

Abstract
Ion charge pairs and hydrogen bonds have been extensively studied for their roles in stabilizing protein complexes and in steering the process of protein association. Recently, it has become clear that some protein complexes are dynamic in that they interconvert between several alternate configurations. We have previously characterized one such system: the EphA2:SHIP2 SAM-SAM heterodimer by solution NMR. Here we carried out extensive all-atom molecular-dynamics simulations on a microsecond time-scale starting with different NMR-derived structures for the complex. Transitions are observed between several discernible configurations at average time intervals of 50–100 ns. The domains reorient relative to one another by substantial rotation and a slight shifting of the interfaces. Bifurcated and intermediary salt-bridge and hydrogen-bond interactions play a role in the transitions in a process that can be described as moving along a “monkey-bar”. We notice an increased density of salt bridges near protein interaction surfaces that appear to enable these transitions, also suggesting why the trajectories can become kinetically hindered in regions where fewer of such interactions are possible. In this context, even microsecond molecular-dynamics simulations are not sufficient to sample the energy landscape unless the structures remain close to their experimentally derived low-energy configurations.
Introduction
Accurate prediction of protein complexes remains a challenge when no structural or sequence homology is available, or in cases where the proteins undergo considerable conformational changes upon association (1,2). Recently, it has also become apparent that some protein complexes are inherently dynamic, fluctuating locally or even at the (sub-)domain level, between different configurations (3–6). We have been characterizing such a protein complex by solution NMR measurements (6), and here by molecular-dynamics simulations. Previous studies have shown that the free-energy landscape of protein-protein complexes can range from simple funnel-like to highly complicated terrains (7,8). How residue-residue and residue-solvent interactions determine the nature of the landscape, and which interactions allow transitions between low energy configurations, are important features of protein interactions that remain to be understood in detail (9,10). In particular, the roles of ion-pair and hydrogen-bond interactions for the stability of the protein complex and for the process of protein association have been heavily studied, indicating sometimes complex and often context-dependent contributions (11,12).
Here we study two features of the ensemble of structures that comprise a dynamic protein complex, as explored by extensive molecular dynamics simulations.
First, the 2.4-μs all-atom molecular dynamics simulations of the complex are used to sample the energy landscape; cluster centers are validated with respect to the experimental NMR restraints. However, the results also suggest that simulations can go off-course, leaving a simple landscape to become trapped in a more restrictive terrain. Second, the trajectories are used to examine transitions between different configurations of the protein-protein complex, especially with respect to the role of ion pairs (salt bridges) and hydrogen-bond interactions in enabling the transitions. Finally, these two findings are proposed to be related, considering the distribution of surface groups.
Computational Methods
Simulations were performed starting from NMR-derived configurations of the EphA2:SHIP2 SAM-SAM complex, using both the software NAMD Vers.2.8 (13) and the ANTON Supercomputer (14) simulation programs. The structures have been determined previously in the laboratory (6) from solution NMR restraints.
Molecular-dynamics simulations
Six simulations were run, each started with different structures. Three simulations began with the lowest energy structures of three different configurations that were derived (Clusters1–3). A fourth structure was the next lowest energy structure of the Cluster3 configuration. The fifth simulation was started with a Cluster1-like configuration that was derived by NMR restraints in a calculation omitting several unambiguous distance restraints. The sixth simulation began with a structure far away from the others (arising as a minor population in the second, control calculation as described in Lee et al. (6)). The starting structures were solvated in a rectangular box of explicitly represented water (TIP3P), minimized and equilibrated for 20 ns using unrestrained all-atom molecular-dynamics (MD) simulation at constant temperature and pressure (300 K and 1 atm). The simulation box contained ∼16,500 TIP3P water molecules, and the box size was around 90 × 70 × 70 Å3. The standard particle-mesh Ewald method was applied to calculate the long-range electrostatic interactions. The system was neutralized by adding ions to a concentration of 0.15 M NaCl. The CHARMM27 all-atom potential function was used including the CMAP correction (15). For nonbonded calculations, a cutoff of 12 Å was used. All bonds, involving hydrogen, were kept rigid using the SHAKE algorithm, allowing 2-fs timesteps. After 20-ns NAMD simulations, we continued the calculations with the ANTON Supercomputer for 2.4 μs. Coordinates were saved every 50 ps.
Trajectory analysis
K-class clustering of the trajectories was carried out using the software WORDOM (16). Solvent-accessible surface calculation used the Lee and Richards algorithm in the CHARMM program (Academic edition from Harvard University) with a probe radius of 1.4 Å. Distances of all protein side-chain hydrogen-bond donors and acceptors to the nearest group across the interface and to water were calculated. The groups involved were identified and intergroup distances were plotted (see Fig. 4). For Table 1, any residue involved in interactions in the four main interface configurations (derived by clustering the four trajectories showing transitions) was counted as an interface residue.
Figure 4.
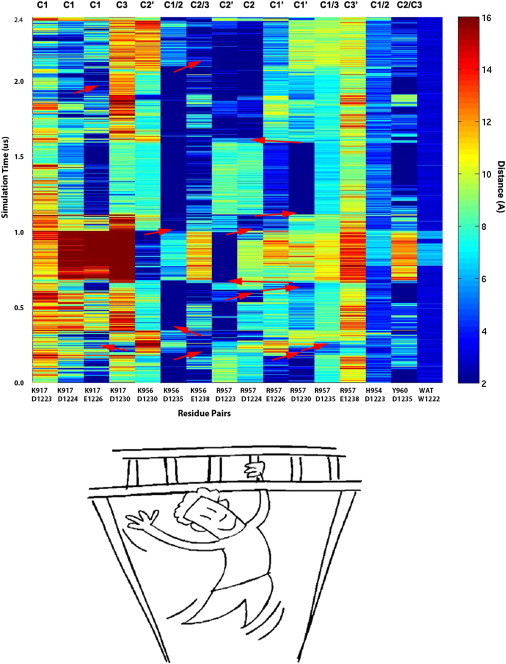
Transitions between residue pairs across the interface as a function of simulation time. (Dark blue) Close contacts; (yellow to red) distant interactions (scale on right). Contact pairs are identified below and associated with cluster/cluster-like configuration above (C#/C#′); see also Fig. S3 in the Supporting Material. It is apparent that in many transitions additional contacts form before old ones are broken (indicated by red arrows). For example, K956-D1238 forms before K956-D1235 is broken at ∼0.25 μs. Similarly, R957 transitions from E1226 to E1230 and then D1235 as partners, before returning to D1224 and then D1223, around the same time. This is conceptually similar to holding onto and traveling along a monkey bar, as depicted below.
Table 1.
Distribution of charged and H-bonding groups on SAM domains
| EphA2 charged | H-bond | SHIP2 charged | H-bond | |
|---|---|---|---|---|
| Interface | 38% | 46% | 47% | 21% |
| Noninterface | 17% | 23% | 27% | 15% |
Results and Discussion
We previously reported the refined structure of the EphA2:SHIP2 SAM-SAM complex (6). While 80% of the NMR-refined structures populated one cluster of conformations (referred to as Cluster1), two additional clusters of structures were observed (Clusters2 and 3; Fig. 1). These structures required further investigation because one of them (Cluster2) was close to the earlier model that had been derived by another group from fewer, and only ambiguous, NMR restraints (17). However, our study also indicated fluctuations toward these alternative states in a 20-ns MD simulation (6). Here we computed lengthy 2.4 μs trajectories on the MD-optimized supercomputer ANTON (15), starting with a structure representative of each of these three clusters as well as other configurations. The simulations were run with standard protocols (see Computational Methods). A typical analysis carried out with the trajectories is shown in Fig. 2. It is apparent that interconversions between Clusters1 and 2 and more rarely with Cluster3-like structures take place on a timescale of 50–100 ns (see Movie S1 in the Supporting Material).
Figure 1.
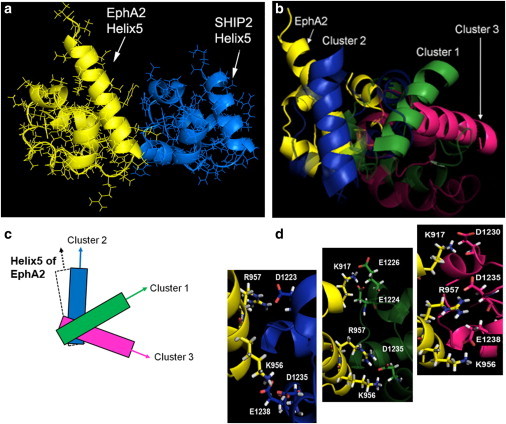
SAM-SAM heterodimer configurations derived from NMR (6) (a) Cluster 2 with fifth helix indicated in each domain; (b) Clusters 1–3, with fifth helix in the SHIP2 SAM domain highlighted; (c) schematic showing different domain orientations; and (d) ion-pairs across the interface in the different configurations.
Figure 2.
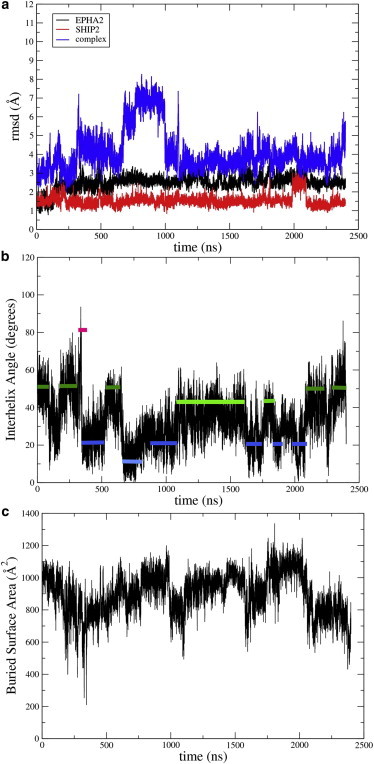
Trajectory analysis for a simulation started from a Cluster-1 conformation. (a) Root-mean-squared deviation from starting structures for EphA2 and SHIP2 SAM domains as well as for the complex; (b) angle between the fifth helix of the two domains (different configurations sampled are indicated by color bars with reference to Fig. 1c); and (c) buried solvent-accessible surface area at the protein-protein interface. To see this figure in color, go online.
Interestingly, the other two trajectories (started with Cluster2 and 3 structures) also sample states near Cluster1, as did a fourth trajectory started with a Cluster1-like structure (see Fig. S1 in the Supporting Material). The four trajectories were combined and clustered (Fig. 3 a), yielding four main clusters of configurations, represented by cluster centers, with occupancies of 53, 17, 15, and 9%, respectively, as well as two minor clusters (<5% occupancy). When these configurations are compared with the experimental NMR data, it is clear that four of six clusters (populating 82% of structures) satisfy the NMR data reasonably well, whereas two other clusters satisfy the NMR data less well (see Table S1 a in the Supporting Material). A similar picture emerges when structures from the two additional trajectories, started with minor configurations (see Computational Methods), are added to the clustering. It is apparent that these two trajectories do not move closer to any of the main configurations, but are unconverged and appear kinetically trapped in a region of the energy landscape (Fig. 3 b, and see Table S1 b, Fig. S2, and Movie S2).
Figure 3.
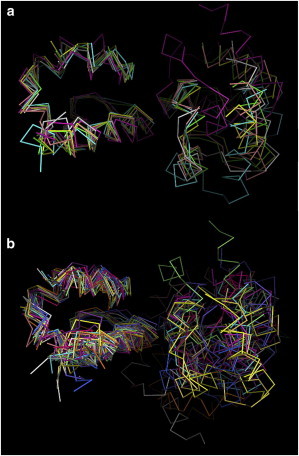
Cluster centers from (a) four and (b) six 2.4-μs trajectories. The clustering was done by side-chain, root-mean-squared deviation (5.5 Å cutoff) of the residues at the Clusters 1–3 SAM-SAM interface. Here the structures are superimposed on the EphA2 SAM domain (left) for visual display. To see this figure in color, go online.
Clustering that includes these latter two trajectories results in 14 cluster centers (only three with populations >10%). Most do not satisfy the experimental data as compared to the clusters from the four initial trajectories (see Table S1). This raises the following question: what features of a protein surface generate a smooth landscape that provides facile but contained transitions, compared to surfaces and interactions that allow protein structures to wander into complex and trapped terrains?
The principal clusters, which are sampled in the initial four well-behaved trajectories, correspond to the experimentally derived structures and show interconversion, as noted above. Geometrically, the structures differ by the alignment of the two SAM domains, both in terms of rotation (measured by the angle between helix 5 of each domain, Fig. 1, a–c) and by a slight shift in the interface (not shown). An explanation for these different configurations are different pairings of charged residues across the protein-protein interfaces, as shown in Fig. 1 d. Several of the pairings overlap, in part, participating in bifurcated bonding with the same acceptor group. The evolution of key interactions in the trajectory started with the Cluster1 configuration is summarized in Fig. 4. It is apparent that, although some of the residue-residue contacts, such as K917-D1224 and K917-D1230, are mutually exclusive for large parts of the trajectory, others display considerable degeneracy; that is, they occur in several of the clusters, as pointed out for the experimental structure determination. For example, in the NMR-derived Cluster2 configuration, K956-D1235 and K956K-E1238, can form simultaneously and these interactions persist for considerable parts of the trajectory, because the two residues (D1235 and E1238) are located close to one another. This interaction is also seen as an intermediate in transitioning to Cluster1-like configurations, where only K956-D1235 can form.
When the closest separation distances are plotted as a function of simulation time, it becomes apparent that many transitions are associated with making and breaking of contacts but in a manner that resembles movement along a monkey bar, i.e., a new contact is formed before the previous one is released. This way the proteins always make some interactions (i.e., in the vernacular sense, at least one of the hands is on the bar). This is true for the other three trajectories that show frequent transitions (see Fig. S3, a–c). Waters may participate in this process by forming bifurcated hydrogen bonding. A detailed trajectory analysis shows that amide and oxygen side-chain groups are close to water molecules throughout the trajectories (data not shown); the only residue side-chain group that becomes fully desolvated in many of the trajectories is SHIP2 W1223, but even this group is desolvated only temporarily (last column of Fig. 4, and similarly in Fig. S3, a–c). Solvation is likely to be important for the transitions, as well as for protein-dissociation and association processes (18). A dissociation of proteins is not seen in these simulations, but detailed in other simulations with a SAM-SAM complex where two key interface residues have been mutated (unpublished data).
The trajectories that move far away from the starting structures show few interconversion events involving ion pairs and hydrogen bonds (see Fig. S4, a and b). Examining the surfaces in contact, there are fewer charged residues (and H-bond donors and acceptors; see Table 1, above). We find the protein-protein complex configurations that are more highly populated also have more ion-pair and hydrogen-bonding residue contacts than others. However, having more interactions does not necessarily mean that the total protein-protein interaction is stronger, and a conclusion in this respect will require detailed free-energy calculations. The suggestion that native protein-protein interfaces have characteristics that are different from other protein surfaces has found support in several structural analyses (9–12). In a followup study, we find that the dissociation of mutant SAM-SAM complexes takes place from configurations in which at least one of the interface regions differs from those utilized by the dynamic and stable configurations near the experimental starting structures.
It should be noted that although the solvent-accessible surface area, which is buried between the proteins, can fluctuate significantly (e.g., Fig. S2 a), the proteins simulated here never truly separate. Interactions, such as those of R957-D1235 and R957-E1238, stay within contact distance (<5 Å) (see Fig. S4, a and b). It is intriguing to note that the latter interaction is not predominant in the four trajectories that show configurational interconversion. Considering the process of protein association, it is possible that such anchoring interactions (19,20) may form easily but might also trap the protein complex in a certain region of the configurational landscape.
Given resources, it would be desirable to run even longer simulations (hundreds of microseconds to milliseconds), but our observations also need to be viewed against a background of studies that have shown that sampling can be influenced by inadequacies in the potential function used in the simulations (e.g., Trbovic et al. (21)), especially when the energy barriers are high, such as in protein refinement (22). However, along with recent reports (e.g., Zeiske et al. (23)), this study suggests that if the protein complex is maintained near experimentally derived configurations, a realistic sampling and transitions may be obtained. Remarkably, we note that surfaces with fewer possible groups for protein interactions appear to lead to slower transition kinetics or even trapped states.
This study extends observations and concepts developed for protein folding and single-domain structural transitions to protein-protein interactions. Specifically, hydrogen-bonding intermediary states are known to play a role in helix-coil transitions (24–26) and in a cell-signaling protein (e.g., Lei et al. (27)). The kinetic role of waters in these transitions is thought to be small: Water moves around the protein much more quickly than even its side-chain transitions (28). More importantly, as seen in the configurational transitions described here, charged/polar side chains that line the protein-protein interface remain at least partially solvated, as found in the great majority of protein interfaces (18,29,30). By contrast, larger-scale desolvation may be expected to slow protein association events (e.g., Camacho et al. (31)).
Finally, experimental studies, examining possible functional differences between the different configurations of the EphA2:SHIP2 SAM-SAM complexes, are needed to show that the different configuration have a biological significance. This will be a considerable undertaking, because the studies need to be carried out for the whole-length receptor/SHIP2 protein as well as in proximity of the plasma membrane, which may influence the equilibrium. For now, however, our study delineates the behavior of the interaction at the SAM-SAM domain level, and the dynamic nature of the interface indicated by our previous NMR study can be seen directly in microsecond simulations.
Acknowledgments
The work was funded by a National Institutes of Health grant to M.B. (No. R01GM092851) and a postdoctoral fellowship to L.Z. (No. T32DK007470). The use of the ANTON Supercomputer at the Pittsburgh Supercomputing Center is supported by a National Institutes of Health award (No. RC2GM093307) to Carnegie Mellon University. Part of this work also used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575.
Supporting Material
References
- 1.Janin J. Protein-protein docking tested in blind predictions: the CAPRI experiment. Mol. Biosyst. 2010;6:2351–2362. doi: 10.1039/c005060c. [DOI] [PubMed] [Google Scholar]
- 2.de Vries S.J., Melquiond A.S., Bonvin A.M. Strengths and weaknesses of data-driven docking in critical assessment of prediction of interactions. Proteins. 2010;78:3242–3249. doi: 10.1002/prot.22814. [DOI] [PubMed] [Google Scholar]
- 3.Hu J., Hu K., Clore G.M. Solution NMR structures of productive and non-productive complexes between the A and B domains of the cytoplasmic subunit of the mannose transporter of the Escherichia coli phosphotransferase system. J. Biol. Chem. 2008;283:11024–11037. doi: 10.1074/jbc.M800312200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boehr D.D., Nussinov R., Wright P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindfors H.E., Drijfhout J.W., Ubbink M. The Src SH2 domain interacts dynamically with the focal adhesion kinase binding site as demonstrated by paramagnetic NMR spectroscopy. IUBMB Life. 2012;64:538–544. doi: 10.1002/iub.1038. [DOI] [PubMed] [Google Scholar]
- 6.Lee H.J., Hota P.K., Buck M. NMR structure of a heterodimeric SAM:SAM complex: characterization and manipulation of EphA2 binding reveal new cellular functions of SHIP2. Structure. 2012;20:41–55. doi: 10.1016/j.str.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai C.J., Kumar S., Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999;8:1181–1190. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schreiber G., Haran G., Zhou H.-X. Fundamental aspects of protein-protein association kinetics. Chem. Rev. 2009;109:839–860. doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones S., Thornton J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo Conte L., Chothia C., Janin J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 11.Xu D., Tsai C.J., Nussinov R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 1997;10:999–1012. doi: 10.1093/protein/10.9.999. [DOI] [PubMed] [Google Scholar]
- 12.Sheinerman F.B., Norel R., Honig B. Electrostatic aspects of protein-protein interactions. Curr. Opin. Struct. Biol. 2000;10:153–159. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 13.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaw D.E., Deneroff M.M., Wang S.C. Anton, a special-purpose machine for molecular dynamics simulation. Commun. ACM. 2008;51:91–97. [Google Scholar]
- 15.Buck M., Bouguet-Bonnet S., MacKerell A.D., Jr. Importance of the CMAP correction to the CHARMM22 protein force field: dynamics of hen lysozyme. Biophys. J. 2006;90:L36–L38. doi: 10.1529/biophysj.105.078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seeber M., Cecchini M., Caflisch A. WORDOM: a program for efficient analysis of molecular dynamics simulations. Bioinformatics. 2007;23:2625–2627. doi: 10.1093/bioinformatics/btm378. [DOI] [PubMed] [Google Scholar]
- 17.Leone M., Cellitti J., Pellecchia M. NMR studies of a heterotypic SAM-SAM domain association: the interaction between the lipid phosphatase SHIP2 and the EPHA2 receptor. Biochemistry. 2008;47:12721–12728. doi: 10.1021/bi801713f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodier F., Bahadur R.P., Janin J. Hydration of protein-protein interfaces. Proteins. 2005;60:36–45. doi: 10.1002/prot.20478. [DOI] [PubMed] [Google Scholar]
- 19.Kortemme T., Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc. Natl. Acad. Sci. USA. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keskin O., Ma B.Y., Nussinov R. Hot regions in protein-protein interactions: the organization and contribution of structurally conserved hot spot residues. J. Mol. Biol. 2005;345:1281–1294. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 21.Trbovic N., Kim B., Palmer A.G., 3rd Structural analysis of protein dynamics by MD simulations and NMR spin-relaxation. Proteins. 2008;71:684–694. doi: 10.1002/prot.21750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raval A., Piana S., Shaw D.E. Refinement of protein structure homology models via long, all-atom molecular dynamics simulations. Proteins. 2012;80:2071–2079. doi: 10.1002/prot.24098. [DOI] [PubMed] [Google Scholar]
- 23.Zeiske T., Stafford K.A., Palmer A.G., 3rd Starting-structure dependence of nanosecond timescale intersubstate transitions and reproducibility of MD-derived order parameters. Proteins. 2013;81:499–509. doi: 10.1002/prot.24209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarek M., Tobias D.J. Role of protein-water hydrogen bond dynamics in the protein dynamical transition. Phys. Rev. Lett. 2002;88:138101. doi: 10.1103/PhysRevLett.88.138101. [DOI] [PubMed] [Google Scholar]
- 25.Armen R., Alonso D.O., Daggett V. The role of α-, 3(10)-, and π-helix in helix→coil transitions. Protein Sci. 2003;12:1145–1157. doi: 10.1110/ps.0240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monticelli L., Tieleman D.P., Colombo G. Mechanism of helix nucleation and propagation: microscopic view from microsecond time scale MD simulations. J. Phys. Chem. B. 2005;109:20064–20067. doi: 10.1021/jp054729b. [DOI] [PubMed] [Google Scholar]
- 27.Lei M., Velos J., Kern D. Segmented transition pathway of the signaling protein nitrogen regulatory protein C. J. Mol. Biol. 2009;392:823–836. doi: 10.1016/j.jmb.2009.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamaneh M.B., Zhang L., Buck M. A direct coupling between global and internal motions in a single domain protein? MD investigation of extreme scenarios. Biophys. J. 2011;101:196–204. doi: 10.1016/j.bpj.2011.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bueno M., Temiz N.A., Camacho C.J. Novel modulation factor quantifies the role of water molecules in protein interactions. Proteins. 2010;78:3226–3234. doi: 10.1002/prot.22805. [DOI] [PubMed] [Google Scholar]
- 30.Reichmann D., Phillip Y., Schreiber G. On the contribution of water-mediated interactions to protein-complex stability. Biochemistry. 2008;47:1051–1060. doi: 10.1021/bi7019639. [DOI] [PubMed] [Google Scholar]
- 31.Camacho C.J., Kimura S.R., Vajda S. Kinetics of desolvation-mediated protein-protein binding. Biophys. J. 2000;78:1094–1105. doi: 10.1016/S0006-3495(00)76668-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.