

Abstract
Staphylococcus saprophyticus is the only species of Staphylococcus that is typically uropathogenic and possesses a gene coding for a d-serine-deaminase (DsdA). As d-serine is prevalent in urine and toxic or bacteriostatic to many bacteria, it is not surprising that the d-serine-deaminase gene is found in the genome of uropathogens. It has been suggested that d-serine-deaminase or the ability to respond to or to metabolize d-serine is important for virulence. For uropathogenic Escherichia coli (UPEC), a high intracellular d-serine concentration affects expression of virulence factors. S. saprophyticus is able to grow in the presence of high d-serine concentrations; however, its d-serine metabolism has not been described. The activity of the d-serine-deaminase was verified by analyzing the formation of pyruvate from d-serine in different strains with and without d-serine-deaminase. Cocultivation experiments were performed to show that d-serine-deaminase confers a growth advantage to S. saprophyticus in the presence of d-serine. Furthermore, in vivo coinfection experiments showed a disadvantage for the ΔdsdA mutant during urinary tract infection. Expression analysis of known virulence factors by reverse transcription-quantitative PCR (RT-qPCR) showed that the surface-associated lipase Ssp is upregulated in the presence of d-serine. In addition, we show that S. saprophyticus is able to use d-serine as the sole carbon source, but interestingly, d-serine had a negative effect on growth when glucose was also present. Taken together, d-serine metabolism is associated with virulence in S. saprophyticus, as at least one known virulence factor is upregulated in the presence of d-serine and a ΔdsdA mutant was attenuated in virulence murine model of urinary tract infection.
INTRODUCTION
Urinary tract infections (UTIs) are common and affect mainly women; it has been estimated that more than 50% of all women will contract a urinary tract infection at least once in their lifetimes (1, 2). Although many bacterial species may cause urinary tract infections in patients predisposed by anatomical or functional abnormalities, indwelling catheters or neurological disorders, only a subset of species/strains may cause urinary tract infections in patients without predisposing conditions (3). These organisms/strains have mostly been found to express virulence factors allowing them to adhere to the uroepithelium (4–6), to invade cells of the urinary tract (7, 8), or to degrade urea (9, 10), a component highly prevalent in urine.
In staphylococci, most species do not cause urinary tract infections in the absence of predisposing factors, and Staphylococcus saprophyticus is the only species that typically causes these infections (11).
We have described a number of virulence factors, such as urease (12, 13), the adhesins Aas (14) and SdrI (15, 16), and the lipase Ssp (17, 18). A role for Ssp and SdrI in virulence was described in a murine model of UTI (19). Since similar proteins are present in many other staphylococci (20, 21), a factor distinguishing S. saprophyticus from other staphylococci has been sought. Interestingly, of all sequenced staphylococcal genomes, only that of S. saprophyticus possesses a gene coding for d-serine-deaminase (22), an enzyme that catabolizes d-serine to pyruvate and ammonia. This enzyme is also present in uropathogenic Escherichia coli (UPEC) but not in enterohemorrhagic E. coli (23). The amino acid d-serine is relatively prevalent in urine (3 to 115 μg/ml) (24, 25) and is toxic or bacteriostatic to many bacteria, probably by inhibiting the synthesis of pantothenic acid (26–29). Therefore, the presence of d-serine may be used as a cue for the presence within the urinary tract of uropathogenic microorganisms. Strains of E. coli expressing the deaminase DsdA can utilize this amino acid as a carbon and energy source (30). The operon dsdCXA comprises the regulator gene, dsdC, a specific transporter gene, dsdX, and the deaminase gene, dsdA (31, 32). In S. saprophyticus, such an operon does not exist. The dsdA gene apparently is under the control of its own promoter, and genes in the vicinity are not related to serine catabolism. Three genes in the genome are annotated as coding for d-serine/d-alanine/glycine transporters but are not located next to the dsdA gene. In UPEC, mutation of the deaminase somewhat surprisingly led to enhanced colonization of bladders and kidneys in experimentally infected mice and caused the mutants to be hyperflagellated and more motile (23). However, only wild-type (WT) UPEC strains possessing dsdA were able to grow on minimal medium containing d-serine as the sole carbon and energy source (23, 30). Later, it was shown that the high intracellular concentration of d-serine caused expression of the hypercolonization phenotype (23, 47). Regulation seems to involve the amino acid, since mutants defective in d-serine uptake showed wild-type colonization capabilities, and mutants not expressing the regulator dsdC showed the same phenotype as dsdA mutants (47).
Here we describe that the d-serine-deaminase of S. saprophyticus contributes to virulence within the urinary tract and that, in contrast to E. coli, the ΔdsdA mutant is attenuated in virulence.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The bacterial strains and plasmids used in this study are listed in Table 1. S. saprophyticus strain 7108, a hemagglutinating, fibronectin- and collagen-binding clinical isolate has been described previously (33–35). E coli DH5α (36) was the host for expression experiments and the intermediate host during construction of the plasmids for allelic replacement. The shuttle plasmid pBT2 (37) contains the temperature-sensitive replicon of pE194, the chloramphenicol resistance of pC194 and the multiple cloning site of pUC18. Plasmid pEC1 (38) was used as the erm(B) source. The pPS44 vector (39) was used for cloning experiments involving Staphylococcus carnosus strain TM300 (40) and for complementation experiments. The plasmid pMB2200, conferring tetracycline resistance, had been isolated from a clinical isolate of S. saprophyticus (41).
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Species or vector | Size (kb) | Descriptiona | Reference or source |
|---|---|---|---|---|
| Strains | ||||
| DH5α | E. coli | F− ϕ80 dlacZΔM15 Δ(lacZA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK−) phoA supE44 λ− thi-1 gyrA96 relA1; cloning host | 36 | |
| M15(pREP4) | E. coli | nalS strS rifS lac ara gal mtl F− recA+ uvr+ (pREP4 lacIq Kanr) | Qiagen | |
| TM300 | S. carnosus | Cloning host | 40 | |
| 7108 | S. saprophyticus | Wild-type isolate, fibronectin binding | 33–35 | |
| TM300(pMB1406) | S. carnosus | Cmr, contains pMB1406, expresses enzyme DsdA | This study | |
| 7108 ΔdsdA | S. saprophyticus | dsdA::erm(B) isogenic knockout mutant of 7108 | This study | |
| 7108 ΔdsdA(pMB1406) | S. saprophyticus | Complemented dsdA knockout mutant of 7108 containing pBM1406 | This study | |
| 7108(pMB2200) | S. saprophyticus | Tetr | 41 | |
| Plasmids | ||||
| pBT2 | 6.97 | Staphylococcal shuttle vector, temp-sensitive replicon of pE194, Cmr of pC194, Apr of pUC18 | 38 | |
| pPS44 | 4.38 | Contains replicon and cat gene of pC194 | ||
| pUC18 | 2.69 | Apr | ||
| pEC1 | pUC18 | 4.14 | Emr Apr donor of erm(B) cassette containing XbaI and PstI sites, tetracycline resistance plasmid from a clinical isolate of S. saprophyticus | 38 |
| pMB1400 | pUC18 | 5.69 | A 3-kb fragment containing dsdA gene with own promoter | This study |
| pMB1401 | pUC18 | 3.39 | Contains 0.7-kb fragment upstream of dsdA gene | This study |
| pMB1402 | pUC18 | 4.84 | Contains insert from pMB1401 ligated with a 1.45-kb erm(B) cassette | This study |
| pMB1403 | pUC18 | 5.44 | Containing insert from pMB1402 ligated with a 0.6-kb fragment downstream of dsdA gene | This study |
| pMB1404 | pBT2 | 9.72 | Contains insert (2.75 kb) of pMB1403 | This study |
| pMB1406 | pPS44 | 3 | Contains dsdA gene with its own promoter | 39 |
Cmr, chloramphenicol resistance; Tetr, tetracycline resistance; Apr, ampicillin resistance; Emr, erythromycin resistance.
Bacterial growth media and antibiotics.
E. coli strains harboring plasmids were grown in lysogeny broth (LB) or on LB agar plates. S. saprophyticus strain 7108 was grown in peptone-yeast extract (PY) broth or tryptic soy broth (TSB) (Oxoid, Wesel, Germany) or on agar plates. Bacteria were usually incubated at 37°C, but in some experiments, temperatures of 30°C and 42°C were also used. Ampicillin (100 μg/ml) was used for the selection of plasmids in E. coli. For selection of plasmids or chromosomal markers in S. saprophyticus, 10 μg/ml chloramphenicol, 5 μg/ml erythromycin, or 10 μg/ml tetracycline was used.
DNA preparation.
Plasmid DNA was prepared using the Qiagen plasmid Midi kit (Qiagen Hilden, Germany) and genomic DNA with the Qiagen DNeasy kit (Qiagen, Hilden, Germany). For DNA preparations from staphylococci, pelleted bacteria were resuspended in lysis buffer (20 mM Tris [pH 8.0], 2 mM EDTA, 1.2% [vol/vol] Triton X-100). To lyse the bacteria, we added 100 μg/ml Amibicin L (Wak Chemie Medici GmbH, Steinbach, Germany) to the lysis buffer.
Construction of an insertion mutant by allelic replacement.
The dsdA gene was deleted by the insertion of the erm(B) resistance gene, and pBT2 (37) was used as the replacement vector. To this end, a 700-bp PCR amplificate of the chromosomal DNA upstream of the dsdA gene generated with the primers Ssa.dsd1seq/BamHI and Ssa.dsd2/2rev/XbaI (Table 2) was digested with BamHI and XbaI and was cloned into pUC18. This plasmid was designated pMB1401. For the construction of pMB1402, we inserted the erm(B) cassette cut from pEC1 into plasmid pMB1401 digested with the enzymes XbaI and PstI. A further 600-bp DNA amplificate downstream of the dsdA gene was generated with the primers Ssa.dsd1rev/HindIII and Ssa.dsd2seq/PstI and was ligated into pMB1402 cut with these enzymes. The resultant plasmid (pMB1403) contained the upstream and downstream flanking regions of the dsdA gene with an erm(B) cassette in the center. The insert of pMB1403 was excised with BamHI and HindIII and ligated into the temperature-sensitive replacement vector pBT2, yielding pMB1404.
Table 2.
Cloning and PCR primers used in this study
| Primer | Sequence (5′→3′)a | Use (bp location)b |
|---|---|---|
| Ssa.dsd1seq/BamHI | CACGGATCCGAAATTGCTGAAGATGCG | Cloning |
| Ssa.dsd1rev/HindIII | GTGAAGCTTTGTACGATGATCGTAAGG | Cloning |
| Ssa.dsd2seq/PstI | CAGCTGCAGTTTACTTTAGTTCGGTAGAG | Cloning |
| Ssa.dsd2/2rev/XbaI | GTGTCTAGAGTTCCTGACTCTTTGTGG | Cloning |
| Ssa.dsd4rev/HindIII | GCGAAGCTTCTATAAGCAAGATTTACC | Cloning |
| Ssa.dsd4seq/BamHI | GTAGGATCCAACGATTTAGCAACACTT | Cloning |
| Ssa.dsd10rev | TCACCTCGCTATATCTGG | PCR |
| Ssa.dsd10seq | TTTCTTCCACTGACGTGC | PCR |
| catseq | GTTACAGTAATATTGACT | PCR, DIG probe synthesis (183–201) |
| catrev | CATAAACAATCCTGCATG | PCR, DIG probe synthesis (872–890) |
| ermB | TCTAGAACTAGTGGATCCC | PCR, DIG probe synthesis |
| ermB | TATTGTCTGCAGccgagagtgattggtctt | PCR, DIG probe synthesis |
| CAT473seq | CAGCAAACTACGTATAGC | PCR, DIG probe synthesis |
| CAT473rev | CAAGGAATCATTGAAATCG | PCR, DIG probe synthesis |
| pPS44rev/SstI | ATAGAGCTGGTGACCACTTGTGATAACG | PCR, cloning |
| pPS44seq/XbaI | TGCTCTAGAACTCGTGTGCATAATTCACGC | PCR, cloning |
Restriction sites in the 5′ extensions of primers are underlined. Lowercase letters indicate nonhomologous sequences with added restriction sites.
DIG, digoxigenin.
The pMB1404 constructs were purified from E. coli DH5α and transformed into S. saprophyticus strain 7108 by protoplast transformation, as previously described (12). Chloramphenicol- and erythromycin-resistant clones were grown in the presence of erythromycin (5 μg/ml at 30°C for 24 h) and used to inoculate 1,000 ml of prewarmed (42°C) broth containing erythromycin. After overnight incubation, appropriate dilutions were plated onto P-agar containing erythromycin. Clones that grew on erythromycin but not on chloramphenicol had lost the plasmid, and correct insertion of the erm(B) cassette was checked by PCR and sequencing. Absence of the cat gene was verified by PCR.
Complementation of the ΔdsdA mutant.
The vector part of pPS44 (39) was amplified by inverse PCR with the primers pPS44BamHI and pPS44HindIII. The dsdA gene with its own promoter was amplified with the primers Ssa.dsdA4seq and Ssa.dsdA4rev, and both were digested with BamHI and HindIII. Ligation and transformation of S. carnosus TM300 (40) were performed as described elsewhere (42). The resulting plasmid (pMB1406) was purified from this strain and introduced into the ΔdsdA mutant by protoplast transformation (12).
Animal experiments.
Eight-week-old female C3H/HeN mice (Harlan) were infected by transurethral catheterization as previously described (43). Static bacterial cultures were started from freezer stocks, grown at 37°C for 18 h in brain heart infusion (BHI) broth and then subcultured at 1:250 or 1:100 into fresh medium. These subcultures were then grown statically at 37°C for 18 h, pelleted, and resuspended in phosphate-buffered saline (PBS), to yield 50-μl inocula containing 1 × 107 to 2 × 107 CFU. For competition experiments, bacteria were diluted appropriately to achieve 107 CFU each of the wild type and dsdA mutant together in a total volume of 50 μl. To quantify bacteria present in mouse organs, bladder and kidneys were aseptically harvested at the indicated times postinfection, homogenized in phosphate-buffered saline, serially diluted, and plated onto BHI agar plates for total CFU enumeration and with erythromycin for mutant CFU (CFUm) enumeration. Wild-type CFU (CFUw) were calculated as total CFU − mutant CFU. In samples where zero CFU were recovered, the limit of detection of the protocol was used. Competition indices (CI) were calculated similarly to the method described by Freter et al. (44) by using the wild type as the reference strain, as follows: CI = (CFUm/CFUw recovered from mice)/(CFUm/CFUw present in the initial inoculum).
Significance was calculated using the nonparametric Wilcoxon signed-rank test (two tailed) comparing the theoretical median CI to 1 (GraphPad Prism 5). All animal studies were performed in accordance with the Committee for Animal Studies at Washington University School of Medicine.
Cocultivation experiments.
Cocultivation experiments were carried out in lysogeny broth (LB) (Invitrogen, Karlsruhe, Germany) supplemented with 2.5, 5, or 10 mg/ml d-serine (AppliChem, Darmstadt, Germany) or l-serine (AppliChem) or without any supplements. Bacteria were precultured overnight in 10 ml medium at 37°C with a flask-to-medium ratio of 100:10 and with shaking at 100 rpm. Cells from overnight cultures were then resuspended in 0.9% NaCl, and the optical density at 600 nm (OD600) was adjusted to 0.5. For inoculation of the coculture, 100 μl of each strain was added to 10 ml of medium. Cultures were then incubated for 24 h at 37°C and 100 rpm. Every 24 h, the mixed populations were diluted 1:100 into fresh medium. Growth was checked by determining CFU every 48 h by serial dilutions (10−3 to 10−9 in 0.9% NaCl). Each dilution was plated onto three LB agar plates (LB agar powder; AppliChem): one without and two with different antibiotics for differentiation of the strains. Erythromycin (10 μg/ml; Sigma-Aldrich) was used for selection of the mutant, chloramphenicol (20 μg/ml; AppliChem) for selection of the complemented mutant, and tetracycline (10 μg/ml; AppliChem) for positive selection of the wild type, which had been transformed with a tetracycline resistance plasmid (pMB2200) isolated from a clinical isolate of S. saprophyticus. Agar plates were incubated overnight at 37°C, and bacterial counts were determined.
Chemically defined medium.
Chemically defined medium contained the following (per liter of water): 7 g K2HPO4 · 3H2O, 2 g KH2PO4, 0.4 g Na3 citrate · 2H2O, 0.05 g MgSO4, 1 g (NH4)2SO4, 20 mg thymine, 0.5 g sodium thiosulfate, 50 mg l-arginine, 100 mg l-glutamine, 50 mg l-glycine, 20 mg l-histidine, 90 mg l-leucine, 50 mg l-lysine, 3 mg l-methionine, 40 mg l-phenylalanine, 80 mg l-proline, 30 mg l-threonine, 10 mg l-tryptophan, 80 mg l-valine, 5 mg FeSO4 · 7H2O, 10 mg CaCl2, 7 mg ZnCl2, 13 mg NiSO4 · 6H2O, 10 mg MnCl2 · 4H2O, 0.5 mg thiamine hydrochloride, 0.6 mg nicotinic acid, 0.125 mg d-pantothenic acid calcium salt, and 0.125 mg biotin, adjusted to pH 7.0. Before use, the medium was filter sterilized. To exclude contaminating constituents from residues in reusable glassware, only new plastic materials were used for preparation and growth. Also, bacterial cultures were grown in plastic tubes (10-ml culture in a 50-ml Falcon tube). Without addition of the carbon and energy source (glucose or d-serine), bacteria were not able to grow.
Pyruvate assay.
The assay was conducted basically as described by McFall (45). It measures the concentration of a colored α-keto acid that is formed as the product of a reaction of pyruvate with 2,4-dinitrophenylhydrazine under alkaline conditions. The strains used in this assay were cultured in 100 ml peptone-yeast extract (PY) broth (10 g/liter peptone, 5 g/liter yeast, 1 g/liter glucose, 5 g/liter NaCl, 1.25 g/liter Na2HPO4 · 2H2O [pH 7.3]) overnight (37°C and 100 rpm). The medium was supplemented with d-serine (1 g/liter) to ensure that the d-serine-deaminase was expressed. Twenty-five milliliters of the overnight culture was pelleted by centrifugation at 4,000 rpm for 10 min. The bacteria were then washed with 0.07 M PBS (pH 7.4) and resuspended in 2 ml of this buffer. The following lysis was done by addition of 10 μl lysostaphin (5 mg/ml; WAK-Chemie, Steinbach, Germany), incubation at 37°C for 15 to 20 min, and subsequent sonication (6 × 30 s; Branson sonifier W185, level 4). Cell debris was removed by centrifugation (13,000 rpm, 1 min), and the lysate was used for the following reaction. Four hundred microliters of lysate was incubated with 100 μl d-serine (10 mg/ml) for 60 min at 37°C. 2,4-Dinitrophenylhydrazine (Acros; 500 μl; 0.3 mg/ml in 1 N NaCl) was then added, and incubation continued at room temperature for 20 min. The reaction was stopped by addition of 1 ml 2.8 N NaOH. The optical density was read at 450 nm in a spectrophotometer against an assay blank to which all components had been added except d-serine. Additionally, a standard curve with pyruvate from 0.5 μg/ml to 10 μg/ml was measured. Analysis of pyruvate generation was done per mg total protein, which was determined by the method of Markwell (46).
RNA preparation from Staphylococcus saprophyticus.
For RNA preparation, wild-type S. saprophyticus or the ΔdsdA mutant was grown in the chemically defined medium supplemented with glucose or glucose and d-serine until an OD600 between 0.450 and 0.500 was reached. For each strain and growth condition, RNA was extracted from three biological replicates. Before harvesting, 4 ml Bacteria Protect reagent (Qiagen) was added to 2 ml bacterial culture, and the mixture was incubated for 5 min at room temperature. By a subsequent centrifugation (5,000 rpm, 10 min, room temperature), bacteria were pelleted, and pellets were stored for not longer than 2 weeks at −20°C. RNA was prepared using the RNeasy minikit (Qiagen). The pelleted bacteria were resuspended in 100 μl TE buffer (30 mM Tris, 1 mM EDTA [pH 8.0]), and 10 μl lysostaphin (5 mg/ml) and 10 μl proteinase K (Applichem) were added. After undergoing vortexing and incubation (10 min, 37°C, 100 rpm), bacteria were disrupted mechanically following the manufacturer's instructions with the modification that a Power Vortex was used instead of a TissueLyser. DNase digestion was done on-column using the Qiagen RNase-free DNase set following the manufacturer's instructions. After elution, aliquots of RNA were frozen in liquid nitrogen and stored at −80°C. The integrity and purity of RNA were checked by gel electrophoresis, and absorbance was measured using a Nanodrop 1000. The ratio of A260 to A280 was between 1.9 and 2.1 for all RNA samples used.
RT-qPCR.
Real-time reverse transcription-quantitative PCR (RT-qPCR) was carried out by using the QuantiFast SYBR green RT-PCR kit (Qiagen) for one-step real-time PCR following the manufacturers' instructions and the LightCycler 1.2 (Roche). The reaction volume was 20 μl. RNA was used as the template at a final concentration of 2.5 ng/reaction. Primers were added to a final concentration of 0.5 pmol/each. After RT for 10 min at 50°C, the following temperature protocol was used: an initial activation step for 5 min at 95°C (temperature transition, 20°C/s), followed by a two-step cycling PCR (40 cycles) consisting of denaturation at 95°C for 10 s (temperature transition of 20°C/s), a combined annealing and extension at 60°C for 30 s (temperature transition of 20°C/s), and fluorescence acquisition at 60°C in single mode. Melting curve analysis was performed at 60°C to 95°C (temperature transition, 0.1°C/s). The specificity of the products was checked by melting point analysis and gel electrophoresis.
Calculation of virulence factor expression.
For each strain and growth condition, RNA was extracted from three biological replicates. Each replicate was analyzed in triplicate. The test genes where either normalized to gyrB or to dpol. Calculations were done by using the formula 2ΔCT test gene/2ΔCT reference gene, in which the change in cycle threshold (ΔCT) is the difference of the CT under glucose conditions − the CT under d-serine conditions. Values of >2.0 were defined as upregulated, and values of <0.5 were defined as downregulated if observed in all three independent biological replicates.
RESULTS
The d-serine-deaminase catabolizes d-serine to pyruvate and ammonia.
To show that in S. saprophyticus the d-serine-deaminase is functional and catabolizes d-serine to pyruvate, we constructed an isogenic mutant of strain 7108 by interrupting the gene with an erythromycin resistance cassette. In addition, the gene was cloned into a staphylococcal vector and expressed in S. carnosus TM300. We prepared cell extracts from the wild-type S. saprophyticus strain, the ΔdsdA mutant, and S. carnosus TM300 as well as S. carnosus TM300(pMB1406) containing the dsdA gene on a plasmid. The extracts were incubated with d-serine, and generation of a keto acid—pyruvate in this case—was measured with the dinitrophenylhydrazone assay (45). The S. saprophyticus wild-type strain 7108 generated 4.6 (standard deviation [SD], 0.9) μg mg−1 min−1 pyruvate, the mutant 0.4 (SD, 0.06) μg mg−1 min−1, the complemented strain 80.2 (SD, 2.8) μg mg−1 min−1, S. carnosus TM300 0.5 (SD, 0.25) μg mg−1 min−1, and S. carnosus TM300(pMB1406) 36.1 (1.04) μg mg−1 min−1. The presence of the d-serine-deaminase led to degradation of d-serine to pyruvate, whereas no degradation was detectable in strains not containing dsdA.
The d-serine-deaminase-negative mutant has a significant disadvantage against the wild type in coinfection experiments.
The wild-type strain, 7108, and its ΔdsdA mutant were used in coinfection experiments in a murine model of urinary tract infection (Fig. 1). The animals were infected transurethrally with a suspension containing ∼1 × 107 CFU of each strain. After 48 h, the animals were sacrificed, bladders and kidneys were aseptically removed, and CFU ml−1 tissue were determined. The competitive index (CI) of the two strains was calculated as follows: CI = (CFU · ml−1 mutant at 0 h/CFU · ml−1 wild type at 0 h)/(CFU · ml−1 of mutant at 48 h/CFU · ml−1 of wild type at 48 h). Competitive indices significantly less than 1 indicate an advantage for the wild type. The CI in the bladders was 0.47, and that for the kidneys was 0.13; both values were statistically different from 1 (P < 0.05, Wilcoxon signed-rank test). These experiments showed that in contrast to E. coli, the ΔdsdA mutation did not lead to increased colonization but conferred a disadvantage.
Fig 1.
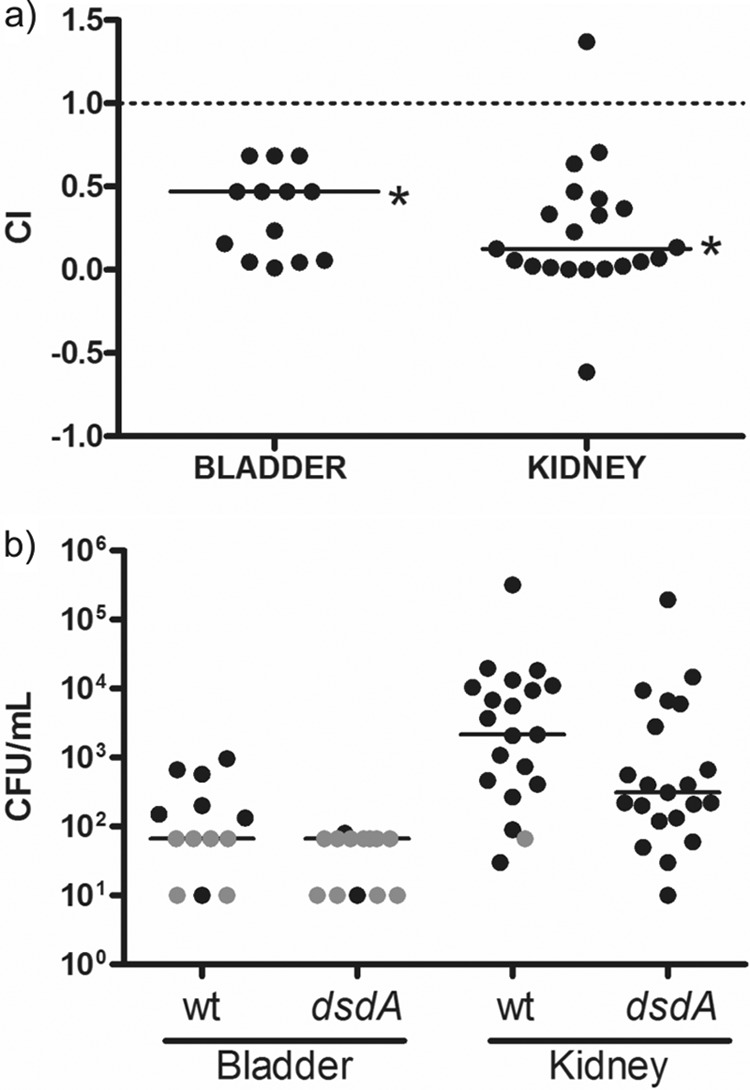
(a) Competitive indices of coinfection experiments. Mice were infected by instillation with a mixture of cells of the wild type and the mutant, and competitive indices (CIs) were determined for bladders and kidneys. The dashed line represents equal numbers of CFU recovered from bladder and kidney. Median CIs, indicated by horizontal lines, were significantly lower than 1 (0.47 in bladders and 0.13 in kidneys), indicating a disadvantage of the mutant in these experiments. n = 13 for bladders and 21 for kidneys. *, P < 0.05 by Wilcoxon signed-rank test. (b) CFU data from coinfection experiments. n = 13 for bladders and 21 for kidneys. Horizontal lines represent the geometric mean titer of each sample (n = 13 for bladders and 21 for kidneys). Gray circles indicate samples in which no CFU were detected and for which the limit of detection of the protocol has been entered.
The d-serine-deaminase-negative mutant is at disadvantage in cocultivation experiments in the presence of d-serine.
Since we wanted to know if the disadvantage of the mutant in animal experiments is solely related to its inability to catabolize d-serine, we used in vitro competition experiments in the presence and absence of d-serine. To allow for selection of the wild type, we transformed this strain with plasmid encoding tetracycline resistance (pMB2200) that had been isolated from a clinical strain of S. saprophyticus (41). When this strain and the ΔdsdA mutant were cocultivated in broth without d-serine, the mutant outcompeted the wild type (Fig. 2a). In cultures containing d-serine, however, the mutant was at disadvantage (Fig. 2b). When the l-enantiomer of serine was used for supplementation, however, the mutant again had an advantage (Fig. 2c). These experiments show that the mutant was only at a disadvantage if d-serine was present, excluding the possibility that the resistance gene on the plasmid caused slower growth. When the ΔdsdA mutant and the complemented mutant were cocultivated in the presence of d-serine, the mutant was not detectable after 8 days (Fig. 2d). The effects were observed with d-serine concentrations as low as 2.5 mg/ml in both media (data not shown).
Fig 2.

(a to d) The ability to degrade d-serine confers a growth advantage. Pairs of strains were grown in full medium in the presence or absence of d-serine. (a) Wild type and mutant grown without d-serine. The mutant outcompetes the wild type. (b) Wild type and mutant in the presence of d-serine. The wild type outcompetes the mutant. (c) Both strains in the presence of l-serine. The mutant again has an advantage. (d) Mutant and the complemented mutant in the presence of d-serine. The complemented mutant outcompetes the mutant. Values are the means of at least three experiments and SD (log CFU/ml).
The Staphylococcus saprophyticus wild-type strain is able to use d-serine as the sole carbon and energy source.
To analyze if S. saprophyticus is able to grow with d-serine as the sole carbon and energy source, like E. coli (30), we conducted growth experiments in a chemically defined medium supplemented with 10 mM d-serine. Whereas the ΔdsdA mutant was not able to grow with d-serine as the sole carbon source, wild-type S. saprophyticus started growing after an extended lag time (∼168 h), and the complemented mutant also grew after a shorter lag time (48 h) compared to the wild type (Fig. 3). Since d-serine is catabolized to pyruvate and ammonia, we also supplemented the medium with 10 mM pyruvate instead of d-serine. In this medium, the wild type, the ΔdsdA mutant, and the complemented mutant grew immediately (lag phase of about 8 h) and reached high cellular densities (data not shown). If no carbon source was added, none of the strains was able to grow.
Fig 3.
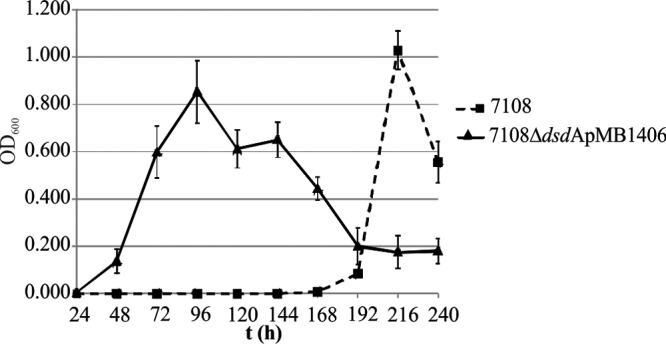
Staphylococcus saprophyticus is able to use d-serine as the sole carbon source. The S. saprophyticus 7108 wild-type strain and the complemented ΔdsdA mutant strain [7108 ΔdsdA(pMB1406)] are able to grow in chemically defined medium supplemented with d-serine (DS) (10 mM) as the sole carbon source. The ΔdsdA mutant was not able to grow. Without any additional supplements, none of the strains could grow. Values are means of at least three experiments and SD.
d-Serine is not only an additional nutrient for Staphylococcus saprophyticus.
To analyze if the growth advantage of wild-type S. saprophyticus in cocultivation experiments was due to the utilization of d-serine as an additional nutrient, we analyzed the influence of d-serine on growth in the S. saprophyticus wild-type strain and the ΔdsdA mutant. To this end, we cultivated S. saprophyticus in our chemically defined medium supplemented with 2.5 mM glucose or 2.5 mM glucose and 5 mM d-serine. Both the wild type and the ΔdsdA mutant grew more slowly in the presence of d-serine (Fig. 4). This indicates that d-serine has an inhibitory effect on growth of S. saprophyticus.
Fig 4.
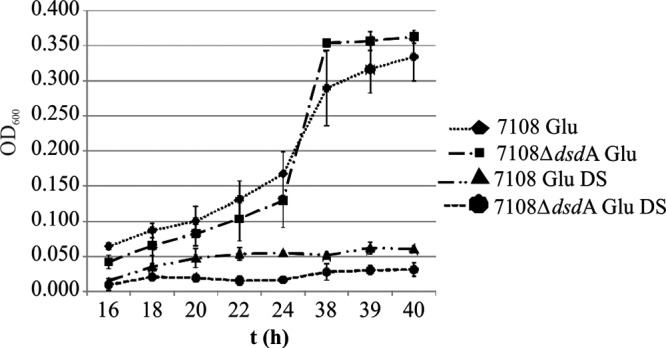
Influence of d-serine on growth of S. saprophyticus. d-Serine (DS) leads to growth impairment of the dsdA-knockout mutant as well as the wild-type strain, 7108, when the chemically defined medium was supplemented with glucose (2.5 mM) and d-serine (5 mM). Values are means of at least three experiments and SD.
Regulation of known virulence factor in the presence of d-serine.
For uropathogenic E. coli, it was shown that a ΔdsdA mutant is hyperflagellated and more motile than the wild type (23). This hyperflagellation leads to hypercolonization; therefore, the ΔdsdA mutant of E. coli is more virulent than the wild type. d-Serine acts as a signal for upregulation of genes encoding fimbriae and some other virulence factors (47, 48). In S. saprophyticus, the ΔdsdA mutant is attenuated in virulence, suggesting that the influence of d-serine and d-serine metabolism on virulence gene expression is different from that in E. coli. To investigate if there is also a connection between virulence and d-serine in S. saprophyticus, we analyzed the expression of known virulence factors in the presence and absence of d-serine in the wild-type S. saprophyticus strain and the ΔdsdA mutant. Using chemically defined medium for these experiments, we were able to study the effect of d-serine without the influence of other, unknown components of the medium. Wild-type strain 7108 of S. saprophyticus and the ΔdsdA mutant were grown in chemically defined medium supplemented with 2.5 mM glucose or with 2.5 mM glucose and 5 mM d-serine to an OD600 between 0.450 and 0.500, which represents the late exponential phase. RNA was prepared, and the expression of known virulence factors was measured by RT-qPCR. The genes and primers used in this study are listed in Table 3.
Table 3.
Primer used for RT-qPCR in this study
| GenBank accession no. | Primer name | Sequence (5′→3′) | Product size (bp) |
|---|---|---|---|
| AF402316 | RNA_sdrI_F1 | GCAGACGCAGACGCAGAC | 147 |
| RNA_sdrI_R1 | CAGCATCTGCATCTGCATCTGAG | ||
| AY551101 | RNA_ssp_F1 | TGGTGCTGCACATGCAGAAAG | 129 |
| RNA-ssp_R1 | ACGGACAGTTTGTCCTCCCATAC | ||
| YP_300522 | RNA_dsdA_F2 | TTACTGAACCAACACATGCCCC | 147 |
| RNA_dsdA_R2 | ATTTGCCCGACTAAGCGAGATG | ||
| AJ000007 | RNA_aas_F1 | GCCGACTACGCAGCAACTAAC | 136 |
| RNA_aas_R1 | CCATGAGGGTCAGAGTGGTCAG | ||
| Q4A0V8 | RNA_uafA_F1 | CGATTACCGTAACGGGTTATCCAG | 131 |
| RNA_uafA_R1 | GAGAGCCAGAATTACCTCCGAATG | ||
| YP_300353 | RNA_ureC_F1 | ACACATATCGGTGGCGGTACAG | 121 |
| RNA_ureC_R1 | GGTTTACAGCTTGCCCTTTACCAG | ||
| YP_300095 | RNA_gyrB_F1 | GCAGAGTCACCCTCGACGATAAAG | 118 |
| RNA_gyrB_R1 | GTGAAGTTACGCGCCGTAAATCAG | ||
| YP_301165 | RNA_dpoI_F1 | TGCGTAGACAAGCGAAAGCAG | 140 |
| RNA_dpoI_R1 | GTTTCACACCAGGGAAACTATCGAG |
We analyzed the expression of the genes by relative quantification using gyrB as an internal reference gene, as previously described for Staphylococcus aureus (49, 50). While it is common practice to use only one reference gene for relative quantification, this practice assumes that the reference gene is not altered under the conditions being examined. However, it has been shown that the conventional normalization strategy based on a single gene can lead to significant errors (51). Therefore, we decided to use a second reference gene, dpol, along with gyrB because their gene products, DNA polymerase and gyrase subunit B, respectively, are not involved in metabolism and therefore should be constantly expressed under the different conditions we were interested in. Accordingly, we observed only minute differences in the expression of both reference genes when strains were grown with glucose compared to d-serine. Fig. 5a to c present the fold changes of each gene under d-serine conditions compared to glucose conditions referenced either to gyrB or to dpol in wild-type S. saprophyticus (Fig. 5a) and the ΔdsdA mutant (Fig. 5b).
Fig 5.

(a to c) Regulation of known virulence factors in the presence of d-serine. Each test gene was analyzed in three biological replicates and referenced to gyrB and dpol as the control genes. The values of the biological replicates are means of two or three experiments and SD. Dashed lines represent the threshold values for up- and downregulation. (a) Regulation of known virulence in wild-type S. saprophyticus. (b) Regulation of known virulence genes in the ΔdsdA mutant. (c) Regulation of ssp in wild-type S. saprophyticus analyzed in three additional biological replicates.
Using our criteria for up- and downregulation, only ssp in wild-type S. saprophyticus was consistently induced in the presence of d-serine. The ability to metabolize d-serine is required for ssp induction, as we did not observe this upregulation in the ΔdsdA mutant. Given the intersample variability in ssp induction, we confirmed the result in three freshly prepared biological samples (Fig. 5c). None of the other genes examined in S. saprophyticus displayed reproducibly altered transcription in the presence of d-serine. None of the genes analyzed in the ΔdsdA mutant was clearly regulated.
DISCUSSION
We have shown previously that S. saprophyticus is the only species of Staphylococcus that possesses a d-serine-deaminase and grows in the presence of high concentrations of d-serine (52). When the dsdA gene is transferred to S. aureus (36) or S. carnosus (this work), which do not naturally express the deaminase, these species can grow in the presence of the same concentration of d-serine as S. saprophyticus. We used the dsdA homologue cloned from S. saprophyticus into S. carnosus to show that its product indeed causes degradation of d-serine. In addition we showed that the S. saprophyticus wild type produces pyruvate from d-serine, whereas the ΔdsdA mutant and S. carnosus TM300 did not.
In uropathogenic E. coli (UPEC), d-serine is used as a cue for the organism's presence in the urinary tract, can be used as the sole carbon source, and regulates the expression of virulence factors (48). Curiously, a dsdA UPEC mutant expressed higher adherence capabilities as well as increased flagellation (23) and outcompeted the wild type in coinfection experiments, which indicates that serine metabolism is important but that there are functions of d-serine besides the pure generation of energy.
In order to analyze the role and relevance of the d-serine-deaminase for virulence of S. saprophyticus, we performed coinfection experiments in mice infected with the wild-type and ΔdsdA mutant strains. In these experiments, the mutant had a distinct disadvantage compared to the wild type. The effect was more pronounced in the bladders of the animals than in the kidneys, although the mutant had a significant disadvantage in both organs. These results clearly indicate that the d-serine-deaminase is important for S. saprophyticus during experimental infection.
To show that the disadvantage of the ΔdsdA mutant in vivo was caused by its inability to metabolize d-serine, we conducted cocultivation experiments in the presence and absence of d-serine. To allow positive selection of the wild-type strain, 7108, we transformed it with a naturally occurring tetracycline resistance plasmid of S. saprophyticus, pMB2200, which we had isolated from a clinical strain (41). When the wild-type strain (Tcr) and the mutant were grown in LB without d-serine, the mutant outcompeted the wild type; in the presence of d-serine, the wild type, however, outcompeted the mutant. When we used the l-enantiomer of serine, the mutant again grew better than the wild type. The advantage of the mutant in the absence of d-serine can be explained by the additional burden the tetracycline plasmid represents; in the presence of d-serine, the advantage of the mutant does not compensate for the effects of d-serine. We therefore conclude that d-serine is the factor that caused the disadvantage of the mutant in these experiments. This conclusion is corroborated by our finding that the wild type and the complemented mutant can grow when d-serine is the only carbon and energy source, whereas the mutant cannot. In these experiments, the lag phases were quite long. When the strains were grown with pyruvate instead of d-serine, all of them grew very fast, suggesting that the velocity of the DsdA enzyme was the limiting factor in these experiments. This is supported by the fact that the complemented mutant grew faster than the wild type. In the presence of glucose, d-serine had a negative effect on growth of the ΔdsdA mutant as well as on the wild type, mainly because of a prolonged lag phase of at least 45 h. Strains start to grow after the prolonged lag phase but apparently also replicate more slowly. We therefore conclude that the growth advantage of the wild type in the presence of d-serine cannot be explained just by an additional nutrient source and that the influence of d-serine on S. saprophyticus is more complex. Obviously, there is an inhibitory effect on growth in the presence of glucose; on the other hand, S. saprophyticus is able to grow with d-serine as the sole carbon and energy source.
DsdA catabolizes d-serine by forming pyruvate and ammonia. Bacteria not able to catabolize d-serine are usually inhibited by the compound, and it is thought that this effect is due to inhibition of the synthesis of pantothenic acid by d-serine, which functions as an analogue of β-alanine (29). In other species, d-serine inhibits pantothenic acid biosynthesis at different steps (26–28, 53). In E. coli, the target of d-serine inhibition is the pantoate-β-alanine ligase, encoded by panC, and it was shown that there is a second target of d-serine inhibition, the l-serine metabolism (29). For growth in our chemically defined medium, S. saprophyticus requires pantothenic acid or β-alanine; therefore, our chemically defined medium contains pantothenic acid. As we saw growth inhibition caused by d-serine in this medium, it is unlikely that growth impairment is caused by this classical mechanism.
In E. coli, d-serine regulates expression of virulence factors (47, 48). Because of our findings that wild-type S. saprophyticus has an advantage over the ΔdsdA mutant in coinfection experiments as well as in cocultivation experiments in the presence of d-serine, we hypothesized that d-serine metabolism affects S. saprophyticus virulence factor expression, albeit in a different way than in E. coli. To test this hypothesis, we analyzed the expression of known S. saprophyticus virulence factors in the presence and absence of d-serine in the wild type and the ΔdsdA mutant by RT-qPCR. Since we observed slight differences in expression of our chosen reference genes under both conditions, we decided to regard only those results as certain that were >2.0 or <0.5 in RNA preparations from three biological replicates referenced to both genes. Using these criteria, only ssp in wild-type S. saprophyticus was induced in the presence of d-serine. This result was confirmed by additional experiments (Fig. 5c). Therefore, we conclude that ssp is upregulated. It has been shown that an ssp-knockout mutant is less virulent in a murine model of UTI (19), although the role of the lipase during infection remains undefined. It has been suggested that lipases may be important for colonization, possibly in terms of nutrition or by release of free fatty acids which may promote adherence (54, 55). We conclude that the d-serine metabolism induces S. saprophyticus to produce greater amounts of lipase, which is needed during infection. Our hypothesis is that d-serine is used as a cue for the presence in the urinary tract and induces a different metabolism, including expression of Ssp in S. saprophyticus. It is not yet clear if d-serine directly or indirectly regulates ssp; these analyses will be the subjects of subsequent studies.
In conclusion, we have shown that S. saprophyticus is able to use d-serine as the sole carbon and energy source, like E. coli (30), but d-serine also has a negative effect on growth of S. saprophyticus. Coinfection experiments and cocultivation experiments showed that the d-serine-deaminase confers an advantage to the wild-type strain. Coinfection experiments demonstrated that the d-serine-deaminase is important for virulence of S. saprophyticus during urinary tract infection. In the presence of d-serine, the virulence-associated lipase Ssp is upregulated in the wild type, which may explain its advantage in experimental infections. We conclude that the d-serine-deaminase acts as a virulence factor in two different ways. First, it catabolizes d-serine, which is toxic or bacteriostatic to many bacteria and other staphylococci. Only strains that expressed this enzyme are able to grow in the presence of d-serine and to catabolize this amino acid. Second, d-serine, the d-serine-deaminase, or d-serine metabolism affects the expression of at least one virulence factor, the lipase Ssp, suggesting that d-serine may serve as a cue to the bacteria for their presence in the urinary tract and to induce adaptation to this environment.
ACKNOWLEDGMENTS
This work was supported by FoRUM grant F572R-2007 from the Faculty of Medicine of the Ruhr-University Bochum.
We thank Susanne Friedrich for performing the pyruvate assays.
Footnotes
Published ahead of print 30 September 2013
REFERENCES
- 1.Foxman B, Barlow R, D'Arcy H, Gillespie B, Sobel JD. 2000. Urinary tract infection: self-reported incidence and associated costs. Ann. Epidemiol. 10:509–515 [DOI] [PubMed] [Google Scholar]
- 2.Griebling TL. 2005. Urologic diseases in America project: trends in resource use for urinary tract infections in women. J. Urol. 173:1281–1287 [DOI] [PubMed] [Google Scholar]
- 3.Ronald A. 2002. The etiology of urinary tract infection: traditional and emerging pathogens. Am. J. Med. 113:14–19 [DOI] [PubMed] [Google Scholar]
- 4.Teti G, Chiofalo MS, Tomasello F, Fava C. 1987. Mediation of Staphylococcus saprophyticus adherence to uroepithelial cells by lipoteichoic acid. Infect. Immun. 55:839–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujita K, Yokota T, Oguri T, Fujime M, Kitagawa R. 1992. In vitro adherence of Staphylococcus saprophyticus, Staphylococcus epidermidis, Staphylococcus haemolyticus, and Staphylococcus aureus to human ureter. Urol. Res. 20:399–402 [DOI] [PubMed] [Google Scholar]
- 6.Lane MC, Mobley HLT. 2007. Role of P-fimbrial-mediated adherence in pyelonephritis and persistence of uropathogenic Escherichia coli (UPEC) in the mammalian kidney. Kidney Int. 72:19–25 [DOI] [PubMed] [Google Scholar]
- 7.Martinez JJ, Mulvey MA, Schilling JD, Pinkner JS, Hultgren SJ. 2000. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 19:2803–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szabados F, Kleine B, Anders A, Kaase M, Sakinç T, Schmitz I, Gatermann S. 2008. Staphylococcus saprophyticus ATCC 15305 is internalized into human urinary bladder carcinoma cell line 5637. FEMS Microbiol. Lett. 285:163–169 [DOI] [PubMed] [Google Scholar]
- 9.McLean RJ, Nickel JC, Cheng KJ, Costerton JW. 1988. The ecology and pathogenicity of urease-producing bacteria in the urinary tract. Crit. Rev. Microbiol. 16:37–79 [DOI] [PubMed] [Google Scholar]
- 10.Mobley HL, Hausinger RP. 1989. Microbial ureases: significance, regulation, and molecular characterization. Microbiol. Rev. 53:85–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raz R, Colodner R, Kunin CM. 2005. Who are you—Staphylococcus saprophyticus? Clin. Infect. Dis. 40:896–898 [DOI] [PubMed] [Google Scholar]
- 12.Gatermann S, Marre R. 1989. Cloning and expression of Staphylococcus saprophyticus urease gene sequences in Staphylococcus carnosus and contribution of the enzyme to virulence. Infect. Immun. 57:2998–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatermann S, John J, Marre R. 1989. Staphylococcus saprophyticus urease: characterization and contribution to uropathogenicity in unobstructed urinary tract infection of rats. Infect. Immun. 57:110–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hell W, Meyer H-GW, Gatermann SG. 1998. Cloning of aas, a gene encoding a Staphylococcus saprophyticus surface protein with adhesive and autolytic properties. Mol. Microbiol. 29:871–881 [DOI] [PubMed] [Google Scholar]
- 15.Sakinc T, Kleine B, Gatermann SG. 2006. SdrI, a serine-aspartate repeat protein identified in Staphylococcus saprophyticus strain 7108, is a collagen-binding protein. Infect. Immun. 74:4615–4623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakinç T, Kleine B, Michalski N, Kaase M, Gatermann SG. 2009. SdrI of Staphylococcus saprophyticus is a multifunctional protein: localization of the fibronectin-binding site. FEMS Microbiol. Lett. 301:28–34 [DOI] [PubMed] [Google Scholar]
- 17.Sakinc T, Woznowski M, Ebsen M, Gatermann SG. 2005. The surface-associated protein of Staphylococcus saprophyticus is a lipase. Infect. Immun. 73:6419–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakinç T, Kleine B, Gatermann SG. 2007. Biochemical characterization of the surface-associated lipase of Staphylococcus saprophyticus. FEMS Microbiol. Lett. 274:335–341 [DOI] [PubMed] [Google Scholar]
- 19.Kline KA, Ingersoll MA, Nielsen HV, Sakinc T, Henriques-Normark B, Gatermann S, Caparon MG, Hultgren SJ. 2010. Characterization of a novel murine model of Staphylococcus saprophyticus urinary tract infection reveals roles for Ssp and SdrI in virulence. Infect. Immun. 78:1943–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCrea KW, Hartford O, Davis S, Eidhin DN, Lina G, Speziale P, Foster TJ, Höök M. 2000. The serine-aspartate repeat (Sdr) protein family in Staphylococcus epidermidis. Microbiology 146:1535–1546 [DOI] [PubMed] [Google Scholar]
- 21.Grüter L, Endres M, Gatermann S. 1992. Cloning and expression of Staphylococcus epidermidis urease gene sequences in Staphylococcus carnosus. FEMS Microbiol. Lett. 93:33–35 [DOI] [PubMed] [Google Scholar]
- 22.Kuroda M, Yamashita A, Hirakawa H, Kumano M, Morikawa K, Higashide M, Maruyama A, Inose Y, Matoba K, Toh H, Kuhara S, Hattori M, Ohta T. 2005. Whole genome sequence of Staphylococcus saprophyticus reveals the pathogenesis of uncomplicated urinary tract infection. Proc. Natl. Acad. Sci. U. S. A. 102:13272–13277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roesch PL, Redford P, Batchelet S, Moritz RL, Pellett S, Haugen BJ, Blattner FR, Welch RA. 2003. Uropathogenic Escherichia coli use D-serine deaminase to modulate infection of the murine urinary tract. Mol. Microbiol. 49:55–67 [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, Nishikawa T, Satoh K, Iwata T, Fukushima T, Santa T, Homma H, Imai K. 1998. Urinary excretion of D-serine in human: comparison of different ages and species. Biol. Pharm. Bull. 21:156–162 [DOI] [PubMed] [Google Scholar]
- 25.Pätzold R, Schieber A, Brückner H. 2005. Gas chromatographic quantification of free D-amino acids in higher vertebrates. Biomed. Chromatogr. 19:466–473 [DOI] [PubMed] [Google Scholar]
- 26.Maas WK, Davis BD. 1950. Pantothenate studies. I. Interference by D-serine and L-aspartic acid with pantothenate synthesis in Escherichia coli. J. Bacteriol. 60:733–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grula MM, Grula EA. 1962. Cell division in a species of Erwinia. IV. Metabolic blocks in panothenate biosynthesis and their relationship to inhibition of cell division. J. Bacteriol. 83:989–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grula EA, Grula MM. 1963. Inhibition in synthesis of beta-alanine by D-serine. Biochim. Biophys. Acta 74:776–778 [DOI] [PubMed] [Google Scholar]
- 29.Cosloy SD, McFall E. 1973. Metabolism of D-serine in Escherichia coli K-12: mechanism of growth inhibition. J. Bacteriol. 114:685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bloom FR, McFall E. 1975. Isolation and characterization of D-serine deaminase constitutive mutants by utilization of D-serine as sole carbon or nitrogen source. J. Bacteriol. 121:1078–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McFall E. 1964. Genetic structure of the D-serine deaminase system of Escherichia coli. J. Mol. Biol. 9:746–753 [DOI] [PubMed] [Google Scholar]
- 32.Norregaard-Madsen M, McFall E, Valentin-Hansen P. 1995. Organization and transcriptional regulation of the Escherichia coli K-12 D-serine tolerance locus. J. Bacteriol. 177:6456–6461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatermann S, Marre R, Heesemann J, Henkel W. 1988. Hemagglutinating and adherence properties of Staphylococcus saprophyticus: epidemiology and virulence in experimental urinary tract infection of rats. FEMS Microbiol. Immunol. 1:179–185 [DOI] [PubMed] [Google Scholar]
- 34.Gatermann S, Meyer HW, Wanner G. 1992. Staphylococcus saprophyticus hemagglutinin is a 160-kilodalton surface polypeptide. Infect. Immun. 60:4127–4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gatermann S, Meyer H. 1994. Staphylococcus saprophyticus hemagglutinin binds fibronectin. Infect. Immun. 62:4556–4563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580 [DOI] [PubMed] [Google Scholar]
- 37.Brückner R. 1997. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol. Lett. 151:1–8 [DOI] [PubMed] [Google Scholar]
- 38.Brückner R. 1992. A series of shuttle vectors for Bacillus subtilis and Escherichia coli. Gene 122:187–192 [DOI] [PubMed] [Google Scholar]
- 39.Wieland B. 1993. Ph.D thesis University of Tübingen, Tübingen, Germany [Google Scholar]
- 40.Wagner E, Doskar J, Götz F. 1998. Physical and genetic map of the genome of Staphylococcus carnosus TM300. Microbiology 144:509–517 [DOI] [PubMed] [Google Scholar]
- 41.Marlinghaus L. 2011. Ph.D thesis Ruhr-University Bochum, Bochum, Germany [Google Scholar]
- 42.Augustin J, Götz F. 1990. Transformation of Staphylococcus epidermidis and other staphylococcal species with plasmid DNA by electroporation. FEMS Microbiol. Lett. 66:203–207 [DOI] [PubMed] [Google Scholar]
- 43.Rosen DA, Hung C-S, Kline KA, Hultgren SJ. 2008. Streptozocin-induced diabetic mouse model of urinary tract infection. Infect. Immun. 76:4290–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freter R, Allweiss B, O'Brien PCM, Halstead SA, Macsai MS. 1981. Role of chemotaxis in the association of motile bacteria with intestinal mucosa: in vitro studies. Infect. Immun. 34:241–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McFall E. 1975. Escherichia coli K-12 mutant forming a temperature-sensitive D-serine deaminase. J. Bacteriol. 121:1074–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Markwell MAK, Haas SM, Tolbert NE. 1978. Determination of the Lowry procedure to simplify protein in membrane and lipoprotein. Anal. Biochem. 87:206–210 [DOI] [PubMed] [Google Scholar]
- 47.Anfora AT, Haugen BJ, Roesch P, Redford P, Welch RA. 2007. Roles of serine accumulation and catabolism in the colonization of the murine urinary tract by Escherichia coli CFT073. Infect. Immun. 75:5298–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haugen BJ, Pellett S, Redford P, Hamilton HL, Roesch PL, Welch RA. 2007. In vivo gene expression analysis identifies genes required for enhanced colonization of the mouse urinary tract by uropathogenic Escherichia coli strain CFT073 dsdA. Infect. Immun. 75:278–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goerke C, Campana S, Bayer MG, Döring G, Botzenhart K, Wolz C. 2000. Direct quantitative transcript analysis of the agr regulon of Staphylococcus aureus during human infection in comparison to the expression profile in vitro. Infect. Immun. 68:1304–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolz C, Goerke C, Landmann R, Zimmerli W, Fluckiger U. 2002. Transcription of clumping factor A in attached and unattached Staphylococcus aureus in vitro and during device-related infection. Infect. Immun. 70:2758–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sakinç T, Michalski N, Kleine B, Gatermann SG. 2009. The uropathogenic species Staphylococcus saprophyticus tolerates a high concentration of D-serine. FEMS Microbiol. Lett. 299:60–64 [DOI] [PubMed] [Google Scholar]
- 53.Durham NN, Milligan R. 1962. A mechanism of growth inhibition by D-serine in a flavobacterium. Biochem. Biophys. Res. Commun. 11:342–345 [DOI] [PubMed] [Google Scholar]
- 54.Gribbon EM, Cunliffe WJ, Holland KT. 1993. Interaction of Propionibacterium acnes with skin lipids in vitro. J. Gen. Microbiol. 139:1745–1751 [DOI] [PubMed] [Google Scholar]
- 55.Longshaw CM, Farrell AM, Wright JD, Holland KT. 2000. Identification of a second lipase gene, gehD, in Staphylococcus epidermidis: comparison of sequence with those of other staphylococcal lipases. Microbiology 146:1419–1427 [DOI] [PubMed] [Google Scholar]