

Abstract
SPOR domains are about 75 amino acids long and probably bind septal peptidoglycan during cell division. We mutagenized 33 amino acids with surface-exposed side chains in the SPOR domain from an Escherichia coli cell division protein named FtsN. The mutant SPOR domains were fused to Tat-targeted green fluorescent protein (TTGFP) and tested for septal localization in live E. coli cells. Lesions at the following 5 residues reduced septal localization by a factor of 3 or more: Q251, S254, W283, R285, and I313. All of these residues map to a β-sheet in the published solution structure of FtsNSPOR. Three of the mutant proteins (Q251E, S254E, and R285A mutants) were purified and found to be defective in binding to peptidoglycan sacculi in a cosedimentation assay. These results match closely with results from a previous study of the SPOR domain from DamX, even though these two SPOR domains share <20% amino acid identity. Taken together, these findings support the proposal that SPOR domains localize by binding to septal peptidoglycan and imply that the binding site is associated with the β-sheet. We also show that FtsNSPOR contains a disulfide bond between β-sheet residues C252 and C312. The disulfide bond contributes to protein stability, cell division, and peptidoglycan binding.
INTRODUCTION
Cell division in Escherichia coli is mediated by a structure called the “septal ring” or “divisome” that assembles at the midcell (1–4). The septal ring contains at least 30 different types of proteins, most of which localize by binding to other septal ring proteins (e.g., see references 5 to 13). But proteins that contain a SPOR domain (Pfam 05036) may localize by a completely different mechanism, because SPOR domains are thought to bind to septal peptidoglycan (PG) (14–16). The details of the SPOR-PG interaction have yet to be elucidated. This is of interest because it is not known how septal PG differs from PG elsewhere in the sacculus, and understanding the SPOR-PG interaction might lead to new insights into PG biogenesis during cell division.
SPOR domains are ∼75 amino acids (aa) long and have been identified using bioinformatic approaches in over 7,000 proteins from over 2,000 bacterial species (17). At least seven of these SPOR domains have been shown to localize to the midcell in vivo and to bind PG in vitro (14–16, 18), and we are not aware of any documented counterexamples, so PG binding is probably a general property of these domains. In contrast, there is good evidence for diversity in the biochemical and physiological functions of the numerous SPOR domain proteins. For starters, most SPOR domain proteins contain additional domains, which are different in different proteins (17). Moreover, although most of the SPOR domain proteins that have been studied are components of the septal ring that mediates cell division (14–16, 19), there are exceptions. Indeed, the name “SPOR” domain arose because the founding member of the family, CwlC of Bacillus subtilis, is a cell wall amidase that helps degrade PG to release the spore from the mother cell (20–22). Another exception is a small Vibrio parahaemolyticus protein, designated VPA1294, that has been implicated in swarmer cell differentiation (23).
The present study is more concerned with the structure-function relationships of SPOR domains than with their physiological roles, so we now summarize what is known about this topic. The solution structures of three SPOR domains have been solved by nuclear magnetic resonance (NMR) spectroscopy. These domains come from two E. coli cell division proteins, FtsN and DamX, and the Bacillus subtilis sporulation protein, CwlC (24–26). Here we refer to these domains as FtsNSPOR, DamXSPOR, and CwlCSPOR, respectively. All three domains exhibit a similar core architecture comprising a βαββαβ secondary structure that folds into a 4-stranded antiparallel β-sheet buttressed on one side by 2 α-helices (Fig. 1). We recently identified three surface-exposed amino acids in the β-sheet of DamXSPOR that are important for septal localization and binding to PG sacculi from whole cells (25) (Fig. 1). These residues are probably part of a binding site for septal PG. Here we address two important questions that were not addressed in our structure-function study of DamXSPOR. First, can the results obtained with DamX be generalized to SPOR domains from other proteins? This is an issue because SPOR domains display less than 20% identity in pairwise comparisons, and it is not known whether all SPOR domains bind the same PG structure. Second, are additional residues, especially residues outside the β-sheet, also important for septal localization? We mutagenized only nine amino acids in DamXSPOR, so important residues might have been overlooked.
Fig 1.
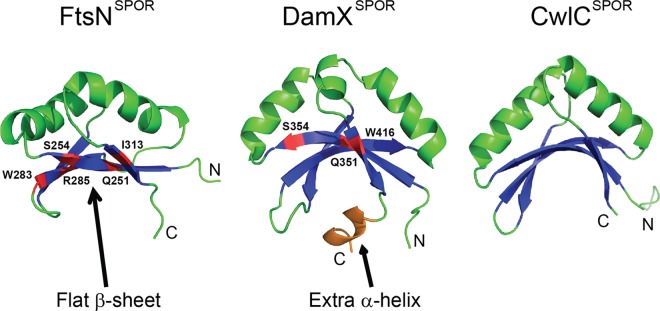
SPOR domains have similar structures. The 4-stranded β-sheet and 2 α-helices common to all SPOR domains are shown in blue and green, respectively. For clarity, turns and coils are also illustrated in green, while the C-terminal helix unique to DamXSPOR is shown in orange. Residues important for septal localization are highlighted in red. Coordinates for these domains were obtained from the Protein Data Bank (PDB entries 1x60, 1UTA, and 2LFV) and rendered using PyMOL.
To address these questions, we undertook a comprehensive mutagenesis of the SPOR domain from FtsN, an essential cell division protein found in a variety of proteobacteria (16, 19). By way of background, E. coli FtsN comprises a short N-terminal cytoplasmic domain (∼30 aa), a single transmembrane helix (∼20 aa), and a comparatively large periplasmic domain (∼265 aa), which is mostly unstructured except for the SPOR domain (residues 243 to 319) at the very C terminus of the protein (26, 27). In vitro, FtsNSPOR binds to intact PG sacculi and to the glycan strands released by digestion of PG with an amidase (18). In vivo, FtsNSPOR localizes to the septal ring in wild-type E. coli but not in a triple amidase mutant (15). Although FtsNSPOR is required for obvious localization of green fluorescent protein (GFP)-FtsN fusion proteins to the septal ring (15, 16), truncated FtsN proteins that lack the SPOR domain support cell division almost as well as the wild type (15, 16, 18). The essential function of FtsN has been mapped to a 35-aa region at the beginning of the periplasmic domain (15). This region of FtsN is thought to interact with some other division protein(s), perhaps a PG synthase, to trigger constriction (13, 15, 26, 28–30). In summary, current thinking is that a portion of FtsN near the start of the periplasmic domain mediates the critical function of FtsN, while the SPOR domain improves the efficiency of this process by targeting FtsN to the septal ring.
The only other well-characterized SPOR domain is from DamX of E. coli. Although mutants lacking DamX have only subtle phenotypic changes (14, 15, 31), the SPOR domain of DamX is very important for these functions (25). This makes DamX a more tractable model protein than FtsN for exploring the physiological importance of the SPOR domain. Nevertheless, as explained below, FtsNSPOR is a good model system for exploring the generality of the structure-function relationships reported for DamXSPOR.
First, the solution structure of FtsNSPOR is available, and assays for studying septal localization and PG binding have been established (15, 18, 26). Second, there are some striking structural differences between FtsNSPOR and DamXSPOR, suggesting that functional differences might exist as well. In FtsNSPOR, the β-sheet is relatively flat, whereas in DamXSPOR it is strikingly curved (Fig. 1). The β-sheet of CwlCSPOR is also rather curved (Fig. 1), making FtsNSPOR the odd one out (24–26). This is notable because residues important for septal localization and PG binding in DamXSPOR map to the β-sheet (25). Thus, unless there are conformational changes, it is doubtful that FtsNSPOR and DamXSPOR could interact with PG by using homologous residues. A third difference is that DamXSPOR contains a short C-terminal α-helix that interacts with the β-sheet and is needed for domain stability (25). No such α-helix exists in FtsNSPOR or CwlCSPOR, as these two domains are slightly shorter than DamXSPOR, but FtsNSPOR possesses a rather flexible C terminus (AAGG), which may destabilize the domain. A final indication that FtsNSPOR and DamXSPOR might not use homologous residues to bind PG is that these domains share <20% amino acid identity. These considerations prompted us to conduct a thorough mutagenesis of FtsNSPOR, the results of which are presented in this report.
MATERIALS AND METHODS
Strains, plasmids, primers, and media.
All strains used in this study are listed in Table S1 in the supplemental material. Unless noted otherwise, E. coli was grown at 30°C in Luria-Bertani (LB) medium containing 1% NaCl or in M9 minimal medium containing 0.2% glucose and thiamine at 1 μg/ml (32). Plates contained 15 g agar per liter. Ampicillin and kanamycin were used at 200 μg/ml and 40 μg/ml, respectively.
Plasmid construction.
Plasmids for localization studies of mutant forms of FtsNSPOR were variants of pDSW992 (ColE1 ori bla lacIq; carries a weakened trc promoter, designated P204, and TTgfp-ftsN residues 240 to 319). Most amino acid substitutions in FtsNSPOR were introduced using degenerate primers and a multistep PCR procedure involving megapriming (33). Plasmids for overproducing hexahistidine (His6)-tagged FtsNSPOR variants were based on pQE80L (Qiagen). See the supplemental information for details of all plasmid constructions, including primer sequences in Table S2.
Protein localization and microscopy.
Cells for localization of TTGFP-FtsNSPOR proteins were grown and visualized essentially as described previously (25), except that overnight cultures were diluted 1:200 and examined when the optical density at 600 nm (OD600) reached ∼0.5. For localization experiments done in the CH34/pMG20 background, the medium included 0.2% l-arabinose and chloramphenicol at 30 μg/ml. Our microscope, camera, and software have been described previously (34).
Purified proteins.
Wild-type and mutant His6-tagged FtsNSPOR proteins were overproduced in E. coli SHuffle T7 (35) (New England BioLabs) and purified at 4°C by cobalt affinity chromatography. Procedures were similar to those described previously (14), except that cells grown in LB medium at 37°C to an OD600 of ∼0.5 were induced overnight at 20°C with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The purified proteins were dialyzed against binding buffer (25 mM Na2HPO4, 150 mM NaCl, pH 7.5) at 4°C. Typical yields were ∼1 mg from a 500-ml culture, and purity was judged to be ∼95% by SDS-PAGE. Aliquots were stored at 4°C, and PG binding assays were conducted within 3 days.
PG binding assay.
Whole PG sacculi were isolated and quantified based on amino sugar content as described previously (14). Binding assays were performed as described previously, using 100-μl reaction mixtures containing 1 nmol of protein and 75 μg of PG sacculi, which corresponds to about 75 nmol of disaccharide units. For tests of the effect of the disulfide bond on PG binding, 1 mM dithiothreitol (DTT) was included in the buffers during dialysis and the binding assay.
Western blotting.
Western blotting was performed as described previously (14). Typically, cells from 1 ml of culture grown to an OD600 of ∼0.5 were pelleted in a microcentrifuge, taken up in 0.1 ml of sample buffer, and boiled for 10 min. Ten microliters of sample was then loaded onto an SDS-polyacrylamide gel. Purified rabbit anti-FtsN and anti-FtsQ were diluted 1:2,000 and 1:4,000, respectively (36). Rabbit anti-GFP serum was obtained from C. Ellermeier and used at a dilution of 1:8,000. The secondary antibody was goat anti-rabbit conjugated to horseradish peroxidase (Thermo Scientific, Rockford, IL), and detection was performed with SuperSignal West Pico chemiluminescent substrate (also from Thermo Scientific). Blots were photographed using a Fujifilm LAS-1000 imager.
RESULTS
Localization-defective mutants of FtsNSPOR map to the β-sheet.
The SPOR domain from FtsN contains 76 residues, 33 of which we judged by visual inspection of the structure in PyMOL to have surface-exposed R groups (37). These 33 residues are distributed throughout the domain (Fig. 2), including many in the two α-helices that were not explored in our previous mutagenesis of DamXSPOR. An alignment of FstN SPOR domains from several species revealed that the residues targeted for mutagenesis are not simply the most highly conserved ones (38). Many of the highly conserved residues are internal to the domain and presumably make important contributions to protein folding. The 33 targeted residues were changed to alanine by using a strategy that involved degenerate primers, allowing other substitutions to be obtained as well, for a total of 92 mutants. The mutant SPOR domains were fused to Tat-targeted GFP (TTGFP) to direct export of the properly folded TTGFP-FtsNSPOR fusion proteins into the periplasm (39).
Fig 2.

Locations of FtsNSPOR amino acids targeted for mutagenesis. (Top) Ribbon diagrams illustrating the locations of all residues (red) where amino acid substitutions were introduced to test for localization defects. (Bottom) Alignment of the SPOR domains from several FtsN proteins. Residues targeted for mutagenesis are shown in red, with arrows pointing to residues where substitutions impaired localization at least 3-fold. The black background highlights the two conserved cysteines proposed to form a disulfide bond. The alignment was prepared in Clustal W (38) with the following FtsN SPOR domain sequences: Escherichia coli MG1655 (locus tag b3833) residues 244 to 319, Citrobacter rodentium ICC168 (ROD_38131) residues 236 to 310, Klebsiella pneumoniae 342 (KPK_5451) residues 245 to 320, Yersinia pestis bv. Antiqua B42003004 (YpAngola_A0114) residues 206 to 281, Proteus mirabilis ATCC 29906 (HMPREF0693_0033) residues 188 to 264, and Haemophilus aegyptius ATCC 11116 (HMPREF9095_0699) residues 185 to 256.
Wild-type E. coli cells harboring the TTGFP-FtsNSPOR plasmids were grown to mid-log phase in LB medium and then immobilized on an agarose pad for fluorescence microscopy. Septal localization was quantified by scoring cells for the presence or absence of a fluorescent band at the midcell, with results reported as the fraction of cells in the population that exhibited septal localization. We used an arbitrary statistical cutoff to identify localization-defective proteins; this cutoff was a 3-fold reduction in localization, which corresponds to ≤23% of the cells in the population being scored positive for a fluorescent band at the midcell. Samples were also taken for Western blotting with anti-GFP antiserum to ascertain whether nonlocalizing TTGFP-FtsNSPOR proteins were produced in normal amounts. The results for our most interesting mutants are presented in Fig. 3. Quantitative localization data for all mutants are shown in Table S3 in the supplemental material.
Fig 3.

Localization-defective mutants of FtsNSPOR. (A) Western blot with anti-GFP antibody demonstrating that wild-type (WT) and mutant TTGFP-FtsNSPOR proteins were produced at similar levels. (−), control strain producing TTGFP not fused to anything. (B) Fluorescence micrographs of cells producing the indicated TTGFP-FtsNSPOR fusion proteins. Numbers in the corners are percentages of cells scored as exhibiting septal localization. See Table S3 in the supplemental material for more detailed quantitative data on localization frequencies. (C) Results of PG binding assay. “Reduced” refers to the presence of 1 mM DTT. Bars represent the means and standard deviations for at least three independent experiments.
A wild-type TTGFP-FtsNSPOR construct localized to the septal ring in ∼70% of the cells, similar to the case in previous reports (14, 15). Lesions at Q251, S254, W283, R285, and I313 reduced septal localization by a factor of at least 3, without destabilizing the protein as judged by Western blotting (Fig. 3). All of these residues map to the β-sheet (Fig. 2). Proteins with substitutions at P290 also localized very poorly, but in this case Western blots revealed folding or stability defects (see Fig. S1 in the supplemental material), so we did not classify P290 as being important for septal localization per se.
If the cutoff used to classify residues as important for septal localization was relaxed to include mutants with a 2-fold defect (i.e., septal localization in ≤35% of the population), some additional amino acids came into play: R256, E262, T263, R265, and F270. These amino acids are in the loop connecting β1 to α1 (R256) or in α1 proper (E262, T263, R265, and F270). Western blotting indicated that these residues are not important for the overall stability of the domain (see Fig. S1 in the supplemental material). We suspect that substitutions at these sites of secondary importance perturb septal localization indirectly by altering the conformation of the β-sheet (see Discussion).
As noted above, these results pertain to a wild-type E. coli host. If the SPOR domain from the native FtsN protein interacts with the SPOR domains produced from the plasmid, either directly by dimerization or indirectly by competition for binding sites in the PG, then use of a wild-type host might lead to misinterpretations. We therefore assayed localization of wild-type and 14 mutant TTGFP-FtsNSPOR constructs in an E. coli strain that produces a truncated FtsN protein with no SPOR domain (15). The mutants chosen included 2 with severe localization defects (Q251E and W283D mutants), 2 with intermediate defects (T263D and F250A mutants), and 10 that localized well when assayed in a wild-type background. Although TTGFP-FtsNSPOR constructs did not localize quite as well in the FtsN SPOR null strain, the trends were very similar to what we observed when the native FtsN protein was present (see Table S4 in the supplemental material). For example, localization of the wild-type construct fell from 70% to 60%, while localization of the W283D mutant fell from 9% to 4%. We concluded that the presence or absence of authentic FtsN produced from the chromosome does not affect the interpretation of our experiments to identify FtsNSPOR residues important for septal localization.
Finally, it should be noted that we did not introduce any SPOR domain amino acid substitutions into full-length FtsN to determine whether these impair cell division. It is unlikely that such mutations would have much effect on cell division, because even a complete deletion of the SPOR domain is surprisingly well tolerated (15, 16, 18). For DamX, whose SPOR domain is critical for the overall function of the protein, we found that SPOR domain point mutations are not as deleterious as SPOR domain deletions (14).
Localization-defective mutants bind PG poorly.
FtsNSPOR probably localizes to the septal ring by binding to septal PG (14, 15, 18, 25). This hypothesis predicts that localization-defective FtsNSPOR mutant proteins will also be defective in PG binding. To test this idea, we subcloned coding sequences for wild-type and three localization-defective SPOR domains (Q251E, S254E, and R285A mutants) into a vector that provides an N-terminal histidine tag to facilitate purification. These constructs also included two C-terminal aspartate residues (DD) to mask a potential recognition signal for the ClpXP protease (40). Addition of DD to the C terminus did not impair septal localization of a TTGFP-FtsNSPOR construct (Fig. 3B; see Table S3 in the supplemental material). (The problems with overproduction that led us to modify the C terminus are described in the supplemental material and shown in Fig. S2.) All proteins were overproduced at 20°C in an E. coli strain engineered to promote disulfide bond formation in the cytoplasm (35).
Approximately 25% ± 5% of the wild-type FtsNSPOR protein cosedimented with purified E. coli PG sacculi upon ultracentrifugation, but for the mutant proteins, this was reduced to 7% ± 3% (Q251E and S254E mutants) or 19% ± 1% (R285A mutant) (Fig. 3C). These values correlate with the relative proficiencies of septal localization: WT > R285A mutant > Q251E mutant ≈ S254E mutant (see Table S1 in the supplemental material). It should be noted that because these experiments employed whole sacculi, it is not known whether the binding observed reflects a general affinity for PG or the specific binding to septal PG inferred from localization studies in vivo.
The β-sheet of FtsNSPOR contains a disulfide bond.
FtsN from E. coli contains two cysteines, C252 and C312, both of which are conserved and located in the β-sheet (Fig. 2). Paired cysteines in E. coli periplasmic proteins usually form disulfide bonds (41, 42). Examination of the published structure of FtsNSPOR revealed that C252 and C312 are adjacent (Fig. 4A) (26), but the two sulfur atoms are separated by about 5.3 ± 0.9 Å (see Table S5 in the supplemental material), which is longer than the ∼2-Å distance expected for a disulfide bond. However, the domain used to determine the structure was overproduced in the cytoplasm and purified in the presence of DTT, so any disulfides normally present in FtsNSPOR might have been reduced. To test for a disulfide in FtsN, we prepared whole-cell extracts in Laemmli sample buffer containing or lacking 5% β-mercaptoethanol (∼700 mM). Western blotting revealed that reducing the thiols in FtsN resulted in slightly lower mobility during polyacrylamide gel electrophoresis, whereas the mobility of FtsQ, which lacks cysteines, was unchanged (Fig. 4B) (because the mobility difference is subtle, a second blot is shown in Fig. S3 to document reproducibility). The location of C252 and C312 suggests that they would form an intramolecular disulfide bond; consistent with this, a larger species indicative of an FtsN dimer was not detected in the unreduced samples (Fig. 4B).
Fig 4.

FtsNSPOR contains a disulfide bond. (A) Closeup view of C252 and C312 (yellow). β-Sheet residues important for septal localization are shown as red stick figures. (B) FtsN contains a disulfide bond. Whole-cell extracts were prepared in Laemmli sample buffer containing (lanes 2 and 4) or lacking (lanes 3 and 5) 5% β-mercaptoethanol and then analyzed by Western blotting with anti-FtsN (top) or anti-FtsQ (bottom). Samples were loaded twice to facilitate visualization of any mobility differences. (C) Western blot showing that FtsN levels are reduced but DamX levels are normal in two dsbA mutants compared to their isogenic wild-type controls. Cells were grown on M9-glucose medium to mid-log phase, and then total cell extracts were prepared and proteins separated by SDS-PAGE (10% polyacrylamide). After transfer to nitrocellulose, proteins were detected with anti-FtsN or anti-DamX antibody. (D) Lengths and widths of wild-type and dsbA mutant strains grown in LB or M9-glucose medium. Cells grown to mid-log phase were fixed with paraformaldehyde, photographed under phase-contrast conditions, and measured using tools in Image-Pro Plus, version 4.1, from Media Cybernetics (Silver Spring, MD). Bars represent the means and standard deviations for at least two (LB medium) or three (M9 medium) experiments in which 170 or more cells were measured.
The major catalyst of disulfide bond formation in the periplasm is DsbA (43). We obtained several dsbA null mutants from Jim Bardwell, along with the corresponding parental wild-type strains. It has been reported that an ftsN SPOR domain deletion mutant exhibits normal cell length in LB medium but is slightly elongated in M9 minimal medium (15). Similarly, we observed that dsbA strains were the same length as wild-type strains when grown in LB medium, but in M9 medium, the dsbA mutants averaged 30% longer (Fig. 4D). There was also a larger fraction of constricting cells, i.e., 30% ± 7% versus 15% ± 9%, suggesting that loss of DsbA retards the rate of cytokinesis. The dsbA mutant strains were also about 10% thinner, on average, than the wild-type strains, and this difference was observed in both LB and M9 media (Fig. 4D). Western blotting revealed that the absence of DsbA reduced steady-state levels of FtsN by about 4-fold in M9 medium (Fig. 4C). The difference was closer to 2-fold in LB medium (see Fig. S4 in the supplemental material). In contrast, the absence of DsbA had no effect on the abundance of DamX, a SPOR domain protein that does not contain any cysteines (Fig. 4C; see Fig. S4).
Cells producing TTGFP-FtsNSPOR were dim, and septal localization was reduced, in a dsbA background (Fig. 5A), but a TTGFP-DamXSPOR construct localized well even in cells lacking DsbA (see Fig. S5 in the supplemental material). Consistent with the localization results, Western blotting revealed about 4-fold less TTGFP-FtsNSPOR in dsbA mutants, but there was no change in the abundance of TTGFP-DamXSPOR (Fig. 5B). Changing the cysteines to alanine, either alone or simultaneously, reduced TTGFP-FtsNSPOR stability so severely that the proteins were essentially undetectable in Western blots (Fig. 5C). Finally, inclusion of DTT in a cosedimentation assay caused a 25% reduction in PG binding by wild-type FtsNSPOR, from 25% ± 5% (n = 4) of the input protein to 19% ± 1% (n = 2) (Fig. 3C).
Fig 5.
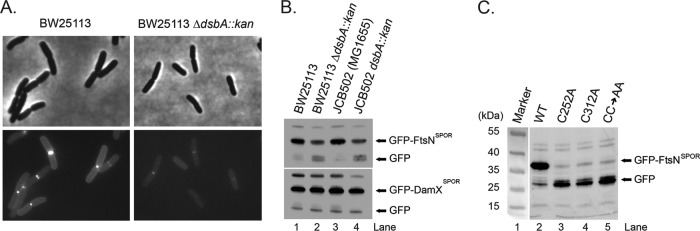
The disulfide bond is important for stability of a TTGFP-FtsNSPOR fusion protein. (A) Reduced septal localization of TTGFP-FtsNSPOR in the absence of DsbA. The indicated strains carrying pDSW992 were grown to mid-log phase, immobilized on an agarose pad, and photographed under phase-contrast (top) and fluorescence (bottom) conditions. (B) Reduced abundance of TTGFP-FtsNSPOR in the absence of DsbA. The indicated strains carrying pDSW992 or pDSW997 were grown to mid-log phase, and levels of TTGFP-FtsNSPOR or TTGFP-DamXSPOR were determined by Western blotting with anti-GFP antibody. (C) Effects of Cys-to-Ala substitutions on the stability of TTGFP-FtsNSPOR. Wild-type strain EC251 carrying plasmids that direct expression of wild-type TTGFP-FtsNSPOR or the indicated mutant derivatives was analyzed as described for panel B.
DISCUSSION
The binding site for septal PG in FtsNSPOR is probably the β-sheet.
A comprehensive mutagenesis of FtsNSPOR residues with surface-exposed side chains revealed only five amino acids that, when mutagenized, reduced localization by a factor of 3 or more: Q251, S254, W283, R285, and I313. All five of these residues are in the β-sheet. Three of the localization-defective mutant proteins (Q251E, S254E, and R285A mutants) were purified and found to bind PG poorly compared to the wild type. Taken together, these findings argue that FtsNSPOR localizes by binding to septal PG and suggest that the binding site is associated with the β-sheet.
Mutations at S254 returned noteworthy results because changes to E and K reduced localization to 13% and 5%, respectively, but the S254A mutant localized as well as the wild type (Fig. 3B; see Table S3 in the supplemental material). Similar results emerged from our analysis of DamXSPOR, where changing S354 to T or K greatly impaired septal localization, but an S354A mutant localized like the wild type (25). Sequence alignments show that S and A occur with roughly equal frequencies at this position (17, 25). We infer that the R group at this position has to be small but does not make a direct contribution to PG binding.
One of the motivations for this study was to determine whether any residues outside the β-sheet are important for septal localization, as this would implicate additional surfaces of FtsNSPOR as potential PG binding sites. Although no critically important residues outside the β-sheet were found, we did identify a few that make modest contributions to septal localization: R256, E262, T263, R265, and F270. We suspect that these residues affect localization indirectly. R256 is in the loop connecting β1 to α1 and interacts with one of the residues critical for septal localization, namely, W283. Residues E262, T263, R265, and F270 are all in α1, which has extensive interactions with the β-sheet, especially strand β2. In particular, parts of the side chain of R265 contact β2 residue I277. Residue E262 also comes close to I277, though whether these two amino acids make direct contact is not clear.
All SPOR domains probably engage PG via the β-sheet.
Residues Q251 and S254 of FtsNSPOR align with the most highly conserved surface-exposed SPOR domain residues, and the corresponding amino acids of DamXSPOR, Q351 and S354, were found to be important for septal localization and PG binding in that domain (25). The SPOR domains from FtsN and DamX have <20% identity and noteworthy differences at the level of tertiary structure, particularly the curvature of the β-sheet and the presence of an extra α-helix at the C terminus of DamXSPOR (Fig. 1). Thus, the fact that we obtained similar results for two such divergent SPOR domains suggests that most, perhaps all, SPOR domains bind PG in a similar fashion.
SPOR domains exhibit a ribonucleoprotein fold (RNP fold) (25, 26). Despite the name, RNP folds are found in a variety of proteins and mediate interactions with a variety of ligands, not just RNAs. There is much precedent for the β-sheet being the primary ligand-binding site in RNP-fold domains. For example, the human spliceosomal protein U1A has two RNP-fold domains, both of which bind RNA via residues in the β-sheet. In particular, U1A residues in β1 and β3 play a critical role in binding to the cognate RNA hairpin (44, 45), which is reminiscent of our finding that the most important residues in FtsNSPOR are in β1 (Q251 and S254) or β3 (W283 and R285).
Although the features of PG recognized by SPOR domains remain to be elucidated, current thinking is that they bind to glycan strands lacking oligopeptide side chains. “Naked” glycan strands are likely enriched at the division site by the action of periplasmic amidases that process septal PG to facilitate separation of daughter cells (39, 46–48). The strongest support for this hypothesis is that FtsNSPOR binds the glycan backbone of PG in vitro and fails to localize in a triple amidase mutant in vivo (15, 18). Stacking of aromatic amino acids, especially tryptophan, with the sugar rings of oligosaccharides is known to be important for many protein-carbohydrate binding interactions (49–52). Both FtsNSPOR and DamXSPOR have a β-sheet tryptophan that plays a key role in septal localization: W283 in FtsN and W416 in DamX. Curiously, however, these residues are not homologous—W283 of FtsN is in β3, whereas W416 of DamX is in β4.
The β-sheet has a disulfide bond.
FtsNSPOR has a disulfide bond that links residue C252 in β1 to C312 in β4. The disulfide is not present in the protein used for structure determination, which was obtained under reducing conditions. An interesting question is whether the absence of the disulfide explains why the β-sheet in FtsNSPOR is relatively flat compared to those of DamXSPOR and CwlCSPOR (Fig. 1). Although we cannot be certain, we doubt that this is the explanation, because modeling the effect of the disulfide bond on the structure of the domain does not change the conformation of the β-sheet very dramatically (see Fig. S6 in the supplemental material). This interesting anomaly deserves further exploration, because it stands to reason that the β-sheets of all three SPOR domains have to adopt the same conformation for homologous residues to bind PG.
Assessing the importance of the disulfide bond for FtsNSPOR function is complicated by the potential pleiotropy of a dsbA null mutation and the fact that FtsN is less stable and is degraded when the disulfide bond is absent. Despite these caveats, multiple observations argue that the disulfide bond is important. First, the cysteines that form this bond are highly conserved in FtsNSPOR sequences from different organisms. Second, FtsN levels were reduced in dsbA mutants, especially when cells were grown in minimal media, where the mutants also exhibited a small division defect. Third, the wild-type TTGFP-FtsNSPOR construct was not well produced and localized poorly in a dsbA background. Fourth, changing the cysteines to alanine, either alone or in combination, destabilized the TTGFP-FtsNSPOR construct so severely that it was difficult to detect it by Western blotting using anti-GFP antiserum. Fifth, overproduction of a histidine-tagged FtsNSPOR construct in the cytoplasm was greatly improved by using an E. coli host engineered to produce disulfide bonds in the cytoplasm (see Fig. S2 in the supplemental material). Finally, inclusion of DTT in the PG binding assay reduced binding by about 25%, similar to the R285A substitution, which caused a 3-fold reduction in septal localization of a TTGFP-FtsNSPOR construct.
To our knowledge, FtsN is the first example of a bacterial cell division protein with a disulfide bond. The Pfam database contains a seed alignment of 136 diverse SPOR domains (http://pfam.janelia.org/family/PF05036#tabview=tab3) (17). Of these, 22 have two cysteines (see Table S6 in the supplemental material), suggesting that about 15% of SPOR domain proteins contain a disulfide bond. We therefore infer that disulfide bonds will prove to be important for the stability or function of many SPOR domains.
Supplementary Material
ACKNOWLEDGMENTS
We thank Craig D. Ellermeier for anti-GFP, Piet de Boer for CH34/pMG20, James Bardwell and Shu Quan for E. coli dsbA mutants, Jon Beckwith for information on the phenotypes of dsbA mutants, Kyle B. Williams for technical assistance, Lokesh Gakhar for help with modeling the structure of the disulfide-bonded SPOR domain from FtsN, and anonymous reviewers for suggestions that improved the manuscript.
This research was supported by National Institutes of Health grant GM083975 to D.S.W.
Footnotes
Published ahead of print 20 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00911-13.
REFERENCES
- 1.Errington J, Daniel RA, Scheffers DJ. 2003. Cytokinesis in bacteria. Microbiol. Mol. Biol. Rev. 67:52–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Boer PA. 2010. Advances in understanding E. coli cell fission. Curr. Opin. Microbiol. 13:730–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goehring NW, Beckwith J. 2005. Diverse paths to midcell: assembly of the bacterial cell division machinery. Curr. Biol. 15:R514–R526 [DOI] [PubMed] [Google Scholar]
- 4.Vicente M, Rico AI. 2006. The order of the ring: assembly of Escherichia coli cell division components. Mol. Microbiol. 61:5–8 [DOI] [PubMed] [Google Scholar]
- 5.Hale CA, de Boer PA. 1997. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell 88:175–185 [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Huang J, Mukherjee A, Cao C, Lutkenhaus J. 1997. Analysis of the interaction of FtsZ with itself, GTP, and FtsA. J. Bacteriol. 179:5551–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gueiros-Filho FJ, Losick R. 2002. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev. 16:2544–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buddelmeijer N, Beckwith J. 2004. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Mol. Microbiol. 52:1315–1327 [DOI] [PubMed] [Google Scholar]
- 9.Villanelo F, Ordenes A, Brunet J, Lagos R, Monasterio O. 2011. A model for the Escherichia coli FtsB/FtsL/FtsQ cell division complex. BMC Struct. Biol. 11:28. 10.1186/1472-6807-11-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masson S, Kern T, Le Gouellec A, Giustini C, Simorre JP, Callow P, Vernet T, Gabel F, Zapun A. 2009. Central domain of DivIB caps the C-terminal regions of the FtsL/DivIC coiled-coil rod. J. Biol. Chem. 284:27687–27700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karimova G, Dautin N, Ladant D. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 187:2233–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosyak L, Zhang Y, Glasfeld E, Haney S, Stahl M, Seehra J, Somers WS. 2000. The bacterial cell-division protein ZipA and its interaction with an FtsZ fragment revealed by X-ray crystallography. EMBO J. 19:3179–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexeeva S, Gadella TW, Jr, Verheul J, Verhoeven GS, den Blaauwen T. 2010. Direct interactions of early and late assembling division proteins in Escherichia coli cells resolved by FRET. Mol. Microbiol. 77:384–398 [DOI] [PubMed] [Google Scholar]
- 14.Arends SJ, Williams K, Scott RJ, Rolong S, Popham DL, Weiss DS. 2010. Discovery and characterization of three new Escherichia coli septal ring proteins that contain a SPOR domain: DamX, DedD, and RlpA. J. Bacteriol. 192:242–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerding MA, Liu B, Bendezu FO, Hale CA, Bernhardt TG, de Boer PA. 2009. Self-enhanced accumulation of FtsN at division sites and roles for other proteins with a SPOR domain (DamX, DedD, and RlpA) in Escherichia coli cell constriction. J. Bacteriol. 191:7383–7401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Möll A, Thanbichler M. 2009. FtsN-like proteins are conserved components of the cell division machinery in proteobacteria. Mol. Microbiol. 72:1037–1053 [DOI] [PubMed] [Google Scholar]
- 17.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res. 40:D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ursinus A, van den Ent F, Brechtel S, de Pedro M, Höltje JV, Löwe J, Vollmer W. 2004. Murein (peptidoglycan) binding property of the essential cell division protein FtsN from Escherichia coli. J. Bacteriol. 186:6728–6737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai K, Xu Y, Lutkenhaus J. 1993. Cloning and characterization of ftsN, an essential cell division gene in Escherichia coli isolated as a multicopy suppressor of ftsA12(Ts). J. Bacteriol. 175:3790–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nugroho FA, Yamamoto H, Kobayashi Y, Sekiguchi J. 1999. Characterization of a new sigma-K-dependent peptidoglycan hydrolase gene that plays a role in Bacillus subtilis mother cell lysis. J. Bacteriol. 181:6230–6237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shida T, Hattori H, Ise F, Sekiguchi J. 2001. Mutational analysis of catalytic sites of the cell wall lytic N-acetylmuramoyl-l-alanine amidases CwlC and CwlV. J. Biol. Chem. 276:28140–28146 [DOI] [PubMed] [Google Scholar]
- 22.Smith TJ, Foster SJ. 1995. Characterization of the involvement of two compensatory autolysins in mother cell lysis during sporulation of Bacillus subtilis 168. J. Bacteriol. 177:3855–3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gode-Potratz CJ, Kustusch RJ, Breheny PJ, Weiss DS, McCarter LL. 2011. Surface sensing in Vibrio parahaemolyticus triggers a programme of gene expression that promotes colonization and virulence. Mol. Microbiol. 79:240–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mishima M, Shida T, Yabuki K, Kato K, Sekiguchi J, Kojima C. 2005. Solution structure of the peptidoglycan binding domain of Bacillus subtilis cell wall lytic enzyme CwlC: characterization of the sporulation-related repeats by NMR. Biochemistry 44:10153–10163 [DOI] [PubMed] [Google Scholar]
- 25.Williams KB, Yahashiri A, Arends SJ, Popham DL, Fowler CA, Weiss DS. 2013. Nuclear magnetic resonance solution structure of the peptidoglycan-binding SPOR domain from Escherichia coli DamX: insights into septal localization. Biochemistry 52:627–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang JC, Van Den Ent F, Neuhaus D, Brevier J, Löwe J. 2004. Solution structure and domain architecture of the divisome protein FtsN. Mol. Microbiol. 52:651–660 [DOI] [PubMed] [Google Scholar]
- 27.Dai K, Xu Y, Lutkenhaus J. 1996. Topological characterization of the essential Escherichia coli cell division protein FtsN. J. Bacteriol. 178:1328–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lutkenhaus J. 2009. FtsN—trigger for septation. J. Bacteriol. 191:7381–7382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Derouaux A, Wolf B, Fraipont C, Breukink E, Nguyen-Distèche M, Terrak M. 2008. The monofunctional glycosyltransferase of Escherichia coli localizes to the cell division site and interacts with penicillin-binding protein 3, FtsW, and FtsN. J. Bacteriol. 190:1831–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller P, Ewers C, Bertsche U, Anstett M, Kallis T, Breukink E, Fraipont C, Terrak M, Nguyen-Distèche M, Vollmer W. 2007. The essential cell division protein FtsN interacts with the murein (peptidoglycan) synthase PBP1B in Escherichia coli. J. Biol. Chem. 282:36394–36402 [DOI] [PubMed] [Google Scholar]
- 31.Lopez-Garrido J, Cheng N, Garcia-Quintanilla F, Garcia-del Portillo F, Casadesus J. 2010. Identification of the Salmonella enterica damX gene product, an inner membrane protein involved in bile resistance. J. Bacteriol. 192:893–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller JH. 1992. A short course in bacterial genetics. Cold Spring Harbor Press, Plainview, NY [Google Scholar]
- 33.Kwok S, Chang SY, Sninsky JJ, Wang A. 1994. A guide to the design and use of mismatched and degenerate primers. PCR Methods Appl. 3:S39–S47 [DOI] [PubMed] [Google Scholar]
- 34.Mercer KL, Weiss DS. 2002. The Escherichia coli cell division protein FtsW is required to recruit its cognate transpeptidase, FtsI (PBP3), to the division site. J. Bacteriol. 184:904–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lobstein J, Emrich CA, Jeans C, Faulkner M, Riggs P, Berkmen M. 2012. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Fact. 11:56. 10.1186/1475-2859-11-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wissel MC, Weiss DS. 2004. Genetic analysis of the cell division protein FtsI (PBP3): amino acid substitutions that impair septal localization of FtsI and recruitment of FtsN. J. Bacteriol. 186:490–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeLano WL. 2008. The PyMOL molecular graphics system, version 1.1. DeLano Scientific, San Carlos, CA [Google Scholar]
- 38.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 39.Bernhardt TG, de Boer PA. 2003. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol. Microbiol. 48:1171–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. 2003. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 11:671–683 [DOI] [PubMed] [Google Scholar]
- 41.Dutton RJ, Boyd D, Berkmen M, Beckwith J. 2008. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. U. S. A. 105:11933–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiniker A, Bardwell JC. 2004. In vivo substrate specificity of periplasmic disulfide oxidoreductases. J. Biol. Chem. 279:12967–12973 [DOI] [PubMed] [Google Scholar]
- 43.Bardwell JC, McGovern K, Beckwith J. 1991. Identification of a protein required for disulfide bond formation in vivo. Cell 67:581–589 [DOI] [PubMed] [Google Scholar]
- 44.Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. 1994. Crystal structure at 1.92 A resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature 372:432–438 [DOI] [PubMed] [Google Scholar]
- 45.Nagai K, Oubridge C, Jessen TH, Li J, Evans PR. 1990. Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A. Nature 348:515–520 [DOI] [PubMed] [Google Scholar]
- 46.Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, de Pedro MA, Höltje JV. 2001. Involvement of N-acetylmuramyl-l-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol. Microbiol. 41:167–178 [DOI] [PubMed] [Google Scholar]
- 47.Priyadarshini R, de Pedro MA, Young KD. 2007. Role of peptidoglycan amidases in the development and morphology of the division septum in Escherichia coli. J. Bacteriol. 189:5334–5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peters NT, Dinh T, Bernhardt TG. 2011. A fail-safe mechanism in the septal ring assembly pathway generated by the sequential recruitment of cell separation amidases and their activators. J. Bacteriol. 193:4973–4983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laederach A, Reilly PJ. 2005. Modeling protein recognition of carbohydrates. Proteins 60:591–597 [DOI] [PubMed] [Google Scholar]
- 50.Pell G, Williamson MP, Walters C, Du H, Gilbert HJ, Bolam DN. 2003. Importance of hydrophobic and polar residues in ligand binding in the family 15 carbohydrate-binding module from Cellvibrio japonicus Xyn10C. Biochemistry 42:9316–9323 [DOI] [PubMed] [Google Scholar]
- 51.Georgelis N, Yennawar NH, Cosgrove DJ. 2012. Structural basis for entropy-driven cellulose binding by a type-A cellulose-binding module (CBM) and bacterial expansin. Proc. Natl. Acad. Sci. U. S. A. 109:14830–14835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spiwok V, Lipovova P, Skalova T, Vondrackova E, Dohnalek J, Hasek J, Kralova B. 2005. Modelling of carbohydrate-aromatic interactions: ab initio energetics and force field performance. J. Comput. Aided Mol. Des. 19:887–901 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.