

Abstract
Background
Proton pump inhibitors (PPIs) are gastric acid suppressing agents widely prescribed for the treatment of gastro-esophageal reflux disease (GERD). Recently, several studies in patients with acute coronary syndrome (ACS) have raised the concern that use of PPIs in these patients may increase their risk of major adverse cardiovascular events (MACE). The mechanism of this possible adverse effect is not known. Whether the general population might also be at risk has not been addressed.
Methods and Results
Plasma ADMA is an endogenous inhibitor of nitric oxide synthase (NOS). Elevated plasma ADMA is associated with increased risk for cardiovascular disease, likely due to its attenuation of the vasoprotective effects of endothelial NOS. We find that PPIs elevate plasma asymmetric dimethylarginine (ADMA) level and reduce nitric oxide (NO) levels and endothelium-dependent vasodilation in a murine model and ex vivo human tissues. PPIs increase ADMA because they bind to, and inhibit dimethylarginine dimethylaminohydrolase (DDAH), the enzyme that degrades ADMA.
Conclusions
We present a plausible biological mechanism to explain the association of PPIs with increased MACE in patients with unstable coronary syndromes. Of concern, this adverse mechanism is also likely to extend to the general population using PPIs. This finding compels additional clinical investigations and pharmacovigilance directed toward understanding the cardiovascular risk associated with use of the PPIs in the general population.
Keywords: asymmetric dimethylarginine, cardiovascular risk, endothelium, nitric oxide, dimethylarginine dimethylaminohydrolase
BACKGROUND
Proton pump inhibitors (PPIs) are effective antagonists of gastric acid secretion, used to treat a number of gastro-esophageal disorders including dyspepsia, gastroesophageal reflux disease (GERD), Zollinger-Ellison syndrome, Barrett’s esophagus and Helicobacter pylori (H. pylori) infection of the upper gastrointestinal tract 1–3. Biologically, PPIs are administered as uncharged prodrugs and require activation by parietal cells of the stomach to form positively charged active (sulfenamide and sulfenic acid) drugs. In this form, the PPIs irreversibly bind to the gastric proton pump and inhibit acid secretion 4, 5. The high oral bioavailability of PPIs, and their efficacy in sustained suppression of gastric acid secretion, has favored their use over other acid-suppressing drugs such as the histamine-receptor (H2-receptor) antagonists. According to the U.S. Food and Drug Administration (FDA), in the US about 21 million people have used one or more prescription PPIs in 2009 6. Most PPIs are now available over-the-counter (OTC), increasing their general usage in the absence of medical supervision. In 2009, sales of PPIs grew to over $13 billion globally 7.
The PPIs are usually well-tolerated when used Intermittently in healthy subjects 3, 8 but may be associated with hypersecretion of gastric acid after their withdrawal 9. When used chronically, they may be associated with bone fracture and low levels of blood magnesium 6, 10. More worrisome are recent reports that PPIs may reduce the benefit of anti-platelet agents in patients with acute coronary syndromes (ACS) 11–14. Initial concern focused on the reduced benefit of clopidogrel in ACS patients taking PPI 11–14. This effect was attributed to the inhibition by PPIs of the hepatic enzyme (CYP2C19), which is required for activation of clopidogrel 12, 15. However, in ACS patients, the PPIs also diminish the benefit of ticagrelor 16, a drug which does not require hepatic activation. Furthermore, recent studies indicate that every member of the PPIs increase CV risk in ACS patients, despite the fact that some of these PPIs do not significantly inhibit CYP2C19 12, 14, 17–19. Accordingly, the mechanism by which the PPIs may increase risk of MACE in ACS patients is unknown. Furthermore, it is not known if the risk might extend to the larger population of ambulatory patients and consumers using PPIs. In this paper we report our finding that PPIs inhibit the activity of dimethylarginine dimethylaminohydrolase (DDAH), an enzyme necessary for cardiovascular health. DDAH metabolizes asymmetric dimethylarginine (ADMA); an endogenous and competitive inhibitor of nitric oxide (NO) synthase (NOS). By inhibiting endothelial NOS, ADMA would be anticipated to increase the risk of vascular inflammation and thrombosis, which may explain the increased risk of MACE in patients taking PPIs. Indeed, elevated plasma ADMA is a risk factor for cardiovascular morbidity and mortality in patients with cardiovascular disease, as well as for healthy individuals 20–26. Here, we provide molecular, cellular, ex-vivo and in vivo data demonstrating direct inhibition of DDAH activity by PPIs. These data compel additional clinical investigations and pharmacovigilance directed toward understanding the cardiovascular risk associated with use of the PPIs in the general population.
Materials and Methods
Molecular and Biochemical Studies
High throughput screening for DDAH inhibitors
Recombinant human DDAH1 (rhDDAH1) was generated and purified as previously described 27. A DDAH activity assay for high throughput chemical screening was used to screen a library of 130,000 small molecules in the Stanford High Throughput Bioscience Center (HTBC 27). Primary hits (inhibitors of DDAH activity) were validated using orthogonal biochemical assays (both colorimetric and fluorimetric activity assays) as we described 27. For the binding study described below, rhDDAH1 purification was modified to include HEPES buffer elution (containing 10mM HEPES in PBS) to avoid competing amine groups during protein coupling.
Enzyme-drug binding studies
To evaluate enzyme-drug binding, and to study the nature of the interaction, we used Surface Plasmon Resonance (SPR). First, rhDDAH1 protein was amine coupled to a CM5 sensor chip. A vehicle (DMSO) or PPI (omeprazole), dissolved in stock DMSO, were diluted in phosphate buffer (100mM Na2HPO4; pH 6.5). The affinity of the PPI to rhDDAH1 was monitored in real time by sensorgrams that reflect the binding of the compound with the coupled protein. The binding study was performed at 4 different biochemically relevant concentrations (12.5–100 μM in serial dilution). The study was performed using Biacore 3000 and the data were analyzed using the BIAevaluation software package.
Reversibility Study
To validate the PPI-DDAH interaction kinetics seen in the SPR study and to evaluate recovery of DDAH enzymatic activity upon inhibitor dilution, we performed reversibility study as described 28. In brief, DDAH (30 μM, at a 100-fold excess to the final concentration used in our enzymatic studies) was pre-incubated with omeprazole (100X, 10X or 1X the IC50 value; IC50 = ~ 60 μM). Inhibition of enzymatic activity and compound reversibility was determined by dilution using a fluorometric assay as described 27. For a reversible inhibitor that binds to a single site of an enzyme (1:1 stoichiometry), it is anticipated that inhibition can be saturated. In this study, a known DDAH1 inhibitor (L-257) 29 was used as a control.
ADMA and NO assays
The effect of the PPIs on ADMA metabolism in cells and in plasma was assessed using an ELISA assay as previously described 30. Production of NO by primary human microvascular endothelial cells (HMVECs) and by human saphenous vein segments (SV) in the presence or absence of the PPI omeprazole was assessed using the standard Griess assay for nitrogen oxides (NOx; Assay Designs, Ann Arbor, MI).
The Effect of PPIs in Human Endothelial Cells
Human microvascular endothelial cells (HMEC-1 31) were plated on cell culture flasks in serum-free Dulbecco’s Modified Eagle Medium (DMEM) and treated with vehicle or PPIs (esomeprazole or lansoprazole at 20 μM final compound concentration), or a known DDAH inhibitor (L-257 29) for 3 hours before switching to fully-supplemented DMEM (supplemented with 10% FBS, 4mM HEPES; and penicillin/streptomycin; pH 7.6) at 37°C/5%CO2 until 24 hours. The cells were then washed, lysed, and the total cell lysate recovered for estimation of protein concentration using the Coomassie Plus protein assay 27 for ADMA measurement. Similarly, intracellular NO level was also measured from cell lysates of primary microvascular endothelial cells using the Griess assay. In addition, the lysate (20 μg each) was also used to study the protein expression of DDAH1 (Abcam, Cambridge, MA), DDAH2 (Abcam), endothelial NOS (eNOS; BD Biosciences, San Jose, CA) and phosphorylated eNOS (peNOS; Cell Signaling, Danvers, MA) 27.
In some studies, NOx were measured in the conditioned medium from human saphenous vein segments harvested during coronary artery bypass graft (CABG) surgery 32. The vessel segment was rinsed in Ringer’s lactated solution (LRS) and then dissected into 0.5 cm sections prior to transferring the pieces into each well of a 12-well plate containing endothelial cell media. The vessels were either kept under basal conditions or stimulated with the calcium ionophore A23187 (0.5 μM) for 24 hours in the presence of omeprazole (3 to 100 μM), L-257 (100 μM) or vehicle. Release of NO into the conditioned media was measured using the Griess assay.
The effect of PPI on vascular reactivity ex-vivo
The effect of omeprazole on vascular reactivity was studied by isolating mouse thoracic aorta for functional studies as described 33. In brief, aorta was isolated from 10-week old C57BL/6J mice and the rings were mounted in myograph at a resting tension. Vessels that manifested reproducible relaxation in response to acetylcholine (ACh) were exposed to omeprazole or vehicle for 24 hours prior to contraction with phenylephrine (PE; 10−12 to 10−4 M) and relaxation with ACh (10−8 to 10−4 M) or sodium nitroprusside (SNP; 10−12 to 10−5 M). Response curves to PE (Force), ACh or SNP (% Relaxation) were plotted.
The effect of a PPI in vivo
The effect of a typical PPI, lansoprazole, on serum ADMA levels was investigated in male C57BL/6J mice (20 weeks old; Jackson Laboratories; Bar Harbor, Maine). Animals were randomized to receive lansoprazole (30 mg/kg/day lansoprazole; n=8) or vehicle (0.5% carboxy-methylcellulose [CMC] n=8); by subcutaneous injection daily for 5 weeks 34. Whole blood was collected by tail clipping at baseline and weekly for ADMA measurement. At sacrifice, the blood sample was collected by cardiac puncture. Serum was separated by centrifugation of the whole blood at 6,000 rpm for 20 min at 4°C for measurement of ADMA as described above. The respective animal studies described above were approved by the Imperial College London and Stanford University Administrative Panel on Laboratory Animal Care.
Statistical Methods
The number of animals needed in each study group was calculated using power and sample size calculation (PS v3.0.14; Vanderbilt University). The in vivo experiment was designed to detect a difference in the experimental and control means (δ) of 0.27 with an estimated standard deviation (σ) of 0.18 at a significance level (α) of 0.05 with 80% power (β). Unless stated otherwise, all other statistical tests described in the study were performed using GraphPad Prism V5 (La Jolla, CA). Data analysis was performed using one-way ANOVA followed by Bonferroni posthoc correction. Unpaired student’s t-test was used when comparing two groups. Statistical significance was noted at p value < 0.05.
Results
High throughput screen identifies PPIs as DDAH inhibitors
We screened approximately 130,000 small molecules in the Stanford HTBC to search for modulators of DDAH activity. The enzymatic activity of DDAH was monitored using colorimetric and fluorometric assays as described 27. This screen identified about 200 small molecules that inhibited DDAH by more than 30%. We were surprised to find amongst our hits four members of the PPI class (omeprazole, pantoprazole, lansoprazole and tenatoprazole). Subsequently, these positive hits and additional members of the class (esomeprazole and rabeprazole) were validated using freshly prepared compounds and orthogonal assays as follows.
PPIs directly inhibit human DDAH1 activity
Using a microplate assay, the enzymatic activity of DDAH was monitored biochemically 27. In this assay, ADMA degradation by DDAH was examined by detecting the product (L-citrulline). In brief, rhDDAH1 was mixed with ADMA in 384-well format and L-citrulline formation was quantified after incubating the enzyme-substrate mix with the PPIs and adding color developing reagent 27. The inhibitory activity of each of the PPIs was confirmed using a full-dose range of the agents. From these data we calculated the half-maximal concentration (IC50) of each agent as shown in Table-1. These studies validated that the direct inhibition of DDAH by the PPIs (Figure-1) was a class effect (Figure-2A). These results were further confirmed using an orthogonal fluorometric assay 27 (Figure-2B).
Table 1.
Determination of the IC50 of PPIs. The half-maximal concentration (IC50) of each of the PPIs was determined using an 8 point concentration range (0.78–100 μM). Data is Mean ± SEM from triplicate values per group.
| Compound | IC50 (μM) |
|---|---|
| Lansoprazole | 51 ± 3 |
| Esomeprazole | 52 ± 5 |
| Rabeprazole | 53 ± 5 |
| Omeprazole | 58 ± 5 |
| Tenatoprazole | 61 ± 1.5 |
| Pantoprazole | 63 ± 5 |
Figure 1.

The ADMA pathway. Asymmetric dimethylarginine (ADMA) is derived from proteins (largely nuclear) containing methylated arginine residues. ADMA is largely (80%) metabolized by dimethylarginine dimethylaminohydrolase (DDAH). ADMA is a competitive inhibitor of nitric oxide synthase (NOS). Endothelial NOS (eNOS) is highly regulated, and produces small amounts of NO locally to effect vascular homeostasis. Increased levels of ADMA (such as through possible inhibition by the PPIs) could impair eNOS activity, reducing NO generation while increasing superoxide anion generation. The vasoprotective action of eNOS is lost, increasing the risk for adverse vascular events. In this setting, inflammatory cells are attracted into the vessel wall, and express inducible NOS (iNOS), which generates superoxide anion and nitric oxide, which combine to form the cytotoxic free radical peroxynitrite anion.
Figure 2.

Proton pump inhibitors (PPIs) inhibit DDAH activity. A) Colorimetric assay showing reduced production of L-citrulline from ADMA. B) Fluorimetric assay showing inhibited signal associated with DDAH enzymatic activity. In A) L-citrulline conc. was calculated from standard curve. In B) ebselen was used as a positive assay control 27. Data is from triplicate experiments (Mean ± SEM) at 50 μM final compound concentration. *p<0.05 when the PPIs are compared to the vehicle control by One-Way ANOVA followed by Bonferroni posttest correction.
PPIs bind to purified human DDAH1 reversibly
The SPR study showed that omeprazole, but not vehicle, generated sensorgram signals indicating a direct interaction between the PPI and DDAH (Figure-3). As expected, the vehicle control, serially diluted DMSO, did not show binding. Moreover, in the enzymatic studies, we found rapid and almost complete reversibility of omeprazole inhibition of DDAH enzymatic activity when serially diluted (Figure-S1). These data are consistent with the SPR study indicating that the PPIs are likely reversible inhibitors of human DDAH1. Meanwhile, the selective and competitive DDAH1 inhibitor L-257 35 also showed complete reversibility upon dilution.
Figure 3.

The PPI omeprazole binds to DDAH. Surface plasmon resonance (SPR) sensorgram data indicating the interaction between omeprazole and human DDAH1. DDAH was coupled to a chip and omeprazole or vehicle was passed over the chip. Binding is shown by the peaks in the sensorgrams at the different concentrations tested (Green = 12.5 μM; Pink = 25 μM; Blue = 50 μM and Purple = 100 μM). Data is representative of duplicate experiments per group.
PPIs increase intracellular ADMA concentration
We next studied the effect of PPIs (esomeprazole and lansoprazole) on intracellular ADMA in human endothelial cells. This study demonstrated that the PPIs increased intracellular ADMA (by ~ 30%) compared to vehicle control. L-257 also increased ADMA as expected (Figure-4A). Notably, this effect of the PPIs was in the absence of any changes in DDAH expression. In brief, HMVECs were exposed to different concentration of the PPI omeprazole (3–100 μM) for 24 hours, and the protein expression of DDAH1 and DDAH2 was examined by Western blot as described 27. In this study, omeprazole neither regulated the expression of DDAH1 nor that of DDAH2 (Figure-S2). These data suggest that PPIs are able to increase intracellular levels of ADMA in endothelial cells, most likely by inhibiting DDAH activity (but not expression).
Figure 4.
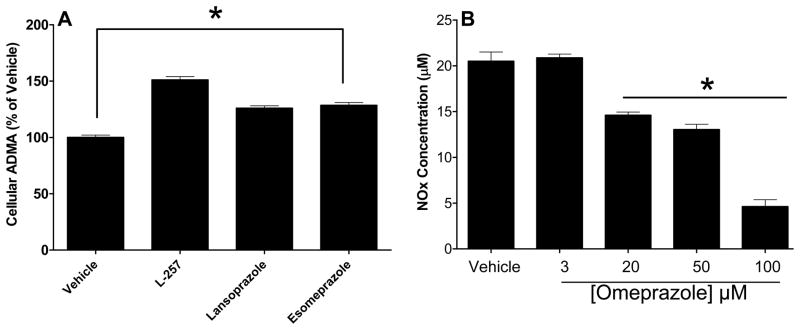
PPIs increase ADMA concentration and reduce NO levels in human endothelial cells. A) Human microvascular endothelial cells (HMVECs) were treated with the indicated small molecules at 20 μM final conc each. DMSO (solvent used to prepare the compounds) was used as a vehicle control. The intracellular [ADMA] was determined after 24 hours. B) The effect of omeprazole on total NO level was assessed by treating HMVECs with vehicle or various concentration of omeprazole for 24 hours. Total nitrite (NOx) was measured from lysates using Griess reaction and was normalized to total protein concentration. Data is Mean ± SEM from duplicate experiments. L-257 is a selective DDAH1 inhibitor and was included as a control. *p<0.05 when each PPI was compared to the vehicle group using unpaired student t-tests.
PPIs reduce intracellular NO level
An increase in cellular ADMA would be expected to reduce the activity of NO synthase. Indeed, omeprazole dose-dependently reduced the levels of nitrogen oxides (NOx) in cultured endothelial cells (Figure-4B). In addition, we found that the expression of total endothelial NOS (eNOS) and active eNOS (phospho-eNOS) in HMVECs was downregulated by omeprazole (Figure-5). Similarly, we found that omeprazole (and L-257) significantly inhibited NOx released from isolated human saphenous veins, in the presence or absence of an activator of NO synthase (Figure-6A and Figure-6B). These data are consistent with a recent report that high levels of omeprazole reduced serum NO levels in an animal model of colorectal cancer 36.
Figure 5.

The PPI omeprazole reduces the expression of total and active endothelial NOS. Regulation of total endothelial NOS (eNOS) and active eNOS (phospho-eNOS) by omeprazole was studied by Western blot using endothelial cell (EC) lysate exposed to omeprazole or vehicle. ECs were treated with VEGF (50 ng/mL) as positive eNOS phosphorylation control. The expression of each protein was normalized to β-actin (ACTB).
Figure 6.

The effect of PPI (Omeprazole) on nitric oxide production of human saphenous vein grafts (SVGs). SVGs were treated with vehicle or omeprazole (3–100 μM) for 24 hours A) at baseline level or B) upon stimulation with the calcium ionophore A23187 (0.5 μM) to increase NO production. Total nitrite (NOx) was measured in the conditioned medium using Griess reaction. Data is Mean ± SEM from duplicate experiments. *p<0.05 when the PPIs are compared to the vehicle control by One-Way ANOVA followed by Bonferroni posttest correction.
The PPI omeprazole impairs vascular function
Omeprazole impaired vascular reactivity in a manner that was consistent with a reduction in NO synthase activity. The PPI enhanced the contraction to phenylephrine (PE); blunted the relaxation to acetylcholine (ACh); but did not effect the (endothelium independent) vasorelaxation to sodium nitroprusside (Figure-7). These findings are consistent with a reduction in endothelial NOS activity, due to accumulation of ADMA 33.
Figure 7.

Omeprazole impairs vascular reactivity in response to the pharmacological agents acetylcholine (ACh) and phenylephrine (PE) on isolated mouse aorta. Concentration response curve to A) PE (10−8 to 10−4 M) and B) ACh (10−12 to 10−4 M) are shown. In C) the percent relaxation in response to the endothelium-independent Sodium Nitroprusside (SNP; 10−12 to 10−5 M) is shown. Drug concentrations in each data set were log transformed and the PE concentration response curve (in Figure A) is expressed as force (mN) applied to the transducer of the myograph for each dose of the compound added. The ACh and SNP induced relaxation (in Figure B and C) is expressed in the concentration response curve as a percentage of the contraction to the 80% of the maximal contraction reached with PE. Differences in best-fit values of selected parameters (force, % relaxation) were compared between the PPI and vehicle groups (n=6 vessels at each drug conc. in each group) using extra sum-of-squares F test and the data are expressed as Mean ± SEM in each panel.
Lansoprazole increases circulating level of ADMA in vivo
We also studied the effect of lansoprazole (LPZ) on serum ADMA levels in mice. As early as one week after LPZ administration, we observed an increase in serum ADMA. This increase was sustained throughout the 5-week study period, an increase of about 20% in the LPZ-treated group compared to the vehicle control (Figure-8). It is known that rodent DDAH shares over 90% homology to the human isoforms 37 and therefore the increase in circulating ADMA is likely mediated by an inhibition of DDAH activity in vivo.
Figure 8.

PPI treatment increases plasma ADMA. Lansoprazole (LPZ) treatment caused a sustained and significant increase in serum ADMA levels (*p<0.05) in mice (n=8 animals per group). Data are from duplicate experiments (Mean ± SEM) at 5-weeks post-treatment.
Discussion
We find that proton pump inhibitors (PPIs) as a class directly bind to, and inhibit the activity of DDAH, the enzyme that degrades ADMA. This effect of the PPIs explains our subsequent observation that PPIs increase ADMA concentration in cultured human endothelial cells, in association with a reduction in NO synthesis. Similarly, the PPI omeprazole reduced NO generated by human saphenous vein segments ex vivo. In addition, PPIs impaired endothelium-dependent vasodilation in isolated murine vessels. Furthermore, lansoprazole 34 administered by subcutaneous injection increased serum ADMA levels in mice by about 20%.
ADMA is an emerging risk factor for cardiovascular (CV) events 20–26. Accordingly, an increase in plasma ADMA induced by PPIs may potentially explain the association of PPIs with cardiovascular events in patients with unstable coronary syndromes (ACS). Of perhaps greater concern, an elevation of plasma ADMA of this magnitude, if the data is translated to humans, might increase the hazard ratio for major adverse cardiovascular events (MACE) and mortality in adults not recognized to have cardiovascular disease. Our study in human endothelial cells showed an increase in ADMA levels by about 30%; an elevation that is reported to significantly increase MACE in humans. However, the dose we used (20 μM) is 5–10 fold higher than the plasma concentration obtained with a typical oral dose of PPI. For example, a single oral dose of 30 mg lansoprazole would produce a maximum plasma concentration (Cmax) of 2 – 6 μM within about 3 hours 38. Neverhtheless, repeated dosing of PPIs to attain consistent suppression of gastric acidity 19 could impair normal vascular endothelial function.
ADMA as a risk factor for cardiovascular disease
Indeed, previous studies have reported that a modest increase in plasma ADMA is associated with an increased risk for MACE and mortality in patients with cardiovascular disease, as well as healthy ambulatory individuals 20–23, 25, 26. In patients with coronary or peripheral arterial disease, individuals in the upper tertile of plasma ADMA are more likely to incur MACE, and have reduced longevity. In the AtheroGene study, ADMA (but not C-reactive peptide; CRP) was predictive of MACE 39. Plasma ADMA is also a risk factor for the general population, as indicated by longitudinal community-based studies. In the Population Study of Women in Gothenburg, the top quintile of ADMA (≥0.71 μM) was associated with a relative risk of 1.75, after adjusting for traditional CV risk factors, renal function, and homocysteine 40. In this study, an increase of plasma ADMA of 0.15 μM (about 20–30%) increased MACE by 30% over 24 years. Similar results were observed in the Framingham Offspring Study 25. Thus the ADMA elevation that we observed in normal mice treated with PPI, is of a magnitude that would significantly increase cardiovascular risk in a human. The PPI-induced elevation in a patient may even be larger, if there is loss of “DDAH reserve” due to the vascular oxidative stress of metabolic perturbations as we have previously described 24. We have initiated a clinical study to determine the effect of PPIs on ADMA levels and endothelial function in healthy subjects and those with cardiovascular disease to directly address these questions.
Mechanisms of ADMA elevation
ADMA is derived from the hydrolysis of proteins containing methylated arginine residues (Figure 1). Subsequently, about 80% of ADMA is degraded by DDAH, and the remainder is excreted in the urine. Individuals with renal insufficiency have elevated plasma ADMA levels, and the magnitude of this elevation is correlated with low estimated glomerular filtration rate (eGFR) and is a predictor for MACE and mortality in individuals with chronic kidney disease 41–44. However, it appears that the most common cause of plasma ADMA elevation is an impairment of DDAH activity. Because it contains a sulfhydryl moiety in its catalytic pocket 45–49, DDAH is highly sensitive to oxidative stress 37. Metabolic perturbations such as hyperlipidemia, hyperglycemia, and hyperhomocysteinemia increase endothelial oxidative stress, impair endothelial DDAH activity, impairing its degradation of ADMA 49–54. Endothelial and systemic levels of ADMA increase, and contribute to impairment in endothelial NOS. The impairment of endothelial NOS is associated with an increase in oxidative stress, and a dysregulation of vasomotor tone 55. Furthermore, given the anti-inflammatory and anti-platelet effects of endothelial-derived NO 56, 57, the impairment of endothelial NOS would be anticipated to increase the risk of MACE, as suggested by studies of coronary and brachial artery vasoreactivity 58. To be sure, iNOS activation in the vessel wall may play a role in the pathophysiology of atherosclerosis, and ADMA would inhibit the pathological activity of this enzyme. Nevertheless, the aggregate effect of an increase in plasma ADMA appears to increase CV risk.
In addition to acquired impairment of DDAH activity, there is emerging evidence for genetic deficiency of the pathway. In the Kuopio Ischemic Heart Disease Risk Factor Study (KIHD), Finnish men with a functional polymorphism of the DDAH1 gene had elevated plasma ADMA and a 50-fold increased risk for coronary heart disease (CHD) as well as a 5-fold increase in the prevalence of hypertension compared to noncarriers 59. Furthermore, retrospective analyses of clinical samples from stroke and CHD patients identified functional genetic polymorphism in DDAH1 promoter resulting in ~40% reduction in the transcriptional activity of DDAH in endothelial cells and subsequent elevation in plasma ADMA (by ~ 30–40%) compared to controls. This loss-of-function has been associated with significantly increased risk of stroke and coronary artery disease (by about 40% each) even after adjusting for traditional risk factors 60. In addition, DDAH polymorphisms have been correlated to ADMA levels in diabetic patients 61 and to susceptibility to preeclampsia 62. Although encouraging, these small studies require confirmation in genome wide association (GWAS) studies.
Proposed mechanism for DDAH inhibition by PPIs
The action of PPIs is dependent on a covalent and irreversible inhibition of the proton (H+/K+ ATPases) pump of parietal cells in the stomach. The PPIs become positively charged (sulfenic acid derivatives) upon interaction with gastric acid in the stomach and covalently and irreversibly bind to active-site cysteines. Interestingly, the biochemical enzymatic assays and cell culture studies in the present study were conducted at nearly physiological pH (6.0 to 7.6). The DDAH inhibitory activity at this non-acidic pH indicates that the PPIs do not necessarily need to be converted to an “activated form”, as seen during the inhibition of gastric pumps, to interfere with the DDAH pathway. DDAH possess a highly conserved catalytic triad containing a critical cysteine (Cys) residue (Cys273 in DDAH1 and Cys276 in DDAH2 37). The reactive Cys residue is crucial in the metabolism of ADMA, forming a transient covalent bond with the carbon in the guanidino residue of the substrate 45. Site-directed mutagenesis study revealed that substitution of the catalytic Cys by alanine abolishes the catalytic activity of the enzyme 48. Because our Biacore and inhibitor-dilution study show rapid and nearly complete reversibility of the PPIs and subsequent recovery of DDAH enzymatic activity, it seems likely that the interaction of DDAH with a PPI is non-covalent. However, increasing substrate concentration of the reaction appears to influence the PPIs inhibitory activity suggesting that although reversible, their mode of inhibition might still be through interaction with an active site of DDAH1 but likely apart from Cys273. To understand the precise mechanism of interaction, we are resolving the structure of DDAH co-crystallized with a PPI.
Role of DDAH inhibition in the potential adverse cardiovascular effects of PPIs
Our proposed biological mechanism for the association between PPIs and MACE is more consistent with the available human data than previously proposed drug-drug interactions. Although several of the PPIs may inhibit the hepatic enzyme (CYP2C19) which activates clopidogrel, other antiplatelet agents not dependent on such activation (eg. ticagrelor) also manifest diminished efficacy when combined with a PPI, even after adjustment for confounding effects 16. Furthermore, it is unlikely that the PPI-induced change in gastric pH is impairing absorption or action of antiplatelet agents as a similar reduction in intragastric pH is achieved with the H2-receptor antagonists without increased CV risk 14, 17.
Thus a PPI-induced impairment of DDAH activity, with subsequent dysregulation of vascular NOS, may be a more likely explanation for the association with MACE and mortality. The current report may heighten the concerns of the FDA regarding the possible association of PPIs with MACE 63. Consistent with this hypothesis, it is worth noting that drugs that reduce circulating levels of ADMA such as angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs) and insulin-sensitizers 24, 64–67 are associated with a reduction in CV risk.
Finally, given the experimental findings, we recently employed a novel data-mining approach to examine the risk of MI in patients with GERD treated with either a PPI or an H2-receptor antagonist, independent of clopidogrel use. These results (replicated in two different datasets) support the hypothesis that PPI use may pose an independent and enhanced risk for myocardial infarction (MI) in the general population (LePendu et al, under review).
Conclusion
In summary, we provide biochemical, cellular, ex-vivo and in vivo data revealing that commonly prescribed PPIs directly interact with and significantly inhibit human DDAH activity, thereby increasing endothelial and serum ADMA levels. The increase in ADMA levels would be anticipated to impair vascular NOS activity, increase oxidative stress, reduce vasodilator function, and impair vasoprotective mechanisms. Such disruption of vascular homeostasis may explain the increased MACE and mortality associated with the prolonged use of PPIs in large clinical trials of patients with ACS 68, 69. Of concern is the effect a chronic elevation of ADMA levels may have on the general population using PPIs. Taken together, our pre-clinical and epidemiological observations raise serious concerns that should be actively and urgently explored so as to delineate the potential cardiovascular risk associated with use of the PPIs in the general population. However, it is important to recognize that the present study cannot establish a cause-and-effect relationship between PPI use and elevation of CV risk in ACS patients or the general population. This is rather a hypothesis-generating observation that warrants further prospective investigation.
Supplementary Material
Acknowledgments
The authors are grateful to Dr James Leiper (Imperial College London) for kindly providing L-257 for the in vitro studies and Dr Michael Eckart for performing the SPR study and analyzing the data. We would also like to thank Dr David Solow-Cordero and Jason Wu of the Stanford high throughput Center for their technical assistance during our high throughput screening effort to discover small molecule modulators of DDAH.
Funding Sources: This work was supported in part by grants to JPC from the NIH (RC2HL103400, 1U01HL100397, and K12HL087746), American Heart Association (AHA) (11IRG5180026), Stanford SPARK Translational Research Program, Stanford Translational Research and Applied Medicine (TRAM) Program and by the Tobacco-Related Disease Research Program of the University of California (18XT-0098). YTG was a recipient of the Stanford School of Medicine Dean’s fellowship (1049528-149- KAVFB) and is currently supported by the Tobacco-Related Disease Research Program (TRDRP) of the University of California (20FT-0090). NHS and PL acknowledge support in part by NIH grant U54HG004028 from the National Center for Biomedical Ontology, seed funding by the Department of Medicine at Stanford University and the Stanford Center for Biomedical Informatics Research. NHS also acknowledges support from U54LM008748 for Informatics for Integrating Biology and the Bedside, and Research Gift Support from Apixio, Inc.
Footnotes
Conflict of Interest Disclosures: JPC and YTG are inventors on patents owned by Stanford University that protect the use of agents that modulate the NOS/DDAH pathway therapeutically.
References
- 1.Berardi RR, Welage LS. Proton-pump inhibitors in acid-related diseases. Am J Health Syst Pharm. 1998;55:2289–2298. doi: 10.1093/ajhp/55.21.2289. [DOI] [PubMed] [Google Scholar]
- 2.Welage LS, Berardi RR. Evaluation of omeprazole, lansoprazole, pantoprazole, and rabeprazole in the treatment of acid-related diseases. J Am Pharm Assoc (Wash) 2000;40:52–62. doi: 10.1016/s1086-5802(16)31036-1. [DOI] [PubMed] [Google Scholar]
- 3.Stedman CA, Barclay ML. Review article: comparison of the pharmacokinetics, acid suppression and efficacy of proton pump inhibitors. Aliment Pharmacol Ther. 2000;14:963–978. doi: 10.1046/j.1365-2036.2000.00788.x. [DOI] [PubMed] [Google Scholar]
- 4.Shin JM, Homerin M, Domagala F, Ficheux H, Sachs G. Characterization of the inhibitory activity of tenatoprazole on the gastric H+,K+-ATPase in vitro and in vivo. Biochem Pharmacol. 2006;71:837–849. doi: 10.1016/j.bcp.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 5.Shin JM, Munson K, Vagin O, Sachs G. The gastric HK-ATPase: structure, function, and inhibition. Pflugers Arch. 2009;457:609–622. doi: 10.1007/s00424-008-0495-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.U.S. Food and Drug Administration (FDA) FDA Drug Safety Podcast for Healthcare Professionals: Low magnesium levels can be associated with long-term use of Proton Pump Inhibitor drugs (PPIs) 2009 http://www.fda.gov/Drugs/DrugSafety/DrugSafetyPodcasts/ucm245455.htm.
- 7.Madanick RD. Proton pump inhibitor side effects and drug interactions: much ado about nothing? Cleve Clin J Med. 2011;78:39–49. doi: 10.3949/ccjm.77a.10087. [DOI] [PubMed] [Google Scholar]
- 8.Simpson IJ, Marshall MR, Pilmore H, Manley P, Williams L, Thein H, Voss D. Proton pump inhibitors and acute interstitial nephritis: report and analysis of 15 cases. Nephrology (Carlton) 2006;11:381–385. doi: 10.1111/j.1440-1797.2006.00651.x. [DOI] [PubMed] [Google Scholar]
- 9.Reimer C, Sondergaard B, Hilsted L, Bytzer P. Proton-pump inhibitor therapy induces acid-related symptoms in healthy volunteers after withdrawal of therapy. Gastroenterology. 2009;137:80–87. doi: 10.1053/j.gastro.2009.03.058. [DOI] [PubMed] [Google Scholar]
- 10.U.S. Food and Drug Administration (FDA) Proton Pump Inhibitors (PPI): Class Labeling Change. 2011 http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm213321.htm.
- 11.Ho PM, Maddox TM, Wang L, Fihn SD, Jesse RL, Peterson ED, Rumsfeld JS. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. Jama. 2009;301:937–944. doi: 10.1001/jama.2009.261. [DOI] [PubMed] [Google Scholar]
- 12.Juurlink DN, Gomes T, Ko DT, Szmitko PE, Austin PC, Tu JV, Henry DA, Kopp A, Mamdani MM. A population-based study of the drug interaction between proton pump inhibitors and clopidogrel. Cmaj. 2009;180:713–718. doi: 10.1503/cmaj.082001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilard M, Arnaud B, Cornily JC, Le Gal G, Lacut K, Le Calvez G, Mansourati J, Mottier D, Abgrall JF, Boschat J. Influence of omeprazole on the antiplatelet action of clopidogrel associated with aspirin: the randomized, double-blind OCLA (Omeprazole CLopidogrel Aspirin) study. J Am Coll Cardiol. 2008;51:256–260. doi: 10.1016/j.jacc.2007.06.064. [DOI] [PubMed] [Google Scholar]
- 14.Charlot M, Grove EL, Hansen PR, Olesen JB, Ahlehoff O, Selmer C, Lindhardsen J, Madsen JK, Kober L, Torp-Pedersen C, Gislason GH. Proton pump inhibitor use and risk of adverse cardiovascular events in aspirin treated patients with first time myocardial infarction: nationwide propensity score matched study. Bmj. 2011;342:d2690. doi: 10.1136/bmj.d2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li XQ, Andersson TB, Ahlstrom M, Weidolf L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab Dispos. 2004;32:821–827. doi: 10.1124/dmd.32.8.821. [DOI] [PubMed] [Google Scholar]
- 16.Goodman SG, Clare R, Pieper KS, Nicolau JC, Storey RF, Cantor WJ, Mahaffey KW, Angiolillo DJ, Husted S, Cannon CP, James SK, Kilhamn J, Steg PG, Harrington RA, Wallentin L. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation. 2012;125:978–986. doi: 10.1161/CIRCULATIONAHA.111.032912. [DOI] [PubMed] [Google Scholar]
- 17.Charlot M, Ahlehoff O, Norgaard ML, Jorgensen CH, Sorensen R, Abildstrom SZ, Hansen PR, Madsen JK, Kober L, Torp-Pedersen C, Gislason G. Proton-pump inhibitors are associated with increased cardiovascular risk independent of clopidogrel use: a nationwide cohort study. Ann Intern Med. 2010;153:378–386. doi: 10.7326/0003-4819-153-6-201009210-00005. [DOI] [PubMed] [Google Scholar]
- 18.Martin de Argila C. Safety of potent gastric acid inhibition. Drugs. 2005;65:97–104. doi: 10.2165/00003495-200565001-00013. [DOI] [PubMed] [Google Scholar]
- 19.Der G. An overview of proton pump inhibitors. Gastroenterol Nurs. 2003;26:182–190. doi: 10.1097/00001610-200309000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Mittermayer F, Krzyzanowska K, Exner M, Mlekusch W, Amighi J, Sabeti S, Minar E, Muller M, Wolzt M, Schillinger M. Asymmetric dimethylarginine predicts major adverse cardiovascular events in patients with advanced peripheral artery disease. Arterioscler Thromb Vasc Biol. 2006;26:2536–2540. doi: 10.1161/01.ATV.0000242801.38419.48. [DOI] [PubMed] [Google Scholar]
- 21.Wilson AM, Shin DS, Weatherby C, Harada RK, Ng MK, Nair N, Kielstein J, Cooke JP. Asymmetric dimethylarginine correlates with measures of disease severity, major adverse cardiovascular events and all-cause mortality in patients with peripheral arterial disease. Vasc Med. 2010;15:267–274. doi: 10.1177/1358863X10364552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu TM, Chung MY, Lin MW, Hsu CP, Lin SJ. Plasma asymmetric dimethylarginine predicts death and major adverse cardiovascular events in individuals referred for coronary angiography. Int J Cardiol. 2011;153:135–140. doi: 10.1016/j.ijcard.2011.06.120. [DOI] [PubMed] [Google Scholar]
- 23.Ari H, Ari S, Erdogan E, Tiryakioglu O, Ustundag Y, Huysal K, Koca V, Bozat T. A novel predictor of restenosis and adverse cardiac events: asymmetric dimethylarginine. Heart Vessels. 2010;25:19–26. doi: 10.1007/s00380-009-1158-x. [DOI] [PubMed] [Google Scholar]
- 24.Cooke JP. Asymmetrical dimethylarginine: the Uber marker? Circulation. 2004;109:1813–1818. doi: 10.1161/01.CIR.0000126823.07732.D5. [DOI] [PubMed] [Google Scholar]
- 25.Boger RH, Sullivan LM, Schwedhelm E, Wang TJ, Maas R, Benjamin EJ, Schulze F, Xanthakis V, Benndorf RA, Vasan RS. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation. 2009;119:1592–1600. doi: 10.1161/CIRCULATIONAHA.108.838268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wanby P, Teerlink T, Brudin L, Brattstrom L, Nilsson I, Palmqvist P, Carlsson M. Asymmetric dimethylarginine (ADMA) as a risk marker for stroke and TIA in a Swedish population. Atherosclerosis. 2006;185:271–277. doi: 10.1016/j.atherosclerosis.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 27.Ghebremariam YT, Erlanson DA, Yamada K, Cooke JP. Development of a Dimethylarginine Dimethylaminohydrolase (DDAH) Assay for High-Throughput Chemical Screening. J Biomol Screen. 2012;17:651–661. doi: 10.1177/1087057112441521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Copeland RA, Basavapathruni A, Moyer M, Scott MP. Impact of enzyme concentration and residence time on apparent activity recovery in jump dilution analysis. Anal Biochem. 2011;416:206–210. doi: 10.1016/j.ab.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 29.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O’Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13:198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 30.Schulze F, Wesemann R, Schwedhelm E, Sydow K, Albsmeier J, Cooke JP, Boger RH. Determination of asymmetric dimethylarginine (ADMA) using a novel ELISA assay. Clin Chem Lab Med. 2004;42:1377–1383. doi: 10.1515/CCLM.2004.257. [DOI] [PubMed] [Google Scholar]
- 31.Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 32.Tsui JC, Souza DS, Filbey D, Karlsson MG, Dashwood MR. Localization of nitric oxide synthase in saphenous vein grafts harvested with a novel “no-touch” technique: potential role of nitric oxide contribution to improved early graft patency rates. J Vasc Surg. 2002;35:356–362. doi: 10.1067/mva.2002.121072. [DOI] [PubMed] [Google Scholar]
- 33.Torondel B, Nandi M, Kelly P, Wojciak-Stothard B, Fleming I, Leiper J. Adenoviral-mediated overexpression of DDAH improves vascular tone regulation. Vasc Med. 2010;15:205–213. doi: 10.1177/1358863X09360264. [DOI] [PubMed] [Google Scholar]
- 34.Matsuzaki J, Suzuki H, Minegishi Y, Sugai E, Tsugawa H, Yasui M, Hibi T. Acid suppression by proton pump inhibitors enhances aquaporin-4 and KCNQ1 expression in gastric fundic parietal cells in mouse. Dig Dis Sci. 2010;55:3339–3348. doi: 10.1007/s10620-010-1167-8. [DOI] [PubMed] [Google Scholar]
- 35.Nandi M, Kelly P, Torondel B, Wang Z, Starr A, Ma Y, Cunningham P, Stidwill R, Leiper J. Genetic and Pharmacological Inhibition of Dimethylarginine Dimethylaminohydrolase 1 Is Protective in Endotoxic Shock. Arterioscler Thromb Vasc Biol. 2012;32:2589–2597. doi: 10.1161/ATVBAHA.112.300232. [DOI] [PubMed] [Google Scholar]
- 36.Kim YJ, Lee JS, Hong KS, Chung JW, Kim JH, Hahm KB. Novel application of proton pump inhibitor for the prevention of colitis-induced colorectal carcinogenesis beyond acid suppression. Cancer Prev Res (Phila) 2010;3:963–974. doi: 10.1158/1940-6207.CAPR-10-0033. [DOI] [PubMed] [Google Scholar]
- 37.Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol. 2007;293:H3227–3245. doi: 10.1152/ajpheart.00998.2007. [DOI] [PubMed] [Google Scholar]
- 38.Hu YR, Qiao HL, Kan QC. Pharmacokinetics of lansoprazole in Chinese healthy subjects in relation to CYP2C19 genotypes. Acta Pharmacol Sin. 2004;25:986–990. [PubMed] [Google Scholar]
- 39.Schnabel R, Blankenberg S, Lubos E, Lackner KJ, Rupprecht HJ, Espinola-Klein C, Jachmann N, Post F, Peetz D, Bickel C, Cambien F, Tiret L, Munzel T. Asymmetric dimethylarginine and the risk of cardiovascular events and death in patients with coronary artery disease: results from the AtheroGene Study. Circ Res. 2005;97:e53–59. doi: 10.1161/01.RES.0000181286.44222.61. [DOI] [PubMed] [Google Scholar]
- 40.Leong T, Zylberstein D, Graham I, Lissner L, Ward D, Fogarty J, Bengtsson C, Bjorkelund C, Thelle D. Asymmetric dimethylarginine independently predicts fatal and nonfatal myocardial infarction and stroke in women: 24-year follow-up of the population study of women in Gothenburg. Arterioscler Thromb Vasc Biol. 2008;28:961–967. doi: 10.1161/ATVBAHA.107.156596. [DOI] [PubMed] [Google Scholar]
- 41.Lu TM, Chung MY, Lin CC, Hsu CP, Lin SJ. Asymmetric dimethylarginine and clinical outcomes in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6:1566–1572. doi: 10.2215/CJN.08490910. [DOI] [PubMed] [Google Scholar]
- 42.Zoccali C, Bode-Boger S, Mallamaci F, Benedetto F, Tripepi G, Malatino L, Cataliotti A, Bellanuova I, Fermo I, Frolich J, Boger R. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet. 2001;358:2113–2117. doi: 10.1016/s0140-6736(01)07217-8. [DOI] [PubMed] [Google Scholar]
- 43.Zoccali C, Benedetto FA, Maas R, Mallamaci F, Tripepi G, Malatino LS, Boger R. Asymmetric dimethylarginine, C-reactive protein, and carotid intima-media thickness in end-stage renal disease. J Am Soc Nephrol. 2002;13:490–496. doi: 10.1681/ASN.V132490. [DOI] [PubMed] [Google Scholar]
- 44.Zoccali C, Mallamaci F, Maas R, Benedetto FA, Tripepi G, Malatino LS, Cataliotti A, Bellanuova I, Boger R. Left ventricular hypertrophy, cardiac remodeling and asymmetric dimethylarginine (ADMA) in hemodialysis patients. Kidney Int. 2002;62:339–345. doi: 10.1046/j.1523-1755.2002.00437.x. [DOI] [PubMed] [Google Scholar]
- 45.Murray-Rust J, Leiper J, McAlister M, Phelan J, Tilley S, Santa Maria J, Vallance P, McDonald N. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat Struct Biol. 2001;8:679–683. doi: 10.1038/90387. [DOI] [PubMed] [Google Scholar]
- 46.Leiper J, Murray-Rust J, McDonald N, Vallance P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc Natl Acad Sci U S A. 2002;99:13527–13532. doi: 10.1073/pnas.212269799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frey D, Braun O, Briand C, Vasak M, Grutter MG. Structure of the mammalian NOS regulator dimethylarginine dimethylaminohydrolase: A basis for the design of specific inhibitors. Structure. 2006;14:901–911. doi: 10.1016/j.str.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 48.Hong L, Fast W. Inhibition of human dimethylarginine dimethylaminohydrolase-1 by S-nitroso-L-homocysteine and hydrogen peroxide. Analysis, quantification, and implications for hyperhomocysteinemia. J Biol Chem. 2007;282:34684–34692. doi: 10.1074/jbc.M707231200. [DOI] [PubMed] [Google Scholar]
- 49.Stuhlinger MC, Tsao PS, Her JH, Kimoto M, Balint RF, Cooke JP. Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine. Circulation. 2001;104:2569–2575. doi: 10.1161/hc4601.098514. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Sim AS, Wang XL, Salonikas C, Naidoo D, Wilcken DE. Relations between plasma asymmetric dimethylarginine (ADMA) and risk factors for coronary disease. Atherosclerosis. 2006;184:383–388. doi: 10.1016/j.atherosclerosis.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 51.Stuhlinger MC, Stanger O. Asymmetric dimethyl-L-arginine (ADMA): a possible link between homocyst(e)ine and endothelial dysfunction. Curr Drug Metab. 2005;6:3–14. doi: 10.2174/1389200052997393. [DOI] [PubMed] [Google Scholar]
- 52.Boger RH, Sydow K, Borlak J, Thum T, Lenzen H, Schubert B, Tsikas D, Bode-Boger SM. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: involvement of S-adenosylmethionine-dependent methyltransferases. Circ Res. 2000;87:99–105. doi: 10.1161/01.res.87.2.99. [DOI] [PubMed] [Google Scholar]
- 53.Abbasi F, Asagmi T, Cooke JP, Lamendola C, McLaughlin T, Reaven GM, Stuehlinger M, Tsao PS. Plasma concentrations of asymmetric dimethylarginine are increased in patients with type 2 diabetes mellitus. Am J Cardiol. 2001;88:1201–1203. doi: 10.1016/s0002-9149(01)02063-x. [DOI] [PubMed] [Google Scholar]
- 54.Altinova AE, Arslan M, Sepici-Dincel A, Akturk M, Altan N, Toruner FB. Uncomplicated type 1 diabetes is associated with increased asymmetric dimethylarginine concentrations. J Clin Endocrinol Metab. 2007;92:1881–1885. doi: 10.1210/jc.2006-2643. [DOI] [PubMed] [Google Scholar]
- 55.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 56.Yao SK, Ober JC, Krishnaswami A, Ferguson JJ, Anderson HV, Golino P, Buja LM, Willerson JT. Endogenous nitric oxide protects against platelet aggregation and cyclic flow variations in stenosed and endothelium-injured arteries. Circulation. 1992;86:1302–1309. doi: 10.1161/01.cir.86.4.1302. [DOI] [PubMed] [Google Scholar]
- 57.Cooke JP, Tsao PS. Cytoprotective effects of nitric oxide. Circulation. 1993;88:2451–2454. doi: 10.1161/01.cir.88.5.2451. [DOI] [PubMed] [Google Scholar]
- 58.Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101:1899–1906. doi: 10.1161/01.cir.101.16.1899. [DOI] [PubMed] [Google Scholar]
- 59.Valkonen VP, Tuomainen TP, Laaksonen R. DDAH gene and cardiovascular risk. Vasc Med. 2005;10:S45–48. doi: 10.1191/1358863x05vm600oa. [DOI] [PubMed] [Google Scholar]
- 60.Ding H, Wu B, Wang H, Lu Z, Yan J, Wang X, Shaffer JR, Hui R, Wang DW. A novel loss-of-function DDAH1 promoter polymorphism is associated with increased susceptibility to thrombosis stroke and coronary heart disease. Circ Res. 2010;106:1145–1152. doi: 10.1161/CIRCRESAHA.109.215616. [DOI] [PubMed] [Google Scholar]
- 61.Abhary S, Burdon KP, Kuot A, Javadiyan S, Whiting MJ, Kasmeridis N, Petrovsky N, Craig JE. Sequence variation in DDAH1 and DDAH2 genes is strongly and additively associated with serum ADMA concentrations in individuals with type 2 diabetes. PLoS One. 2010;5:e9462. doi: 10.1371/journal.pone.0009462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akbar F, Heinonen S, Pirskanen M, Uimari P, Tuomainen TP, Salonen JT. Haplotypic association of DDAH1 with susceptibility to pre-eclampsia. Mol Hum Reprod. 2005;11:73–77. doi: 10.1093/molehr/gah116. [DOI] [PubMed] [Google Scholar]
- 63.U.S. Food and Drug Administration (FDA) Update of Safety Review - Follow-up to the August 9, 2007, Communication about the Ongoing Safety Review of Omeprazole and Esomeprazole. 2007 http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm143258.htm.
- 64.Chen JW, Hsu NW, Wu TC, Lin SJ, Chang MS. Long-term angiotensin-converting enzyme inhibition reduces plasma asymmetric dimethylarginine and improves endothelial nitric oxide bioavailability and coronary microvascular function in patients with syndrome X. Am J Cardiol. 2002;90:974–982. doi: 10.1016/s0002-9149(02)02664-4. [DOI] [PubMed] [Google Scholar]
- 65.Sakurada M, Shichiri M, Imamura M, Azuma H, Hirata Y. Nitric oxide upregulates dimethylarginine dimethylaminohydrolase-2 via cyclic GMP induction in endothelial cells. Hypertension. 2008;52:903–909. doi: 10.1161/HYPERTENSIONAHA.108.114207. [DOI] [PubMed] [Google Scholar]
- 66.Yazici D, Yavuz DG, Unsalan S, Toprak A, Yuksel M, Deyneli O, Aydin H, Tezcan H, Rollas S, Akalin S. Temporal effects of low-dose ACE inhibition on endothelial function in Type 1 diabetic patients. J Endocrinol Invest. 2007;30:726–733. doi: 10.1007/BF03350809. [DOI] [PubMed] [Google Scholar]
- 67.Ozgurtas T, Oktenli C, Dede M, Tapan S, Kenar L, Sanisoglu SY, Yesilova Z, Yenen MC, Erbil MK, Baser I. Metformin and oral contraceptive treatments reduced circulating asymmetric dimethylarginine (ADMA) levels in patients with polycystic ovary syndrome (PCOS) Atherosclerosis. 2008;200:336–344. doi: 10.1016/j.atherosclerosis.2007.12.054. [DOI] [PubMed] [Google Scholar]
- 68.Kwok CS, Jeevanantham V, Dawn B, Loke YK. No consistent evidence of differential cardiovascular risk amongst proton-pump inhibitors when used with clopidogrel: Meta-analysis. Int J Cardiol. 2012 doi: 10.1016/j.ijcard.2012.03.085. http://www.ncbi.nlm.nih.gov/pubmed/22464478. [DOI] [PubMed]
- 69.Maggio M, Corsonello A, Ceda GP, Cattabiani C, Lauretani F, Butto V, Ferrucci L, Bandinelli S, Abbatecola AM, Spazzafumo L, Lattanzio F. Proton pump inhibitors and risk of 1-year mortality and rehospitalization in older patients discharged from acute care hospitals. JAMA Intern Med. 2013;173:518–523. doi: 10.1001/jamainternmed.2013.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.