

Catalytic enantioselective addition of vinyl groups to C-based electrophiles constitutes a versatile class of C–C bond forming processes.[1–3] We recently reported that vinylaluminum reagents, generated through reactions of alkyl- or alkenyl-substituted terminal alkynes with commercially available and inexpensive diisobutylaluminum hydride (dibal-H), can be used in site- and enantioselective Cu-catalyzed allylic alkylations.[4] The above protocol, however, suffers from two notable deficiencies. First, cis vinylmetals cannot be accessed (>98% trans product formed due to syn addition of Al–H to terminal alkynes). Second, aryl-substituted vinylaluminums cannot be utilized since the corresponding hydroaluminations are inefficient.[5] Herein, we present solutions to the above shortcomings through efficient, site- and stereoselective preparation of vinylaluminum reagents by reactions of Si-substituted alkynes with dibal-H. The vinylmetals can be used in situ in site- and enantioselective Cu-catalyzed allylic alkylations.[6] Enantiomerically enriched vinylsilanes are proto-desilylated with trifluoroacetic acid (TFA), affording the derived Z or E alkenes typically as a single stereoisomer in up to >99:1 enantiomeric ratio (e.r.).
The present investigations originated partly from considerations regarding application of the enantioselective allylic alkylations of vinylaluminum reagents,[4] catalyzed by chiral bidentate N-heterocyclic carbene (NHC) Cu complexes,[7] to synthesis of nyasol.[8] The proposed plan (Scheme 1) demands the efficient and selective formation of a Z vinylaluminum from an aryl-substituted alkyne. Consequently, addressing the aforementioned drawbacks, namely identification of a protocol allowing efficient access to a cis vinylmetal derived from an aryl-substituted alkyne, was required.
Scheme 1.
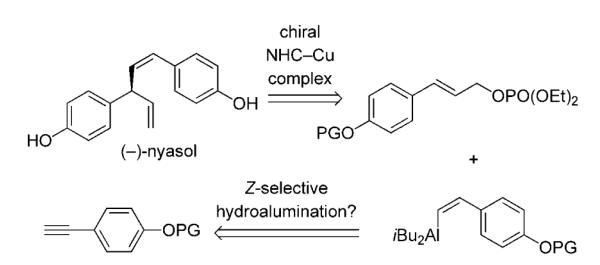
Projected diastereo- and enantioselective synthesis of nyasol through Cu-catalyzed allylic alkylation with a cis vinylaluminum reagent. NHC=N-heterocyclic carbene, PG=protecting group.
We surmised that a silyl substituent at the alkyne terminus (I, Scheme 2) would allow hydroaluminations of aryl-substituted substrates to proceed readily and with minimal byproduct formation. Efficiency would arise from stabilization of the incipient electron density at the carbon of the C–Al bond by the d orbitals of silicon, or through hyperconjugation with the low-lying σ* orbital of the adjacent C–Si.[9] Accordingly, site- and stereoselective hydrometallation of a silyl-substituted alkyne[10] (I, Scheme 2), reaction of the resulting vinylmetal (II) with an electrophile (→III), followed by protonation of the C–Si bond (→IV) would deliver the desired product bearing a trans alkene. The corresponding Z olefin could be obtained stereoselectively (via V–VII, Scheme 2) if the initial hydroalumination adduct (II) could be induced to undergo isomerization, positioning the large aryl and silyl substituent trans to one another.
Scheme 2.
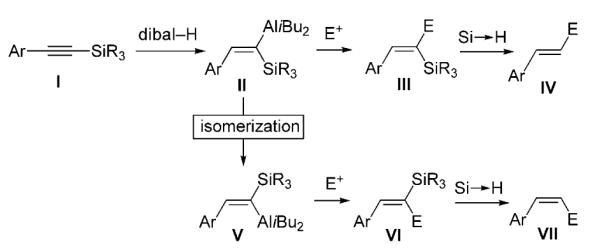
Synthesis of a cis- or trans-disubstituted alkene may be accomplished through hydroalumination/desilylation of a Si-substituted alkyne.
To access the requisite cis vinylmetals, we turned to the pioneering investigations of Eisch, disclosed nearly four decades ago, in connection with hydroaluminations of silyl-substituted alkynes.[11] Treatment of trimethylsilyl-substituted phenylacetylene 1 with dibal-H in 5:1 hexanes:THF (55 °C, 2 h) leads to vinylaluminum 2 in >98% Z-selectivity (Scheme 3). When hydrometallation is performed in hexanes, 3 is formed with >98% E-selectivity (>98% conv.). A mechanistic scheme, related to that suggested by Eisch,[11] is illustrated in Scheme 3. In the absence of Lewis basic THF, alkene isomerization occurs readily (VIII→IX→X) due to the energetically accessible and unoccupied p orbital of the Al metal. When THF is present, its coordination to the Lewis acidic Al inhibits isomerization of the initially generated Z-vinylmetal. In both cases, hydroaluminations proceed with >98% site-selectivity.
Scheme 3.
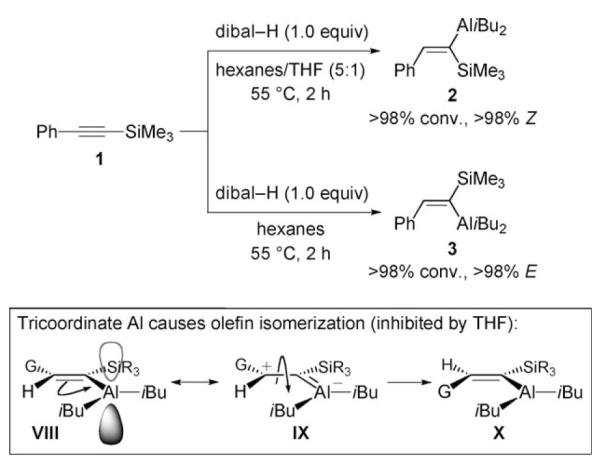
Stereoselective hydroalumination of trimethylsilylphenylacetylene with dibal-H, and a rationale for the effect of a Lewis base (THF). dibal-H=(iBu)2AlH.
Next, we examined the related NHC-Cu-catalyzed enantioselective allylic alkylations. Subjection of 1.5 equivalents of vinylaluminum 3, prepared and used in situ, to aryl and alkyl-substituted allylic phosphates 4a–k, in the presence of 1.0 mol% NHC-AgI complex 5 and 2.0 mol% CuCl2·2H2O, results in complete substrate consumption (Table 1). 3-Substituted 1,4-dienes 6a–k are generated with >98% site-selectivity (SN2′), in 82% to >98% yield and in 94:6 to >99:1 e.r. Selectivities are high, regardless of the steric or electronic attributes of aryl substituents of the allylic phosphates. Alkyl-substituted substrates (entries 10–11, Table 1) can be used effectively in the hydroalumination/allylic alkylation process, albeit with somewhat lower enantioselectivity (94:6 e.r.). In all the transformations examined, there is >98% E-selectivity, and products derived from the addition of an iBu unit of the Al-based reagent are not detected (<2% by 400 MHz 1H NMR analysis). It is thus a notable attribute of this class of transformations that the overall process, beginning with the silyl-substituted alkyne and ending with the enantiomerically enriched dienes, requires five distinct issues of selectivity to be addressed: site- and stereoselectivity in generation of the vinylaluminum (Scheme 3), followed by site- (SN2′ vs. SN2), group- (vinyl- vs. iBu addition) and, finally, enantioselectivity of allylic alkylation. In reactions summarized in Table 1, there is complete (>98%) control of selectivity in all aspects except product enantiomeric purity, which varies between 94:6 and >99:1 e.r.
Table 1.
Catalytic enantioselective allylic alkylations with vinylaluminum reagent 3 promoted by NHC–Cu complex derived from 5.[a]
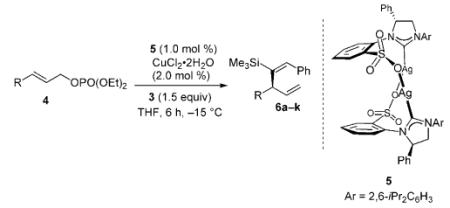
| Entry | Substrate [R] | Yield [%][b] | SN2′:SN2[c] | E:Z[c] | e.r.[d] |
|---|---|---|---|---|---|
| 1 | 4a [Ph] | 93 | >98:2 | >98:2 | 99:1 |
| 2 | 4b [oMeC6H4] | 96 | >98:2 | >98:2 | 99:1 |
| 3 | 4c [oMeOC6H4] | 82 | >98:2 | >98:2 | 98:2 |
| 4 | 4d [oNO2C6H4] | 89 | >98:2 | >98:2 | >99:1 |
| 5 | 4e [mBrC6H4] | 95 | >98:2 | >98:2 | 99:1 |
| 6 | 4 f [pClC6H4] | 94 | >98:2 | >98:2 | 98.5:1.5 |
| 7 | 4g [pNO2C6H4] | 87 | >98:2 | >98:2 | 97.5:2.5 |
| 8 | 4h [pTsOC6H4] | >98 | >98:2 | >98:2 | 99:1 |
| 9 | 4i [pMeC6H4] | 92 | >98:2 | >98:2 | >99:1 |
| 10 | 4j [(CH2)2C6H5] | 88 | >98:2 | >98:2 | 94:6 |
| 11 | 4k [Cy] | 88 | >98:2 | >98:2 | 94:6 |
Reactions were performed under N2 atmosphere; >98% conversion and <2% iBu addition product in all cases (400 MHz 1H NMR analysis of unpurified product mixtures).
Yields of isolated purified products.
Site-selectivities and alkene stereoselectivities established by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification.
Enantiomer ratios determined by HPLC analysis; see the Supporting Information for details.
Sequential hydroalumination/Cu-catalyzed allylic alkylation can be performed with silyl-substituted alkynes bearing different aryl units (Table 2). Products are isolated with complete site- and group-selectivity and in 92:8–99:1 e.r. Hydroaluminations proceed with exceptional site-selectivity, irrespective of the steric or electronic characteristics of the alkyne substituent. Only in one instance is the final product isolated as a mixture of E and Z isomers (entry 4, 88:12 E:Z). The incomplete vinylaluminum isomerization may be due to destabilization of the zwitterionic structure (IX in Scheme 3) by the electron withdrawing p-CF3 unit.
Table 2.
Catalytic enantioselective allylic alkylations with various vinylaluminum reagents promoted by NHC–Cu complex derived from 5.[a]
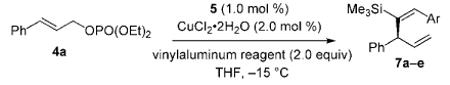
The representative example, illustrated in Equation (1), underlines the exceptional efficiency of this single-vessel class of transformations. Approximately 0.5 gram of 6a (90% yield) can be synthesized in 98.5:1.5 e.r. and 95:5 site-selectivity (SN2′:SN2) through a reaction that proceeds with otherwise complete (>98%) selectivity, requiring only 0.05 mol% (1.1 mg) of chiral NHC-AgI complex 5.
The first enantioselective synthesis of nyasol was subsequently realized (Scheme 2). Pd-catalyzed cross-coupling[12] of commercially available trimethylsilylacetylene with 4-iodophenol delivers the substrate (8) required for site- and
 |
(1) |
stereoselective hydroalumination in 76% yield (Scheme 4).[13] Treatment of 8 with dibal-H (55 °C, 2 h) delivers vinylaluminum 9, which is used in Cu-catalyzed allylic alkylation of allylic phosphate 10. The one-pot process furnishes silyl-substituted 1,4-diene 11 with complete control of alkene selectivity (>98% E), as well as >98% group- (<2% iBu addition) and site-selectivity (<2% SN2) in 76% yield and 98.5:1.5 e.r. (−)-Nyasol is obtained in 73% yield from 11 by a three-step procedure that includes proto-desilylation with trifluoroacetic acid (42% overall yield from commercially available trimethylsilylacetylene).[14]
Scheme 4.
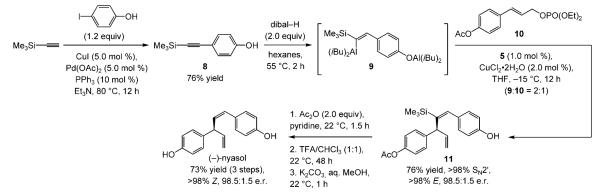
Enantioselective synthesis of (−)-nyasol, featuring a one-pot alkyne hydroalumination/Cu-catalyzed enantioselective allylic alkylation. TFA=trifluoroacetic acid.
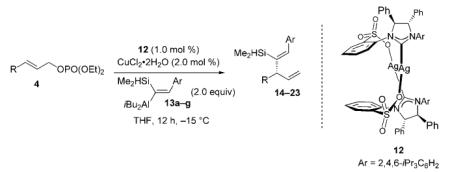
The Z-vinylaluminum reagents, such as 2 (Scheme 3), obtained site-selectively through syn aluminum hydride addition to silyl-substituted aryl alkynes, allow us to obtain products that cannot be efficiently accessed by direct hydroalumination of aryl-substituted alkynes.[5] An assortment of enantiomerically enriched 1,4-dienes can be obtained through enantioselective Cu-catalyzed allylic alkylation with this set of stereochemically defined organometallic reagents (Table 3). Similar to reactions with E vinylaluminum reagents (Tables 1–2 and Scheme 4), when 13a–g are used (Table 3), high site- (> 98% SN2′) and enantioselectivity are observed (95.5:4.5–98.5:1.5 e.r.).[15] In all instances except one (Z:E = 90:10, entry 7),[16] there is >98% Z-selectivity.
Table 3.
Catalytic enantioselective allylic alkylations with Z-vinylaluminum reagents 13 promoted by NHC–Cu complex derived from 12.[a]
| Entry | 4 [R] | 13 [Ar] | Product | Yield [%][b] |
Vinyl:iBu addition[c] |
SN2′:SN2[c] | E:Z[c] | e.r.[d] |
|---|---|---|---|---|---|---|---|---|
| 1 | 4a [Ph] | 13a [Ph] | 14 | 78 | 91:9 | >98:2 | >98:2 | 97:3 |
| 2 | 4a [Ph] | 13b [oMeOC6H4] | 15 | 68 | 72:28 | >98:2 | >98:2 | 97.5:2.5 |
| 3 | 4a [Ph] | 13c [oMeC6H4] | 16 | 61 | 76:24 | >98:2 | >98:2 | 97:3 |
| 4 | 4a [Ph] | 13d [mCF3C6H4] | 17 | 86 | 91:9 | >98:2 | >98:2 | 95.5:4.5 |
| 5 | 4a [Ph] | 13e [pCF3C6H4] | 18 | 71 | 75:25 | >98:2 | >98:2 | 96.5:3.5 |
| 6 | 4a [Ph] | 13 f [pFC6H4] | 19 | 85 | 90:10 | >98:2 | >98:2 | 98:2 |
| 7 | 4a [Ph] | 13g [pMeOC6H4] | 20 | 75 | 80:20 | >98:2 | >98:2 | 98.5:1.5 |
| 8 | 4g [pNO2C6H4] | 13a [Ph] | 21 | 81 | 93:7 | >98:2 | >98:2 | 97:3 |
| 9 | 4h [pTsOC6H4] | 13a [Ph] | 22 | 81 | 90:10 | >98:2 | >98:2 | 98:2 |
| 10 | 4i [pMeC6H4] | 13a [Ph] | 23 | 84 | 87:13 | >98:2 | >98:2 | 98:2 |
Several points regarding the data in Table 3 merit further discussion:
In contrast to reactions of E-vinylaluminums (Tables 1–2 and Scheme 4), 7–28% of the products derived from addition of an isobutyl group are observed (Table 3). The origin of such group-selectivity differences is likely due to subtle variations in equilibria involving the formation of vinyl- versus isobutylcopper complexes as well as the relative facility with which such entities undergo allylic alkylation (i.e., only C–C bond formation may be irreversible).
Allylic alkylations with Ag complex 5, used in reactions of E vinylaluminum reagents (Tables 1–2 and Scheme 4), furnish similar efficiency and site-selectivity (vs. 12). With complex 12, however, enantioselectivity is improved significantly. As an example, the transformation shown in entry 1 of Table 3 generates 14 in 86.5:13.5 e.r. when 5 is used, whereas in the presence of 12 the desired 1,4-diene is isolated in 97:3 e.r. The amount of product derived from isobutyl addition is slightly lower with complex 5 (96:4 vs. 91:9 with 12). Similarly, when 12 is used to promote addition of E-vinylsilane 3 to 4a, under otherwise identical conditions, only 53% conversion is observed and 6a is obtained in 94.5:5.5 e.r. (vs. >98% conv. and >99:1 e.r. with 5). The rationale for such selectivity differences is unclear.
Vinylaluminums 13a–g, bearing a dimethylsilyl hydride (vs. trimethylsilyl), are employed because the corresponding Cu-catalyzed allylic alkylations proceed with a stronger preference for the transfer of the vinyl unit (vs. iBu group). For example, when trimethylsilyl 2 (see Scheme 3) is used for the reaction in entry 1 of Table 3, 40% isobutyl addition is observed (vs. 9% with 13a).
Proto-desilylation of the enantiomerically enriched 1,4-dienes shown in Table 3, obtained from the single-vessel process, can be utilized to access the products expected from a sequence beginning with hydroalumination of aryl-substituted terminal alkynes, which, as mentioned before,[5] undergo hydrometallation inefficiently.[5] The example provided in Equation (2) is a case in point.
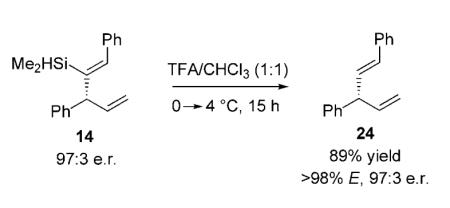 |
(2) |
We therefore demonstrate that trisubstituted vinylaluminum reagents, accessed by hydroalumination of silyl-substituted alkynes, offer a practical protocol for stereoselective synthesis of alkenes. The application to Cu-catalyzed enantioselective allylic alkylation represents one of several other possible C–C bond forming processes feasible through the use of these vinylmetal reagents. Such considerations, together with continuing advances in catalytic cross-coupling reactions of vinylsilanes,[17] augur well for upcoming developments regarding diastereo- and enantioselective synthesis of molecules bearing a trisubstituted alkene.
Supplementary Material
Footnotes
The NIH (Grant GM-47480) provided financial support. Mass spectrometry facilities at Boston College are supported by the NSF (CHE-0619576).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200905223.
References
- [1].For examples of catalytic enantioselective vinyl additions to carbonyls, see: Oppolzer W, Radinov RN. J. Am. Chem. Soc. 1993;115:1593–1594. Miller KM, Huang W-S, Jamison TF. J. Am. Chem. Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y. Li H, Walsh PJ. J. Am. Chem. Soc. 2004;126:6538–6539. doi: 10.1021/ja049206g. Tomita D, Wada R, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2005;127:4138–4139. doi: 10.1021/ja0507362. Yang Y, Zhu S-F, Zhou C-Y, Zhou Q-L. J. Am. Chem. Soc. 2008;130:14052–14053. doi: 10.1021/ja805296k. Kerrigan MH, Jeon S-J, Chen YK, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2009;131:8434–8445. doi: 10.1021/ja809821x. Biradar DB, Gau H-M. Org. Lett. 2009;11:499–502. doi: 10.1021/ol801999u.
- [2].For examples of catalytic enantioselective vinyl additions to aldimines, see: Patel SJ, Jamison TF. Angew. Chem. 2004;116:4031–4034. Angew. Chem. Int. Ed. 2004;43:3941–3944. doi: 10.1002/anie.200460044. Kong J-R, Cho C-W, Krische MJ. J. Am. Chem. Soc. 2005;127:11269–11276. doi: 10.1021/ja051104i. Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:12644–12645. doi: 10.1021/ja075438e. Lou S, Schaus SE. J. Am. Chem. Soc. 2008;130:6922–6923. doi: 10.1021/ja8018934. Nakao Y, Takeda M, Chen J, Salvi L, Hiyama T, Ichikawa Y, Shintani R, Hayashi T. Chem. Lett. 2008;37:290–291.
- [3].For examples of catalytic enantio-selective vinyl conjugate additions to unsaturated carbonyls, see: Oi S, Taira A, Honma Y, Inoue Y. Org. Lett. 2003;5:97–99. doi: 10.1021/ol0272904. Oi S, Sato T, Inoue Y. Tetrahedron Lett. 2004;45:5051–5055. Otomaru Y, Hayashi T. Tetrahedron: Asymmetry. 2004;15:2647–2651. Nicolaou KC, Tang W, Dagneau P, Faraoni R. Angew. Chem. 2005;117:3942–3947. doi: 10.1002/anie.200500789. Angew. Chem. Int. Ed. 2005;44:3874–3879. doi: 10.1002/anie.200500789. Nakao Y, Chen J, Imanaka H, Hiyama T, Ichikawa Y, Duan W-L, Shintani R, Hayashi T. J. Am. Chem. Soc. 2007;129:9137–9143. doi: 10.1021/ja071969r. Vuagnoux-d Augustin M, Alexakis A. Chem. Eur. J. 2007;13:9647–9662. doi: 10.1002/chem.200701001. Lee K.-s., Hoveyda AH. J. Org. Chem. 2009;74:4455–4462. doi: 10.1021/jo900589x.
- [4].Lee Y, Akiyama K, Gillingham DG, Brown MK, Hoveyda AH. J. Am. Chem. Soc. 2008;130:446–447. doi: 10.1021/ja0782192. [DOI] [PubMed] [Google Scholar]
- [5].Treatment of aryl-substituted terminal alkynes with dibal-H leads to the generation of significant amounts (≈30%) of the derived alkynylalanes. See: Zweifel G, Snow JT, Whitney CC. J. Am. Chem. Soc. 1968;90:7139–7141.
- [6].For reviews on catalytic allylic alkylation reactions with “hard” alkyl- or arylmetal-based reagents, see: Hoveyda AH, Hird AW, Kacprzynski MA. Chem. Commun. 2004:1779–1785. doi: 10.1039/b401123f. Yorimitsu H, Oshima K. Angew. Chem. 2005;117:4509–4513. Angew. Chem. Int. Ed. 2005;44:4435–4439. doi: 10.1002/anie.200500653. Falciola CA, Alexakis A. Eur. J. Org. Chem. 2008:3765–3780.
- [7].For other examples of catalytic enantioselective allylic alkylations promoted by chiral bidentate NHC-Cu complexes, see: Larsen AO, Leu W, Nieto-Oberhuber C, Campbell JE, Hoveyda AH. J. Am. Chem. Soc. 2004;126:11130–11131. doi: 10.1021/ja046245j. Van Veldhuizen JJ, Campbell JE, Giudici RE, Hoveyda AH. J. Am. Chem. Soc. 2005;127:6877–6882. doi: 10.1021/ja050179j. Lee Y, Hoveyda AH. J. Am. Chem. Soc. 2006;128:15604–15605. doi: 10.1021/ja067456m. Kacprzynski MA, May TL, Kazane SA, Hoveyda AH. Angew. Chem. 2007;119:4638–4642. doi: 10.1002/anie.200700841. Angew. Chem. Int. Ed. 2007;46:4554–4558. doi: 10.1002/anie.200700841. Gillingham DG, Hoveyda AH. Angew. Chem. 2007;119:3934–3938. doi: 10.1002/anie.200700501. Angew. Chem. Int. Ed. 2007;46:3860–3864. doi: 10.1002/anie.200700501. For reactions promoted by the corresponding ZnII- and AlIII-based complexes, see: Lee Y, Li B, Hoveyda AH. J. Am. Chem. Soc. 2009;131:11625–11633. doi: 10.1021/ja904654j.
- [8].For a previous enantioselective synthesis of di-O-methyl ether of nyasol, involving a Wittig olefination that proceeds with 3:2 Z:E selectivity, and related natural products, see: Quan W-G, Yu B-X, Zhang J-Y, Liang Q-R, Sun Y-Q, She X-G, Pan X-F. Chin. J. Chem. 2007;25:688–693.
- [9].Both mechanisms, delocalization through Si d orbitals and hyperconjugation with a neighboring σ* C–Si bond, have been put forth to account for stabilization of electron density adjacent to a Si substituent. See: Brook MA. Silicon in Organic, Organometallic, and Polymer Chemistry. Wiley-VCH; Weinheim: 2006. pp. 31–34. and 500–503, and references therein.
- [10].For a review regarding stereoselective synthesis through the use of Si-containing compounds, see: Fleming I, Barbero A, Walter D. Chem. Rev. 1997;97:2063–2192. doi: 10.1021/cr941074u.
- [11] a).Eisch JJ, Foxton MW. J. Org. Chem. 1971;36:3520–3526. Eisch JJ, Rhee S-G. J. Am. Chem. Soc. 1975;97:4673–4682. For a related review, see: Eisch JJ. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Schreiber SL, editors. Vol. 8. Pergamon; Oxford: 1991. pp. 733–761.
- [12].For a review of Sonogashira-type coupling processes, see: Chinchilla R, Nájera C. Chem. Rev. 2007;107:874–922. doi: 10.1021/cr050992x.
- [13].Yashima E, Huang S, Matsushima T, Okamoto Y. Macromolecules. 1995;28:4184–4193. [Google Scholar]
- [14].A plausible rationale for the retention of stereochemistry might involve hyperconjugative assistance of the protonation process by the C–Si bond. It is likely that a stepwise mechanism that includes the formation of a carbocation would lead to isomerization and generation of substantial amounts of the lower energy trans alkene. For a similar explanation provided to account for the stereochemical outcome of the reaction of deuterium chloride with trans-β-trimethylsilylstyrene, a process which proceeds with retention of stereochemistry, see: Koenig KE, Weber WP. J. Am. Chem. Soc. 1973;95:3416–3418.
- [15].The absolute stereochemistry of major enantiomers in reactions in Table 3 are opposite to those shown in Tables 1 and 2 because of the stereochemical identity of the chiral NHC ligands in 12 versus 5.
-
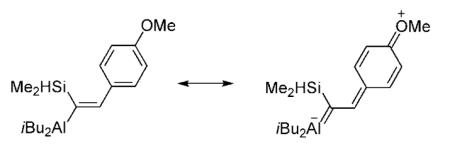
[16].Generation of a minor amount of E alkene (20 formed with 90:10 Z:E selectivity) in the reaction shown in entry 7 of Table 3 might be caused by olefin isomerization facilitated by the p-methoxy substituent, as shown below. The inability of the substrate in entry 2 (Table 3), containing an o-methoxyphenyl group (15 formed with >98% Z selectivity) might be because the corresponding resonance structure suffers from destabilization as a result of the attendant allylic strain.
- [17].For representative examples, see: Hatanaka Y, Hiyama T. J. Org. Chem. 1988;53:918–920. Tamao K, Kobayashi K, Ito Y. Tetrahedron Lett. 1989;30:6051–6054. Denmark SE, Pan W. Org. Lett. 2003;5:1119–1122. doi: 10.1021/ol0342002. Denmark SE, Tymonko SA. J. Am. Chem. Soc. 2005;127:8004–8005. doi: 10.1021/ja0518373. Nakao Y, Imanaka H, Sahoo AK, Yada A, Hiyama T. J. Am. Chem. Soc. 2005;127:6952–6953. doi: 10.1021/ja051281j. Denmark SE, Baird JD. Chem. Eur. J. 2006;12:4954–4963. doi: 10.1002/chem.200600034.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.