

Abstract
Influenza virus is a worldwide global health concern causing seasonal morbidity mortality and economic burden. Chemotherapeutics is available; however, rapid emergence of drug-resistant influenza virus strains has reduced its efficacy. Thus, there is a need to discover novel antiviral agents. In this study, RNA interference (RNAi) was used to screen host genes required for influenza virus replication. One pro-influenza virus host gene identified was dual-specificity phosphatase cell division cycle 25 B (CDC25B). RNAi screening of CDC25B resulted in reduced influenza A virus replication, and a CDC25B small-molecule inhibitor (NSC95397) inhibited influenza A virus replication in a dose-dependent fashion. Viral RNA synthesis was reduced by NSC95397 in favor of increased beta interferon (IFN-β) expression, and NSC95397 was found to interfere with nuclear localization and chromatin association of NS1, an influenza virus protein. As NS1 has been shown to be chromatin associated and to suppress host transcription, it is likely that CDC25B supports NS1 nuclear function to hijack host transcription machinery in favor of viral RNA synthesis, a process that is blocked by NSC95397. Importantly, NSC95397 treatment protects mice against lethal influenza virus challenge. The findings establish CDC25B as a pro-influenza A virus host factor that may be targeted as a novel influenza A therapeutic strategy.
INTRODUCTION
Influenza A virus (IAV) causes yearly epidemics and periodic pandemics in humans with recent estimates that 20% of the world population is affected yearly (1). IAV undergoes antigenic drift, a feature requiring the need for new vaccines to be developed annually to confer protection against heterovariant strains. Periodically, multiple IAV strains simultaneously infect a “mixing vessel,” such as swine, leading to viruses with new gene segments and an antigenic shift that may cause a pandemic. Although IAV vaccines are generally safe and effective, they cannot always meet the population coverage demands, and due to the short time frame between identification of a pandemic strain and need for vaccination, they are not always available or efficacious. An option to control influenza virus infection in influenza-afflicted or at-risk people is the use of antiviral drugs. Current FDA-approved antiviral drugs are the M2-ion channel inhibitor adamantanes and the neuraminidase inhibitors zanamivir and oseltamivir (2–4). Despite the utility of these antiviral drugs, new and novel antivirals are being sought due to the development of drug resistance (5–9). Several reports have demonstrated a host factor requirement for influenza A virus replication (10–15). Thus, targeting host genes offers an innovative and refractory approach to drug resistance because IAV requires host gene pathways for replication (11, 15), and host gene targets are stable. Several host factors have been previously identified to promote IAV replication in different stages of the virus life cycle. Among these, organic anion transporter 3 (OAT3) and vacuolar ATPase (vATPase) have recently been shown to facilitate IAV entry into host cells, while other host factors such as importin-α and calcium/calmodulin-dependent protein kinase II β (CAMK2B) have postentry roles (15–17). IAV also utilizes host factors, such as cellular P58IPK, which has been implicated in inhibition of the host double-stranded RNA (dsRNA)-dependent protein kinase R (PKR) response, to modulate antiviral responses (18). Numerous host genes have been identified as potential drug targets for blocking key events required for influenza virus replication in host cells using a genome-wide small interfering RNA (siRNA) assay platform (13–16, 19, 20). Among the provirus genes identified was cell division cycle 25 B (CDC25B), which is a member of the CDC25 family of phosphatases. CDC25B dephosphorylates cyclin-dependent kinases (CDKs) and regulates the cell division cycle. Removal of inhibitory phosphates from threonine and tyrosine residues on CDK1 and CDK2 allow these factors to promote cell cycle progression from G2 to mitosis (21).
IAV is a single-strand negative-sense segmented RNA virus whose genome includes nucleoproteins (NP) and a viral polymerase complex (PA, PB1, and PB2) that is enclosed in a host-derived envelope containing hemagglutinin (HA), neuraminidase (NA), and M2 ion channel proteins to facilitate viral binding and fusion for subsequent release of the negative-stranded viral RNA (vRNA) genome into the cells (reviewed in references 22 and 23). Other viral proteins such as matrix protein (M1) and nuclear export protein (NEP) are incorporated into the virion (24). Nonstructural protein, NS1, is not incorporated into the virion but is required for efficient virus replication. NS1 protein is known to antagonize host antiviral interferon (IFN) responses during IAV infection by inhibiting functions of retinoic acid inducible gene I (RIG-I), PKR, and 2′5′-oligoadenylate synthetase (OAS) (25–34). NS1 also has been implicated in inhibition of host mRNA maturation and nuclear export and promotion of translation of viral mRNA (28, 29, 31, 35, 36). A recent study also demonstrated strain-specific NS1 targeting to host chromatin, specifically, to transcriptionally active loci (e.g., viral inducible gene), by mimicking the histone tail to block active host transcription elongation (37). NS1 protein is shuttled between cytoplasmic and nuclear compartments throughout infection to facilitate these actions (17). Another recent study described NS1 phosphorylation by CDK1 and extracellular signal-regulated kinase 1/2 (ERK1/2) (38), both direct downstream targets of CDC25B, and showed that this phosphorylation is required for NS1 nuclear localization (38).
In this study, NSC95397, a specific inhibitor of CDC25B phosphatase (39, 40), was evaluated for its ability to modulate IAV infection in human bronchial epithelial (BEAS2B) cells. It was shown that NSC95397 limits IAV replication in a dose-dependent manner and acts to block CDC25B-mediated NS1 function in the nucleus by limiting viral RNA synthesis while upregulating IFN-β expression. The effect of this compound was also tested in a mouse model of IAV infection. The results show that NSC95397 inhibition of CDC25B phosphatase activity effectively controls IAV replication in the lungs of IAV-infected mice and protects mice from lethal IAV infection, suggesting that targeting CDC25B phosphatase could be a valuable therapeutic approach to treat IAV infection.
MATERIALS AND METHODS
Cells and viruses.
Human type II respiratory epithelial (A549) cells (ATCC CCL-185) and Madin-Darby canine kidney (MDCK) cells (ATCC CCL-34) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% heat-inactivated fetal bovine serum (FBS) (HyClone) in a 37°C incubator with 5% CO2. Human bronchoepithelial (BEAS2B) cells (ATCC CRL-9609) were maintained in bronchial epithelial basal medium (BEBM; Lonza) supplemented with 30 μg/ml bovine pituitary extract, 0.5 μg/ml hydrocortisone, 0.5 ng/ml human recombinant epidermal growth factor, 0.5 μg/ml epinephrine, 10 μg/ml transferrin, 5 μg/ml insulin, 0.1 ng/ml retinoic acid, and 6.5 ng/ml triiodothyronine (BEGM SingleQuots; Lonza) at 37°C in a 5% CO2 incubator. Representative strains of influenza A virus, A/WSN/33(H1N1) (WSN), and influenza B virus, B/Florida/04/06, were propagated in 9-day-old embryonated chicken eggs obtained from a specific-pathogen-free leghorn chicken flock (Sunrise Farms). The allantoic fluid from this parental stock was tested for hemagglutin (HA) activity. HA-positive allantoic fluids were pooled, divided into aliquots, and stored at −80°C until use. A mutant influenza A/WSN/33 virus expressing defective RNA-binding NS1 (NS1 R38AK41A) was a kind gift from Adolfo García-Sastre (Mount Sinai School of Medicine) and was passaged once in MDCK cells. Mutations were confirmed by sequence analysis. MDCK cells were used to determine the titer of the A/WSN/33 stock virus and culture supernatant of infected cells as previously described (41, 42).
RNAi transfection.
A primary RNA interference (RNAi) screen was performed using four pooled siRNAs to target each gene of the 4,795 genes in the human drug target library (SMARTpool; Dharmacon ThermoFisher) using A549 cells infected with influenza A/WSN/33 virus as previously described (15, 20). For a CDC25B validation study, individual siRNAs targeting human CDC25B and a nontargeting siRNA (siNEG) were used (Dharmacon ThermoFisher). A549 cells were reverse transfected with siRNA using DharmaFECT-1 reagent (Dharmacon) as previously described (20). Transfections were carried out for 48 h to allow maximal expression knockdown before cells were infected with influenza A/WSN/33 virus at an a multiplicity of infection (MOI) of 0.001. The level of infectious virus was measured 48 h postinfection (hpi) by titration of A549 cell supernatants on MDCK cells (43). In addition, A549 cell monolayers on culture plates were fixed and analyzed for the presence of influenza NP by immunofluorescence staining as described below. Transfected cells were also collected to assess CDC25B gene expression knockdown using the quantitative real-time PCR (qRT-PCR) method described below.
In vitro inhibition assays.
NSC95697 (2,3-bis-[2-hydroxyethylsulfanyl]-[1,4]naphthoquinone) (TOCRIS Bioscience) was dissolved in dimethyl sulfoxide (DMSO) and serially diluted in BEBM. For dose-response virus inhibition experiments, cells were washed with phosphate-buffered saline (PBS) once prior to titration of NSC95607 using a Hewlett-Packard (HP) D300 Digital Dispenser (Tecan) (44) 1 h before infection. For time-of-addition experiments, 2 μM NSC95397 was added at different time points pre- or postinfection. Where indicated, cells were subsequently infected with influenza A/WSN/33 virus at MOI = 0.05 (multistep growth) or 1 (single-step growth). At the indicated time points, cells were fixed with 4% formaldehyde for subsequent immunostaining, collected for total RNA isolation using a Qiagen RNAeasy kit (Qiagen) for gene expression analyses, or collected for protein analyses using immunoblotting. Furthermore, culture supernatant was collected for IAV titration in MDCK cells and cytotoxic analysis. Cellular toxicity was determined by measuring adenylate kinase release using a ToxiLight Bioassay kit (Lonza).
Gene expression analyses.
For measurement of influenza A viral copy number, total RNAs collected from infected A549 cells, BEAS2B cells, or lungs of infected mice were used for a quantitative real-time PCR (qRT-PCR) assay using a OneStep RT-PCR kit (Qiagen). A universal influenza primer-probe set (InfA forward primer, InfA reverse primer, and InfA probe; Bioresearch Technologies, Inc.) was used for amplification and detection of influenza A virus RNA as previously described (16).
For strand-specific IAV qRT-PCR analyses, primers specific for IAV segment 5 cRNA, mRNA, and vRNA containing an additional 18-to-20-nucleotide tag unrelated to IAV at the 5′ end were used for increased specificity to distinguish the three different IAV RNA species as described previously (45). Briefly, equal amounts of total RNA from infected cells were used to synthesize cDNA complementary to the three types of IAV RNA using a Verso cDNA synthesis kit (Thermo Scientific). Quantitative PCR analysis was performed using RT2 SYBR green qPCR Master Mix (SABioscience) and primer sets specific to the corresponding IAV RNA species in an MX3005P thermocycler. To assess CDC25B and IFN-β gene expression, cDNA were synthesized using random hexamers as primers (Thermo Scientific). cDNAs were subsequently used for quantitative PCR amplifications using CDC25B, IFN-β, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene-specific primers and RT2 SYBR green qPCR Master Mix (SABioscience) in an MX3005P thermocycler as previously described (16). The levels of viral RNA, CDC25B, and IFN-β gene expression were normalized to that of GAPDH, and their expression levels relative to mock-treated samples were calculated using the 2(−ΔΔCt) formula.
Immunofluorescence staining.
Cells were fixed with 4% formaldehyde for 10 min, blocked in 3% bovine serum albumin (BSA), and incubated with the primary antibodies mouse anti-NP and rabbit anti-NS1 (Pierce antibodies; Thermo Scientific), followed by incubation with the appropriate secondary antibodies Alexa 488-conjugated goat anti-mouse and Alexa 546-conjugated goat anti-rabbit (1 μg/ml; Invitrogen) and with DAPI (4′,6-diamidino-2-phenylindole) counterstain (2 μg/ml; Invitrogen). Cells were visualized using an EVOS fluorescent imaging system (Advanced Microscopy Group). For high-content imaging, cells were visualized and counted using a Cellomics ArrayScan system (Thermo Scientific) with proprietary image and analytical software.
Protein isolation, subcellular fractionation, and immunoblot analysis.
To evaluate total protein expression following NSC95397 treatment, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris HCl [pH 7.5], 150 mM NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 1 mM EDTA, and 0.1% sodium dodecyl sulfate [SDS]) supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Scientific), followed by 4°C centrifugation at 16,000 × g for 10 min to clarify lysates. For protein fractionation experiments, cell pellet were subjected to subcellular fractionation (Pierce/Thermo Scientific) to biochemically isolate cytoplasmic, soluble nuclear, and chromatin-bound proteins. Briefly, for isolation of chromatin proteins, nonsoluble nuclear pellets were digested with micrococcal nuclease, supplied in the manufacturer's kit, to digest chromatin and extract chromatin-bound proteins.
Equivalent protein amounts were diluted in SDS sample buffer (for 4× buffer, 40% glycerol, 240 mM Tris/HCl [pH 6.8], 8% SDS, 0.04% bromophenol blue, 5% β-mercaptoethanol), boiled, and resolved by SDS-polyacrylamide gel electrophoresis followed by immunoblotting. The primary antibodies used for immunoblot analyses were mouse anti-NP, rabbit anti-NS1 (Pierce antibodies; Thermo Scientific), mouse anti-NS1 (Santa Cruz Biotechnology), mouse anti-CDK1 and rabbit anti-CDK1 pY15 (Abcam), rabbit anti-ERK1/2, ERK1/2 pT202/Y204, interferon regulatory factor 3 (IRF3) pS396, and histone H3 (Cell Signaling Technology), mouse anti-IRF3 (a kind gift from Michael Gale, Jr., University of Washington) (46), and rabbit anti-GAPDH (Millipore). Horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse antibodies (Sigma-Aldrich) were used as secondary antibodies. Protein bands were visualized following addition of SuperSignal West Dura Extended Duration substrate (Pierce/Thermo Scientific) and using a FluorChem-E Western imaging system (ProteinSimple).
Mice and infections.
BALB/c female mice (8 to 10 weeks old) were obtained from NCI. All experiments and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Georgia. Mice were treated with DMSO or NSC95397 orally by gavage at 24 h pre- or postinfection. Prior to virus inoculation, mice were anesthetized with Avertin and intranasally infected with a lethal (103 PFU) or sublethal (70 PFU) dose of influenza A/WSN/33 virus–50 μl PBS. Body weight and survival were evaluated daily for 14 days. For assessment of lung viral burden, lungs of infected mice were collected at 72 hpi and homogenized in TRizol (Invitrogen) for total RNA isolation. Five nanograms of total RNA was used for assessment of IAV copy number by qRT-PCR analysis as described above. Experiments were performed with 10 mice per group.
Statistical analyses.
The Student t test was used throughout, except for the survival curves where one-way analysis of variance (ANOVA) and the Mantel-Cox test were employed. P values < 0.05 are considered significant.
RESULTS
CDC25B is a pro-influenza virus host factor.
The RNAi screening that was performed identified numerous host cell genes that are required for IAV replication (16, 19, 20). One of the genes identified was CDC25B, a member of the dual-specificity CDC25 phosphatases that has been shown to dephosphorylate CDK1 and ERK1/2 (21, 47). To validate this finding, individual nonpooled siRNAs targeting CDC25B (siCDC25B) were used for transfection of A549 cells 48 h prior to infection with influenza A/WSN/33 virus (MOI = 0.05). siCDC25B transfection resulted in a reduction of influenza A virus NP-positive cell numbers at 48 hpi compared to nontargeting siRNA (siNEG) transfection as visualized and quantified by high-content imaging (Fig. 1A; quantified in the bottom panel). Similarly, siCDC25B-transfected cells also had significant (P < 0.05) reductions of influenza A/WSN/33 virus titers in culture supernatant collected at 48 hpi compared to siNEG-transfected cells (Fig. 1B). To verify knockdown of CDC25B gene expression following siRNA transfection, mock- or A/WSN/33-infected, siRNA-transfected cells were collected for total RNA isolation and qRT-PCR analysis. A549 cells transfected with siCDC25B showed an 80% or 45% reduction of CDC25B relative to GAPDH mRNA levels for mock-infected cells (P < 0.01) or A/WSN/33-infected cells (P < 0.05), respectively, compared to siNEG-transfected, mock-infected cells (Fig. 1C). Interestingly, influenza A/WSN/33 virus infection resulted in upregulation of CDC25B gene expression, further indicating a role for virus replication.
Fig 1.

CDC25B is a pro-influenza A virus host factor. To evaluate the role of CDC25B during IAV infection, A549 cells were transfected with nontargeting siRNA (siNEG) or siRNA targeting CDC25B (siCDC25B). (A and B) At 48 h posttransfection, cells were infected with influenza A/WSN/33 virus at MOI = 0.05 and fixed (A) or harvested (B) for RNA isolation at 48 hpi. (A) Following fixation, cells were stained for influenza virus NP and nuclei (DAPI). Cells were visualized using a high-content imaging system. Three representative images for each condition are shown (top), and percentages of NP-positive cells were quantified from 10 fields (bottom). (B) Culture supernatants from infected cells were used for virus titration in MDCK cells. (C) Knockdown of CDC25B mRNA expression was verified using qRT-PCR normalized to GAPDH. *, P < 0.05; **, P < 0.01.
NSC95397 modulates CDC25B phosphatase activity during IAV infection.
To further determine if CDC25B is a viable target to limit IAV replication, a selective and irreversible inhibitor of CDC25B phosphatase, i.e., NSC95397, was evaluated (39, 40, 48). CDC25B has been found to be overexpressed in carcinoma cells (49, 50); thus, nonneoplastic human bronchoepithelial BEAS2B cells were employed in the subsequent studies. To demonstrate that NSC95397 inhibits CDC25B activity in the context of virus infection, levels of phosphorylated CDK1 and ERK1/2, both known targets of CDC25B phosphatase activity, were evaluated in mock- or influenza A/WSN/33 virus-infected BEAS2B cells in the presence or absence of NSC95397. BEAS2B cells were pretreated with DMSO or increasing doses of NSC95397 for 1 h prior to mock or IAV infection at an MOI = 1, and protein lysates were collected at 8 hpi for immunoblot analyses (Fig. 2A). Mock- or IAV-infected cells pretreated with NSC95397 displayed higher levels of phosphorylated CDK1 and ERK1/2 proteins in a dose-dependent fashion. Levels of total CDK1 and ERK1/2, as detected by immunoblot assays, appeared to be diminished as levels of phosphorylated CDK1 and ERK1/2 increased. This is presumably due to a reduced affinity of CDK1 and ERK1/2 antibodies for phosphorylated CDK1 and ERK1/2, respectively. Consistent with Fig. 1C, the CDC25B protein level was upregulated in IAV-infected cells (Fig. 2A, lane 5) compared to mock-infected cells (lane 1) in the absence of treatment. However, the CDC25B protein level was reduced at high concentrations of NSC95397 regardless of infection status (lanes 3 and 4 and lanes 7 and 8). Importantly, NSC95397 treatment was able to reduce the level of influenza NP protein in a dose-dependent manner (lanes 5 to 8), with 71.8% and 81.5% reductions of NP expression for 1 μM and 5 μM NSC95397, respectively.
Fig 2.
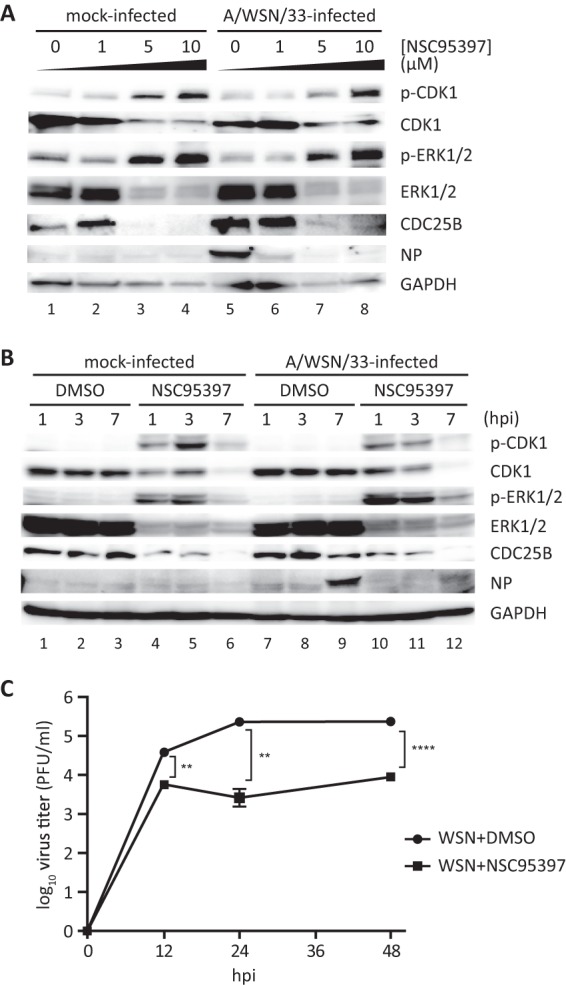
NSC95397 treatment resulted in increased levels of phosphorylated CDK1 and ERK1/2. (A) BEAS2B cells were treated with DMSO (0 μM) or increasing doses of NSC95397 (1, 5, or 10 μM). At 1 h posttreatment, cells were mock infected or infected with influenza A/WSN/33 virus at MOI = 1. Cells were harvested for protein analyses at 7 hpi. (B and C) Cells were treated with DMSO or 5 μM NSC95397. At 1 h posttreatment, cells were mock infected or infected with influenza A/WSN/33 virus at MOI = 1 (B) or MOI = 0.05 (C). (B) Cells were harvested for protein analyses at 1, 3, or 7 hpi. To determine effects of NSC95397 treatment in dephosphorylation of CDC25B targets CDK1 and ERK1, protein lysates were subjected to immunoblot analysis using phosphospecific and total CDK1 and ERK1/2 antibodies. Levels of cellular CDC25B, GAPDH, and viral NP proteins were also evaluated. (C) At 12, 24, and 48 hpi, culture supernatants were collected and titrated on MDCK cells. **, P < 0.01; ****, P < 0.0001.
To further evaluate the kinetic of NSC95397 activity toward CDC25B during IAV infection, BEAS2B cells were pretreated with DMSO or 5 μM NSC95397 1 h prior to mock or influenza A/WSN/33 virus infection at MOI = 1 (Fig. 2B). Protein lysates were collected for immunoblotting at 1, 3, or 7 hpi. Levels of phosphorylated CDK1 and ERK1/2 were readily observable at 2 h following NSC95397 treatment (1 hpi) in both mock- or IAV-infected cells and diminished by 8 h posttreatment (7 hpi) (Fig. 2B, lanes 4 to 6 and lanes 10 to 12) but not in DMSO-treated cells (lanes 1 to 3 and lanes 7 to 9). Cells treated with NSC95397 maintained a CDC25B protein level for up to 4 h posttreatment, but the level diminished by 8 h (lanes 4 to 6 and lanes 10 to 12). It is important that the increase in CDK1 and ERK1/2 phosphorylation preceded the downregulation of the CDC25B protein level, suggesting that NSC95397 acted to inhibit the activity of CDC25B to dephosphorylate CDK1 and ERK1/2. However, this modulation of CDC25B by NSC95397 would ultimately result in reduction of CDC25B protein abundance, presumably by affecting its stability. Importantly, while NP protein can be detected at 8 hpi in DMSO-treated cells (lane 9), NSC95397 treatment resulted in diminished NP expression (lane 12), in agreement with the finding represented in Fig. 2A, demonstrating the ability of NSC95397 to limit IAV growth as indicated by viral protein expression.
To further evaluate NSC95397 inhibition of the IAV growth kinetic, BEAS2B cells were treated with DMSO or 5 μM NSC95397 prior to infection with influenza A/WSN/33 virus at MOI = 0.05 (Fig. 2C). Culture supernatants were collected at 12, 24, and 48 hpi for virus titration by plaque assays. Cells pretreated with NSC95397 displayed a significant reduction of virus titers compared to DMSO-treated cells at 12, 24, and 48 hpi (P < 0.01, P < 0.01, and P < 0.005, respectively). Together, these findings demonstrate that inhibition of CDC25B activity by NSC95397 ultimately led to a reduction of IAV growth and replication.
NSC95397 limits IAV replication in BEAS2B cells.
To further determine if CDC25B is a druggable target for inhibiting influenza virus replication, the efficacy of NSC95397 against influenza A and B viruses was evaluated (Fig. 3). In agreement with the findings represented in Fig. 2, BEAS2B cells pretreated for 1 h with NSC95397 and subsequently infected with A/WSN/33 (MOI = 0.05) displayed reduced virus titers at 24 hpi (Fig. 3A). NSC95397 was able to limit influenza A/WSN/33 virus replication in BEAS2B cells in a dose-dependent manner, with a 50% inhibitory concentration (IC50) of 5.73 μM (Fig. 3B). Similarly, NSC95397 treatment was also able to reduce influenza B/Florida/04/06 virus titers in a dose-dependent manner, with an IC50 of 8.41 μM (Fig. 3C and D). NSC95397-treated BEAS2B cells displayed minimal cytotoxicity up to 302 μM, where 50% cellular cytotoxicity (CC50) was yet to be observed (CC50 > 302 μM; Fig. 3E). These results demonstrated the efficacy of NSC95397 against representative strains of both influenza A and B viruses, with selectivity indices (S.I.) of >52.7 and >35.9, respectively (Fig. 3F). Taken together, these results further confirmed that CDC25B has a proviral role during IAV replication and that its inhibition by RNAi or by a small-molecule inhibitor limits IAV infection in vitro.
Fig 3.

CDC25B inhibitor NSC95397 limits influenza A and B infections in vitro. (A to D) BEAS2B cells were infected with influenza A/WSN/33 virus (A and B) or B/Florida/04/06 virus (C and D) at MOI = 0.05 in the presence of increasing NSC95397 concentrations (in 1/4-log increments). At 24 hpi, cell supernatants were harvested for virus titration in MDCK cells. Dotted lines indicate 50% inhibition of virus titer (IC50). TCID50, 50% tissue culture infective dose. (E) BEAS2B cells were treated with increasing doses of NSC95397 (in 1/2-log increments), and cellular cytotoxicity was assessed at 24 h posttreatment using a ToxiLight bioassay kit. Percent cell viability of NSC95397-treated cells was determined relative to nontreated cells (100% viability) and lysed cells (0% viability). The dotted line indicates 50% cytotoxicity (CC50). (D) IC50 and CC50 values were determined using the nonlinear regression method. Selectivity indices were calculated as the ratio of CC50 to IC50.
NSC95397 inhibits IAV RNA synthesis and promotes type I IFN expression.
To identify a mechanism of action for NSC95397 inhibition of IAV replication, the point in the virus life cycle inhibited by NSC95397 was determined. To address this, BEAS2B cells were treated with 2 μM NSC95397 at different time points pre- or post-influenza A/WSN/33 virus infection at MOI = 1 (Fig. 4A). Culture supernatant of infected cells was collected at 24 hpi for virus titration in MDCK cells (Fig. 4B). Different periods of 2 μM NSC95397 treatment over 24 hpi had no effect on host cell viability as assessed by ToxiLight bioassay and by phase-contrast microscopy (data not shown). However, a significant reduction in virus titer was evident in cells treated with NSC95397 before 6 hpi, indicating that NSC95397 inhibits IAV replication midcycle, i.e., when virus RNA replication is occurring in the nucleus. To determine if viral RNA synthesis was inhibited by NSC95397, strand-specific qRT-PCR was employed to evaluate the abundance of viral cRNA, mRNA, and vRNA at 7 hpi (Fig. 4C). The levels of positive-sense [(+)sense] viral cRNA and mRNA were reduced 55% and 80%, respectively, by NSC95397 compared to DMSO-treated cells (P < 0.01). Although a slight reduction of (−)sense vRNA was also observed in the presence of NSC95397, this difference was not statistically significant. Importantly, while the abundance of viral RNA was reduced, higher IFN-β expression was observed following NSC95397 treatment compared to DMSO-treated cell results (Fig. 4D). A/WSN/33-infected cells treated with DMSO did not show s significant increase of IFN-β expression at 4 and 7 hpi, consistent with previous findings on antagonism of the host IFN response by IAV. Remarkably, cells infected with A/WSN/33 in the presence of 2 μM NSC95397 displayed 23-fold and 10-fold increases of IFN-β mRNA expression at 4 and 7 hpi, respectively. This was a significant increase compared to DMSO-treated cells at the corresponding time points postinfection (P < 0.01 and P < 0.05 for 4 and 7 hpi, respectively). In the absence of infection, neither DMSO nor NSC95397 exposure resulted in upregulation of IFN-β expression. Higher expression of type III IFN (IFN-λ1) was also detected in the presence of NSC95397 at 4 hpi (data not shown). Together, these results suggest that NSC95397 inhibits IAV (+)sense RNA synthesis while inducing a higher level of host type I and type III IFN gene expression. Since influenza virus is known to antagonize host IFN responses by inhibiting the RIG-I-like receptor (RLR) signaling pathway through the action of viral NS1 protein (32–34), the signaling event downstream of RLR activation, i.e., phosphorylation of interferon regulatory factor 3 (IRF3) during the course of IAV infection, was evaluated in the absence or presence of NSC95397 treatment. BEAS2B cells were pretreated with DMSO or 5 μM NSC95397 1 h prior to mock or influenza A/WSN/33 virus infection (MOI = 1). Cells were harvested for protein isolation at 1, 3, or 7 hpi, and levels of phosphorylated and total IRF3 were evaluated by immunoblotting (Fig. 5A). Despite the higher level of IFN-β expression observed in NSC95397-treated cells relative to DMSO-treated cells at 4 hpi (Fig. 4D), the level of phosphorylated IRF3 was lower in NSC95397-treated cells relative to DMSO-treated cells infected with IAV at 1 and 3 hpi. This finding suggests that NSC95397 acts to upregulate IFN expression by means other than relieving virus inhibition of cytoplasmic RLR signaling.
Fig 4.

NSC95397 reduced the abundance of viral RNA and increased IFN-β expression. BEAS2B cells were mock infected or infected with influenza A/WSN/33 virus at MOI = 1. (A and B) To assess when NSC95397 acts to block IAV infection, 2 μM NSC95397 was added at different time points pre- or postinfection. Culture supernatants were collected for virus titration by plaque assay at 24 hpi, and the results are presented as percentages of virus inhibition relative to DMSO-treated cells, set as 100%. (C to E) Cells were pretreated with DMSO or 2 μM NSC95397 1 h prior to infection. (C) To determine whether the abundance of specific viral RNA species was reduced in the presence of NSC95397, total RNAs were isolated at 7 hpi and subjected to qRT-PCR analysis using primers specific to cRNA, mRNA, and vRNA of IAV segment 5. Viral RNA abundance was normalized to GAPDH, and the abundance in NSC95397 relative to DMSO-treated cells was graphed. (D) Total RNAs were isolated at 4 or 7 hpi and subjected to IFN-β qRT-PCR. IFN-β mRNA abundance was normalized to GAPDH, and the abundance in DMSO- or NSC95397-treated cells relative to noninfected/nontreated BEAS2B cells was graphed. n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. (E) To evaluate IRF3 phosphorylation during infection, protein lysates collected at 1, 3, or 7 hpi were used for immunoblot analysis using antibodies against phosphorylated IRF3 (p-IRF3), total IRF3, and GAPDH as loading controls.
Fig 5.

NSC95397 modulates NS1 nuclear localization and its association with cellular chromatin. (A and B) BEAS2B cells were infected with influenza A/WSN/33 virus at MOI = 1 in the presence of DMSO or NSC95397 and fixed or harvested at 4 or 7 hpi. (A) Fixed cells were stained for NS1 (red), NP (green), and nuclei (DAPI; blue). White arrowheads signify nuclear NS1 staining. (B) Protein extracts from 7 hpi were subjected to subcellular fractionation. Total cell lysates and the chromatin-bound protein fraction were used for immunoblot analyses using antibodies against viral NS1 and NP proteins and using cellular GAPDH, histone H3, and transcription factor SP1 as fractionation controls. At 7 hpi, viral NS1 and NP was found associated with cellular chromatin in DMSO-treated but not in NSC95397-treated cells. (C) BEAS2B cells were infected with wild-type (wt) or reconstructed influenza A/WSN/33 virus expressing NS1 R38AK41A (NS1 RK) in the presence of DMSO or 2 μM NSC95397 at MOI = 0.1. Culture supernatants were collected for virus titration by a plaque assay at 24 hpi, and the results are presented as percent inhibition compared to DMSO-treated cells for each respective virus, set as 100%. n.s., not significant; ***, P < 0.001.
NSC95397 modulates NS1 nuclear localization and its association with cellular chromatin.
In addition to its known role in the cytoplasm to inhibit RLR signaling, a pool of influenza NS1 protein is also found in the nucleus of infected cells which is thought to modulate transcription of the host's antiviral genes (17, 51, 52). Nuclear NS1 has been shown to inhibit transcription elongation in addition to maturation and export of host mRNA (28, 37, 53). Additionally, a report has previously shown that IAV expressing nonphosphorylated NS1 protein is attenuated in vitro, displaying slower growth, smaller plaque size, and delayed localization into discrete intranuclear foci (38). Therefore, the effect of NSC95397 on nuclear NS1 function was further evaluated. To determine if NS1 localization is modulated in the presence of NSC95397, BEAS2B cells were infected with IAV in the presence of DMSO or 2 μM NSC95397, fixed at 7 hpi, and stained for viral NP and NS1 proteins. Consistent with previous findings (17, 54), NP and NS1 proteins were nuclear at 7 hpi in DMSO-treated cells (Fig. 5A). However, NSC95397-treated cells displayed diffused NS1 staining but nuclear NP staining, suggesting that NS1 protein is no longer retained in the nucleus in NSC95397-treated cells. Since NS1 nuclear foci are observed in certain IAV strains (17), NS1 chromatin association was also determined using subcellular fractionation and nucleases to extract chromatin-bound proteins from an insoluble nuclear pellet. In DMSO-treated BEAS2B cells infected with A/WSN/33, NS1 and NP proteins were found to be associated with cellular chromatin (Fig. 5B). This finding is in agreement with previous findings that NS1 is chromatin bound to prevent transcription elongation of antiviral genes and that vRNP is found associated with cellular chromatin (37, 55). In contrast, NS1 and NP proteins are not found in the chromatin fraction of infected cells treated with NSC95397. GAPDH (a cytoplasmic protein) and SP1 (a soluble nuclear protein) were not found in the chromatin fraction, whereas histone H3 was enriched in the chromatin fraction.
To confirm that NSC95397 acts to block NS1 function, growth of recombinant influenza A/WSN/33 virus expressing defective NS1 protein (NS1 R38AK41A [NS1 RK]) was assessed in the presence of DMSO or NSC95397 (Fig. 5C). In agreement with our previous findings, the wild-type influenza A/WSN/33 virus titer was significantly lower at 24 hpi in cells treated with NSC95397 (P < 0.001). However, growth of A/WSN/33 NS1 RK virus was not affected by NSC95397 treatment, demonstrating that NSC95397 acts to limit IAV replication by inhibition of NS1 action.
NSC95397 protects mice against lethal IAV infection.
To assess if NSC95397 can be used to limit IAV replication in vivo, mice were treated with NSC95397 (2.5 mg/kg of body weight) at 24 h pre- or postchallenge with a lethal dose (103 PFU) of A/WSN/33 virus. Mice were monitored daily for 14 days to observe survival (Fig. 6A) and weight loss (Fig. 6B). Mice treated with NSC95397 pre- or postinfection were fully protected against lethal A/WSN/33 infection (P < 0.001) and displayed less-severe weight loss than DMSO-treated mice (P < 0.001 or P < 0.5). To assess lung viral burden, mice were treated with DMSO or increasing doses of NSC95397 preinfection (prophylactic) or postinfection (therapeutic) and infected with a sublethal dose (70 PFU) of A/WSN/33 (Fig. 6C). At 72 hpi, mice treated prophylactically with NSC95397 at 2.5 mg/kg (24 h preinfection; P < 0.01) and at 5 mg/kg (two administrations of 2.5 mg/kg each at 24 and 12 h preinfection; P < 0.001) displayed a significant reduction of lung virus copy number. Additionally, 5 mg/kg NSC95397 administered therapeutically (two administrations of 2.5 mg/kg dose at 12 and 24 hpi) also significantly reduced lung virus copy number (P < 0.001). A suboptimal NSC95397 dose (0.5 mg/kg) administered prophylactically or therapeutically did not significantly reduce virus copy number. In sublethal WSN infection (70 PFU), a single 2.5 mg/kg dose of NSC95397 administered at 24 hpi also did not significantly reduce the lung virus burden, although this dose was protective against mortality associated with lethal WSN infection (103 PFU). However, this treatment regimen provided only partial but statistically significant protection from weight loss during lethal infection (P < 0.05). Taken together, these results demonstrate that inhibition of CDC25B function by its small-molecule inhibitor NSC95397 can potentially be used as a novel influenza antiviral therapeutic strategy.
Fig 6.
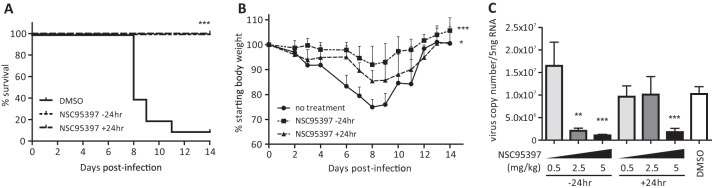
NSC95397 limits the pathogenesis of influenza A/WSN/33 virus infection in vivo. BALB/c female mice were infected with a lethal dose (103 PFU) (A and B) or a sublethal dose (70 PFU) (C) of influenza A/WSN/33 virus and treated with DMSO or 2.5 mg/kg NSC95397 orally at 24 h pre- or postinfection. Mouse survival (A) and weight loss (B) were monitored daily for 14 days according to guidelines from the IACUC of the University of Georgia. (C) RNA was isolated from lungs of infected mice at 72 hpi, and virus copy numbers were evaluated by qRT-PCR. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
There are limited influenza drugs available and few new drug therapies or approaches reported to control influenza virus replication (10). However, as investigators have begun to harness the power of RNAi, a greater understanding of how influenza viruses co-opt host cell pathways to facilitate replication is being uncovered (11, 15), and this is opening new avenues for drug targeting and repurposing for specific host cell pathways. This present report represents a continuation of earlier work that identified and validated mammalian host genes in A549 type II respiratory epithelial cells required for A/WSN/33 replication using a high-throughput siRNA screening approach similar to that described by others (12–16, 19, 20, 56, 57). One gene in the phosphatase family, i.e., the CDC25B gene, is critical for A/WSN/33 replication in A549 cells (Fig. 1). In the present study, inhibition of CDC25B using NSC95397 was shown to prevent IAV replication in BEAS2B cells. NSC95397 (p-naphthoquinone) is a small molecule previously identified to inhibit CDC25B activity in vitro through a screen of 10,070 compounds against recombinant human CDC25B and is the most potent CDC25 inhibitor described to date (39, 40). CDC25B is a proto-oncogene as it facilitates mitotic entry during the cell cycle progression. Thus, overexpression of CDC25B has been reported in several cancers and is associated with a poor prognosis, and its inhibition has been suggested for anticancer therapeutics (21, 49, 50, 58). NSC95397 is thought to inhibit CDC25B phosphatase activity function by covalently modifying serine residues on the active site of CDC25 and has been shown to increase levels of phosphorylated CDC25B targets such as CDK1, CDK2, and ERK (Fig. 2) (47, 48). In this study, pretreatment of BEAS2B cells with NSC95397 effectively reduced replication of influenza A/WSN/33 and B/Florida/04/06 viruses, which are representative strains of influenza A virus and B virus, respectively (Fig. 3). Furthermore, mice treated with NSC95397, administered prophylactically (preinfection) or therapeutically (postinfection), were completely protected against lethal A/WSN/33 virus challenge (Fig. 6), which suggests the potential use of NSC95397 as an IAV therapeutic.
Although NSC95397 was found to be effective to limit IAV infection, its mechanism of action against IAV is unclear. One possibility is that a decrease of CDC25B phosphatase activity could result in inhibition of its target, i.e., the CDK/cyclin complexes affecting influenza virus replication. Many RNA and DNA viruses depend on the host cell cycle for replication, with some such as simian virus 40 (59) and adenovirus (60) encoding proteins that promote cell cycle progression to support viral replication. In contrast, human immunodeficiency virus type 1 (HIV-1) encodes viral proteins that induce cell cycle arrest in the G2/M phase (61). A recent study demonstrated that upregulation of cell cycle molecules, including CDC25B and CDK2, may be linked to disease severity associated with IAV infection (62). It is likely that more than one viral protein is involved in host cell cycle modulation; however, IAV NS1 protein was recently shown to be phosphorylated by CDK1 at its threonine-215 residue (38). Recombinant IAV expressing nonphosphorylatable NS1 protein was attenuated in vitro and displayed slower growth, smaller plaque size, and slower nuclear localization of NS1 protein (38).
To determine when and where CDC25B is involved in IAV replication, NSC95397 was added to BEAS2B cells before or after IAV infection. NSC95397 was found to be effective only when added prior to 6 hpi (Fig. 4), suggesting that CDC25B has a role in support of IAV replication at midcycle. Regarding the IAV replication cycle, vRNP has been shown to be imported into the nucleus between 1.5 and 2 h post-virus binding and uncoating (23, 63). Once in the nucleus, (+)sense viral mRNA is synthesized using the incoming (−)sense genomic vRNA for subsequent viral protein translation in the cytoplasm (64, 65). Additionally, (+)sense viral cRNA is also synthesized from vRNA as the templates for nascent vRNA, followed by vRNP export to the cytoplasm at approximately 8 hpi, for packaging and release of new viral progenies to complete the viral replication cycle, which is approximately 12 hpi (22–24). Based on this outline of IAV replication, the effect of NSC95397 in altering the abundance of specific viral RNA species was evaluated. NSC95397 was found to specifically reduce the abundance of (+)sense viral RNAs, i.e., mRNA and cRNA, while upregulating expression of host type I and type III IFNs (Fig. 4D). Various reports have demonstrated influenza virus NS1 protein's action to block the RIG-I-like receptor (RLR) signaling upstream of IRF3 activation to suppress the host's IFN expression (26, 32–34). Interestingly, the presence of NSC95397 did not result in increased IRF3 phosphorylation (Fig. 4E). This suggests that NSC95397 did not act to upregulate IFN expression by means of inhibiting NS1 action in the cytoplasm to block intracellular pattern recognition receptor signaling (i.e., RLR signaling).
Due to diverse roles of NS1 that include modulating host processes and to the evidence that NS1 phosphorylation by CDC25B target CDK1 was shown to modulate its nuclear localization, NSC95397 activity on NS1 function was evaluated. One interesting finding is the demonstration that NS1 is chromatin bound to prevent transcription elongation of antiviral genes (37, 55). In this study, NSC95397 treatment was found to modulate nuclear localization of NS1 protein and its association with cellular chromatin (Fig. 5). Based on this finding and previously reported functions, we postulated that CDC25B promotes IAV replication by activating CDK1 and ERK kinases, a step that is blocked by CDC25B inhibitor NSC95397, to phosphorylate NS1 at the threonine-215 residue, which ultimately resulted in repression of host antiviral gene expression, such as that by type I and III IFNs.
Taken together, the results of this study show that a better understanding of the host genes required for IAV replication can provide critical information about host cell pathways co-opted by influenza virus, and this in turn can be used to repurpose or reposition existing drugs to inhibit functions of these host factors and limit virus replication. The studies performed here utilized BEAS2B cells that are biosimilar to normal bronchial epithelium cells and corroborated findings in a mouse model. Importantly, this report demonstrates that whole-genome siRNA screens (such as the siGENOME screen) can be used to identify host genes critical for IAV replication, which can then be translated to other cell culture systems as well as in vivo murine studies, features that should hasten novel antiviral drug discovery for IAV.
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases (HHSN266200700006C), and by funding from the Georgia Research Alliance.
We thank Geraldine Saavedra and Leslie Jones for sequence analysis assistance, Cheryl Jones for virus propagation assistance, Kate Oakley for critical reading of the manuscript, Michael Gale, Jr., and Arjun Rustagi for the IRF3 (AR-1) antibody, and Adolfo García-Sastre and Randy Albrecht for kindly providing us with the influenza A/WSN/33 NS1 R38AK41A virus. We also thank the RNAi Global Team, specifically, Jon Karpilow, for providing guidance for this study.
Footnotes
Published ahead of print 9 October 2013
REFERENCES
- 1.Girard MP, Tam JS, Assossou OM, Kieny MP. 2010. The A (H1N1) influenza virus pandemic: a review. Vaccine 28:4895–4902 [DOI] [PubMed] [Google Scholar]
- 2.Monto AS. 2003. The role of antivirals in the control of influenza. Vaccine 21:1796–1800 [DOI] [PubMed] [Google Scholar]
- 3.McKimm-Breschkin JL. 2005. Management of influenza virus infections with neuraminidase inhibitors: detection, incidence, and implications of drug resistance. Treat Respir. Med. 4:107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinto LH, Holsinger LJ, Lamb RA. 1992. Influenza virus M2 protein has ion channel activity. Cell 69:517–528 [DOI] [PubMed] [Google Scholar]
- 5.Boltz DA, Aldridge JR, Jr, Webster RG, Govorkova EA. 2010. Drugs in development for influenza. Drugs 70:1349–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Layne SP, Monto AS, Taubenberger JK. 2009. Pandemic influenza: an inconvenient mutation. Science 323:1560–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simonsen L, Clarke MJ, Williamson GD, Stroup DF, Arden NH, Schonberger LB. 1997. The impact of influenza epidemics on mortality: introducing a severity index. Am. J. Public Health 87:1944–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deyde VM, Xu X, Bright RA, Shaw M, Smith CB, Zhang Y, Shu Y, Gubareva LV, Cox NJ, Klimov AI. 2007. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J. Infect. Dis. 196:249–257 [DOI] [PubMed] [Google Scholar]
- 9.Nguyen JT, Hoopes JD, Le MH, Smee DF, Patick AK, Faix DJ, Blair PJ, de Jong MD, Prichard MN, Went GT. 2010. Triple combination of amantadine, ribavirin, and oseltamivir is highly active and synergistic against drug resistant influenza virus strains in vitro. PLoS One 5:e9332. 10.1371/journal.pone.0009332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Min JY, Subbarao K. 2010. Cellular targets for influenza drugs. Nat. Biotechnol. 28:239–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe T, Watanabe S, Kawaoka Y. 2010. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 7:427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y. 2008. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454:890–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karlas A, Machuy N, Shin Y, Pleissner K-P, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, Maurer AP, Muller E, Wolff T, Rudel T, Meyer TF. 2010. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463:818–822 [DOI] [PubMed] [Google Scholar]
- 15.König R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, Garcia-Sastre A, Young JAT, Palese P, Shaw ML, Chanda SK. 2010. Human host factors required for influenza virus replication. Nature 463:813–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perwitasari O, Yan X, Johnson S, White C, Brooks P, Tompkins SM, Tripp RA. 5 November 2012. Targeting the organic anion transporter-3 (OAT3) with probenecid as a novel anti-influenza A virus strategy. Antimicrob. Agents Chemother. 10.1128/AAC.01532-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melén K, Kinnunen L, Fagerlund R, Ikonen N, Twu KY, Krug RM, Julkunen I. 2007. Nuclear and nucleolar targeting of influenza A virus NS1 protein: striking differences between different virus subtypes. J. Virol. 81:5995–6006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodman AG, Smith JA, Balachandran S, Perwitasari O, Proll SC, Thomas MJ, Korth MJ, Barber GN, Schiff LA, Katze MG. 2007. The cellular protein P58IPK regulates influenza virus mRNA translation and replication through a PKR-mediated mechanism. J. Virol. 81:2221–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meliopoulos VA, Andersen LE, Birrer KF, Simpson KJ, Lowenthal JW, Bean AG, Stambas J, Stewart CR, Tompkins SM, van Beusechem VW, Fraser I, Mhlanga M, Barichievy S, Smith Q, Leake D, Karpilow J, Buck A, Jona G, Tripp RA. 2012. Host gene targets for novel influenza therapies elucidated by high-throughput RNA interference screens. FASEB J. 26:1372–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meliopoulos VA, Andersen LE, Brooks P, Yan X, Bakre A, Coleman JK, Tompkins SM, Tripp RA. 2012. MicroRNA regulation of human protease genes essential for influenza virus replication. PLoS One 7:e37169. 10.1371/journal.pone.0037169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kristjánsdóttir K, Rudolph J. 2004. Cdc25 phosphatases and cancer. Chem. Biol. 11:1043–1051 [DOI] [PubMed] [Google Scholar]
- 22.Nayak DP, Hui EK, Barman S. 2004. Assembly and budding of influenza virus. Virus Res. 106:147–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samji T. 2009. Influenza A: understanding the viral life cycle. Yale J. Biol. Med. 82:153–159 [PMC free article] [PubMed] [Google Scholar]
- 24.Paterson D, Fodor E. 2012. Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog. 8:e1003019. 10.1371/journal.ppat.1003019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Min J-Y, Krug RM. 2006. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. U. S. A. 103:7100–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hale BG, Randall RE, Ortín J, Jackson D. 2008. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89:2359–2376 [DOI] [PubMed] [Google Scholar]
- 27.Krug RM, Yuan W, Noah DL, Latham AG. 2003. Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology 309:181–189 [DOI] [PubMed] [Google Scholar]
- 28.Nemeroff ME, Barabino SM, Li Y, Keller W, Krug RM. 1998. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol. Cell 1:991–1000 [DOI] [PubMed] [Google Scholar]
- 29.Qiu Y, Krug RM. 1994. The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J. Virol. 68:2425–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu Y, Wambach M, Katze MG, Krug RM. 1995. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology 214:222–228 [DOI] [PubMed] [Google Scholar]
- 31.Garfinkel MS, Katze MG. 1993. Translational control by influenza virus. Selective translation is mediated by sequences within the viral mRNA 5′-untranslated region. J. Biol. Chem. 268:22223–22226 [PubMed] [Google Scholar]
- 32.Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M, Jr, Garcia-Sastre A. 2007. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 81:514–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Opitz B, Rejaibi A, Dauber B, Eckhard J, Vinzing M, Schmeck B, Hippenstiel S, Suttorp N, Wolff T. 2007. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 9:930–938 [DOI] [PubMed] [Google Scholar]
- 34.Guo Z, Chen LM, Zeng H, Gomez JA, Plowden J, Fujita T, Katz JM, Donis RO, Sambhara S. 2007. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 36:263–269 [DOI] [PubMed] [Google Scholar]
- 35.Satterly N, Tsai PL, van Deursen J, Nussenzveig DR, Wang Y, Faria PA, Levay A, Levy DE, Fontoura BM. 2007. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. U. S. A. 104:1853–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park YW, Katze MG. 1995. Translational control by influenza virus: identification of cis-acting sequences and trans-acting factors which may regulate selective viral mRNA translation. J. Biol. Chem. 270:28433–28439 [DOI] [PubMed] [Google Scholar]
- 37.Marazzi I, Ho JS, Kim J, Manicassamy B, Dewell S, Albrecht RA, Seibert CW, Schaefer U, Jeffrey KL, Prinjha RK, Lee K, Garcia-Sastre A, Roeder RG, Tarakhovsky A. 2012. Suppression of the antiviral response by an influenza histone mimic. Nature 483:428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hale BG, Knebel A, Botting CH, Galloway CS, Precious BL, Jackson D, Elliott RM, Randall RE. 2009. CDK/ERK-mediated phosphorylation of the human influenza A virus NS1 protein at threonine-215. Virology 383:6–11 [DOI] [PubMed] [Google Scholar]
- 39.Lazo JS, Nemoto K, Pestell KE, Cooley K, Southwick EC, Mitchell DA, Furey W, Gussio R, Zaharevitz DW, Joo B, Wipf P. 2002. Identification of a potent and selective pharmacophore for Cdc25 dual specificity phosphatase inhibitors. Mol. Pharmacol. 61:720–728 [DOI] [PubMed] [Google Scholar]
- 40.Park H, Carr BI, Li M, Ham SW. 2007. Fluorinated NSC as a Cdc25 inhibitor. Bioorg. Med. Chem. Lett. 17:2351–2354 [DOI] [PubMed] [Google Scholar]
- 41.Gaush CR, Smith TF. 1968. Replication and plaque assay of influenza virus in an established line of canine kidney cells. Appl. Microbiol. 16:588–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szretter KJ, Balish AL, Katz JM. 2005. Influenza: propagation, quantification, and storage. Curr. Protoc. Microbiol. 3:15G.1.1–15G.1.22. 10.1002/0471729256.mc15g01s3 [DOI] [PubMed] [Google Scholar]
- 43.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27:493–497 http://aje.oxfordjournals.org/content/27/3/493.full.pdf+html [Google Scholar]
- 44.Jones RE, Zheng W, McKew JC, Chen CZ. 24 May 2013. An alternative direct compound dispensing method using the HP D300 digital dispenser. J. Lab. Autom. [Epub ahead of print.]. 10.1177/2211068213491094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, Kawaoka Y. 2011. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J. Virol. Methods 173:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rustagi A, Doehle BP, McElrath MJ, Gale M., Jr 2013. Two new monoclonal antibodies for biochemical and flow cytometric analyses of human interferon regulatory factor-3 activation, turnover, and depletion. Methods 59:225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nemoto K, Vogt A, Oguri T, Lazo JS. 2004. Activation of the Raf-1/MEK/Erk kinase pathway by a novel Cdc25 inhibitor in human prostate cancer cells. Prostate 58:95–102 [DOI] [PubMed] [Google Scholar]
- 48.Peyregne VP, Kar S, Ham SW, Wang M, Wang Z, Carr BI. 2005. Novel hydroxyl naphthoquinones with potent Cdc25 antagonizing and growth inhibitory properties. Mol. Cancer Ther. 4:595–602 [DOI] [PubMed] [Google Scholar]
- 49.Bugler B, Schmitt E, Aressy B, Ducommun B. 2010. Unscheduled expression of CDC25B in S-phase leads to replicative stress and DNA damage. Mol. Cancer 9:29. 10.1186/1476-4598-9-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boutros R, Lobjois V, Ducommun B. 2007. CDC25 phosphatases in cancer cells: key players? Good targets? Nat. Rev. Cancer 7:495–507 [DOI] [PubMed] [Google Scholar]
- 51.Greenspan D, Palese P, Krystal M. 1988. Two nuclear location signals in the influenza virus NS1 nonstructural protein. J. Virol. 62:3020–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, Yamakita Y, Krug RM. 1998. Regulation of a nuclear export signal by an adjacent inhibitory sequence: the effector domain of the influenza virus NS1 protein. Proc. Natl. Acad. Sci. U. S. A. 95:4864–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fortes P, Beloso A, Ortin J. 1994. Influenza virus NS1 protein inhibits pre-mRNA splicing and blocks mRNA nucleocytoplasmic transport. EMBO J. 13:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elton D, Simpson-Holley M, Archer K, Medcalf L, Hallam R, McCauley J, Digard P. 2001. Interaction of the influenza virus nucleoprotein with the cellular CRM1-mediated nuclear export pathway. J. Virol. 75:408–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chase GP, Rameix-Welti MA, Zvirbliene A, Zvirblis G, Götz V, Wolff T, Naffakh N, Schwemmle M. 2011. Influenza virus ribonucleoprotein complexes gain preferential access to cellular export machinery through chromatin targeting. PLoS Pathog. 7:e1002187. 10.1371/journal.ppat.1002187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shapira SD, Gat-Viks I, Shum BOV, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, Hill DE, Regev A, Hacohen N. 2009. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139:1255–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sui B, Bamba D, Weng K, Ung H, Chang S, Van Dyke J, Goldblatt M, Duan R, Kinch MS, Li W-B. 2009. The use of random homozygous gene perturbation to identify novel host-oriented targets for influenza. Virology 387:473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lyon MA, Ducruet AP, Wipf P, Lazo JS. 2002. Dual-specificity phosphatases as targets for antineoplastic agents. Nat. Rev. Drug Discov. 1:961–976 [DOI] [PubMed] [Google Scholar]
- 59.DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsilio E, Paucha E, Livingston DM. 1988. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54:275–283 [DOI] [PubMed] [Google Scholar]
- 60.Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, Livingston DM. 1994. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 8:869–884 [DOI] [PubMed] [Google Scholar]
- 61.Goh WC, Rogel ME, Kinsey CM, Michael SF, Fultz PN, Nowak MA, Hahn BH, Emerman M. 1998. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: a mechanism for selection of Vpr in vivo. Nat. Med. 4:65–71 [DOI] [PubMed] [Google Scholar]
- 62.Parnell G, McLean A, Booth D, Huang S, Nalos M, Tang B. 2011. Aberrant cell cycle and apoptotic changes characterise severe influenza A infection—a meta-analysis of genomic signatures in circulating leukocytes. PLoS One 6:e17186. 10.1371/journal.pone.0017186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boulo S, Akarsu H, Ruigrok RW, Baudin F. 2007. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 124:12–21 [DOI] [PubMed] [Google Scholar]
- 64.Neumann G, Brownlee GG, Fodor E, Kawaoka Y. 2004. Orthomyxovirus replication, transcription, and polyadenylation. Curr. Top. Microbiol. Immunol. 283:121–143 [DOI] [PubMed] [Google Scholar]
- 65.Vreede FT, Brownlee GG. 2007. Influenza virion-derived viral ribonucleoproteins synthesize both mRNA and cRNA in vitro. J. Virol. 81:2196–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]