

Abstract
The spliceosome is a dynamic macromolecular machine that catalyzes the removal of introns from pre-mRNA, yielding mature message. Schizosaccharomyces pombe Cwf10 (homolog of Saccharomyces cerevisiae Snu114 and human U5-116K), an integral member of the U5 snRNP, is a GTPase that has multiple roles within the splicing cycle. Cwf10/Snu114 family members are highly homologous to eukaryotic translation elongation factor EF2, and they contain a conserved N-terminal extension (NTE) to the EF2-like portion, predicted to be an intrinsically unfolded domain. Using S. pombe as a model system, we show that the NTE is not essential, but cells lacking this domain are defective in pre-mRNA splicing. Genetic interactions between cwf10-ΔNTE and other pre-mRNA splicing mutants are consistent with a role for the NTE in spliceosome activation and second-step catalysis. Characterization of Cwf10-NTE by various biophysical techniques shows that in solution the NTE contains regions of both structure and disorder. The first 23 highly conserved amino acids of the NTE are essential for its role in splicing but when overexpressed are not sufficient to restore pre-mRNA splicing to wild-type levels in cwf10-ΔNTE cells. When the entire NTE is overexpressed in the cwf10-ΔNTE background, it can complement the truncated Cwf10 protein in trans, and it immunoprecipitates a complex similar in composition to the late-stage U5.U2/U6 spliceosome. These data show that the structurally flexible NTE is capable of independently incorporating into the spliceosome and improving splicing function, possibly indicating a role for the NTE in stabilizing conformational rearrangements during a splice cycle.
INTRODUCTION
Eukaryotic pre-mRNA splicing involves the precise removal of introns from pre-mRNA to form protein-coding mature messages (mRNA). This essential step in gene regulation is catalyzed by the spliceosome, a dynamic machine composed of four snRNPs (small nuclear ribonucleoproteins), U1, U2, U5, and U4/U6, and additional pre-mRNA splicing factors (1, 2). In vitro studies have led to a stepwise model of spliceosome assembly whereby the formation of an active spliceosome involves a series of regulated steps requiring the assembly and disassembly of large multiprotein complexes (1, 3). In this model, spliceosome assembly begins with the recognition of the 5′ and 3′ splice sites by the U1 snRNP and U2AF, respectively, while additional components in the U2 snRNP recognizes the branch point sequence. The subsequent engagement of the U4/U6/U5 tri-snRNP triggers the unwinding of the U4/U6 snRNA duplex that is then replaced with the U2/U6 snRNA duplex. Furthermore, the U1 snRNA base pairing at the 5′ splice site is disrupted and exchanged for base pairing between the 5′ splice site and the U6 snRNA. The subsequent release of the U1 and U4 snRNPs marks the transition from the inactive-to-active spliceosome, which contains only the U2, U5, and U6 snRNPs. Following activation, the spliceosome undergoes two-step catalysis, mRNA release, and disassembly. The conformational changes required for spliceosome function are facilitated by an assortment of enzymes, including evolutionarily conserved kinases, phosphatases, DEAD box (DExD/H) ATPase helicases, and a GTPase (3–5). Although there are now comprehensive lists of spliceosomal components associated with each splicing intermediate (2, 6–11), the roles of many of these proteins in the splicing reaction are not well understood.
The sole spliceosomal GTPase is highly conserved across species (32% identity between Saccharomyces cerevisiae Snu114 and human U5-116K) and is a core component of the U5 snRNP (10, 12, 13). The Snu114 family of proteins is required for spliceosome activation and disassembly (14–16), as well as for the integrity of the U5 snRNP and tri-snRNP (14, 15). S. cerevisiae Snu114 and its orthologs interact both physically and genetically with Prp8 and Brr2 (17–20), two highly conserved U5 core components that are essential for facilitating the splicing reaction. Prp8 is located at the “heart” of the spliceosome, since it physically contacts the 5′ and 3′ splice sites and branch point sequence on the pre-mRNA transcript and interacts with the U5 and U6 snRNAs (21–23). Brr2, a U5 snRNP helicase, is required for spliceosome remodeling events, specifically the disruption of U4/U6 interactions (24) and the release of U6 from U2 in spliceosome disassembly (16). In vitro, the GTPase activity of Snu114 is required for Brr2 function, modulating Brr2's ATPase activity (16, 24). Thus, both the physical and functional interactions of Snu114 with Prp8 and Brr2 place it in a key position to facilitate and/or regulate rearrangements near the catalytic center; however, the molecular details of how Snu114 carries out these potential functions are not well understood. Schizosaccharomyces pombe Snu114 homolog Cwf10 (Complexed with Cdc5) has been investigated only briefly and noted for roles in RNA interference (RNAi)-directed centromere repeat silencing and in splicing (25).
The Snu114 family of proteins shares homology with the eukaryotic translation elongation factor EF2 (12) but is also predicted to contain regions of intrinsic disorder (26, 27). We and others have taken advantage of crystal structures of EF2 (28) to predict the EF2-like domain boundaries in the sequences of S. cerevisiae Snu114 (19) and S. pombe Cwf10 (Fig. 1A). By homology, there are six domains that define the “EF2-like” portion of Cwf10 (I, G', II, III, IV, and V in Fig. 1A). Extensive mutagenic analysis of S. cerevisiae Snu114 has demonstrated that altering residues in all six EF2-like domains impairs protein function (19, 29).
Fig 1.
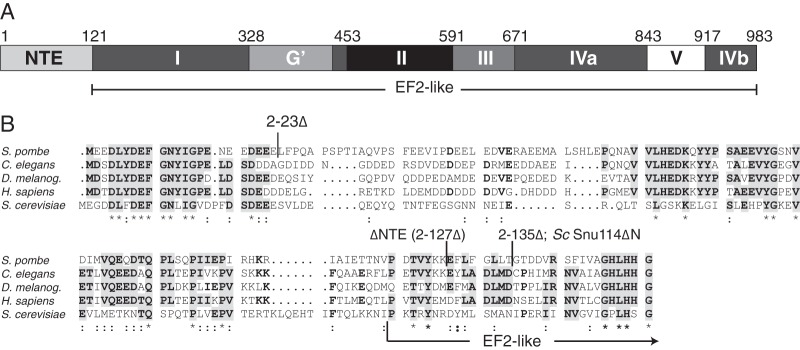
The N-terminal extension is conserved in Snu114/Cwf10 family members. (A) Domain map of S. pombe Cwf10, as defined by three-dimensional modeling (Modeller [95]) of S. pombe Cwf10 onto the crystal structure of S. cerevisiae EF2 (PDB 1N0V). Domains are named as defined by homology with EF2, and the residue numbers listed above the domains refer to S. pombe Cwf10. The NTE domain is not present in EF2. (B) ClustalW (96) sequence alignment of the N termini of Snu114/Cwf10 homologs (from S. pombe, Caenorhabditis elegans, Drosophila melanogaster, Homo sapiens, and S. cerevisiae). Residues that are identical in at least three of the five homologs are in bold. Clusters of identical residues are highlighted in gray. Boundaries of the various Cwf10 truncations and the S. cerevisiae Snu114ΔN truncation are noted above the alignment. Symbols: *, identical; : (colon), similar. An arrow marks the transition from the NTE to EF2-like domain I, as assigned in panel A.
The Snu114/Cwf10 proteins differ significantly from EF2 in that they contain a conserved N-terminal extension (NTE) (Fig. 1A). The NTE is approximately 120 amino acids (aa) long and is rich in acidic residues, with 39% of the first 56 residues being aspartate or glutamate in S. pombe Cwf10 (Fig. 1B). The human and S. pombe NTEs are 43% identical, a much larger percentage than the human and S. cerevisiae NTEs (26% identical), suggesting that studies in S. pombe may also yield relevant insights into the activity of the human NTE.
Functional data implicate the NTE in spliceosome activation and catalysis; however, very little is known about the structure and binding partners of the NTE. In human in vitro splicing assays, addition of antibodies against the U5-116K NTE partially blocks the second step of splicing (12). Removal of S. cerevisiae Snu114's NTE (S. cerevisiae snu114ΔN) causes a temperature-sensitive (ts) phenotype in vivo, while in vitro splicing extracts prepared from S. cerevisiae snu114ΔN cells are unable to efficiently unwind the U4/U6 snRNAs prior to the first catalytic step of pre-mRNA splicing (14). These extracts also contain a destabilized U5 snRNP (14). Finally, the S. cerevisiae snu114ΔN allele shows synthetic lethal and synthetic sick interactions with mutations in the U5 snRNA loop 1 and internal loop 1 (IL-1), as well as with U6 snRNA alleles that disrupt U2/U6 base pairing (30). These results led the authors to speculate that the S. cerevisiae Snu114 NTE may be involved in facilitating U5 and U6 snRNA interactions near the 5′ splice site. Interestingly, no physical interactions have been determined for the NTE in any organism, leaving open the question of how the NTE is spatially oriented within the spliceosome. Additionally, the only structural information about the NTE is a bioinformatics prediction that the NTE of human U5-116K is composed of two disordered regions of equal length (26).
In this study, we characterize the S. pombe Cwf10 NTE both in vivo and biochemically. We show that although the NTE is not essential in S. pombe, deleting this region leads to a general splicing defect at all temperatures. We define a small region of the NTE required for efficient splicing and demonstrate the presence of both structural order and disorder within the NTE. Finally, we show that when the NTE is overexpressed in vivo it stably associates with a protein complex similar to the S. pombe U5.U2/U6 spliceosomal complex and rescues the splicing defect caused by deletion of the NTE from endogenous cwf10+. Taken together, these findings suggest that the NTE is a semiordered domain that has the ability to function in trans to the EF2-like portion of Cwf10.
MATERIALS AND METHODS
Strains, yeast methods, and molecular biology.
Strains used in this study are listed in Table S1 in the supplemental material. Yeast strains were grown in yeast extract (YE) medium or Edinburgh minimal medium with appropriate supplements. The spp41-1 (31) and spp42-1 (31) open reading frames (ORFs) were tagged endogenously at the 3′ end with kanMX6 for genetic analyses as previously described (32). Transformations were done as described previously (33) for all tag insertions, gene replacements, and introduction of plasmids. Integration of tags was verified using whole-cell PCR and immunoblot analysis as appropriate. Crossing of tagged and mutated loci into other strains was accomplished using standard S. pombe mating, sporulation, and tetrad dissection techniques. For spot assays, cells were grown to mid-log phase at 25°C and resuspended in water to achieve an optical density at 595 nm (OD595) of 0.2 (Fig. 2) or 0.6 (see Fig. 5). Tenfold serial dilutions were made, and 2.5 μl of each dilution was plated on YE. Plates were incubated at the indicated temperatures for 2 to 9 days before imaging.
Fig 2.

Characterization of the S. pombe cwf10-ΔNTE allele. (A) Growth of serial dilutions of wild-type (cwf10+) and cwf10-ΔNTE. Serial dilutions of equal numbers of cells were spotted on solid YE medium and grown at 18°C, 25°C, 32°C, and 36°C. (B) Growth of cwf10+ and cwf10-ΔNTE cells in liquid YE at 25°C. Samples were collected at 2-h intervals to determine cell numbers. (C) The NTE is required for efficient pre-mRNA splicing. RT-PCR was used to characterize RNA extracted from cwf10+ and cwf10-ΔNTE cells. Total RNA was isolated from cells grown at 25°C. RT-PCR was performed with random hexamer primers. Portions of the tbp1 (containing introns) and act1 (intronless) transcripts were amplified from the RT-PCR products. Mature (m) and pre-mRNA (p) forms are labeled. No bands were detected in any RT-PCR samples in which the reverse transcriptase enzyme was omitted (−RT). (D to H) Immunoblots demonstrating relative protein levels of pre-mRNA splicing factors in either cwf10+ or cwf10-ΔNTE backgrounds. An asterisk (*) marks the position of each protein on the blot. (D and E) Lysates from cwf10+ and cwf10-ΔNTE cells were probed with anti-PSTAIR and either anti-Cwf10 (D) or anti-Cdc5 (E) antibodies. (F) Lysates from brr2-HA and brr2-HA cwf10-ΔNTE cells probed with anti-HA and anti-PSTAIR. (G) Lysates from prp1-myc and prp1-myc cwf10-ΔNTE cells probed with anti-myc and anti-PSTAIR. (H) Lysates from prp3-V5 and prp3-V5 cwf10-ΔNTE cells probed with anti-V5 and anti-PSTAIR. Anti-PSTAIR detects Cdc2. Numbers to the left of each blot indicate molecular mass in kilodaltons. (I) Comparison of protein levels of each pre-mRNA splicing factor in cwf10-ΔNTE cell lysates relative to the cwf10+ value (100%), normalized to Cdc2, quantitated from Western analysis. Values were calculated using three biological replicates. Error bars indicate standard errors of the means. The P value was determined using the Student t test.
Fig 5.
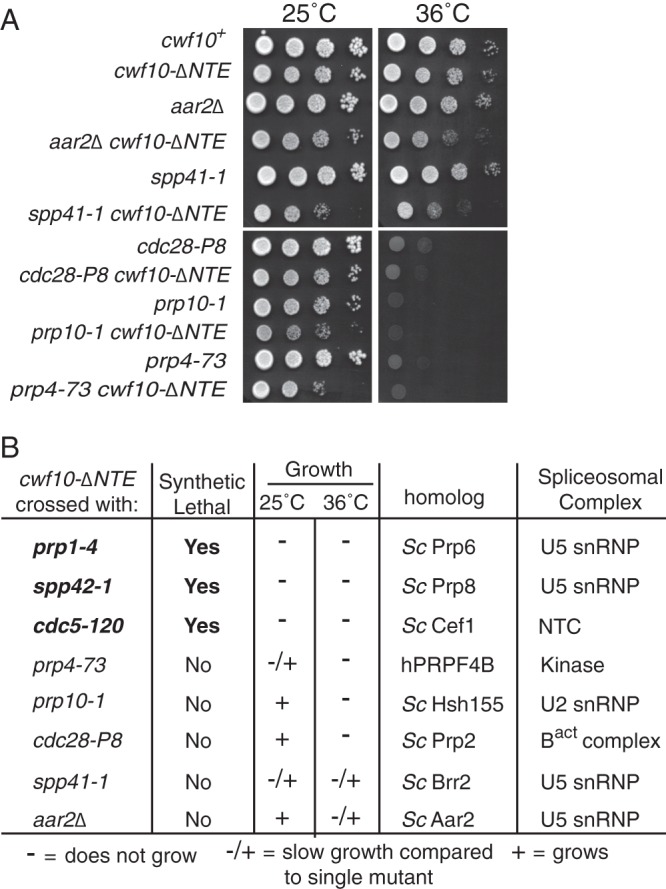
Genetic interactions of cwf10-ΔNTE with alleles of other pre-mRNA splicing factors. (A) Serial dilutions of single and double mutants grown at 25°C and 36°C were used to examine growth. (B) Table summarizing results of crossing cwf10-ΔNTE with other pre-mRNA splicing factor mutants. The column labeled “Growth” indicates growth of the double mutant compared with that of the single mutant allele in a wild-type (cwf10+) background at 25°C and 36°C. −, no growth; −/+, slow growth; +, growth; Sc, S. cerevisiae; h, human.
For induction of the nmt3X promoter (34), the cells were first grown overnight in medium containing 5 μg/ml of thiamine and then washed three times with medium lacking thiamine and allowed to grow for at least 16 h in thiamine-free medium. For harvesting smaller cultures, the growth in 5 μg/ml of thiamine was omitted. Overexpression plasmids used include pREP3X cwf10 1–135 (pOHI1038), pREP3X cwf10 1–107 (pOHI1037), pREP41 NTAP (35) (pOHI756), pREP41 NTAP-cwf10 2–135 (pOHI1039), and pREP41 NTAP-cwf10 2–26 (pOHI1076).
All plasmids were generated by standard molecular biology techniques. For gene replacements at the endogenous cwf10+ locus, mutant open reading frames were generated using QuikChange II or QuikChange Lightning Multi technologies (Agilent Technologies, Santa Clara, CA). The mutant ORF and at least 500 bp of 5′- and 3′-flanking nucleotides were subcloned into the pIRT2 plasmid containing the LEU2 marker. A diploid cwf10+/cwf10::ura4+ strain was transformed with pIRT2-cwf10 mutant constructs and grown on minimal medium lacking leucine, adenine, and uracil. Transformants were allowed to sporulate, and stable haploid integrants were selected based on resistance to 5-fluoroorotic acid (5-FOA). Mutants were validated by whole-cell PCR with primers outside the 5′- and 3′-flanking regions. pIRT2-cwf10 mutant plasmids used include pOHI1035 (2–135Δ), pOHI978 (2–127Δ), pOHI1071 (2–23Δ), and pOHI1070 (E/D-A).
Liquid culture cell numbers were counted with a Z1 Coulter Counter (Beckman Coulter, Brea, CA) as previously described (36).
Antibody preparation.
His6-Cwf10 (673–983) was expressed in Escherichia coli BL21(DE3) cells (EMD Millipore, Battymarch Park, MA), purified with HisPur Cobalt agarose (Pierce/Thermo Scientific, Rockford, IL), and used to immunize rabbits (Cocalico Biologicals, Reamstown, PA), as approved by the Vanderbilt Institutional Biosafety Committee. Cwf10-specific antibodies were affinity purified over N-hydroxysuccinimide (NHS)-activated Sepharose Fast Flow 4 (GE Healthcare Life Sciences, Piscataway, NJ) covalently linked to His6-Cwf10 (673-983).
Immunoprecipitations, immunoblotting, and sucrose gradients.
“Native” and “denatured” whole-cell lysates were prepared as previously described, with leupeptin omitted from lysis buffers (37). For immunoblots, proteins were resolved by 10% SDS-PAGE (all lysates except Brr2-HA), 4 to 12% Bis-Tris PAGE (Brr2-HA lysates), or 8% SDS-PAGE (lysate gradients) and transferred by electroblotting to a Protran nitrocellulose membrane (Whatman, GE Healthcare, Piscataway, NJ). Primary antibodies/antisera used included anti-Cdc5 (1/5,000) (37) and anti-Asp1 (1/5,000) rabbit polyclonal antisera, anti-Cwf10 affinity purified rabbit polyclonal antibody (0.84 μg/ml), and anti-PSTAIR (detects S. pombe Cdc2) monoclonal antibody (1/10,000) (Sigma, St. Louis, MO). For some of the Cwf10 quantification (Fig. 2), the antiserum was used instead of the antibody. Anti-myc (9E10, 0.3 μg/ml) and antihemagglutinin (anti-HA) (12CA5, 1 μg/ml) mouse monoclonal antibodies were used to detect Myc- and HA-tagged proteins, respectively. Primary antibodies were detected by secondary antibodies Alexa Fluor 700 (Invitrogen, Life Technologies, Grand Island, NY) or IRDye 800CW (LI-COR Biosciences, Lincoln, NE) (1/10,000 dilution) and visualized and quantified using an Odyssey scanner and software (LI-COR Biosciences).
For gradients, a 15-OD pellet was lysed and a 200-μl volume corresponding to 25% of the lysate was layered onto a 10 to 30% sucrose gradient and centrifuged at 25,000 rpm at 4°C for 16 h in a SW55Ti rotor (Beckman). Fractions from the gradients were collected manually and either (i) trichloroacetic acid (TCA) precipitated and resuspended in SDS sample buffer to detect protein or (ii) extracted with 5:1 acid phenol-chloroform and resuspended in Tris-borate-EDTA (TBE)-urea sample buffer (Life Technologies, Grand Island, NY) to detect RNA. Parallel standard gradients contained thyroglobulin (19S) and catalase (11.35S) (HMW calibration kit; GE Healthcare, Piscataway, NJ) or 20% of lysate from a 20-OD pellet of FAS2-V5-tagged S. cerevisiae (strain OHI375) (40S marker).
Coimmunoprecipitations for Western blot analysis were performed as described in reference 38. Tandem affinity purifications (TAPs) were performed as described previously (39), with the following modifications: native lysis buffer was as described earlier in this section, calmodulin binding volume was reduced by 3 ml, calmodulin binding buffer (CBB) washes were 10 ml and 2 ml, respectively, and the final CBB buffer and the calmodulin elution buffer (CEB) contained no detergent. For TAPs analyzed in Table 1, purifications were done using 75 mM NaCl.
Table 1.
pre-mRNA splicing factors copurifying with Cdc5-TAP in either a cwf10-ΔNTE or a wild-type background identified using 1D liquid chromatography-tandem mass spectrometryabcd
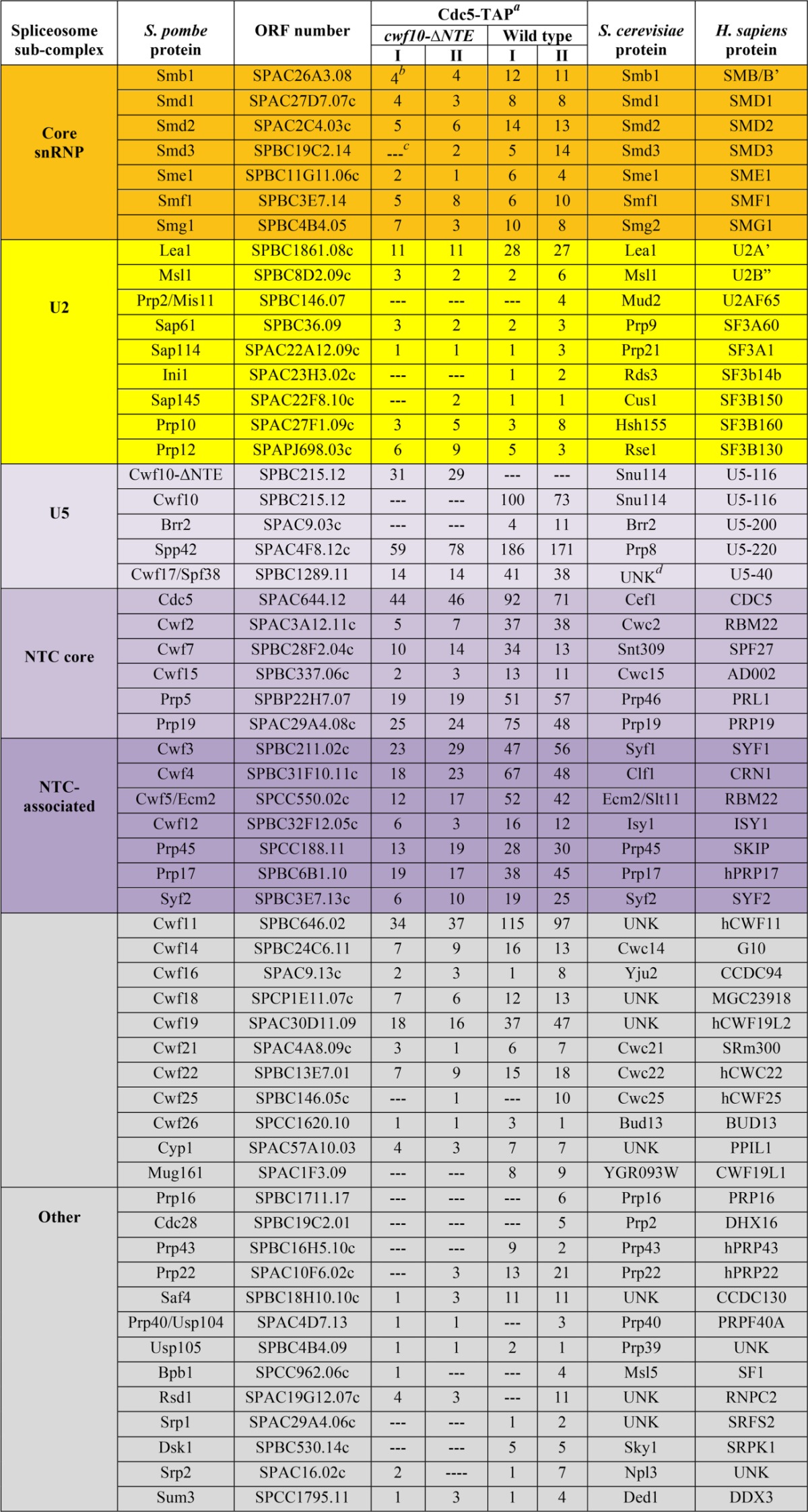
Purifications done using 75 mM NaCl.
Peptide counts of identified proteins in each biological replicate (I and II).
—, no peptides identified.
UNK, an ortholog is unknown or not present.
RNA isolation, Northern blotting, and RT-PCR.
Total RNA was prepared from cells by extraction with hot acidic phenol as described previously (40). To visualize snRNAs found in fractions collected either from sucrose gradients or from TAP-NTE and anti-snRNA cap (antitrimethylguanosine [m3G]; Millipore) pulldowns, RNAs were resolved using 6% TBE-urea gels (Life Technologies). RNA was transferred to a Duralon-UV membrane (Agilent Technologies, Santa Clara, CA), UV cross-linked using energy setting 700 (UVC500 cross-linker; GE Healthcare, Piscataway, NJ), and detected by using [γ-32P]ATP (PerkinElmer, Waltham, MA)-labeled oligonucleotides complementary to S. pombe U1, U2, U4, U5, and U6 (for sequences, see Table S2 in the supplemental material). Blots were exposed to phosphorimager screens for 14 to 18 h and visualized using Typhoon 9200 or FLA-7000IP instruments (GE Healthcare, Piscataway, NJ). Quantification for snRNAs found in fractions collected from sucrose gradients was performed using ImageQuant TL 8.1 (GE Healthcare). For reverse transcription (RT)-PCR analysis, RNA was treated with DNase I (Life Technologies) and reverse transcribed according to the manufacturer's directions using random hexamers for priming (SuperScript; Life Technologies). Eight hundred nanograms of RNA was used for each reaction. Three technical samples per genotype were processed. The resulting cDNA was PCR amplified with tbp1_a or mrps16_b primers (see Table S2 in the supplemental material) for 27 cycles, and ethidium bromide-stained gels were imaged within a linear range and quantified with ImageQuant TL 8.1 (GE Healthcare).
RNA sequencing and analyses.
Replicates were grown together and processed separately for all following steps. The Vanderbilt Technologies for Advanced Genomics Core Facility (Vantage, Nashville, TN) used the TruSeq Stranded mRNA sample preparation kit (Illumina, San Diego, CA) to convert the mRNA in 100 ng of total RNA into a library of template molecules suitable for subsequent cluster generation and sequencing on the Illumina HiSeq 2500. The input total RNA was quality checked by running an aliquot on the Agilent Bioanalyzer to confirm integrity. The Qubit RNA fluorometry assay was used to measure concentration. The input to library prep was 50 μl of 2 ng/μl DNase-treated total RNA. The total RNA underwent enrichment of the poly(A)-containing mRNA molecules using poly(T) oligoattached magnetic beads. Following purification, the eluted poly(A) RNA was cleaved into small fragments of 120 to 210 bp using divalent cations under elevated temperature. The cleaved RNA fragments were copied into first-strand cDNA using SuperScript II reverse transcriptase and random primers. This was followed by second-strand cDNA synthesis using DNA Polymerase I and RNase H. The cDNA fragments were then put through an end repair process, the addition of a single A base, and ligation to the Illumina multiplexing adapters. The products were then purified and enriched with PCR to create the final cDNA sequencing library. The cDNA library underwent quality control by running on the Agilent Bioanalyzer HS DNA assay to confirm the final library size and on the Agilent Mx3005P qPCR machine using the KAPA Illumina library quantification kit to determine the concentration. A 2 nM stock was created, and samples were pooled by molarity for multiplexing. From the pool, 12 pM was loaded into each well for the flow cell on the Illumina cBot for cluster generation. The flow cell was then loaded onto the Illumina HiSeq 2500 utilizing v3 chemistry and HTA 1.8. The raw sequencing reads in BCL format were processed through CASAVA-1.8.2 for FASTQ conversion and demultiplexing. The RTA chastity filter was used, and only the PF (passfilter) reads were retained for further analysis. Raw expression data files are available from Gene Expression Omnibus (GEO accession number GSE47573; http://www.ncbi.nlm.nih.gov/geo/).
Genome level alignments and annotation.
Paired-end reads of 76-base length (each end) originating from each sample were aligned using Bowtie 0.12.7 (41) to the S. pombe genome sequence (Ensembl S. pombe, Build EF1, version 13) (42) as well as to the corresponding exon-exon junctions database (only the first part of the paired-end reads was considered). Up to 3 base pair mismatches were allowed. Reads that matched multiple loci were removed from further analysis, and the resultant alignment files were processed to generate “pile-ups” against each chromosome.
Exon-exon junctions.
Searches were performed against the genome sequence combined with a data set of known exon-exon junctions as defined by Ensembl S. pombe release 13. To ensure that a 76-base read mapped to a splice junction, only the last 70 bases of the first exon and the first 70 bases of the second exon were considered (if the exon exceeded a length of 70 bases). In this way, reads that overlapped a junction by <6 nucleotides were excluded. Reads that matched to more than one junction or elsewhere in the genome were also discarded.
Defining intronic and intergenic regions.
The known annotated set of S. pombe genes (7022; Ensembl version 13, as before) was used to define unambiguous antisense transcripts (i.e., those that exactly mirror known annotated genes without overlapping nearby genes), and unique accessions were assigned (“anti_xxx”; 3097). Using this augmented annotation, intergenic regions were defined (i.e., regions between known annotated and unambiguous antisense regions on each strand), and unique accessions were assigned (“inter_xxx”; 8810). Unique accessions were also assigned to all known introns (“int_xxx”; 5361). Thus, in total 21,193 regions were interrogated across the 4 samples (annotated genes, introns, and intergenic regions; see Data set S1 in the supplemental material).
Normalization, fold changes, and differential expression.
Differential expression between samples was determined using the DESeq Bioconductor package (43). A cutoff of ±2-fold change and corrected P value of <0.05 were applied to derive a list of differentially expressed genes, introns, and intergenic regions.
RNA-seq expression level.
For RNA sequence expression levels, normalized expression levels (E) for individual exons and introns were calculated using the following formula as described in references 44 and 45 (RPKM measure): E = log2[C(Ri/TiL)]. Briefly, the number of reads (R) detected across a given region at a given sample (i) was multiplied by a constant (C = 1 × 109) and divided by the total number of reads at that sample (Ti) multiplied by the region's length (L).
A small constant was added (10−5) to all expression values to avoid taking logarithms of zero. Gene level expression values were summarized using exon data. Sample-specific expression levels for all regions interrogated in this study are provided in Data set S1 in the supplemental material.
SE and differential splicing significance.
Splicing efficiency (SE) reflects the proportion of spliced mRNA signal relative to pre-mRNA signal. Splicing efficiency is computed by dividing junction reads (JR; also known as trans-reads) by reads that straddle an exon-intron boundary (EI; only the upstream 5′ exon relative to the intron was considered) according to the following formula: SE = log2(JR/EI).
A Cochran-Mantel-Haenszel (CMH) chi-square test for repeated test of independence, which accounts for biological replicates (46), was applied to identify statistically significant introns (i.e., those that display differences in their splicing efficiency between samples; mantelhaen.test command in R). The false-discovery rate (q-value) was computed using the Bioconductor q-value package (47), and a cutoff for q of <0.05 was applied.
Expression and purification of Cwf10(1–135).
The N-terminal sequence of amino acids 1 to 135 of Cwf10 with 6 C-terminal histidine residues [Cwf10(1–135)His6] was cloned into pET15b (NcoI/BamHI; plasmid pOHI1020) (EMD Millipore) and transformed into E. coli Rosetta 2(DE3)pLysS cells (EMD Millipore). Cells were grown in Terrific broth (Invitrogen, Grand Island, NY) to an OD595 of ∼0.8 and cold shocked for 20 min on ice. Upon addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside), the plasmid was overexpressed for 20 h at 15°C. Cells were lysed in phosphate-buffered saline (PBS) (pH 7.0), 350 mM NaCl, 0.1% Triton X-100, and one SigmaFAST protease tablet (Sigma-Aldrich, St. Louis, MO). Cwf10(1–135)His6 was purified using two 5-ml Histrap HP columns (GE Healthcare, Waukesha, WI) and a 2.5 to 500 mM imidazole linear gradient. Cwf10(1–135)His6 was further purified using anion-exchange (Uno Q1; Bio-Rad) chromatography in buffer A (50 mM Tris-HCl, pH 8.0) using a 0 to 1 M NaCl linear gradient, followed by gel filtration (Superdex 200; GE Healthcare) in 25 mM Tris-HCl (pH 7.3), 100 mM NaCl, and 1 mM EDTA.
CD.
For circular dichroism (CD) spectrometry, purified Cwf10(1–135)His6 was analyzed using a Jasco J-810 spectropolarimeter (Jasco Analytical Instruments, Easton, MD). Far-UV data were collected at a protein concentration of 0.18 mg/ml in a 1-mm quartz cuvette. Spectra were collected with an average time of 4 s for each point and a step size of 20 nm/min from 198 to 260 nm. Far-UV spectra were collected in duplicate and background corrected against a buffer blank. Spectra were analyzed using the program K2D2 (48) to estimate secondary structure. Near-UV data were collected at a protein concentration of 2.01 mg/ml in a 1-cm quartz cuvette. Spectra were collected with an average time of 4 s for each point and a step size of 10 nm/min from 250 to 330 nm. For both Cwf10(1–135)His6 and denatured Cwf10(1–135)His6 in 6 M guanidine-HCl, five spectra were collected for near-UV data and background corrected against a buffer blank. Data were converted to mean residue ellipticity [θ]m (degrees cm2 dmol−1) using the formula [θ]m = θ/(10lcn), where θ is the measured ellipticity, l is the cell path length in cm, c is the molar concentration of protein in mol/liter, and n is the number of residues/chain.
Analytical ultracentrifugation.
Purified Cwf10(1–135)His6 was run in an Optima XLI ultracentrifuge (Beckman Coulter, Brea, CA) equipped with a four-hole An-60 Ti rotor at 42,000 rpm at 4°C at 0.45 mg/ml. Samples were loaded into double-sector cells (path length of 1.2 cm) with charcoal-filled Epon centerpieces and sapphire windows. Sedfit (version 12.0) (49) was used to analyze velocity scans using every five scans from a total of 167 scans. Approximate size distributions were determined for a confidence level set at P values of 0.95, a resolution of n = 300, and sedimentation coefficients between 0 and 5S.
NMR analysis.
Cwf10(1–135)His6 was purified as above, except that cells were grown and expressed in M9 medium supplemented with 15N-labeled ammonium chloride (Cambridge Isotopes, Andover, MA) as the only nitrogen source and 10% D2O was added to the final sample. During purification, one half of the sample was left in gel filtration buffer (25 mM Tris-HCl [pH 7.3], 100 mM NaCl, and 1 mM EDTA) and the other half was buffer exchanged into gel filtration buffer plus 6 M guanidine-HCl. Standard sensitivity-enhanced echo/antiecho 15N-1H heteronuclear single-quantum correlation (HSQC) nuclear magnetic resonance (NMR) data were collected at 25°C for both samples and at 50°C for the gel filtration buffer sample (no guanidine-HCl) using a 600 MHz Bruker AVIII spectrometer (Bruker, Billerica, MA) with a CPQCI probe and z-axis gradient. The spectra were processed using Topspin 3.2 (Bruker, Billerica, MA). The indirect dimension was four times zero filled to a final matrix of 2,048 × 1,024 data points, and 720 and 900 squared sine bell apodization was applied in the F2 and F1 dimensions, respectively. Spectra were further analyzed with Sparky (T. D. Goddard and D. G. Kneller, University of California, San Francisco).
MS.
TAP elutions were trichloroacetic acid (TCA) precipitated, resolubilized in 8 M urea–100 mM Tris (pH 8.5), reduced, alkylated, and then diluted back to 2 M urea and digested overnight with trypsin as described previously (50). Resulting peptides (corresponding to about 5% of the TAP eluate) were analyzed by a 70-min data-dependent liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis in the Vanderbilt Mass Spectrometry core. In brief, peptides were autosampled onto a 200-mm by 0.1-mm (Jupiter 3 micron, 300 Å) self-packed analytical column coupled directly to an LTQ (ThermoFisher) using a nanoelectrospray source and resolved using an aqueous to organic gradient. A series of a full-scan mass spectrum followed by five data-dependent tandem mass spectra was collected throughout the run, and dynamic exclusion was enabled to minimize acquisition of redundant spectra. Tandem mass spectra were searched via Sequest against an S. pombe database (UniprotKB taxon 284812 reference proteome set) that also contained a reversed version for each of the entries (51). Identifications were filtered and collated at the protein level using Scaffold (Proteome Software). Data sets for one-dimensional (1D) LC-MS/MS analyses are provided in Data sets S2 and S3 in the supplemental material.
Negative-stain EM.
Uranyl formate-stained samples were prepared for electron microscopy (EM) as described previously (52). In brief, 2.5 μl of sample was absorbed to a glow-discharged 400-mesh copper grid covered with carbon-coated collodion film. The grid was washed in two drops of water and then stained with two drops of uranyl formate (0.75%). Samples were imaged on a Morgagni electron microscope (FEI, Hillsboro, OR) operated at an acceleration voltage of 100 kV. Images were recorded at a magnification of 28,000 and collected using a 1K × 1K charge-coupled-device (CCD) camera (AMT, Woburn, MA).
RESULTS
Deleting the S. pombe Cwf10 NTE impairs splicing in vivo.
To more closely examine the in vivo role of the Cwf10 NTE in pre-mRNA splicing, we prepared an S. pombe cwf10 construct corresponding to S. cerevisiae snu114ΔN (14). Although cwf10 2–135Δ aligns with snu114ΔN (Fig. 1B), the S. pombe allele did not support growth at either 25°C or 32°C when integrated at the endogenous locus (data not shown), while the S. cerevisiae allele is viable at temperatures below 40°C (30). Upon closer examination of the alignment of Snu114 family members with EF2, it appears that Cwf10 2–135Δ lacks residues corresponding to domain I (Fig. 1B), which may affect the folding of this essential domain. Therefore, we deleted eight fewer amino acids and successfully integrated cwf10 2–127Δ (here, cwf10-ΔNTE) at the endogenous locus. This result suggests that some of the defects seen in the S. cerevisiae snu114ΔN strain may be partly attributable to a defect in the folding of domain I in the EF2-like region of Snu114.
We further characterized cwf10-ΔNTE to examine the NTE's contribution to in vivo splicing. The cwf10-ΔNTE strain formed slightly smaller colonies on solid media at all temperatures tested (Fig. 2A) and exhibited slow growth in liquid culture at 25°C and 36°C compared with wild-type cells (Fig. 2B and data not shown). Conversely, overexpression of the NTE (cwf10 1–135 or 1–107) under the high-strength nmt (no message in thiamine) promoter did not impair cell viability, indicating its expression is not dominant negative (data not shown). RT-PCR analysis of tbp1_a, an mRNA intron highly sensitive to splicing defects (53), indicated that cwf10-ΔNTE cells grown at 25°C accumulate unspliced transcript (Fig. 2C). Thus, removal of the Cwf10 NTE reduces splicing efficiency in vivo.
In our experience, yeast spliceosome mutants often have a lower steady-state mutant protein level that correlates with the splicing defect. To determine whether the deletion of the Cwf10 NTE simply decreases the amount of Cwf10 in cells, thus reducing splicing efficiency, antibodies were generated against Cwf10 amino acid residues 673 to 983 (domains IV and V). Immunoblotting of S. pombe lysate with α-Cwf10 antiserum detected a single protein at the anticipated molecular mass of ∼116 kDa (Fig. 2D, cwf10+ lane). In cwf10-ΔNTE lysates, the antiserum detected a band ∼20 kDa smaller than the wild-type protein, consistent with the predicted molecular mass of the deletion (Fig. 2D). Using quantitative Western blotting, we found that levels of Cwf10 were consistently higher in cwf10-ΔNTE cells than in wild-type cells when quantified against the loading control Cdc2 (Cdk1) (Fig. 2I). These results show that low levels of Cwf10 do not cause the splicing defect in cwf10-ΔNTE cells. To examine whether levels of other splicing components might be affected in this background, we analyzed the protein levels of S. pombe Cdc5 (homolog of S. cerevisiae Cef1) (Fig. 2E), a core component of the Nineteen complex (NTC) (54, 55), Brr2 (S. cerevisiae Brr2) (Fig. 2F), and a core U5 snRNP member (13), as well as Prp1 (S. cerevisiae Prp6) and Prp3 (S. cerevisiae Prp3) (Fig. 2G and H), both members of the U4/U6/U5 tri-snRNP and B-complex (2, 56). In the cwf10-ΔNTE background, levels of Brr2 and Prp3 were similar to levels seen in wild-type cells; however, both Cdc5 and Prp1 levels were reduced to ∼70% and ∼65%, respectively (Fig. 2I). Since the prp1 transcript does not contain introns, we attribute the lower level of these proteins to reduced protein stability rather than a general reduction in transcript levels.
RNA-seq demonstrates a general splicing defect in cwf10-ΔNTE cells.
Previously it has been demonstrated that mutations in core spliceosome proteins affect splicing of introns to different degrees (57–59). That is, one mutant can have a profile of inefficiently spliced introns that is significantly different from the profile of another mutant. To examine the extent of splicing changes in cwf10-ΔNTE cells, we performed transcriptome analysis. RNA was extracted from cwf10+ and cwf10-ΔNTE cells grown at 25°C, and two replicates for each genetic background were sequenced on the Illumina HiSeq platform. Two methods were employed to evaluate the effect of NTE deletion on splicing efficiency using the RNA sequencing data. A Cochran-Mantel-Haenszel (CMH) test assessed the significance of the change in the ratio of spliced to unspliced transcripts between cwf10-ΔNTE and cwf10+ (see Materials and Methods), while DESeq (43) revealed differentially expressed introns between the two conditions. Splicing efficiency was highly reproducible between replicates, with a tight distribution along the diagonal (see Fig. S1 in the supplemental material), but displayed a nearly global shift when cwf10-ΔNTE and cwf10+ were compared; this can be seen as a shift of most points toward the right of the diagonal in Fig. 3A. This suggests an extensive splicing defect in the NTE-deleted strain that affected most introns. Using the splicing efficiency (SE) approach under a stringent threshold (q-value, <0.05; fold change, >2), we identified 1,193 introns (of 5,361 possible introns) whose splicing efficiency was significantly compromised in the mutant. Similarly, using DESeq, 883 introns were more highly expressed in cwf10-ΔNTE relative to cwf10+, indicating that their splicing was significantly less efficient in the mutant strain (adjusted P value of <0.05 and fold change of >2; Fig. 3B). There was a good correspondence between the two methods (P < 2.2e−226, hypergeometric test [Fig. 3C]), which was even stronger when no fold change cutoff was applied (P < 2.7e−249 [data not shown]). Overall, the splicing of 2,352 introns (approximately 44% of all introns) was significantly compromised by the NTE deletion, as indicated by at least one method without the fold change cutoff. The effect of the NTE deletion was not dependent on intron length, and neither was the magnitude of the reduction in splicing efficiency (Fig. 3D, top and bottom panels, respectively). Moreover, the effect of the mutation was not dependent on intron GC content, branch site sequence, 5′ intron sequence, or the position of the intron within the transcript (data not shown). For a small set of introns (total, 17), splicing efficiency was significantly improved by the deletion, but there was no agreement between the two methods under these criteria (14 were found by DESeq and 3 by SE). However, when the magnitude of the fold change was ignored, the splicing efficiency of 223 introns was significantly improved (209 by DESeq and 24 by SE, of which 10 were identified by both methods; P < 3.9e−10). We did not find any clear sequence features that could explain why a small percentage of introns were spliced with an efficiency that was either the same as or better than that in the NTE deletion background. Taken together, the RNA sequence (RNA-seq) data indicate that the NTE of Cwf10 is required for efficient splicing of most S. pombe introns.
Fig 3.

RNA-seq data reveal a nearly global splicing defect in cwf10-ΔNTE cells. (A) Global reduction in splicing efficiency in the cwf10-ΔNTE strain (y axis; log2 scale) compared to the wild type (x axis; log2 scale). Each point represents an intron (5,361 in total), while red dots represent the 1,193 introns for which splicing efficiency was significantly compromised in the mutant compared to the wild type (CMH test, q-value, <0.05, and absolute fold change cutoff, >2). Note that this graph compares one replicate of each genotype to the wild type. (B) Differentially expressed genes and intergenic regions (blue) and introns (red) identified by DESeq (adjusted P value, <0.05, and absolute fold change, >2; x axis, normalized mean expression; y axis, log2 fold change). A total of 21,193 regions (genes, introns, and intergenic regions) were interrogated, of which 883 introns were upregulated in the mutant. (C) Venn diagram illustrating the overlap between the two methods used to assess the extent of the splicing defect (SE is in light blue and DESeq in light pink). The probability of observing the same number of overlaps or more by chance assuming a hypergeometric distribution was determined by the “phyper” function in R. (D) Top panel, proportion of significant and nonsignificant introns within a given intron length bin (2,352 introns identified by SE or DESeq, with q-value or adjusted P value of <0.05, respectively; for a given intron, splicing efficiency was compromised in both replicates; light gray, significant; dark gray, nonsignificant; the exact numbers are indicated). Bottom panel, splicing efficiency of the significant intron set as a function of their length (straight and dashed lines with circles, biological replicates of the wild type; straight and dashed lines with diamonds, biological replicates of mutant; line with X's, mean log10 of mutant/wild-type SE fold change).
The lack of Cwf10-NTE changes spliceosome sedimentation patterns.
In in vitro splicing assays, deletion of the S. cerevisiae Snu114 NTE inhibits U4/U6 snRNA unwinding, thus impairing activation of the spliceosome (14). Since S. pombe does not have a robust in vitro splicing extract system, likely due to the stability of the U5.U2/U6 spliceosomal complex (60, 61), we could not directly test whether S. pombe Cwf10-NTE plays a similar role in fission yeast. However, we postulated that if the S. pombe Cwf10-NTE shares a function similar to that of its S. cerevisiae ortholog, cells that lack the Cwf10-NTE might have altered sedimentation patterns of spliceosomal complexes and/or snRNAs. Thus, to test the effect of deleting Cwf10-NTE in vivo, we compared the sedimentation patterns of Cwf10, Cdc5 (S. cerevisiae Cef1), and the five snRNAs in sucrose gradients using native lysates made from either wild-type or cwf10Δ-NTE cells (Fig. 4). It has been previously shown that in lysates from asynchronously growing wild-type S. pombe cells, the majority of spliceosomes sediment as a stable ∼37S U5.U2/U6 complex (10, 37, 60, 61), although less abundant U5/U6/U4/U2/U1 and U5/U6/U4/U2 complexes have also been characterized (62, 63). Unlike what is found in other organisms, S. pombe lysates lack detectable quantities of a preassembled U5.U4/U6 tri-snRNP (61).
Fig 4.

Sedimentation of spliceosome components is altered in cwf10-ΔNTE cells. Lysates from cwf10+ and cwf10-ΔNTE cells grown at 25°C were run on 10 to 30% sucrose gradients. Fractions were collected, and either they were TCA precipitated for immunoblot analysis or RNA was extracted for Northern analysis. (A and C) Immunoblots of fractions collected from 10 to 30% sucrose gradients of lysates from cwf10+ prp1-myc and cwf10-ΔNTE prp1-myc using anti-Cwf10 (A) and anti-Cdc5 (C). (B and D) Intensities of the bands of the anti-Cwf10 blot (B) and anti-Cdc5 (D) were quantified and plotted as a percentage of the sum of the signal from all fractions. The bands were quantified by near-infrared detection and Odyssey software (Licor). (E, G, I, K, and M) RNA was isolated from fractions collected from 10 to 30% sucrose gradients of cwf10+ and cwf10-ΔNTE lysates. Blots were probed with 32P-labeled oligonucleotides complementary to the S. pombe U2 (E), U5 (G), U6 (I), U1 (K), and U4 (M) snRNAs. (F, H, J, L, and N) Intensities of the bands of the U2 (F), U5 (H), U6 (J), U1 (L), and U4 (N) probes were quantified and plotted as a percentage of the sum of the signal from all fractions. The bands were quantified by phosphorimaging and ImageQuant TL 8.1 software (GE Healthcare). The asterisk (*) in panels H and L highlights the peak of the U5 and U1 snRNAs found in a small-molecular-mass fraction in cwf10-ΔNTE. Column I indicates where the U5.U2/U6 spliceosomal complex sediments in the gradients. The migration of fatty acid synthase (FAS) (40S), thyroglobulin (19S), and catalase (11.3S) collected from parallel gradients is indicated.
As expected, in wild-type lysates a majority of Cwf10 sediments at an ∼37S peak, corresponding to the sedimentation pattern of the U5.U2/U6 complex (Fig. 4A and B, fraction 9 [10]); however, the sedimentation pattern of Cwf10-ΔNTE in cwf10-ΔNTE lysates changes, with a portion of Cwf10-ΔNTE shifting to higher-molecular-mass fractions (Fig. 4A and B, fractions 11 and 12). The sedimentation pattern of Cdc5, a core NTC component, does not appear grossly altered in lysates from cwf10-ΔNTE cells (Fig. 4C and D). We next compared the sedimentation profiles of spliceosomal snRNAs in both wild-type and cwf10-ΔNTE lysates. As expected in the wild-type background, the U2, U5, and U6 snRNAs cosedimented at ∼37S as described before (10, 37, 60, 61) (Fig. 4E to J). The sedimentation pattern of U4, U5, and U6 snRNAs from wild-type cells (Fig. 4G to J, M, and N) are consistent with previous reports that S. pombe appears to lack an independently sedimenting U5.U4/U6 tri-snRNP (61). Another apparent difference between fission yeast and other organisms is the abundance of the U2 snRNP at 12S (Fig. 4E and F), which most likely represents the core U2 snRNP rather than the larger 17S complex that contains the SF3a and SF3b subcomplexes (64–67). However, when comparing wild-type and cwf10-ΔNTE lysates, the sedimentation patterns of all the snRNAs were altered to some degree (Fig. 4E to N). The differences include a small but consistent shift of some of the U2, U4, U5, and U6 snRNAs to higher-molecular-mass fractions (>40S, fractions 11 to 13 [Fig. 4E to J, M, and N]), as well as increases in the amount of the U5 and U1 snRNAs found at lower-molecular-mass fractions (<11.3S [Fig. 4G and H and K and L). Thus, from these analyses we conclude that deleting the S. pombe Cwf10 NTE does alter the distribution of spliceosomes in vivo, although in a complex pattern with shifts to both higher and lower molecular masses.
cwf10-ΔNTE synthetically interacts with factors predicted to be involved in spliceosome activation.
As another approach to investigate whether S. pombe NTE function can be correlated to a specific step of the pre-mRNA splicing reaction or stage of spliceosomal organization, we tested for genetic interactions between cwf10-ΔNTE and eight previously characterized S. pombe pre-mRNA splicing alleles (Fig. 5). Our analysis revealed that cwf10-ΔNTE is synthetic lethal when combined with prp1-4 (68) (S. cerevisiae PRP6), spp42-1 (31) (S. cerevisiae PRP8), and cdc5-120 (69) (S. cerevisiae CEF1). When cwf10-ΔNTE is combined with either prp4-73 (70) (hPRPF4B) or spp41-1 (31) (S. cerevisiae BRR2) the cells are synthetically sick. Conversely, synthetic interactions are weak or nonexistent between cwf10-ΔNTE and aar2Δ (71) (S. cerevisiae AAR2), prp10-1 (68) (S. cerevisiae HSH155), or cdc28-P8 (72) (S. cerevisiae PRP2). Two of the three synthetic lethal interactions, prp1-4 and spp42-1, are with genes previously shown to be important for spliceosome activation (62, 73). The third synthetic lethal interaction, cdc5-120, is interesting because Cdc5 is likely involved in spliceosome activation as a component of the NTC (74) and in modulating the transition between first- and second-step catalysis (75). We conclude that the S. pombe Cwf10 NTE is likely important for a specific stage in spliceosome activation, and possibly also for a later stage(s) in the splicing reaction.
To further address whether the NTE may be involved in facilitating the transition from an inactive to activated spliceosome, we analyzed the protein composition of the S. pombe U5.U2/U6 spliceosomal complex purified from either a cwf10-ΔNTE or a wild-type background using one-dimensional (1D) liquid chromatography-tandem mass spectrometry (LC-MS/MS), a technique well suited for detecting peptides that are stoichiometrically present in a purification. Because the NTE is not essential and deletion of this region does not cause a temperature-sensitive phenotype (Fig. 1), we reasoned that any changes seen in the U5.U2/U6 complex purified from cwf10-NTE cells would likely be subtle, and thus we did the purifications for this analysis using mild salt (75 mM NaCl) conditions. Cdc5-TAP (S. cerevisiae Cef1), which associates with the S. pombe U5.U2/U6 complex (10), was used to purify the U5.U2/U6 complex. Peptide counts for spliceosomal proteins found in each 1D LC-MS/MS run are included in Table 1 to provide a semiquantitative indication of protein amounts. From this analysis, the major differences between the two purifications were the lack of Brr2 (S. cerevisiae Brr2), Mug161 (human CWF19L1), and Prp43 (S. cerevisiae Prp43) peptides detected in the Cdc5-TAP from cwf10-ΔNTE cells. Brr2 is an essential U5 snRNP component that forms a salt-stable complex with Prp8 (S. pombe Spp42) and Snu114 (S. pombe Cwf10) (13). Mug161 (human CWF19L1) is a protein of unknown function that has been isolated in spliceosomes purifications from both S. pombe (38) and mammalian cells (76), and Prp43 is required for spliceosome disassembly (77–79). Because this analysis was done using 1D LC-MS/MS rather than the more-sensitive MudPit (multidimensional protein identification technology) MS, the lack of detected peptides for particular proteins such as Brr2 could indicate either a complete absence of the protein in the sample or that the protein is at substoichiometric levels in the purification. Overall, the differences that we detect between U5.U2/U6 complexes purified from either wild-type or cwf10-ΔNTE backgrounds suggests that NTE may play a role in stabilizing Brr2's interaction with the U5 snRNP during spliceosome activation, a model supported by the synthetic sick interaction shown between cwf10-ΔNTE and spp41-1 (S. cerevisiae Brr2) (Fig. 5A).
The Cwf10 NTE is partially folded but contains regions of disordered coiled coil.
The structural characteristics of the NTE were of interest to us for several reasons. First, the only published data on NTE structure consist of a bioinformatics predication that the region is unfolded (26). Second, the NTE does not carry strong homology to known protein domains or to primary sequences in other proteins (data not shown). To more carefully examine the structural characteristics of this domain, we used Disopred (80), a program that predicts structure disorder, to precisely map potential regions of intrinsic disorder in the Cwf10 NTE. This analysis showed that while a majority of the first N-terminal 60 amino acids are strongly predicted to not adopt any secondary structure, the C-terminal 75 amino acids are predicted to be structurally ordered (Fig. 6A), perhaps as β-strands as suggested by analysis with Psipred version 3.3 (81, 82), a program that predicts secondary structure (data not shown). To experimentally test this model, we expressed and purified recombinant Cwf10(1–135)His6 from E. coli (see Fig. S2A in the supplemental material). Analysis by sedimentation analytical ultracentrifugation (SVAU) shows that the Cwf10 NTE sediments as a monomer (s = 0.8; predicted molecular mass, ∼19 kDa; root mean square deviation [rsmd] = 0.09) with a frictional ratio of 2.13 (Fig. 6B). Cwf10(1–135)His6 was then analyzed by circular dichroism (CD) spectrometry using near-UV wavelengths, which can be used to detect tertiary structure. Analysis of this spectrum shows a strong signal between 260 and 290 nm, suggesting that there are some aromatic residues found in a folded environment (Fig. 6C). Importantly, this signal is no longer seen when the protein is completely denatured in 6 M guanidine-HCl (Fig. 6D). The NTE contains six tyrosines, two in the first 12 amino acids and the remaining four between residues 78 and 125 (Fig. 1B). Interestingly, these regions both correspond to predicted regions of order (Fig. 6A). Next, the protein was analyzed by CD spectrometry using far-UV wavelengths (Fig. 6E), which can be used to predict secondary structure. Analysis of this spectrum using the program K2D2 (48) predicts that Cwf10(1–135)His6 in solution is composed of ∼10% α-helix and ∼31% β-sheet, leaving over 50% of the NTE likely disordered (see Fig. S2B in the supplemental material).
Fig 6.

Biophysical characterization shows that the Cwf10 NTE contains regions of order and disorder. (A) Residues 18 to 61 of the NTE are predicted to be intrinsically disordered in solution using Disopred (80), a program that predicts structure disorder. The false-positive rate (FPR) was set at 5%. (B) Cwf10(1–135)His6 is monomeric by SVAU. The calculated s [c(s)] is plotted versus the sedimentation coefficient (s) for Cwf10(1–135)His6. The sedimentation velocity profiles were fitted to a continuous sedimentation distribution. (C) Near-UV CD spectrum of Cwf10(1–135)His6. (D) Near-UV CD spectrum of Cwf10(1–135)His6 in 6 M guanidine-HCl. (E) Far-UV CD spectrum of Cwf10(1–135)His6. (F) 15N-1H HSQC spectra of Cwf10(1–135)His6. (G) The first 23 amino acids of the Cwf10-NTE are required for efficient splicing. RNA was prepared from cwf10+, cwf10-ΔNTE, and cwf10 2–23Δ cells grown at 25°C, and RT-PCR was done using random hexamer primers. (H) A portion of the tbp1 transcript was amplified from the RT-PCR products, and ratios of mRNA/pre-mRNA were calculated. Quantification represents the average of three biological replicates per genotype. Error bars indicate standard errors of the means.
To more carefully examine the tertiary structure of the NTE, we 15N labeled Cwf10(1–135)His6 and collected a two-dimensional 15N-1H heteronuclear single-quantum correlation (HSQC) experiment using nuclear magnetic resonance (NMR) spectroscopy. By this analysis, we found that the Cwf10 NTE contains 103 well-dispersed resonances (of a total of 141 residues) (Fig. 6F). When Cwf10(1–135)His6 is either heated to 50°C or treated with 6 M Guanidine-HCl in order to cause complete denaturation of any folded domain(s), the resonance peaks collapse and are no longer dispersed (see Fig. S2C and D in the supplemental material). Well-dispersed resonances in HSQC spectra result from the variable environment of the amines in a folded protein. Thus, the NMR analysis confirms both the computational and CD analyses indicating that the Cwf10 NTE contains regions of disorder, most likely in the first 60 amino acids (Fig. 6A), as well as regions that adopt a well-ordered secondary structure.
Interestingly, amino acids 1 to 23 of the NTE are 70% identical between S. pombe and human orthologs (Fig. 1B), and most of these residues (amino acids 1 to 17) are not predicted to be disordered (Fig. 6A). To test if this patch of conserved residues is required for NTE function, we replaced the chromosomal copy of cwf10+ with cwf10 2–23Δ to observe whether this smaller truncation would also cause a pre-mRNA splicing defect. RNAs from wild-type, cwf10 2–23Δ, and cwf10-ΔNTE cells were extracted, and RT-PCR was performed (Fig. 6G). Measures were taken to improve the quantitative nature of the PCR (83), including reverse transcribing similar amounts of RNA, reducing PCR cycles, and quantifying against an inherent internal control (the spliced and unspliced forms are amplified in the same reaction). The ratio of mature to premature signal for the tbp1_a intron was almost identical for the two truncations (2.1 ± 0.2 for cwf10-ΔNTE versus 2.0 ± 0.3 for cwf10 2–23Δ), while the ratio in the wild-type strain was 9.7 ± 1.5 (Fig. 6H). This suggests that the conserved, extreme N terminus (residues 1 to 23) is required for NTE function.
The Cwf10 NTE copurifies with splicing factors.
The spliceosomal binding partners of the NTE have not been determined in any organism. We attempted to screen interactions by a yeast two-hybrid assay; however, the assay was hindered by the high self-activation of the GAL binding domain when fused to the acidic NTE. Therefore, we fused cwf10 2–135 to the N-terminal tandem affinity purification (TAP) tag in a pREP41 NTAP vector (35). We overexpressed NTAP-NTE in cwf10-ΔNTE cells, performed a two-step purification at 150 mM NaCl, and analyzed the eluate using negative-stain electron microscopy (EM). Although the purification was clearly dilute, we did see particles that were reminiscent of negative-stain images of the S. pombe U5.U2/U6 complex (Fig. 7A) (10, 60), suggesting that the Cwf10 NTE is able to interact with spliceosomal complexes on its own. As an initial characterization of the TAP-NTE complex, we analyzed the snRNA content of the TAP-NTE purification from cwf10-ΔNTE cells. As seen in Cdc5-TAP (10), TAP-NTE is associated with the U2, U5, and U6 snRNAs (Fig. 7B). While it is possible that a small amount of U1 snRNA is present in the purification (Fig. 7B), since no U1 snRNP components were found in the 1D LC-MS/MS analysis (Table 2) this interaction would have to be substoichiometric.
Fig 7.

Cwf10 NTE can incorporate into spliceosomal complexes and partially rescues the splicing defect in cwf10-ΔNTE cells. (A) Gallery of particles present in TAP-NTE purifications from cwf10-ΔNTE cells as seen by negative-stain EM. Scale bar, 20 nm. (B) TAP-NTE associates with the U2, U5, and U6 snRNAs. RNA was isolated from TAP-NTE purified from cwf10-ΔNTE cells and anti-snRNA cap (antitrimethylguanosine [m3G]) purified from wild-type cells. Blots were probed with 32P-labeled oligonucleotides complementary to the S. pombe U1, U2, U4, U5, and U6 snRNAs. (C) Overexpression of cwf10-NTE in cwf10-ΔNTE cells rescues splicing defects. Total RNA was isolated from cells grown at 32°C. RT-PCR was performed with random hexamer primers. Portions of the tbp1 and mrps16 transcripts were amplified from the RT-PCR products. Mature (m) and pre-mRNA (p) forms are indicated. RT, reverse transcriptase enzyme. (D and E) Quantification of mRNA/pre-mRNA ratios for three biological replicates per genotype for RT-PCR analysis of tbp1 (D) and mrps16 (E) transcripts. Error bars indicate standard errors of the means. (F) Overexpression of cwf10 2–23 in cwf10-ΔNTE cells does not rescue splicing defects. Total RNA was isolated from cells grown at 32°C. RT-PCR was performed with random hexamer primers. Portions of the tbp1 and mrps16 transcripts were amplified from the RT-PCR products. (G and H) Quantification of mRNA/pre-mRNA ratios for three biological replicates per genotype for RT-PCR analysis of tbp1 (G) and mrps16 (H) transcripts. ns, not significant.
Table 2.
pre-RNA splicing factors copurifying with TAP-NTE in a cwf10-ΔNTE background and Cdc5-TAP in a wild-type background identified using 1D liquid chromatography-tandem mass spectrometryabcd
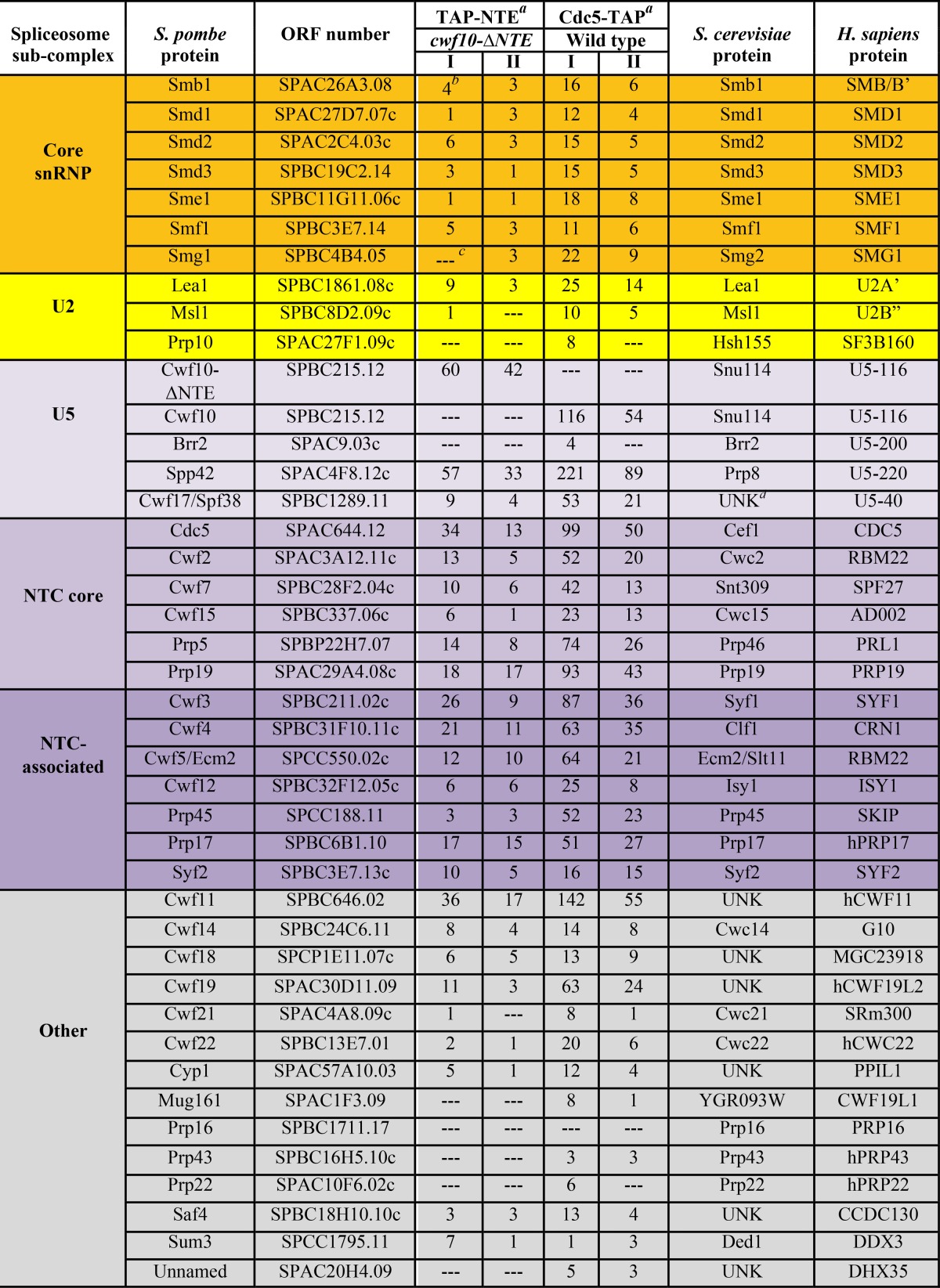
Purifications done at 150 mM NaCl.
Peptide counts of identified proteins in each biological replicate (I and II).
—, no peptides identified.
UNK, an ortholog is unknown or not present.
To further characterize the composition of the TAP-NTE purification, we analyzed the protein content of the eluate using 1D LC-MS/MS and compared it to Cdc5-TAP from wild-type cells under similar salt conditions. Peptide counts for spliceosomal proteins found in each LC-MS/MS run are included in Table 2 to provide a semiquantitative indication of protein amounts. After heat shock proteins (see Data set S3 in the supplemental material), the next-highest group of proteins identified in the purification comprised pre-mRNA splicing factors that are similar to the S. pombe U5.U2/U6 spliceosomal complex (10, 38) (Table 2). Highly represented in this group are the Sm proteins, components of the U5 snRNP (although Brr2 peptides were not detected), and components of and related to the hPrp19/Cdc5L complex. Importantly, the EF2-like portion of Cwf10 (from the genomic cwf10-ΔNTE allele) was also highly represented, confirming the ability of Cwf10-ΔNTE to incorporate into higher-order complexes, as seen by sucrose gradient sedimentation (Fig. 4A). The presence of U2 snRNP proteins Lea1 and Msl1 indicates at least the partial presence of the U2 snRNP; however, the SF3 U2 snRNP components were not detected. Thus, the compositions of Cdc5-TAP and TAP-NTE are almost identical (Table 2) and show that the NTE can associate with a complex similar in composition to the late-stage U5.U2/U6 spliceosome.
To address whether we could detect Brr2 and SF3 peptides under less stringent purification conditions, the TAP-NTE purifications were repeated using 75 mM NaCl and analyzed by 1D LC-MS/MS (see Table S3 in the supplemental material). Once again, no peptides for either Brr2 or SF3 U2 snRNP components were detected (see Table S3 in the supplemental material), even though Brr2 peptides were detected in U5.U2/U6 complexes purified from wild-type cells using 75 mM NaCl (Table 1) and SF3 peptides were detected in U5.U2/U6 purifications from both wild-type and cwf10-ΔNTE cells using 75 mM NaCl (Table 1).
In regard to the SF3 U2 snRNP components, our data suggest that while they may not be as stably associated with the U5.U2/U6 complex as Lea1 (S. cerevisiae Lea1, human U2A'), peptides for SF3 proteins were detected in Cdc5-TAP purifications from both wild-type and cwf10-ΔNTE cells under mild salt conditions (Table 1). Conversely, no SF3 peptides were detected in the TAP-NTE purifications using either 150 or 75 mM salt (see Table S3 in the supplemental material). This shows that while the presence or absence of the NTE does not affect how SF3 components interact with the U5.U2/U6 complex, either the TAP-NTE complex does not contain SF3 proteins or they are present at substoichiometric levels.
To more closely examine whether the NTE associates with Brr2, we asked whether TAP-NTE can coimmunoprecipitate Brr2 in cwf10-ΔNTE cells. To this end, either NTAP or NTAP-NTE was overexpressed in a cwf10-ΔNTE brr2-3XHA strain. Immunoprecipitations were done using IgG-Sepharose beads and then immunoblotted with anti-HA antibodies to detect Brr2-HA. TAP-NTE was able to pull down Brr2-HA, while TAP alone was not (see Fig. S3 in the supplemental material), showing that Brr2 can associate with the TAP-NTE complex. However, one caveat of this experiment is that it does not replicate the two-step TAP protocol that was used for purifying the sample for mass spectrometry analysis. Therefore, it is possible that Brr2 is present after the first step of the purification but dissociates during the second step of the TAP. Combined, our analyses show that while TAP-NTE can coimmunoprecipitate Brr2 as detected by Western blot analysis, Brr2 is likely present at only substoichiometric levels in both TAP-NTE and Cdc5-TAP purifications from cwf10-ΔNTE cells. Overall, our results demonstrate that TAP-NTE is able to bind to a surface(s) of the U5.U2/U6 core without being covalently linked to the EF2-like portion of Cwf10 (residues 128 to 983) and that the protein(s) and/or RNA(s) that creates the binding surface(s) for the Cwf10 NTE is present in the purified complex of 32 detected spliceosome components.
Because Cwf10-NTE copurified with spliceosomal components, we next asked whether the domain could restore pre-mRNA splicing efficiency in cwf10-ΔNTE, i.e., function in trans. To test this, we performed semiquantitative RT-PCR on RNA extracted from cwf10-ΔNTE cells overexpressing either NTAP or NTAP-NTE and looked at the splicing efficiency of two introns. Analysis of the tbp1_a and mrps16_b introns revealed that overexpression of NTAP-NTE in the cwf10-ΔNTE background improved the mature/premature ratio over the NTAP control by ∼25% and 30%, respectively, returning splicing efficiency close to wild-type levels (Fig. 7C to E). Although only semiquantitative, the statistical significances of these data support a model in which the NTE can incorporate into spliceosomal complexes independent of any covalent connection with the C-terminal EF2-like portion of Cwf10, partially rescuing the splicing deficiency seen in cwf10-ΔNTE cells.
Given the previous result that cwf10 2–127Δ (cwf10-ΔNTE) and cwf10 2–23Δ have almost identical splicing defects (Fig. 6G and H), we wondered whether overexpressing just the first 26 amino acids of the NTE would be sufficient to restore the pre-mRNA splicing efficiency of cwf10-ΔNTE. Therefore, we repeated the RT-PCR experiment, using RNA extracted from cwf10-ΔNTE cells overexpressing either NTAP or NTAP-cwf10 2–26. Although Western blotting confirmed the expression of the TAP-Cwf10 2–26 protein (data not shown), the smaller region was unable to complement deletion of the entire NTE (Fig. 7F to H). Thus, the conserved region of amino acids 2 to 23 is necessary for splicing but not sufficient to complement cwf10-ΔNTE in trans.
DISCUSSION
Deletion of the NTE and its effects on transcripts.
In this study, we have investigated the function and structural characteristics of the Cwf10 NTE. Although this domain is not essential in fission yeast, deletion of the NTE causes a general splicing defect, a change in the sedimentation patterns of in vivo spliceosomal complexes, and synthetic lethal and synthetic sick interactions with mutant alleles of other pre-mRNA splicing factors.
Because cwf10-ΔNTE cells show robust growth at all temperatures, we were surprised to detect a pre-mRNA splicing defect and postulated that the lack of the NTE domain may affect the splicing of a specific subset of pre-mRNAs rather than cause a global pre-mRNA splicing defect. To test if this was the case, we used deep sequencing analysis to comprehensively determine which transcripts are affected by this mutation. Interestingly, most introns show a tendency toward reduced splicing efficiency (all points to the right of the diagonal in Fig. 3A), and the reduced splicing is statistically significant for about 44% of total introns. It is not surprising, then, that there is no obvious type of transcript (i.e., specific splice sites, number of introns, and/or size of introns) that is specifically affected. Thus, the splicing defect in cwf10-ΔNTE cells appears to be global in nature and widespread in scope. This suggests that the NTE is important for a general (not transcript-specific) process in the splicing reaction.
Because functions have not been assigned for any subregion of the NTE, we removed just the first 23 amino acids from the NTE in Cwf10 and found a similar splicing defect between cwf10-ΔNTE and cwf10 2–23Δ. We speculate that loss of this small, highly conserved, acidic region undermines the NTE's ability to bridge interactions or support conformational rearrangements in the spliceosome critical for the NTE's function (see “Model of Cwf10 NTE interactions in the spliceosome” below).
The NTE's roles in the splicing cycle.
A previous study using S. cerevisiae snu114ΔN demonstrated a role for the Snu114/Cwf10 NTE in U4/U6 snRNA unwinding (14). However, a study using human splicing extracts and antibodies directed against the human U5-116K NTE suggested that the NTE may also be involved in the first- to second-step transition (12). This NTE function, uncovered in the human system, is further supported by negative genetic interactions (30) between S. cerevisiae snu114ΔN and both a substitution at snRNA U6-A59, which inhibits the second step of splicing (84), and several alleles of the U5 loop 1, which helps position the exons for ligation in the second step (85, 86).
Our genetic findings are consistent with a role for the NTE in both spliceosome activation and second-step catalysis. cwf10-ΔNTE interacts with several alleles of genes known to be involved in spliceosome activation (Fig. 5), including spp42-1 (S. cerevisiae PRP8), prp1-4 (S. cerevisiae PRP6), and cdc5-120 (S. cerevisiae CEF1). Additionally, the cdc5-120 mutation, with which cwf10-ΔNTE is synthetically lethal, may play an additional role in second-step chemistry. First, cdc5-120 is lethal with S. pombe prp17Δ (38), a known second-step splicing factor in S. cerevisiae (87). Second, cdc5-120 is a point mutation in one of the two conserved Myb repeats (88), a region important for first-step to second-step modulation in ortholog S. cerevisiae Cef1 (75). The fact that the NTE does not immunoprecipitate a preactivation spliceosome (Table 2), but rather a complex similar to U5.U2/U6, could indicate that the NTE becomes stably “locked” into the spliceosome following activation and is positioned to act near the catalytic core to help modulate that transition.
Our analysis is consistent with the NTE playing a role in stabilizing U5 snRNP integrity during spliceosome transitions, a model supported by the lack of Brr2 peptides found in both Cdc5-TAP and TAP-NTE purifications from the cwf10-ΔNTE backgrounds (Tables 1 and 2; see also Table S3 in the supplemental material) and the increase in the amount of smaller U5 snRNP complexes in cwf10-ΔNTE cells (Fig. 4G and H). It has been proposed that in S. cerevisiae Snu114 acts as a transducer that signals and regulates Brr2 activity throughout the splicing cycle (16), although the mechanism(s) of how Snu114 could modulate Brr2 activity is not yet understood on a molecular level. One possibility is that the NTE acts as a flexible scaffold stabilizing Brr2's interaction with the U5.U2/U6 complex during the structural rearrangements that occur during spliceosome activation. When the NTE is missing, Brr2 is not able to remain as tightly associated with the spliceosome during these transitions, affecting the overall integrity of the U5 snRNP and leading to reduced pre-mRNA splicing efficiency.
Alterations in sizes of snRNA-containing complexes.
Analysis of snRNAs in cells lacking the NTE shows that loss of this domain changes the sedimentation patterns of spliceosomal complexes. Although all the snRNA sedimentation patterns show some degree of change in the ΔNTE background (Fig. 4E to N), for both the U1 and U5 snRNAs there is a shift away from higher-molecular-mass fractions into lower-molecular-mass fractions (<11.3S [Fig. 4H and L]). For the U5 snRNA, this shift is too high in the gradient (<11.3S [Fig. 4H]) to contain the full complement of U5-snRNP specific proteins, as both human and S. cerevisiae U5 snRNPs sediment at 15 to 20S when bound to the U5-specific proteins (89, 90). Similar results with U5 snRNA sedimentation in the S. cerevisiae snu114ΔN background led the authors to speculate that the NTE is required for U5 snRNP stability (14), and our results are consistent with that hypothesis. For the U1 snRNA, it is unclear whether this lower-molecular-mass sedimenting fraction represents free U1 snRNA or the snRNA complexed with at least some of the U1 snRNP proteins.
Sucrose gradient analysis also revealed that a small fraction of the U2, U5, U6, and U4 snRNAs, as well as Cwf10, shifted to high-molecular-mass fractions that are more pronounced in cwf10-ΔNTE (fractions 11 and 12 [Fig. 4B, F, H, J, and N]). The presence of this peak could suggest that a preactivation spliceosome is stalled and accumulating. Alternatively, this peak could represent a different multi-snRNP complex or aggregates of spliceosome components that are not able to organize properly.
Although the sedimentation data support a model in which the in vivo cwf10-ΔNTE splicing complexes that are slow to activate or complete catalysis may be accumulating or aggregating, other possible fates for stalled spliceosomes include degradation by the proteasome and/or disassembly. The lower steady-state levels of Cdc5 and Prp1-myc (Fig. 2I), which sediment mainly as part of large complexes in cwf10-ΔNTE (Fig. 4C and D and data not shown), could favor the hypothesis that these slow-to-splice complexes are degraded.
The NTE's structure.
Although the NTE was predicted to be intrinsically unstructured in solution (26), our modeling and biophysical analysis unexpectedly showed that approximately one-half of NTE residues are in a folded environment in solution. Because the Disopred program (Fig. 6A) predicted disorder to exist mostly in the N terminus (aa 18 to 61) of the NTE and the ordered amino acids to exist at the extreme N terminus (aa 1 to 17) and the C terminus (aa 62 to 120), it is tempting to begin thinking of the NTE as three subregions with separate characteristics. The extreme N terminus, which is not predicted to be disordered, is required for function, since deleting 23 conserved residues in the N terminus fully recapitulates the splicing defect seen when the entire NTE is deleted (Fig. 6G and H). However, this small region cannot complement the cwf10 mutant lacking the NTE in trans, suggesting that the C-terminal NTE regions are also important. The region spanning amino acids 1 to 17 is physically linked to aa 18 to 61, which is predicted to be disordered and has a high content of acidic residues, as does the aa 1 to 17 region. The acidic charge of the NTE is likely essential since we were unable to integrate a cwf10 mutant that had all negatively charged residues in aa 1 to 61 replaced with alanine residues (data not shown). Possibly, this unstructured subregion of the NTE becomes folded in the context of the dynamic spliceosome when it comes into contact with a binding partner(s). This idea has precedent in pre-mRNA splicing. A 70-amino-acid intrinsically disordered region of the NTC-associated protein human SKIP undergoes a disordered-to-ordered transition upon binding to cyclophilin PPIL1 (91). Similarly, a 31-amino-acid, predominantly random coil region of human U4/U6-60K adopts structure when bound to U4/U6-20K (another cyclophilin) (92). However, the disordered regions of human SKIP or U4/U6-60K do not share the same high acidic content of Cwf10-NTE aa 1 to 61. Next, a predicted structured portion of the NTE, amino acids 62 to 120, links the N-terminal NTE regions to the “EF2-like” portion of the protein. Further studies are needed to delineate the boundaries of order and disorder within the NTE, as well as to determine binding partners of these subregions.
Potential binding environment in the spliceosome.
When overexpressing TAP-NTE (Cwf10 residues 2 to 135) in cwf10-ΔNTE cells, we found that this fragment is able to incorporate into a spliceosomal complex that is similar in composition to the S. pombe U5.U2/U6 complex. From this observation, we conclude that the Cwf10-NTE recognizes and interacts with other spliceosomal components independently of its polypeptide linkage to the main Cwf10 “EF2-like” portion. What does the NTE bind to in the context of the late-stage spliceosome? Likely, the binding site does not involve Brr2, because it is not present at stoichiometric amounts in the purification (Table 2), although other U5-specific proteins, i.e., Spp42 (S. cerevisiae Prp8), Spf38 (human U5-40), and the EF2-like portion of Cwf10, are highly represented. Therefore, the binding partner(s) is likely one of the above U5-specific proteins, the U5 Sm core, U2 snRNP protein Lea1, or one or more of the NTC and NTC-related proteins. Additionally, the specific genetic interactions of S. cerevisiae snu114ΔN with snRNA alleles (30) may also support an orientation of the NTE near U5 loop 1 and U6. Indeed, the U5 snRNA loop 1 has been shown to be a binding platform for S. cerevisiae Snu114, Brr2, and Prp8 during U5 snRNP assembly (93). Importantly, NTE binding partners may change as protein-protein, protein-RNA, and RNA-RNA rearrangements occur during the splicing reaction. The combination of structured and unstructured regions within the NTE may allow the domain to bridge transient conformational states adopted by the spliceosome as splicing occurs. However, regardless of the exact mechanism, the presence of the Cwf10 NTE in trans is sufficient to partially rescue the splicing defect seen in cwf10-ΔNTE cells. There are other spliceosome proteins that can function when their domains are expressed separately, highlighting the multiple interactions between spliceosome components. Examples include the large, 280-kDa S. cerevisiae Prp8 (18) and first-step factor S. cerevisiae Yju2 (94).
Model of Cwf10 NTE interactions in the spliceosome.
Based on our data and the work of others outlined above, we propose a model of NTE function that includes (i) stabilization of the U5 snRNP structure and (ii) facilitation of certain conformations among U5 snRNP components or between the U5 snRNP and other spliceosome proteins/RNAs. The NTE is able to independently incorporate into the spliceosome and likely makes contact with more than one splicing component through its predicted structured and unstructured regions. Although we were not able to determine specific binding partners for the Cwf10 NTE, these partners must be in the vicinity of the EF2-like portion of Cwf10. It is tempting to speculate that these contacts promote cohesion within the U5 snRNP and may transmit a signal or form a stabilizing bridge, first within the context of the preactivation B complex and then near the catalytic center of the C complex.
Supplementary Material
ACKNOWLEDGMENTS
We thank N. F. Käufer for sharing S. pombe strains, Kathy Gould and the Gould lab for generous contributions of strains, reagents, protocols, and discussion, Liping Ren and Anna Feoktistova for valuable technical assistance, and Hayes McDonald for assistance with mass spectrometry analysis. We thank Ohi lab members and Kate Mittendorf for helpful discussions and comments on the manuscript.
This work was supported by T32 CA009582 to S.B.L., T32 GM08320 to S.E.C., and NIH DP2OD004483 to M.D.O., as well as by a Wellcome Trust Senior Investigator Award to J.B. Recombinant His6-Cwf10 (673-983) and anti-Cwf10 antibodies were produced and purified by the Vanderbilt Antibody and Protein Resource, which is supported by the Vanderbilt Institute of Chemical Biology and the Vanderbilt Ingram Cancer Center (P30 CA68485).
Footnotes
Published ahead of print 6 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00140-13.
REFERENCES
- 1. Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136:701–718 [DOI] [PubMed] [Google Scholar]
- 2. Fabrizio P, Dannenberg J, Dube P, Kastner B, Stark H, Urlaub H, Lührmann R. 2009. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol. Cell 36:593–608 [DOI] [PubMed] [Google Scholar]
- 3. van der Feltz C, Anthony K, Brilot A, Pomeranz Krummel DA. 2012. Architecture of the spliceosome. Biochemistry 51:3321–3333 [DOI] [PubMed] [Google Scholar]
- 4. Staley JP, Guthrie C. 1998. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 92:315–326 [DOI] [PubMed] [Google Scholar]
- 5. Will CL, Lührmann R. 2011. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 3(7):pii:a003707. 10.1101/cshperspect.a003707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jurica MS, Moore MJ. 2003. Pre-mRNA splicing: awash in a sea of proteins. Mol. Cell 12:5–14 [DOI] [PubMed] [Google Scholar]
- 7. Lardelli RM, Thompson JX, Yates JR, III, Stevens SW. 2010. Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA 16:516–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Agafonov DE, Deckert J, Wolf E, Odenwälder P, Bessonov S, Will CL, Urlaub H, Lührmann R. 2011. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol. Cell. Biol. 31:2667–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bessonov S, Anokhina M, Will CL, Urlaub H, Lührmann R. 2008. Isolation of an active step I spliceosome and composition of its RNP core. Nature 452:846–850 [DOI] [PubMed] [Google Scholar]
- 10. Ohi MD, Link AJ, Ren L, Jennings JL, McDonald WH, Gould KL. 2002. Proteomics analysis reveals stable multiprotein complexes in both fission and budding yeasts containing Myb-related Cdc5p/Cef1p, novel pre-mRNA splicing factors, and snRNAs. Mol. Cell. Biol. 22:2011–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Warkocki Z, Odenwälder P, Schmitzova J, Platzmann F, Stark H, Urlaub H, Ficner R, Fabrizio P, Lührmann R. 2009. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat. Struct. Mol. Biol. 16:1237–1243 [DOI] [PubMed] [Google Scholar]
- 12. Fabrizio P, Laggerbauer B, Lauber J, Lane WS, Lührmann R. 1997. An evolutionarily conserved U5 snRNP-specific protein is a GTP-binding factor closely related to the ribosomal translocase EF-2. EMBO J. 16:4092–4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Achsel T, Ahrens K, Brahms H, Teigelkamp S, Lührmann R. 1998. The human U5-220kD protein (hPrp8) forms a stable RNA-free complex with several U5-specific proteins, including an RNA unwindase, a homologue of ribosomal elongation factor EF-2, and a novel WD-40 protein. Mol. Cell. Biol. 18:6756–6766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bartels C, Klatt C, Lührmann R, Fabrizio P. 2002. The ribosomal translocase homologue Snu114p is involved in unwinding U4/U6 RNA during activation of the spliceosome. EMBO Rep. 3:875–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brenner TJ, Guthrie C. 2006. Assembly of Snu114 into U5 snRNP requires Prp8 and a functional GTPase domain. RNA 12:862–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Small EC, Leggett SR, Winans AA, Staley JP. 2006. The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Mol. Cell 23:389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu S, Rauhut R, Vornlocher HP, Lührmann R. 2006. The network of protein-protein interactions within the human U4/U6.U5 tri-snRNP. RNA 12:1418–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boon KL, Norman CM, Grainger RJ, Newman AJ, Beggs JD. 2006. Prp8p dissection reveals domain structure and protein interaction sites. RNA 12:198–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brenner TJ, Guthrie C. 2005. Genetic analysis reveals a role for the C terminus of the Saccharomyces cerevisiae GTPase Snu114 during spliceosome activation. Genetics 170:1063–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang L, Xu T, Maeder C, Bud LO, Shanks J, Nix J, Guthrie C, Pleiss JA, Zhao R. 2009. Structural evidence for consecutive Hel308-like modules in the spliceosomal ATPase Brr2. Nat. Struct. Mol. Biol. 16:731–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dix I, Russell CS, O'Keefe RT, Newman AJ, Beggs JD. 1998. Protein-RNA interactions in the U5 snRNP of Saccharomyces cerevisiae. RNA 4:1675–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grainger RJ, Beggs JD. 2005. Prp8 protein: at the heart of the spliceosome. RNA 11:533–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Turner IA, Norman CM, Churcher MJ, Newman AJ. 2006. Dissection of Prp8 protein defines multiple interactions with crucial RNA sequences in the catalytic core of the spliceosome. RNA 12:375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maeder C, Kutach AK, Guthrie C. 2009. ATP-dependent unwinding of U4/U6 snRNAs by the Brr2 helicase requires the C terminus of Prp8. Nat. Struct. Mol. Biol. 16:42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bayne EH, Portoso M, Kagansky A, Kos-Braun IC, Urano T, Ekwall K, Alves F, Rappsilber J, Allshire RC. 2008. Splicing factors facilitate RNAi-directed silencing in fission yeast. Science 322:602–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Korneta I, Magnus M, Bujnicki JM. 2012. Structural bioinformatics of the human spliceosomal proteome. Nucleic Acids Res. 40:7046–7065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coelho Ribeiro ML, Espinosa J, Islam S, Martinez O, Thanki JJ, Mazariegos S, Nguyen T, Larina M, Xue B, Uversky VN. 2013. Malleable ribonucleoprotein machine: protein intrinsic disorder in the Saccharomyces cerevisiae spliceosome. PeerJ 1:e2. 10.7717/peerj.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jørgensen R, Ortiz PA, Carr-Schmid A, Nissen P, Kinzy TG, Andersen GR. 2003. Two crystal structures demonstrate large conformational changes in the eukaryotic ribosomal translocase. Nat. Struct. Biol. 10:379–385 [DOI] [PubMed] [Google Scholar]
- 29. Bartels C, Urlaub H, Lührmann R, Fabrizio P. 2003. Mutagenesis suggests several roles of Snu114p in pre-mRNA splicing. J. Biol. Chem. 278:28324–28334 [DOI] [PubMed] [Google Scholar]
- 30. Frazer LN, Lovell SC, O'Keefe RT. 2009. Analysis of synthetic lethality reveals genetic interactions between the GTPase Snu114p and snRNAs in the catalytic core of the Saccharomyces cerevisiae spliceosome. Genetics 183:497–515, 1SI–4SI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmidt H, Richert K, Drakas RA, Käufer NF. 1999. spp42, identified as a classical suppressor of prp4-73, which encodes a kinase involved in pre-mRNA splicing in fission yeast, is a homologue of the splicing factor Prp8p. Genetics 153:1183–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, III, Steever AB, Wach A, Philippsen P, Pringle JR. 1998. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14:943–951 [DOI] [PubMed] [Google Scholar]
- 33. Keeney JB, Boeke JD. 1994. Efficient targeted integration at leu1-32 and ura4-294 in Schizosaccharomyces pombe. Genetics 136:849–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maundrell K. 1993. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 123:127–130 [DOI] [PubMed] [Google Scholar]
- 35. Tasto JJ, Carnahan RH, McDonald WH, Gould KL. 2001. Vectors and gene targeting modules for tandem affinity purification in Schizosaccharomyces pombe. Yeast 18:657–662 [DOI] [PubMed] [Google Scholar]
- 36. Ohi R, McCollum D, Hirani B, Den Haese GJ, Zhang X, Burke JD, Turner K, Gould KL. 1994. The Schizosaccharomyces pombe cdc5+ gene encodes an essential protein with homology to c-Myb. EMBO J. 13:471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McDonald WH, Ohi R, Smelkova N, Frendewey D, Gould KL. 1999. Myb-related fission yeast cdc5p is a component of a 40S snRNP-containing complex and is essential for pre-mRNA splicing. Mol. Cell. Biol. 19:5352–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ren L, McLean JR, Hazbun TR, Fields S, Vander Kooi C, Ohi MD, Gould KL. 2011. Systematic two-hybrid and comparative proteomic analyses reveal novel yeast pre-mRNA splicing factors connected to Prp19. PLoS One 6:e16719. 10.1371/journal.pone.0016719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gould KL, Ren L, Feoktistova AS, Jennings JL, Link AJ. 2004. Tandem affinity purification and identification of protein complex components. Methods 33:239–244 [DOI] [PubMed] [Google Scholar]
- 40. Collart MA, Oliviero S. 2001. Preparation of yeast RNA. Curr. Protoc. Mol. Biol. Chapter 13:Unit13.12. 10.1002/0471142727.mb1312s23 [DOI] [PubMed] [Google Scholar]
- 41. Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kersey PJ, Lawson D, Birney E, Derwent PS, Haimel M, Herrero J, Keenan S, Kerhornou A, Koscielny G, Kähäri A, Kinsella RJ, Kulesha E, Maheswari U, Megy K, Nuhn M, Proctor G, Staines D, Valentin F, Vilella AJ, Yates A. 2010. Ensembl Genomes: extending Ensembl across the taxonomic space. Nucleic Acids Res. 38:D563–D569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106. 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bradford JR, Hey Y, Yates T, Li Y, Pepper SD, Miller CJ. 2010. A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling. BMC Genomics 11:282. 10.1186/1471-2164-11-282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621–628 [DOI] [PubMed] [Google Scholar]
- 46. Agresti A. 1990. Categorical data analysis. Wiley, New York, NY [Google Scholar]
- 47. Storey JD. 2002. A direct approach to false discovery rates. J. Royal Stat. Soc. Ser. B 64:479–498 [Google Scholar]
- 48. Perez-Iratxeta C, Andrade-Navarro MA. 2008. K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct. Biol. 8:25. 10.1186/1472-6807-8-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78:1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM, Tasto JJ, Gould KL, Wolters D, Washburn M, Weiss A, Clark JI, Yates JR., III 2002. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc. Natl. Acad. Sci. U. S. A. 99:7900–7905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yates JR, III, Eng JK, McCormack AL, Schieltz D. 1995. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67:1426–1436 [DOI] [PubMed] [Google Scholar]
- 52. Ohi M, Li Y, Cheng Y, Walz T. 2004. Negative staining and image classification—powerful tools in modern electron microscopy. Biol. Proced. Online 6:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Habara Y, Urushiyama S, Shibuya T, Ohshima Y, Tani T. 2001. Mutation in the prp12+ gene encoding a homolog of SAP130/SF3b130 causes differential inhibition of pre-mRNA splicing and arrest of cell-cycle progression in Schizosaccharomyces pombe. RNA 7:671–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tsai WY, Chow YT, Chen HR, Huang KT, Hong RI, Jan SP, Kuo NY, Tsao TY, Chen CH, Cheng SC. 1999. Cef1p is a component of the Prp19p-associated complex and essential for pre-mRNA splicing. J. Biol. Chem. 274:9455–9462 [DOI] [PubMed] [Google Scholar]
- 55. Ohi MD, Gould KL. 2002. Characterization of interactions among the Cef1p-Prp19p-associated splicing complex. RNA 8:798–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stevens SW, Abelson J. 1999. Purification of the yeast U4/U6.U5 small nuclear ribonucleoprotein particle and identification of its proteins. Proc. Natl. Acad. Sci. U. S. A. 96:7226–7231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Romfo CM, Lakhe-Reddy S, Wise JA. 1999. Molecular genetic analysis of U2AF59 in Schizosaccharomyces pombe: differential sensitivity of introns to mutational inactivation. RNA 5:49–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. 2007. Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol. 5:e90. 10.1371/journal.pbio.0050090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sridharan V, Heimiller J, Singh R. 2011. Genomic mRNA profiling reveals compensatory mechanisms for the requirement of the essential splicing factor U2AF. Mol. Cell. Biol. 31:652–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ohi MD, Ren L, Wall JS, Gould KL, Walz T. 2007. Structural characterization of the fission yeast U5.U2/U6 spliceosome complex. Proc. Natl. Acad. Sci. U. S. A. 104:3195–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huang T, Vilardell J, Query CC. 2002. Pre-spliceosome formation in S. pombe requires a stable complex of SF1-U2AF(59)-U2AF(23). EMBO J. 21:5516–5526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lützelberger M, Bottner CA, Schwelnus W, Zock-Emmenthal S, Razanau A, Käufer NF. 2010. The N-terminus of Prp1 (Prp6/U5-102 K) is essential for spliceosome activation in vivo. Nucleic Acids Res. 38:1610–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Carnahan RH, Feoktistova A, Ren L, Niessen S, Yates JR, III, Gould KL. 2005. Dim1p is required for efficient splicing and export of mRNA encoding lid1p, a component of the fission yeast anaphase-promoting complex. Eukaryot. Cell 4:577–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Behrens SE, Galisson F, Legrain P, Lührmann R. 1993. Evidence that the 60-kDa protein of 17S U2 small nuclear ribonucleoprotein is immunologically and functionally related to the yeast PRP9 splicing factor and is required for the efficient formation of prespliceosomes. Proc. Natl. Acad. Sci. U. S. A. 90:8229–8233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Behrens SE, Tyc K, Kastner B, Reichelt J, Lührmann R. 1993. Small nuclear ribonucleoprotein (RNP) U2 contains numerous additional proteins and has a bipartite RNP structure under splicing conditions. Mol. Cell. Biol. 13:307–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brosi R, Hauri HP, Kramer A. 1993. Separation of splicing factor SF3 into two components and purification of SF3a activity. J. Biol. Chem. 268:17640–17646 [PubMed] [Google Scholar]
- 67. Pauling MH, McPheeters DS, Ares M., Jr 2000. Functional Cus1p is found with Hsh155p in a multiprotein splicing factor associated with U2 snRNA. Mol. Cell. Biol. 20:2176–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Urushiyama S, Tani T, Ohshima Y. 1996. Isolation of novel pre-mRNA splicing mutants of Schizosaccharomyces pombe. Mol. Gen. Genet. 253:118–127 [DOI] [PubMed] [Google Scholar]
- 69. Nurse P, Thuriaux P, Nasmyth K. 1976. Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 146:167–178 [DOI] [PubMed] [Google Scholar]
- 70. Rosenberg GH, Alahari SK, Kaufer NF. 1991. prp4 from Schizosaccharomyces pombe, a mutant deficient in pre-mRNA splicing isolated using genes containing artificial introns. Mol. Gen. Genet. 226:305–309 [DOI] [PubMed] [Google Scholar]
- 71. Kim DU, Hayles J, Kim D, Wood V, Park HO, Won M, Yoo HS, Duhig T, Nam M, Palmer G, Han S, Jeffery L, Baek ST, Lee H, Shim YS, Lee M, Kim L, Heo KS, Noh EJ, Lee AR, Jang YJ, Chung KS, Choi SJ, Park JY, Park Y, Kim HM, Park SK, Park HJ, Kang EJ, Kim HB, Kang HS, Park HM, Kim K, Song K, Song KB, Nurse P, Hoe KL. 2010. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 28:617–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nasmyth K, Nurse P. 1981. Cell division cycle mutants altered in DNA replication and mitosis in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 182:119–124 [DOI] [PubMed] [Google Scholar]
- 73. Kuhn AN, Li Z, Brow DA. 1999. Splicing factor Prp8 governs U4/U6 RNA unwinding during activation of the spliceosome. Mol. Cell 3:65–75 [DOI] [PubMed] [Google Scholar]
- 74. Chan SP, Kao DI, Tsai WY, Cheng SC. 2003. The Prp19p-associated complex in spliceosome activation. Science 302:279–282 [DOI] [PubMed] [Google Scholar]
- 75. Query CC, Konarska MM. 2012. CEF1/CDC5 alleles modulate transitions between catalytic conformations of the spliceosome. RNA 18:1001–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rappsilber J, Ryder U, Lamond AI, Mann M. 2002. Large-scale proteomic analysis of the human spliceosome. Genome Res. 12:1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Arenas JE, Abelson JN. 1997. Prp43: an RNA helicase-like factor involved in spliceosome disassembly. Proc. Natl. Acad. Sci. U. S. A. 94:11798–11802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Martin A, Schneider S, Schwer B. 2002. Prp43 is an essential RNA-dependent ATPase required for release of lariat-intron from the spliceosome. J. Biol. Chem. 277:17743–17750 [DOI] [PubMed] [Google Scholar]
- 79. Tsai RT, Fu RH, Yeh FL, Tseng CK, Lin YC, Huang YH, Cheng SC. 2005. Spliceosome disassembly catalyzed by Prp43 and its associated components Ntr1 and Ntr2. Genes Dev. 19:2991–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. 2004. The DISOPRED server for the prediction of protein disorder. Bioinformatics 20:2138–2139 [DOI] [PubMed] [Google Scholar]
- 81. Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. 2005. Protein structure prediction servers at University College London. Nucleic Acids Res. 33:W36–W38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195–202 [DOI] [PubMed] [Google Scholar]
- 83. Marone M, Mozzetti S, De Ritis D, Pierelli L, Scambia G. 2001. Semiquantitative RT-PCR analysis to assess the expression levels of multiple transcripts from the same sample. Biol. Proced. Online 3:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fabrizio P, Abelson J. 1990. Two domains of yeast U6 small nuclear RNA required for both steps of nuclear precursor messenger RNA splicing. Science 250:404–409 [DOI] [PubMed] [Google Scholar]
- 85. Newman AJ, Norman C. 1992. U5 snRNA interacts with exon sequences at 5′ and 3′ splice sites. Cell 68:743–754 [DOI] [PubMed] [Google Scholar]
- 86. O'Keefe RT, Newman AJ. 1998. Functional analysis of the U5 snRNA loop 1 in the second catalytic step of yeast pre-mRNA splicing. EMBO J. 17:565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jones MH, Frank DN, Guthrie C. 1995. Characterization and functional ordering of Slu7p and Prp17p during the second step of pre-mRNA splicing in yeast. Proc. Natl. Acad. Sci. U. S. A. 92:9687–9691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ohi R, Feoktistova A, McCann S, Valentine V, Look AT, Lipsick JS, Gould KL. 1998. Myb-related Schizosaccharomyces pombe cdc5p is structurally and functionally conserved in eukaryotes. Mol. Cell. Biol. 18:4097–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Neubauer G, Gottschalk A, Fabrizio P, Séraphin B, Lührmann R, Mann M. 1997. Identification of the proteins of the yeast U1 small nuclear ribonucleoprotein complex by mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 94:385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bach M, Winkelmann G, Lührmann R. 1989. 20S small nuclear ribonucleoprotein U5 shows a surprisingly complex protein composition. Proc. Natl. Acad. Sci. U. S. A. 86:6038–6042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wang X, Zhang S, Zhang J, Huang X, Xu C, Wang W, Liu Z, Wu J, Shi Y. 2010. A large intrinsically disordered region in SKIP and its disorder-order transition induced by PPIL1 binding revealed by NMR. J. Biol. Chem. 285:4951–4963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Reidt U, Wahl MC, Fasshauer D, Horowitz DS, Lührmann R, Ficner R. 2003. Crystal structure of a complex between human spliceosomal cyclophilin H and a U4/U6 snRNP-60K peptide. J. Mol. Biol. 331:45–56 [DOI] [PubMed] [Google Scholar]
- 93. Nancollis V, Ruckshanthi JP, Frazer LN, O'Keefe RT. 16 July 2013. The U5 snRNA internal loop 1 is a platform for Brr2, Snu114 and Prp8 protein binding during U5 snRNP assembly. J. Cell. Biochem. [Epub ahead of print] 10.1002/jcb.24625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chiang TW, Cheng SC. 2013. A weak spliceosome-binding domain of Yju2 functions in the first step and bypasses Prp16 in the second step of splicing. Mol. Cell. Biol. 33:1746–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815 [DOI] [PubMed] [Google Scholar]
- 96. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.