemistry as a prescreening method for the diagnosis of hereditary nonpolyposis colorectal cancer (HNPCC)–results of an international collaborative study. Fam Cancer. 2001;1:87–92. doi: 10.1023/a:1013840907881. [DOI] [PubMed] [Google Scholar]
- 119.Overbeek LI, Ligtenberg MJ, Willems RW, et al. Interpretation of immunohistochemistry for mismatch repair proteins is only reliable in a specialized setting. Am J Surg Pathol. 2008;32:1246–51. doi: 10.1097/pas.0b013e31816401bb. [DOI] [PubMed] [Google Scholar]
- 120.Matsubara N. Diagnostic application of hMLH1 methylation in hereditary non-polyposis colorectal cancer. Dis Markers. 2004;20:277–82. doi: 10.1155/2004/371941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Deng G, Chen A, Hong J, et al. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999;59:2029–33. [PubMed] [Google Scholar]
- 122.Esteller M, Fraga MF, Guo M, et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet. 2001;10:3001–7. doi: 10.1093/hmg/10.26.3001. [DOI] [PubMed] [Google Scholar]
- 123.Rahner N, Friedrichs N, Steinke V, et al. Coexisting somatic promoter hypermethylation and pathogenic MLH1 germline mutation in Lynch syndrome. J Pathol. 2008;214:10–6. doi: 10.1002/path.2263. [DOI] [PubMed] [Google Scholar]
- 124.Cunningham JM, Christensen ER, Tester DJ, et al. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998;58:3455–60. [PubMed] [Google Scholar]
- 125.Kuismanen SA, Holmberg MT, Salovaara R, et al. Epigenetic phenotypes distinguish microsatellite-stable and -unstable colorectal cancers. Proc Natl Acad Sci USA. 1999;96:12661–6. doi: 10.1073/pnas.96.22.12661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuismanen SA, Holmberg MT, Salovaara R, et al. Genetic and epigenetic modification of MLH1 accounts for a major share of microsatellite-unstable colorectal cancers. Am J Pathol. 2000;156:1773–9. doi: 10.1016/S0002-9440(10)65048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wheeler JM, Loukola A, Aaltonen LA, et al. The role of hypermethylation of the hMLH1 promoter region in HNPCC versus MSI+ sporadic colorectal cancers. J Med Genet. 2000;37:588–92. doi: 10.1136/jmg.37.8.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Potocnik U, Glavac D, Golouh R, et al. Causes of microsatellite instability in colorectal tumors: implications for hereditary non-polyposis colorectal cancer screening. Cancer Genet Cytogenet. 2001;126:85–96. doi: 10.1016/s0165-4608(00)00399-x. [DOI] [PubMed] [Google Scholar]
- 129.Ollikainen M, Hannelius U, Lindgren CM, et al. Mechanisms of inactivation of MLH1 in hereditary nonpolyposis colorectal carcinoma: a novel approach. Oncogene. 2007;26:4541–9. doi: 10.1038/sj.onc.1210236. [DOI] [PubMed] [Google Scholar]
- 130.Ramsoekh D, Van Leerdam ME, Wagner A, et al. Review article: detection and management of hereditary non-polyposis colorectal cancer (Lynch syndrome) Aliment Pharmacol Ther. 2007;26:101–11. doi: 10.1111/j.1365-2036.2007.03492.x. [DOI] [PubMed] [Google Scholar]
- 131.Ferreira AM, Westers H, Wu Y, et al. Do microsatellite instability profiles really differ between colorectal and endometrial tumors. Genes Chromosomes Cancer. 2009;48:552–7. doi: 10.1002/gcc.20664. [DOI] [PubMed] [Google Scholar]
- 132.De Leeuw WJ, Dierssen J, Vasen HF, et al. Prediction of a mismatch repair gene defect by microsatellite instability and immunohistochemical analysis in endometrial tumours from HNPCC patients. J Pathol. 2000;192:328–35. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH701>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 133.Hewitt MJ, Wood N, Quinton ND, et al. The detection of microsatellite instability in blind endometrial samples–a potential novel screening tool for endometrial cancer in women from hereditary nonpolyposis colorectal cancer families. Int J Gynecol Cancer. 2006;16:1393–400. doi: 10.1111/j.1525-1438.2006.00604.x. [DOI] [PubMed] [Google Scholar]