

Abstract
Rotavirus nonstructural protein 4 (NSP4) induces dramatic changes in cellular calcium homeostasis. These include increased endoplasmic reticulum (ER) permeability, resulting in decreased ER calcium stores and activation of plasma membrane (PM) calcium influx channels, ultimately causing a 2- to 4-fold elevation in cytoplasmic calcium. Elevated cytoplasmic calcium is absolutely required for virus replication, but the underlying mechanisms responsible for calcium influx remain poorly understood. NSP4 is an ER-localized viroporin, whose activity depletes ER calcium, which ultimately leads to calcium influx. We hypothesized that NSP4-mediated depletion of ER calcium activates store-operated calcium entry (SOCE) through activation of the ER calcium sensor stromal interaction molecule 1 (STIM1). We established and used a stable yellow fluorescent protein-expressing STIM1 cell line (YFP-STIM1) as a biosensor to assess STIM1 activation (puncta formation) by rotavirus infection and NSP4 expression. We found that STIM1 is constitutively active in rotavirus-infected cells and that STIM1 puncta colocalize with the PM-localized Orai1 SOCE calcium channel. Expression of wild-type NSP4 activated STIM1, resulting in PM calcium influx, but an NSP4 viroporin mutant failed to induce STIM1 activation and did not activate the PM calcium entry pathway. Finally, knockdown of STIM1 significantly reduced rotavirus yield, indicating STIM1 plays a critical role in virus replication. These data demonstrate that while rotavirus may ultimately activate multiple calcium channels in the PM, calcium influx is predicated on NSP4 viroporin-mediated activation of STIM1 in the ER. This is the first report of viroporin-mediated activation of SOCE, reinforcing NSP4 as a robust model to understand dysregulation of calcium homeostasis during virus infections.
INTRODUCTION
Calcium (Ca2+) is a ubiquitous secondary messenger, and the concentration of intracellular Ca2+ is tightly regulated. As obligate intracellular parasites, viruses subvert host cell pathways to support robust virus replication. Many viruses disrupt host Ca2+ homeostasis in order to establish a cellular environment conducive for virus replication and assembly (1). One well-established hallmark of rotavirus (RV) infection is dramatic changes in cellular Ca2+ homeostasis, including increased permeability of the endoplasmic reticulum (ER), resulting in decreased ER Ca2+ stores and activation of Ca2+ influx channels in the plasma membrane (PM), ultimately resulting in an elevated cytoplasmic Ca2+ concentration ([Ca2+]cyto) (2–4). While both ER Ca2+ stores and extracellular Ca2+ contribute to the increased [Ca2+]cyto, the extracellular pool is much greater than the ER stores; therefore, Ca2+ influx through the PM likely accounts for the bulk of the increase in [Ca2+]cyto in RV-infected cells. Using expression of individual recombinant RV proteins, nonstructural protein 4 (NSP4) was identified as the sole RV protein responsible for the elevation in [Ca2+]cyto levels in Sf9 insect cells and a variety of mammalian cell lines, and NSP4 recapitulates all of the changes in Ca2+ homeostasis observed in RV-infected cells (5, 6). Because the NSP4-induced rapid and sustained increase in [Ca2+]cyto is absolutely required for RV replication, several studies have sought to define the underlying mechanisms responsible for the elevation in [Ca2+]cyto (4, 5, 7). These studies largely agreed that NSP4 functions in the ER to elevate [Ca2+]cyto, and we recently determined that NSP4 elevates [Ca2+]cyto by functioning as a viroporin, which is a member of a diverse class of virus-encoded pore-forming and ion channel proteins (8).
Although different viroporins target a range of subcellular compartments and ions, they all have similar structural motifs, including being oligomeric, having a cluster of basic residues, and having an amphipathic alpha-helix that upon oligomerization form the aqueous channel through a membrane (8). NSP4 is an ER-localized glycoprotein with pleiotropic functions during RV replication (9). The NSP4 viroporin domain is comprised of amino acids (aa) 47 to 90, and this domain is critical for elevation of [Ca2+]cyto, since mutation of either the cluster of basic residues or amphipathic alpha-helix abolishes the observed elevation in [Ca2+]cyto (8). Therefore, viroporin activity in the ER is the primary NSP4 function that initiates the global disruption in cellular Ca2+ homeostasis (8). However, the mechanism by which NSP4 viroporin activity in the ER membrane is linked to activation of Ca2+ uptake through the PM has not been defined.
The coordinated regulation of Ca2+ release from the ER and subsequent Ca2+ entry across the PM to replenish ER stores was first identified by Putney and termed “capacitative Ca2+ entry” (10) This model has been refined to show that activation of these PM Ca2+ entry channels is a direct consequence of ER Ca2+ store depletion and is now termed “store-operated calcium entry” (SOCE) (11, 12). SOCE is a homeostatic cellular mechanism by which the ER Ca2+ store levels are measured and maintained to ensure proper Ca2+-mediated signaling (12). ER Ca2+ levels are sensed by stromal interacting molecule 1 (STIM1). STIM1 is an ER single transmembrane glyco/phosphoprotein that senses ER Ca2+ levels through a low-affinity EF-hand Ca2+ binding site located within its N-terminal domain in the ER lumen. Depletion of ER Ca2+ stores, by inosital-1,4,5-triphosphate- or ryanodine receptor-mediated release through ER Ca2+ release channels or thapsigargin (TG)-mediated inhibition of the sarcoendoplasmic reticulum ATPase (SERCA) pump, reduces ER Ca2+ levels and causes Ca2+ to dissociate from the STIM1 EF-hand.
Loss of Ca2+ from the EF-hand causes a conformational change in STIM1 that induces the formation of STIM1 oligomers, representing the active form of this molecule. STIM1 oligomers remain embedded in the ER membrane but rapidly move to ER-PM junctions, which are areas of the cell where the ER membrane is closely juxtaposed to the PM (e.g., junctional ER). It is in these ER-PM junctions that the extended cytoplasmic C terminus of STIM1 binds to Ca2+-release-activated Ca2+ (CRAC) channels in the PM (13). The canonical CRAC channel is Orai1, a pentameric Ca2+-selective ion channel that is gated (i.e., the channel is open) by a direct interaction with the CRAC activation domain (CAD) found within the STIM1 cytoplasmic tail (14). Although there are three members of the Orai channel family (Orai1 to Orai3), because STIM1 and Orai1 are sufficient for generation of CRAC Ca2+ currents, Orai1 is widely considered the primary channel activated by Ca2+ store release and STIM1 activation. In addition to the Orai channel family, members of the transient receptor potential canonical (TRPC) ion channel family (primarily TRPC1) have also been implicated in Ca2+ entry through the PM in response to ER store depletion (15). TRPC1 is a nonselective cation channel that is permeable to both Ca2+ and Na+. While STIM1 also interacts with and activates TRPC1, recent data show that activation of TRPC1 by STIM1 first requires the formation of a functional STIM1/Orai1 complex before TRPC1 can be recruited and activated by STIM1 (16, 17). Furthermore, upon ER store depletion, STIM1 can indirectly activate L-type voltage-activated Ca2+ channels through TRPC-mediated membrane depolarization or downregulate/inhibit Ca2+ entry through the CaV1.2 L-type channel (18–20), as well as inactivate the plasma membrane Ca2+ pump (PMCA) (21).
Although it has been shown that RV infection and NSP4 expression lead to Ca2+ entry across the PM, the contribution of STIM1 as a Ca2+ sensor in the ER- and PM-localized CRAC channels to the elevated [Ca2+]cyto is not known. The ultimate role of SOCE in the RV-induced increase in [Ca2+]cyto has been controversial, since the magnitude of Ca2+ entry observed in RV-infected cells is 4- to 6-fold greater than can be induced by full ER store depletion of mock-inoculated cells using TG. Furthermore, an Orai1 CRAC channel blocker, 2-aminoethoxydiphenyl borate (2-APB), only partially blocked RV/NSP4-induced Ca2+ entry (7). Thus, the role SOCE plays in the NSP4-induced Ca2+ entry pathway remains unclear. The purpose of these studies was to examine whether RV induces SOCE through STIM1 activation due to NSP4 viroporin activity in the ER and to determine the contribution of SOCE to the elevation of [Ca2+]cyto in RV-infected cells.
MATERIALS AND METHODS
Cell culture and rotavirus propagation.
Human embryonic kidney cells of the HEK293 line, obtained from ATCC, and MA104 African green monkey kidney cells (22) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum and maintained in a humidified 95% air–5% CO2 incubator at 37°C. The SA114F strain of rotavirus was grown in MA104 cells as previously described (23) and trypsin activated with 10 μg/ml Worthington trypsin for 30 min at 37°C prior to use.
Antibodies and plasmids.
Anti-STIM1 monoclonal antibody was purchased from AbCam (Cambridge, MA) and used as shown in Fig. 1. An anti-STIM1 polyclonal antibody was purchased from Sigma-Aldrich (St. Louis, MO) and used as shown in Fig. 7. Anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) and anti-enhanced green fluorescent protein (anti-EGFP) monoclonal antibodies were purchased from Clontech (Mountain View, CA). The antibody to rotavirus NSP4 was the rabbit anti-NSP4114–135 peptide described previously (24). Guinea pig hyperimmune sera prepared against NSP2 and NSP5, purified from Escherichia coli as previously described (25), were generated by Cocalico Biologicals, Inc. (Reamstown, PA). Yellow fluorescent protein (YFP)-tagged STIM1 plasmid was a kind gift from Tobias Meyer (Stanford University) (26). NSP4 with a C-terminal red fluorescent protein tag (TagRFP) (Evrogen, Moscow, Russia) was previously described (27), and PCR-based mutagenesis of this plasmid was used to generate the viroporin-deficient mutant ASDASA, as previously described (8). The NSP4 ASDASA mutant is referred to here as “NSP4-Mut.” Vectors encoding myc-tagged Orai1 (plasmid 21638) and the genetically encoded Ca2+ sensor GCaMP5G (plasmid 31788) were purchased from Addgene (Cambridge, MA). Orai1-RFP was generated by inserting the TagRFP sequence in frame into the XhoI restriction site of the polylinker.
Fig 1.

Stable expression of YFP-STIM1 in HEK293 cells. (A) Cell lysates from parental HEK293 cells (H) or HEK293 cells stably expressing YFP-STIM1 (S) were analyzed by immunoblotting with antibodies specific for STIM1 (left), EGFP for the detection of enhanced YFP (center), and GAPDH as a loading control (right). Endogenous STIM1 (arrowhead) and YFP-STIM1 (*) are both detected by STIM1 antibodies, but only YFP-STIM1 (*) is detected by EGFP antibodies. Molecular mass marker sizes (kDa) are listed to the left of the respective blots. (B) Immunofluorescence analysis by confocal microscopy shows the YFP-STIM1 completely colocalized with anti-STIM1 detected with Alexa Fluor 596.
Fig 7.
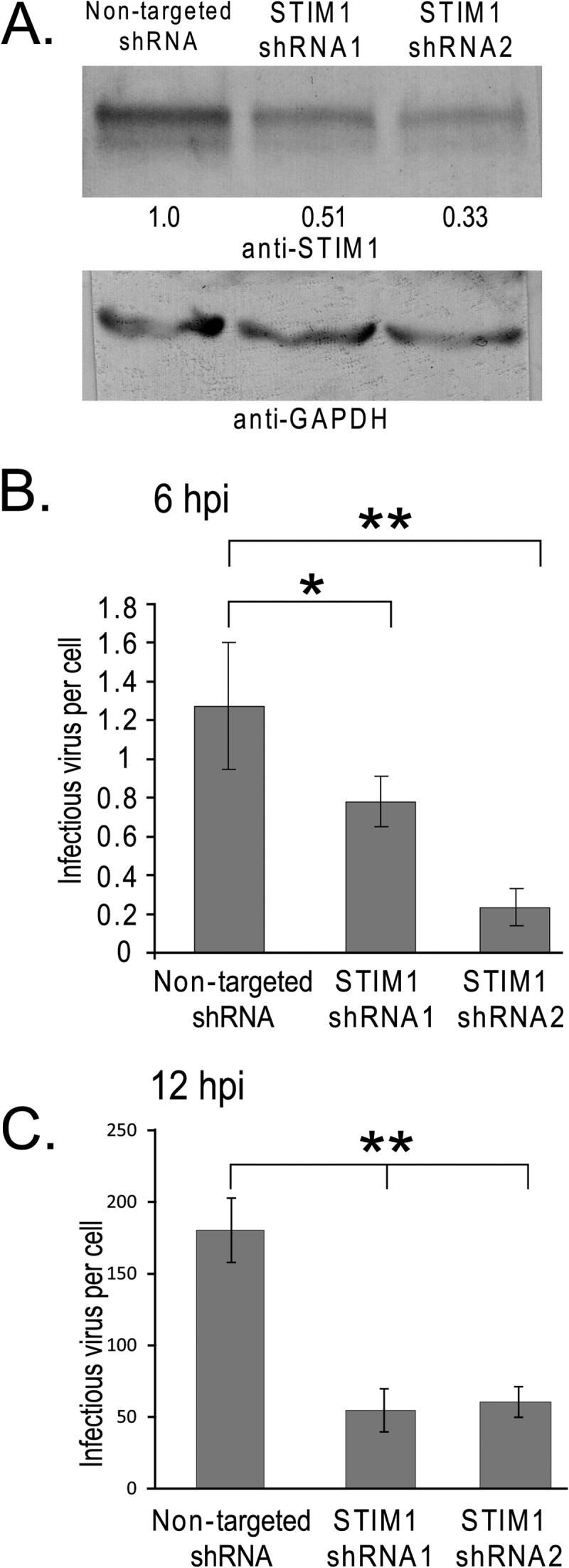
STIM1 knockdown inhibits rotavirus replication. Lentivirus vectors expressing short hairpin RNAs were used to generate three MA104 cell lines stably expressing a nontargeted shRNA or two different STIM1-targeted shRNAs. (A) Knockdown of STIM1 expression was validated by immunoblot analysis of whole-cell lysates from each cell line using a STIM1-specific antibody (upper panel) and a GAPDH-specific antibody as a loading control (bottom panel). The percentage of STIM1 in the STIM1 knockdown cell lines relative to the nontargeted shRNA cell line was determined by densitometry using STIM1 antibody. Densitometry was determined by ImageJ and normalized to GAPDH levels detected with a GAPDH antibody. (B and C) The stable shRNA-expressing MA104 cell lines were infected with SA114F (MOI of 1) and harvested at 6 hpi (B) or 12 hpi (C), and titers of virus yields were determined by fluorescence focus assay. Each experiment was performed in quadruplicate. The final RV yield from nontargeted shRNA-expressing cells was 4.71 × 105 ± 1.41 × 105 focus-forming units (FFU)/ml. *, P < 0.05; **, P < 0.01.
Generation of the YFP-STIM1 stable cell line.
HEK293 cells were grown to 70 to 80% confluence in a 25-cm2 flask and then transfected with the YFP-STIM1 expression vector using Lipofectamine 2000 (Life Technologies), according to the manufacturer's instructions. Transfected cells were selected using 600 μg/ml Geneticin (G418; Life Technologies). Selected cells were then enriched for YFP signal by fluorescence-activated cell sorting (FACS) using a BD FACSAria sorter in the Cytometry and Cell Sorting Core at the Baylor College of Medicine. Two separate FACS enrichment steps were performed, and single-cell cloned cell lines stably expressing STIM-YFP were isolated using limiting dilution in 96-well culture plates. Cell viability, growth rate, and the YFP-STIM1 response to TG were used to validate each clone. The cloned cell line, designated “YFP-STIM1 B1,” had a good growth rate (data not shown), YFP-STIM1 signal, and response to TG and was therefore expanded to be used as our STIM1 biosensor cell line.
Generation of shRNA knockdown cell lines.
Lentivirus vectors encoding either a nontargeted short hairpin RNA (shRNA) or two different STIM1-targeted shRNAs (GIPZ lentivirus shRNA vectors) were purchased from Thermo Scientific (Rockford, IL). MA104 cells were seeded at ∼2 × 105 cells per well in a 24-well plate and the following day were transduced overnight with the GIPZ lentivirus vectors (multiplicity of infection [MOI] of 10) in DMEM with 6 μg/ml Polybrene (Sigma-Aldrich). At 24 h postransduction, the cells were washed in DMEM and then cultured in 10% DMEM supplemented with 8 μg/ml puromycin (Life Technologies) to select for transduced cells. Cells were visualized for EGFP expression to confirm outgrowth of only lentivirus-transduced cells. Cells were maintained in 8 μg/ml puromycin for normal culturing, but puromycin was removed 24 h prior to rotavirus infection. shRNA-expressing cells were infected with trypsin-activated SA114F (MOI of 1) and harvested at 6 or 12 h postinfection (hpi). Progeny virus was recovered by three freeze-thaw cycles and trypsin activated (10 μg/ml) for 30 min at 37°C, and the virus yield was determined by fluorescent focus assay (see below). For each experiment, quadruplicate infections were performed.
Immunoblot analysis.
Samples were analyzed by immunoblot analysis as previously described (24). The following antibodies and dilutions were used: anti-STIM1 (AbCam or Sigma) at a dilution of 1:1,000, anti-GAPDH at a dilution of 1:1,000, or anti-EGFP monoclonal antibody at a dilution of 1:1,000 (Clontech) in 0.5% Blotto with 0.02% sodium azide. For detection of STIM1 and GAPDH, membranes were developed using alkaline phosphatase (AP) detection solution (50 mM Tris, 3 mM MgCl2, 0.1 mg/ml p-nitroblue tetrazolium chloride, 0.05 mg/ml 5-bromo-4-chloro-3-indolyl phosphate). For anti-EGFP analysis, the membrane was developed with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) according to the manufacturer's instructions, and the blot was exposed to film for detection.
Confocal and deconvolution microscopy.
Cells were seeded onto sterile poly-d-lysine-coated 12-mm-diameter glass coverslips (no. 1.5). At the indicated time posttransfection/infection, cells were fixed with 4% paraformaldehyde in 1× phosphate-buffered saline (PBS) for 30 min and permeabilized with 0.5% Triton X-100 for 10 min. After three washes with PBS, cells were subjected to TOPRO-3 (Invitrogen) staining (1 μM) in PBS for 10 min. After being washed with PBS, cells on coverslips were mounted using ProLong Gold antifade reagents (Life Technologies). Mounted slides were observed using a Carl Zeiss LSM 510 META microscope with a 63× oil immersion objective (Carl Zeiss, Inc., Thornwood, NY) or a Nikon A1 confocal microscope with a 60× oil immersion objective (Nikon Instruments, Melville, NY) in multitrack mode. For EGFP/YFP imaging, cells were excited using a 488-nm laser line, and emissions were collected between 500 and 540 nm. For excitation of TagRFP, a 543 laser line was used, and emissions were collected between 595 and 620 nm. For excitation of TOPRO-3, a 633 laser line was used, and emissions were collected between 650 and 680 nm. Optical slice thickness was kept equal for all channels by varying the pinhole diameter, which was kept as close to 1 absorbance unit (AU) as possible, and pixel dwell time was set at 3.20 μs with 8 frames averaged for each slice. Collected images were processed using Zeiss AIM software (Carl Zeiss, Inc.) or Nikon NIS Element (Nikon Instruments) and saved in 12-bit tagged-image format. Deconvolution microscopy was performed on a DeltaVision Core image restoration microscope (Applied Precision, Issaquah, WA). Orthogonal views (z-stacks) were imaged at a 0.3-μm separation and a frame size of 1,024 by 1,024 pixels at 1-by-1 binning with an Olympus IX70 microscope using a 40×, 0.95-numerical aperture (NA) Plan Apochromat objective (Olympus, Center Valley, PA) and a Photometrics CoolSnap HQ2 charge-coupled device (CCD) camera. Deconvolution was performed with dedicated software (SoftwoRx version 4.0; Applied Precision), using 5 iterations of the conservative deconvolution method. All observations were carried out in the Integrated Microscopy Core Laboratory at the Baylor College of Medicine.
YFP-STIM1 puncta formation assay.
YFP-STIM1 cells were seeded into ΔT Dishes (Bioptics, Butler, PA) and transfected with plasmids expressing RFP-tagged wild-type NSP4 (NSP4-WT) or mutant NSP4 (NSP4-Mut) RFP-labeled proteins. We quantitated the number of cells containing diffuse or punctate YFP-STIM1 similar to a previously described NSP4/LC3 puncta formation assay (28). At ∼20 h posttransfection, random fields from two experiments were imaged by deconvolution microscopy, and the images were collected. Cells with reticular YFP-STIM1 were scored as having no puncta. The majority of cells with NSP4-WT contained between 10 and 30 puncta. However, cells transfected with NSP4-Mut with the presence of even a single YFP-STIM puncta were scored as having puncta.
FFA.
Determination of infectious rotavirus yield by fluorescent focus assay (FFA) was performed as previously described (28). Results are reported as the number of infectious virus per cell.
Calcium imaging.
MA104 cells were seeded into ΔT dishes (Bioptics, Butler, PA) and the next day cotransfected (as described above) with expression vectors for either wild-type or the ASDASA viroporin-deficient mutant NSP4-RFP and the GCaMP5G genetically encoded calcium sensor. GCaMP5G is a next-generation EGFP-based calcium sensor with low fluorescence under basal [Ca2+]cyto that dramatically increases its fluorescence upon an increase in [Ca2+]cyto (29). At 24 h posttransfection, the culture medium was replaced with Live Cell Imaging solution (Life Technologies) and imaged using an Olympus IX70 epifluorescence inverted microscope. Baseline GCaMP5G fluorescence was determined, and then extracellular Ca2+ was increased from 1.8 mM to 10 mM while imaging the GCaMP5G signal. ImageJ software was used to define regions of interest and measure the GCaMP5G fluorescence signal. The change in GCaMP5G fluorescence was calculated as the maximum fluorescence divided by the basal fluorescence (Fmax/F0).
Statistical analysis.
Microsoft Excel or GraphPad Prism software was used for statistical analysis. P < 0.05 was considered a significant difference.
RESULTS
YFP-STIM1 biosensor cell line.
We recently showed NSP4 viroporin activity mediates an increase in [Ca2+]cyto during RV infection (8, 27). To determine whether reduction of the ER Ca2+ store by RV or NSP4 is the ER trigger for SOCE, we generated a cell line stably expressing YFP-STIM1 as a fluorescent biosensor for STIM1 activation. Human embryonic kidney 293 (HEK293) cells were transfected with a vector encoding YFP-STIM1, and stable lines were isolated by G418 drug selection and fluorescence-activated cell sorting (FACS) to positively select for YFP-STIM1-expressing cells. A clonal cell line was established and used as the biosensor cell line in further analyses. This cloned cell line showed stable YFP-STIM1 expression over many cell passages, with a normal growth rate and good cell viability (data not shown). Immunoblot analysis of STIM1 levels from cell lysates of parental HEK293 (H) and YFP-STIM1 (S) cells (Fig. 1A, left) show that both cells express similar levels of endogenous STIM1 (arrowhead), using GAPDH as a loading control (Fig. 1A, right). In contrast, the YFP-STIM1 cells expressed much lower levels of YFP-STIM1 (asterisk) than endogenous STIM1 (arrowhead), which we confirmed was YFP-tagged STIM1 by immunoblot analysis using an EGFP-specific monoclonal antibody (Fig. 1A, middle). Finally, we showed that YFP-STIM1 is distributed similarly to endogenous STIM1 by confocal microscopy of cells stained with a STIM1-specific antibody (Fig. 1B).
ER Ca2+ store depletion induces oligomerization of STIM1, which is visualized as the formation of STIM1 puncta, and movement of STIM1 oligomers to ER-PM junctions to engage CRAC channels in the PM (13, 26, 30). We used the SERCA pump inhibitor TG to reduce ER luminal Ca2+ levels and determine whether YFP-STIM1 puncta were induced in the biosensor cell line (Fig. 2). Prior to stimulation, YFP-STIM1 showed a diffuse/reticular distribution indicative of ER localization (Fig. 2A), but within 5 min posttreatment with 5 μM TG, YFP-STIM1 puncta were observed in most cells (Fig. 2B, arrows and inset). YFP-STIM1 puncta formation continued to increase (15 min) (Fig. 2C), and by 30 min, nearly all of the YFP-STIM1 localized into the puncta structures (Fig. 2D). The rapid accumulation of YFP-STIM1 puncta was verified in real time by live cell microscopy after TG treatment, with the first YFP-STIM1 puncta forming at approximately 3 min posttreatment (see Movie S1 in the supplemental material). Next, we assessed the redistribution of YFP-STIM1 to ER-PM junctions upon activation. Mock- or TG-treated YFP-STIM1 cells were stained with wheat germ agglutinin conjugated to Texas Red (WGA-TxRed) to label the PM and were examined by confocal microscopy. In mock-treated cells, few YFP-STIM1 puncta were observed, and orthogonal reconstruction (z-stacks) showed localization of YFP-STIM1 within the cell and no colocalization with the PM (Fig. 3A); however, in TG-treated cells, YFP-STIM1 formed puncta that localized in close proximity to the PM, as expected for activated STIM1 (Fig. 3B, arrows). Lastly, we examined the colocalization of YFP-STIM1 puncta with PM CRAC channels.
Fig 2.

Activation of YFP-STIM1 puncta formation by thapsigargin. Shown are still images of YFP-STIM1 fluorescence taken prior to addition of 5 μM thapsigargin (A) or at 5 min (B), 15 min (C), or 30 min (D) post-thapsigargin treatment. Rapid formation of YFP-STIM1 puncta is observed by 5 min post-thapsigargin treatment (B, arrows) and is shown in greater detail in the inset. Scale bar, 50 μm.
Fig 3.

Thapsigargin induces YFP-STIM1 puncta translocation to the plasma membrane. (A and B) YFP-STIM1 cells were mock treated (A) or treated with thapsigargin (B) and then fixed and stained with Texas Red-conjugated wheat germ agglutinin (red). The cellular distribution and localization of YFP-STIM1 were determined by confocal microscopy. Orthogonal views (z-stacks) of each image are shown at the top (xz planes) and right (yz planes). Colocalization of YFP-STIM1 puncta with the plasma membrane is marked by arrowheads. (C and D) YFP-STIM1 cells were transfected with a vector expressing RFP-tagged human Orai1, the canonical CRAC channel. Cells either were mock treated (C) or treated with thapsigargin (D) and then fixed, and the cellular distribution and localization of YFP-STIM1 and Orai1-RFP were determined by confocal microscopy. Colocalization of the activated YFP-STIM1 puncta and Orai1-RFP is marked with arrowheads (D). Scale bar, 10 μm.
Previous studies on the mechanics of SOCE have established that colocalization of STIM1 puncta with Orai CRAC channels equates with activation of calcium entry (12, 13, 26, 30–33). Furthermore, while there are CRAC channels other than Orai1 to Orai3, such as select members of the TRPC family, activation of TRPC channels first requires colocalization of STIM1 and an Orai channel before the TRPC channel can be recruited and activated (16, 17). YFP-STIM1 cells were transfected with a vector encoding RFP-tagged Orai1, the canonical CRAC channel, and were mock or TG treated, fixed, and imaged using confocal microscopy. In mock-treated cells, Orai1-RFP localized to the PM, and the few YFP-STIM1 puncta—likely induced through normal cellular functions—colocalized with Orai1-RFP (Fig. 3C). In TG-treated cells, activated YFP-STIM1 colocalized with Orai1-RFP in punctate structures at ER-PM junctions, indicative of strong activation of SOCE through TG (Fig. 3D, arrowheads). Thus, the YFP-STIM1 biosensor cell line responded as expected to ER Ca2+ store depletion and engaged CRAC channels at ER-PM junctions, as has been well characterized in previous studies (30, 32). These cells are a useful tool to examine whether rotavirus disrupts ER Ca2+ homeostasis and activates SOCE.
Activation of YFP-STIM1 during rotavirus infection.
We proposed that the elevation of cytoplasmic Ca2+ in RV-infected cells is due to NSP4 viroporin activity in the ER decreasing ER Ca2+ levels, and the reduced ER Ca2+ level is sensed by STIM1, the cell's endogenous sensor for maintaining a full ER Ca2+ store, which subsequently activates SOCE. We used the YFP-STIM1 biosensor cell line to determine whether STIM1 becomes activated during RV infection. The YFP-STIM1 cells were infected with SA114F rotavirus at a low MOI (MOI of 0.12) to have a limited number of infected cells. At 8 h postinfection (hpi), YFP-STIM1 puncta were observed only in RV-infected cells but not in adjacent uninfected cells, indicating STIM1 is specifically activated during RV infection (Fig. 4A and D). RV-infected cells were identified by staining with an NSP5-specific antisera (Fig. 4B, arrows) and WGA-TxRed labeling of the PM (Fig. 4C). We next examined the subcellular localization of the YFP-STIM1 puncta in RV-infected cells (Fig. 4E, asterisk), and as was observed after TG treatment, the YFP-STIM1 puncta localized to near the PM at the periphery of the cell (Fig. 4E, arrowheads). To confirm that the YFP-STIM1 puncta in RV-infected cells were localized to ER-PM junctions and colocalized with CRAC channels, we transfected the STIM1 biosensor cells with the Orai1-RFP expression vector and then infected the cells as described above (Fig. 4F to I). Cells were fixed, stained for NSP2 to identify RV-infected cells, and imaged by confocal microscopy. As before, YFP-STIM1 puncta were only observed in RV-infected cells but not in adjacent uninfected cells (Fig. 4F and G), even those expressing Orai1-RFP (Fig. 4F and H, asterisks). Furthermore, in RV-infected cells, Orai1-RFP also forms clusters (Fig. 4H) that colocalized with the STIM1 puncta at the PM (Fig. 4I, arrowheads). Thus, similar to TG treatment, RV infection induces activation of STIM1 and redistribution of STIM1 oligomers to regions of the ER juxtaposed to the PM (ER-PM junctions) to colocalize with PM-localized CRAC channels.
Fig 4.

Activation of YFP-STIM1 in rotavirus-infected cells. (A to D) YFP-STIM1 (green) cells were infected with SA114F and at 8 hpi fixed and stained for rotavirus NSP5 (Alexa Fluor 633 secondary [blue]) and with Texas Red-wheat germ agglutinin (WGA-TxRed [red]). Confocal microscopy shows YFP-STIM1 puncta (i.e., activation) only in rotavirus-infected cells (arrows). (E) Orthogonal views of the xz planes (above) and yz planes (right) from the same field as panels A to D, showing two rotavirus-infected cells. YFP-STIM1 puncta colocalize with WGA-TxRed at the plasma membrane (arrowheads). (F) YFP-STIM1 (green) cells were transfected with the Orai1-RFP (red) expression vector and then infected as described above. Cells were fixed and stained for rotavirus NSP2 (Alexa Fluor 633 secondary [blue]) and then imaged by confocal microscopy. Activated YFP-STIM1 forms puncta that colocalized with Orai1-RFP (arrowheads), but YFP-STIM1 is inactive and does not colocalize with Orai1-RFP in the transfected but uninfected cell (*).
Previous studies showed the formation of NSP4 punctate structures is dependent on elevation of [Ca2+]cyto (8, 28, 34). Therefore, we hypothesize activation of STIM1 should correlate with NSP4 puncta formation because STIM1 activation would activate Ca2+ entry, which would then promote NSP4 puncta formation due to elevated [Ca2+]cyto. Using confocal microscopy of RV-infected cells, identified by staining with NSP5-specific antisera, we quantitated the number of infected cells (>100 cells per time point) with both YFP-STIM1 puncta and NSP4 puncta from 6 to 24 hpi (Fig. 5A). The background percentage of YFP-STIM1 puncta ranged from 5 to 12% (average, 8.7% ± 2.7%) in uninfected cells (>750 cells per time point) (Fig. 5A, gray line). Early during infection, both YFP-STIM1 and NSP4 were primarily reticular (82% [open circles]), with only a few cells showing both punctate YFP-STIM1 and NSP4 (13% [filled circles]). Over the course of the infection, there was a concomitant increase in the number of RV-infected cells with punctate YFP-STIM1 and NSP4 and a decrease in reticular YFP-STIM1 and NSP4 (Fig. 5A); however, only minimal colocalization between YFP-STIM1 puncta and NSP4 puncta was observed, despite substantial colocalization between STIM1 and Orai1 (Fig. 5B). Furthermore, while [Ca2+]cyto was elevated by 6 to 8 hpi, there was no observed decrease in the percentage of cells with activated YFP-STIM1, suggesting that YFP-STIM1 was constitutively activated and not returning to an inactive reticular resting state. The number of cells with punctate YFP-STIM1 but diffuse NSP4 or diffuse YFP-STIM1 but punctate NSP4 remained low (<3%) (Fig. 5A, dashed lines) throughout the infection, further supporting that activation of STIM1 to induce Ca2+ entry underlies the correlation between YFP-STIM1 and NSP4 puncta formation.
Fig 5.

Kinetics of YFP-STIM1 activation in rotavirus-infected cells. (A) Twenty-four-hour time course analysis of YFP-STIM1 activation in rotavirus-infected cells. YFP-STIM1 distribution in uninfected and rotavirus-infected cells was scored as reticular (inactive) or punctate (activate). The YFP-STIM1 distribution was then correlated with NSP4 distribution (reticular versus puncta) in rotavirus-infected cells. The degree of YFP-STIM1 puncta in uninfected cells (gray squares) remained ∼10% throughout the experiment. In rotavirus-infected cells, cells with reticular YFP-STIM1 and reticular NSP4 (open circles) decreased during the infection, while cells with punctate YFP-STIM1 and punctate NSP4 (filled circles) increased. The number of rotavirus-infected cells with reticular YFP-STIM1 but punctate NSP4 (open squares) or punctate YFP-STIM1 but reticular NSP4 (filled squares) remained consistently low throughout the experiment. The percentage of rotavirus-infected cells with punctate YFP-STIM1 was significantly greater than that of uninfected cells starting from 7 hpi (P < 0.01). (B) YFP-STIM1 (green) cells were transfected with the Orai1-RFP (red) expression vector and then infected as described above. Cells were fixed and stained for rotavirus NSP4 (Alexa Fluor 405 secondary [blue]) and imaged by confocal microscopy. The merged image shows that while YFP-STIM1 puncta colocalize with Orai1 (yellow), they did not colocalize with NSP4 puncta, which would generate a white signal.
NSP4 viroporin activity is required for YFP-STIM1 activation.
Induction of STIM1 puncta by RV infection suggests that ER Ca2+ store levels are reduced during RV infection. We have previously shown that expression of recombinant NSP4 in Sf9 insect cells increases Ca2+ permeability of the ER membrane, but not expression of VP7, the only other RV ER-localized protein (4). Recently we identified and characterized a membrane-associated viroporin domain (amino acids [aa] 47 to 90) in NSP4 and showed that mutation of the viroporin domain abolished NSP4-induced elevation in [Ca2+]cyto (8). We hypothesized that direct permeabilization of the ER membrane by NSP4 releases ER luminal Ca2+, causing STIM1 activation. To test this, we transfected the YFP-STIM1 biosensor cells with expression vectors encoding RFP-tagged wild-type SA11 NSP4 (NSP4-WT) or a viroporin-deficient NSP4 mutant (NSP4-Mut), and at 24 h posttransfection, the cells were fixed and imaged by confocal microscopy. Both NSP4-WT and NSP4-Mut are expressed (Fig. 6B versus E) and are stable in transfected cells, as was previously described (8). However, while NSP4-WT spontaneously forms puncta due to elevated [Ca2+]cyto, the NSP4-Mut protein remains reticular because it does not elevate [Ca2+]cyto (8). YFP-STIM1 puncta were observed (Fig. 6A) only in cells expressing NSP4-WT (Fig. 6B) but not in the adjacent cells lacking NSP4-WT expression (Fig. 6C). In contrast, no YFP-STIM1 puncta were observed (Fig. 6D) in cells expressing NSP4-Mut (Fig. 6E), but instead these molecules colocalized in a reticular pattern, indicating a failure of NSP4-Mut to activate STIM1 (Fig. 6F). The occurrence of YFP-STIM1 puncta with expression of NSP4-WT or NSP4-Mut were quantitated, and while expression of NSP4-WT induced YFP-STIM1 puncta in 83% of cells, NSP4-Mut-expressing cells had only a 10% incidence of puncta (Fig. 6G). Thus, the NSP4 viroporin mutant is deficient in triggering YFP-STIM1 activation.
Fig 6.

NSP4 viroporin activity is required for activation of YFP-STIM1 and calcium influx. (A to F) YFP-STIM1 cells were transfected with expression vectors encoding either wild-type NSP4 (NSP4-WT) (A to C) or viroporin-deficient (NSP4-Mut) (D to F) NSP4-RFP proteins. At 24 h posttransfection, cells were fixed and imaged by deconvolution microscopy for YFP-STIM1 and NSP4 localization, with representative images shown in panels A and B. NSP4-WT but not NSP4-Mut activates YFP-STIM1 to form puncta. (G) Quantitation of the number of NSP4-expressing cells with YFP-STIM1 puncta shows that NSP4-Mut is deficient in activating YFP-STIM1 puncta. (H) Extracellular calcium influx into MA104 cells expressing RFP and either RFP-tagged NSP4-WT or the viroporin mutant vector, NSP4-Mut. MA104 cells were transfected with vectors for the GCaMP5G calcium sensor and either wild-type or the viroporin mutant NSP4-RFP vectors. At 24 h posttransfection, baseline GCaMP5G fluorescence (F0) was determined, and then the extracellular calcium level was raised from 1.8 mM to 10 mM. The maximum level in GCaMP5G fluorescence (Fmax) was determined by ImageJ, and the results are reported as ΔF (Fmax/F0). The calcium-induced ΔF was significantly greater (*) in cells expressing NSP4-WT than those expressing NSP4-Mut (P < 0.01). NS, not significant.
To correlate NSP4 activation of STIM1 with induction of the PM Ca2+ entry pathway and to confirm that the NSP4-Mut failed to induce Ca2+ entry, we performed fluorescence Ca2+ imaging in MA104 cells expressing NSP4-RFP. Cells were cotransfected with vectors encoding RFP alone, as a negative-control vector, RFP-tagged NSP4-WT, or NSP4-Mut and the EGFP-based Ca2+ sensor GCaMP5G, which increases its fluorescent emission upon elevation of cytoplasmic Ca2+, similar to small molecule Ca2+ sensors like Fluo-4 (29). At ∼24 h posttransfection, basal GCaMP5G fluorescence was measured, and then extracellular Ca2+ was increased from 1.8 mM to 10 mM while GCaMP5G fluorescence was monitored to assess the change in cytoplasmic Ca2+. In cells expressing RFP alone, a 1.5-fold average increase in GCaMP5G fluorescence was observed after extracellular Ca2+ was increased to 10 mM (Fig. 6H). Expression of wild-type NSP4, known to induce a PM Ca2+ entry pathway, resulted in a 2.1-fold average increase in GCaMP5G fluorescence upon elevation of bath Ca2+ (Fig. 6H). In contrast, cells expressing the viroporin-deficient NSP4-Mut showed no or an attenuated increase in GCaMP5G fluorescence, with a 1.6-fold average increase (range, 1.0- to 3.0-fold), representing a significantly decreased Ca2+ entry compared to that of NSP4-WT (Fig. 6H) (P < 0.001), but no significant difference from cells expressing RFP (P > 0.25). Thus, the viroporin-deficient NSP4 mutant fails to activate STIM1 and does not induce the Ca2+ entry pathway typical of wild-type NSP4-expressing cells. These data indicate a direct correlation between NSP4 viroporin activity and STIM1 activation of Ca2+ entry through CRAC channels, and viroporin activity is the primary mechanism through which NSP4-induced Ca2+ entry is triggered.
RNA interference knockdown of STIM1 impairs rotavirus replication.
The above data suggest STIM1 is activated upon sensing the NSP4 viroporin-mediated depletion of ER Ca2+ stores, thus triggering Ca2+ influx through PM channels. Gaining access to extracellular Ca2+ is necessary for robust RV replication; therefore, we wondered whether STIM1 is a critical cellular cofactor for RV replication. To test this, we generated STIM1 knockdown cell lines by transduction of MA104 cells with lentivirus vectors expressing either a nontargeted short hairpin RNA (shRNA) or two different shRNAs targeting STIM1. All three cell lines showed normal cell viability by trypan blue exclusion, which was 99% in all three cases (data not shown). Partial knockdown of STIM1 expression was confirmed by immunoblot analysis of cell lysates, showing approximately 49% and 66% decreases in STIM1 in cells expressing shRNA1 and shRNA2, respectively, compared to the nontargeted control cell line (Fig. 7A, upper panel). An immunoblot for GAPDH was used as a loading control (Fig. 7A, lower panel). The lack of complete STIM1 knockdown was not surprising since it has been shown to be an essential protein for cell viability (35). To determine whether knockdown of STIM1 expression negatively affected RV replication, the cells were infected with SA114F (MOI of 1) and virus yield was determined at 6 and 12 h postinfection. Virus yields were significantly reduced in both STIM1 shRNA cell lines compared to the control nontargeted shRNA cell line at both 6 and 12 hpi (Fig. 7B and C). At 6 hpi, virus yield was reduced ∼40% and ∼80% for STIM1-targeted shRNA1 and shRNA2, respectively. At 12 hpi, RV yield was reduced by ∼70% and ∼65% for shRNA1 and shRNA2, respectively. These data demonstrate that even moderate reduction in STIM1 levels significantly reduces RV replication, making STIM1 a critical cellular cofactor for RV replication.
DISCUSSION
We previously showed that NSP4 viroporin activity is the underlying function responsible for the elevation of [Ca2+]cyto in RV-infected cells (8). However, the mechanism by which viroporin activity in the ER activates Ca2+ entry across the PM remained unknown. While insertion of the NSP4 viroporin in the ER seems well suited to directly release ER luminal Ca2+ and activate SOCE, the contribution of SOCE to the elevation in [Ca2+]cyto was unclear. To determine whether RV, via NSP4 viroporin activity, reduces ER luminal Ca2+ to activate SOCE, we generated a cell line stably expressing YFP-STIM1 and used this protein as a biosensor to monitor ER Ca2+ store depletion through YFP-STIM1 puncta formation. In both RV-infected and NSP4-WT-expressing cells, YFP-STIM1 was activated and formed punctate structures; however, expression of a viroporin domain mutant failed to substantially activate YFP-STIM1. These data demonstrate that NSP4 viroporin activity in the ER reduces Ca2+ stores below the STIM1 activation threshold, a 35 to 40% decrease in ER Ca2+ store levels (13), and this is consistent with the ∼4-fold decrease in agonist-releasable Ca2+ from the ER observed in NSP4-expressing cells (5). Furthermore, cells expressing the NSP4 viroporin-deficient mutant had no or significantly reduced Ca2+ entry through the PM, indicating that NSP4 viroporin activity in the ER is the initial trigger for the activation of PM Ca2+ influx channels. Finally, STIM1 remained activated throughout the course of RV replication, indicating a chronic depletion in ER Ca2+. The chronic permeabilization of the ER membrane, by NSP4 viroporin activity, kept ER Ca2+ low, resulting in constitutive activation of YFP-STIM1 and increased Ca2+ influx to maintain the elevated [Ca2+]cyto level, which remains elevated for up to 16 h in infected Caco-2 cells (36). Depletion of ER Ca2+ needed to sustain chronic STIM1 activation seems incompatible with the need for ER Ca2+ to assemble VP7 onto RV particles during morphogenesis. Our recent data showing that NSP4 viroporin activity induces autophagy to traffic NSP4 and VP7 to viroplasms (28) suggests that final virus morphogenesis takes place at discrete ER microdomains, which are likely isolated from the ER-PM junctions that are the sites of Ca2+ entry and resequestration into the ER. This predicts that RV may generate a Ca2+ microdomain within membranes that surround viroplasms, but further investigation is needed to determine how reduced ER Ca2+, induction of SOCE, and RV assembly are integrated during infection.
This is the first report that provides mechanistic insight into virus-induced SOCE and the first demonstration of SOCE activation by a viroporin releasing ER Ca2+. Previous studies demonstrated that both hepatitis B virus X protein and Epstein-Barr virus latent membrane protein 1 (LMP-1) increase Ca2+ influx through the Orai1 CRAC channel, but in contrast to NSP4, ER Ca2+ stores are not depleted, suggesting a mechanism of activation not involving viroporin activity (37, 38). Nevertheless, several other viruses both elevate [Ca2+]cyto and encode ER-localized viroporins, suggesting that activation of STIM1 and SOCE through release of ER Ca2+ stores may be a broadly used mechanism to disrupt cellular Ca2+ levels. Several studies on the picornavirus 2B viroporin, particularly poliovirus and coxsackievirus, have shown decreased Ca2+ contents in both the ER and Golgi apparatus and Ca2+ influx through the PM, very similar to the changes seen in RV-infected cells. As with NSP4, mutation of the 2B amphipathic alpha-helix prevents the elevation of [Ca2+]cyto, and van Kuppeveld et al. postulated 2B viroporin could activate SOCE (39). As detailed below, SOCE requires STIM1-mediated activation of Orai1 for Ca2+ entry, and prevention of initial Ca2+ influx significantly impairs elevation of [Ca2+]cyto (16). Since elevation of [Ca2+]cyto is critical for both RV and picornaviruses, the recent development of specific and selective Orai1 blockers raises the possibility that drugs targeting CRAC channels, rather than individual viral targets, may be effective and broadly active antiviral drugs (40).
The full contribution of SOCE in the RV-induced Ca2+ influx pathways has been actively debated for a number of years. Early studies showed that the Ca2+ entry pathway has many characteristics of SOCE, including being permeable to other divalent cations (e.g., Ba2+, Sr2+, and Mn2+) and blocked by La3+ (41). However, the magnitude of Ca2+ entry in RV-infected or NSP4-expressing cells is up to 6-fold greater than that in control cells treated with agonists (e.g., ATP or TG) to fully deplete ER luminal Ca2+ stores (7). Thus, the ultimate contribution of SOCE to the elevated [Ca2+]cyto was unknown, and the involvement of an alternative store-independent Ca2+ entry pathway directly activated by RV was hypothesized (42). Here we show that RV induces SOCE, since activated STIM1 puncta at the PM colocalized with the Orai1 CRAC channel in RV-infected cells, which is similar to standard pharmacologically mediated activation of SOCE by treatment of cells with TG or other agonists (12, 26, 30). These data support early studies on NSP4 (4) and more recent results by Diaz et al., who showed that an Orai1 CRAC channel blocker (2-APB) partially inhibited Ca2+ entry in RV-infected cells (7). However, since only partial inhibition of Ca2+ entry was achieved by blocking Orai1, other non-store-operated channels may also be activated (7). Our results showed that the viroporin mutant NSP4 failed to activate STIM1, suggesting no depletion of ER Ca2+ stores, and essentially no PM Ca2+ entry was observed compared to that in NSP4-WT-expressing cells. These results suggest STIM1 is central to sensing the NSP4-mediated decrease in ER Ca2+ levels and subsequent activation of PM Ca2+ entry. We confirmed that STIM1 is critical for RV replication, since shRNA-mediated knockdown of STIM1 significantly reduced RV yield from MA104 cells. Thus, if store-independent Ca2+ entry channels are activated by NSP4, the viroporin-mediated depletion of ER Ca2+ and STIM1 activation appears to be a necessary initial trigger in the activation of subsequent Ca2+ entry pathways.
Based on our current data and the Ca2+ channel blockers previously shown to inhibit NSP4-induced Ca2+ entry, we propose a model (Fig. 8) that integrates NSP4 viroporin activity in the ER to Ca2+ influx through several different host PM Ca2+ channels. This model predicts that initial ER Ca2+ release, caused by NSP4 viroporin activity, activates STIM1 due to store depletion and thereby triggers SOCE through Orai1, which we have demonstrated in the present study. Previous work on SOCE suggests that activated STIM1/Orai1 subsequently activates other Ca2+ entry channels (16, 17).
Fig 8.

Model for induction of Ca2+ entry by viroporin-mediated activation of STIM1. NSP4 viroporin activity in the ER (gray box) reduces the luminal Ca2+ concentration, activating STIM1. STIM1 traffics to ER-PM junctions (white box), where the cytoplasmic tail of STIM1 binds to and opens CRAC channels, such as Orai1 (shown in boldface). Other downstream effects (shown in italics) may involve recruitment of TRPC channels that could increase the membrane voltage (Vm); this in turn could activate voltage-activated Ca2+ channels (VACC) and the sodium/calcium exchanger in reverse mode (NCXrev). STIM1 may also potentiate NCXrev activation. Drugs that block these Ca2+ entry channels and block NSP4-induced Ca2+ entry are shown. Steps in this model that are not yet demonstrated are indicated with dashed lines and question marks.
Our model of NSP4 viroporin-induced Ca2+ entry (Fig. 8) is further supported by previous pharmacological studies showing that different small molecule ion channel inhibitors block Ca2+ entry (to various extents) into RV-infected cells, including 2-APB, KB-R7943, and D600 (7, 41). As stated above, 2-APB is a widely used Orai1 channel blocker that inhibits SOCE when used at a high concentration (>10 μM), so the ability of 2-APB to block NSP4-induced Ca2+ entry (even partially) supports our data that STIM1 activates Orai1 in RV-infected cells to induce Ca2+ entry (7). The same study also suggested that the sodium-calcium exchanger (NCX) is an important route of Ca2+ entry because Ca2+ entry is blocked by KB-R7943, a commonly used blocker of NCX functioning in “reverse mode.” Under normal conditions, NCX pumps Ca2+ out of cells using the Na+ gradient as the driving force, but if intracellular Na+ is elevated, as seen in RV-infected cells, NCX will pump Na+ out and bring Ca2+ into the cytoplasm. However, interpretation of KB-R7942-mediated inhibition of Ca2+ entry is complicated by the fact that KB-R7943 also potently blocks TRPC channels with a 50% inhibitory concentration (IC50) similar to that for NCX (43). Like Orai1, TRPC channels are also gated by STIM1 and are activated in response to ER Ca2+ store depletion, but TRPC activation first requires activation of Orai1 by STIM1 and Ca2+ influx through Orai1 (17). Since both intracellular Ca2+ and Na+ are elevated in RV-infected cells (2), depolarizing the PM, activation of STIM1/Orai1 by NSP4 viroporin activity may also recruit membrane-potential driven Ca2+ channel, such as NCXrev and voltage-activated Ca2+ channels (Fig. 8) (44). The drug D600 is an inhibitor of L-type voltage-activated Ca2+ channels that blocks RV-induced Ca2+ entry (41), and the L-type α1D (CaV1.3) Ca2+ channel subunit is upregulated 2.1-fold in rhesus rotavirus (RRV)-infected Caco-2 cells (45) (Fig. 8). Finally, it is likely that the physiological characteristics of Ca2+ entry will vary from cell to cell due to differences in the repertoire of Ca2+ channels expressed by the cell; however, STIM1 and Orai1 are ubiquitously expressed, and, therefore, this central axis of SOCE is available to be activated by NSP4 viroporin activity in all RV-susceptible cells.
Recruitment of additional Ca2+ channels due to persistent STIM1 activation may explain the exaggerated Ca2+ entry seen in RV-infected cells. NSP4 viroporin activity causes chronic store depletion and/or STIM1 activation, perhaps activating additional Ca2+ SOCE channels (e.g., TRPC) or typically non-SOCE Ca2+ influx pathways (e.g., NCX and L-type channels) to try to replenish Ca2+ stores. This augmented, pathophysiological form of SOCE could explain why Ca2+ entry in RV-infected cells is much greater than normal with SOCE induced by short-term TG treatment (7). Long-term treatment of cells with TG would more accurately mimic the chronic store depletion caused by NSP4, and previous studies using prolonged treatment (>12 h) with 1 μM TG showed a large sustained [Ca2+]cyto increase, similar to what is observed in RV-infected cells (46). Interestingly, prolonged TG treatment ultimately results in Ca2+-triggered apoptosis, and a recent study showed that elevation in [Ca2+]cyto by NSP4 contributes to the induction of proapoptotic proteins, but progression to apoptosis early during infection is blocked by NSP1 (47).
This report establishes NSP4 viroporin-mediated activation of STIM1 as the initiating trigger for the induction of PM Ca2+ influx into RV-infected cells and demonstrates that NSP4 viroporin activity underlies all of the observed disruptions in cellular Ca2+ homeostasis. Our results also highlight that while multiple Ca2+ entry channels are likely activated in RV-infected cells, activation of these Ca2+ entry pathways is predicated on STIM1 activation by NSP4 viroporin-mediated depletion of the ER Ca2+ stores. This makes NSP4 and other ER-localized Ca2+ viroporins potential tools for dissecting the complexities of SOCE dynamics present during virus infections.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by NIH grants K01DK093657 and R01AI080656, NIH Research Training in Pediatric Gastroenterology grant T32DK007664, Public Health Service grant P30DK56338, which funds the Texas Medical Center Digestive Diseases Center (TMCDDC), and a TMCDDC Pilot and Feasibility Grant. This project was supported by the BCM Integrated Microscopy Core (U54HD007495 and P30CA125123), as well as the BCM Cytometry and Cell Sorting Core with funding from the NIH (AI036211, CA125123, and RR024574).
This project was also supported by the expert assistance of Joel M. Sederstrom.
Footnotes
Published ahead of print 9 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02629-13.
REFERENCES
- 1.Zhou Y, Frey TK, Yang JJ. 2009. Viral calciomics: interplays between Ca2+ and virus. Cell Calcium 46:1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Michelangeli F, Ruiz MC, del Castillo JR, Ludert JE, Liprandi F. 1991. Effect of rotavirus infection on intracellular calcium homeostasis in cultured cells. Virology 181:520–527 [DOI] [PubMed] [Google Scholar]
- 3.Zambrano JL, Diaz Y, Pena F, Vizzi E, Ruiz MC, Michelangeli F, Liprandi F, Ludert JE. 2008. Silencing of rotavirus NSP4 or VP7 expression reduces alterations in Ca2+ homeostasis induced by infection of cultured cells. J. Virol. 82:5815–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian P, Estes MK, Hu Y, Ball JM, Zeng CQ, Schilling WP. 1995. The rotavirus nonstructural glycoprotein NSP4 mobilizes Ca2+ from the endoplasmic reticulum. J. Virol. 69:5763–5772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diaz Y, Chemello ME, Pena F, Aristimuno OC, Zambrano JL, Rojas H, Bartoli F, Salazar L, Chwetzoff S, Sapin C, Trugnan G, Michelangeli F, Ruiz MC. 2008. Expression of nonstructural rotavirus protein NSP4 mimics Ca2+ homeostasis changes induced by rotavirus infection in cultured cells. J. Virol. 82:11331–11343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian P, Hu Y, Schilling WP, Lindsay DA, Eiden J, Estes MK. 1994. The nonstructural glycoprotein of rotavirus affects intracellular calcium levels. J. Virol. 68:251–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diaz Y, Pena F, Aristimuno OC, Matteo L, De Agrela M, Chemello ME, Michelangeli F, Ruiz MC. 2012. Dissecting the Ca(2)(+) entry pathways induced by rotavirus infection and NSP4-EGFP expression in Cos-7 cells. Virus Res. 167:285–296 [DOI] [PubMed] [Google Scholar]
- 8.Hyser JM, Collinson-Pautz MR, Utama B, Estes MK. 2010. Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. mBio 1(5):e00265–10. 10.1128/mBio.00265-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu L, Crawford SE, Hyser JM, Estes MK, Prasad BV. 2012. Rotavirus non-structural proteins: structure and function. Curr. Opin. Virol. 2:380–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Putney JW., Jr 1986. A model for receptor-regulated calcium entry. Cell Calcium 7:1–12 [DOI] [PubMed] [Google Scholar]
- 11.Takemura H, Hughes AR, Thastrup O, Putney JW., Jr 1989. Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. Evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. J. Biol. Chem. 264:12266–12271 [PubMed] [Google Scholar]
- 12.Soboloff J, Rothberg BS, Madesh M, Gill DL. 2012. STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 13:549–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. 2008. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 454:538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou X, Pedi L, Diver MM, Long SB. 2012. Crystal structure of the calcium release-activated calcium channel Orai. Science 338:1308–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ong HL, Ambudkar IS. 2011. The dynamic complexity of the TRPC1 channelosome. Channels (Austin) 5:424–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng KT, Ong HL, Liu X, Ambudkar IS. 2013. Contribution and regulation of TRPC channels in store-operated Ca(2+) entry. Curr. Top. Membr. 71:149–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng KT, Ong HL, Liu X, Ambudkar IS. 2011. Contribution of TRPC1 and Orai1 to Ca(2+) entry activated by store depletion. Adv. Exp. Med. Biol. 704:435–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, Gill DL. 2010. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330:105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park CY, Shcheglovitov A, Dolmetsch R. 2010. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330:101–105 [DOI] [PubMed] [Google Scholar]
- 20.Soboloff J, Spassova M, Xu W, He LP, Cuesta N, Gill DL. 2005. Role of endogenous TRPC6 channels in Ca2+ signal generation in A7r5 smooth muscle cells. J. Biol. Chem. 280:39786–39794 [DOI] [PubMed] [Google Scholar]
- 21.Krapivinsky G, Krapivinsky L, Stotz SC, Manasian Y, Clapham DE. 2011. POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc. Natl. Acad. Sci. U. S. A. 108:19234–19239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitaker AM, Hayward CJ. 1985. The characterization of three monkey kidney cell lines. Dev. Biol. Stand. 60:125–131 [PubMed] [Google Scholar]
- 23.Ciarlet M, Crawford SE, Estes MK. 2001. Differential infection of polarized epithelial cell lines by sialic acid-dependent and sialic acid-independent rotavirus strains. J. Virol. 75:11834–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyser JM, Zeng CQ, Beharry Z, Palzkill T, Estes MK. 2008. Epitope mapping and use of epitope-specific antisera to characterize the VP5* binding site in rotavirus SA11 NSP4. Virology 373:211–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang X, Jayaram H, Kumar M, Ludtke SJ, Estes MK, Prasad BV. 2006. Cryoelectron microscopy structures of rotavirus NSP2-NSP5 and NSP2-RNA complexes: implications for genome replication. J. Virol. 80:10829–10835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. 2005. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15:1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hyser JM, Utama B, Crawford SE, Estes MK. 2012. Genetic divergence of rotavirus nonstructural protein 4 results in distinct serogroup-specific viroporin activity and intracellular punctate structure morphologies. J. Virol. 86:4921–4934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crawford SE, Hyser JM, Utama B, Estes MK. 2012. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-beta signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. U. S. A. 109:E3405–E3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, Shigetomi E, Khakh BS, Baier H, Lagnado L, Wang SS, Bargmann CI, Kimmel BE, Jayaraman V, Svoboda K, Kim DS, Schreiter ER, Looger LL. 2012. Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 32:13819–13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liou J, Fivaz M, Inoue T, Meyer T. 2007. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. U. S. A. 104:9301–9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fahrner M, Muik M, Derler I, Schindl R, Fritsch R, Frischauf I, Romanin C. 2009. Mechanistic view on domains mediating STIM1-Orai coupling. Immunol. Rev. 231:99–112 [DOI] [PubMed] [Google Scholar]
- 32.Derler I, Madl J, Schutz G, Romanin C. 2012. Structure, regulation and biophysics of I(CRAC), STIM/Orai1. Adv. Exp. Med. Biol. 740:383–410 [DOI] [PubMed] [Google Scholar]
- 33.Cahalan MD. 2009. STIMulating store-operated Ca(2+) entry. Nat. Cell Biol. 11:669–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berkova Z, Crawford SE, Trugnan G, Yoshimori T, Morris AP, Estes MK. 2006. Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J. Virol. 80:6061–6071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida J, Iwabuchi K, Matsui T, Ishibashi T, Masuoka T, Nishio M. 2012. Knockdown of stromal interaction molecule 1 (STIM1) suppresses store-operated calcium entry, cell proliferation and tumorigenicity in human epidermoid carcinoma A431 cells. Biochem. Pharmacol. 84:1592–1603 [DOI] [PubMed] [Google Scholar]
- 36.Brunet JP, Cotte-Laffitte J, Linxe C, Quero AM, Geniteau-Legendre M, Servin A. 2000. Rotavirus infection induces an increase in intracellular calcium concentration in human intestinal epithelial cells: role in microvillar actin alteration. J. Virol. 74:2323–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang B, Bouchard MJ. 2012. The hepatitis B virus X protein elevates cytosolic calcium signals by modulating mitochondrial calcium uptake. J. Virol. 86:313–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dellis O, Arbabian A, Papp B, Rowe M, Joab I, Chomienne C. 2011. Epstein-Barr virus latent membrane protein 1 increases calcium influx through store-operated channels in B lymphoid cells. J. Biol. Chem. 286:18583–18592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Kuppeveld FJ, Hoenderop JG, Smeets RL, Willems PH, Dijkman HB, Galama JM, Melchers WJ. 1997. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 16:3519–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Derler I, Schindl R, Fritsch R, Heftberger P, Riedl MC, Begg M, House D, Romanin C. 2013. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 53:139–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez JF, Ruiz MC, Chemello ME, Michelangeli F. 1999. Characterization of a membrane calcium pathway induced by rotavirus infection in cultured cells. J. Virol. 73:2481–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruiz MC, Diaz Y, Pena F, Aristimuno OC, Chemello ME, Michelangeli F. 2005. Ca2+ permeability of the plasma membrane induced by rotavirus infection in cultured cells is inhibited by tunicamycin and brefeldin A. Virology 333:54–65 [DOI] [PubMed] [Google Scholar]
- 43.Kraft R. 2007. The Na+/Ca2+ exchange inhibitor KB-R7943 potently blocks TRPC channels. Biochem. Biophys. Res. Commun. 361:230–236 [DOI] [PubMed] [Google Scholar]
- 44.Liu B, Peel SE, Fox J, Hall IP. 2010. Reverse mode Na+/Ca2+ exchange mediated by STIM1 contributes to Ca2+ influx in airway smooth muscle following agonist stimulation. Respir. Res. 11:168. 10.1186/1465-9921-11-168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cuadras MA, Feigelstock DA, An S, Greenberg HB. 2002. Gene expression pattern in Caco-2 cells following rotavirus infection. J. Virol. 76:4467–4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jackisch C, Hahm HA, Tombal B, McCloskey D, Butash K, Davidson NE, Denmeade SR. 2000. Delayed micromolar elevation in intracellular calcium precedes induction of apoptosis in thapsigargin-treated breast cancer cells. Clin. Cancer Res. 6:2844–2850 [PubMed] [Google Scholar]
- 47.Bhowmick R, Halder UC, Chattopadhyay S, Chanda S, Nandi S, Bagchi P, Nayak MK, Chakrabarti O, Kobayashi N, Chawla-Sarkar M. 2012. Rotaviral enterotoxin nonstructural protein 4 targets mitochondria for activation of apoptosis during infection. J. Biol. Chem. 287:35004–35020 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.