

Abstract
Alzheimer’s disease is hypothesized to be caused by an over-production or reduced clearance of amyloid-beta (Aβ) peptide. Autosomal Dominant Alzheimer’s Disease (ADAD) caused by mutations in the presenilin (PSEN) gene have been postulated to result from increased production of Aβ42 compared to Aβ40 in the central nervous system (CNS). This has been demonstrated in rodent models of ADAD but not in human mutation carriers We used compartmental modeling of stable isotope labeling kinetic (SILK) studies in human carriers of PSEN mutations and related non-carriers to evaluate the pathophysiological effects of PSEN1 and PSEN2 mutations on the production and turnover of Aβ isoforms. We compared these findings by mutation status and amount of fibrillar amyloid deposition as measured by positron emission tomography (PET) using the amyloid tracer, Pittsburgh compound B (PiB). CNS Aβ42 to Aβ40 production rates were 24% higher in mutation carriers compared to non-carriers and this was independent of fibrillar amyloid deposits quantified by PET PiB imaging. The fractional turnover rate of soluble Aβ42 relative to Aβ40 was 65% faster in mutation carriers and correlated with amyloid deposition, consistent with increased deposition of Aβ42 into plaques leading to reduced recovery of Aβ42 in cerebrospinal fluid (CSF). Reversible exchange of Aβ42 peptides with pre-existing unlabeled peptide was observed in the presence of plaques. These findings support the hypothesis that Aβ42 is overproduced in the CNS of humans with presenilin mutations that cause AD, and demonstrate that soluble Aβ42 turnover and exchange processes are altered in the presence of amyloid plaques, causing a reduction in Aβ42 concentrations in the CSF.
Introduction
The pathogenic causes of Alzheimer’s disease (AD) are not fully understood, partly due to the difficulty in demonstrating the steps that lead to dementia in humans. However, rare autosomal dominant AD (ADAD) can be predicted with nearly 100% certainty in individuals with specific mutations in the genes encoding presenilin 1 (PSEN1), presenilin 2 (PSEN2), or the amyloid precursor protein (APP)(1). Recent findings suggest that a series of pathophysiological changes occur in the brains of ADAD mutation carriers ecades before clinical dementia manifests (2).
The amyloid hypothesis (3) predicts that AD is caused by increased production or decreased clearance of amyloid-beta (Aβ) in the brain, resulting in amyloidosis and a pathological hallmark of AD, amyloid plaques. Aβ is produced from the C-terminal fragment of amyloid precursor protein (APP) by cleavage of APP by β-secretase to form C99, followed by cleavage of C99 by PSEN1 or PSEN2, the enzymatic components of γ-secretase (4). The principal peptide produced by γ-secretase is a 40-amino acid peptide (Aβ40), but this enzymatic cleavage lacks specificity and peptides ranging in length from 38 to 43 amino acids are produced (5). Aβ42 has attracted considerable attention because it has greater amyloidgenic properties and is the principal component of amyloid plaques (6). In support of the amyloid hypothesis, an APP mutation that reduces Aβ production is associated with a strong protective effect against AD (7), whereas duplication of APP or mutations that are thought to increase total Aβ species or Aβ42 relative to Aβ40 cause dominantly inherited AD (8–10). In cell culture and in plasma from human ADAD participants, PSEN mutations have been reported to increase the Aβ42:Aβ40 ratio (11–14), which is hypothesized to increase the risk of amyloidosis (15). However, others have found that neither the Aβ42:Aβ40 ratio nor Aβ42 concentrations are increased in vitro (16). Furthermore, findings of reduced Aβ42 concentrations in the cerebrospinal fluid (CSF) of ADAD participants (13, 17) do not directly support the hypothesis that increased Aβ42 production is an etiological mechanism in dominantly inherited AD.
We previously used stable isotope labeling kinetics (SILK) to examine the turnover kinetics of Aβ in sporadic late onset AD (18). Both sporadic AD and ADAD are associated with lower CSF Aβ42 concentrations and Aβ42:Aβ40 ratios (13, 17, 19–21). However, PSEN mutations in ADAD are hypothesized to cause increased Aβ42 production (4, 12, 22), although direct evidence for increased in vivo production of Aβ42 in humans has not been reported.
We hypothesized that the CNS Aβ42:Aβ40 production rate ratio is increased in PSEN1 and PSEN2 mutation carriers compared to non-carriers. To address this hypothesis, we performed in vivo SILK studies in participants with ADAD mutations and sibling non-carrier controls. A comprehensive compartmental model was developed to determine steady state metabolic kinetic parameters including fractional turnover rates and production rates of Aβ38, Aβ40, and Aβ42 for each participant. The Aβ kinetic parameters were compared to the presence of a PSEN mutation and insoluble amyloid deposition, which was measured by positron emission tomography (PET) imaging of Pittsburgh compound B (PiB-PET)
Results
Stable isotope labeling kinetics (SILK) was performed in 11 individuals who carry mutations in PSEN associated with ADAD, and in 12 sibling control persons who do not carry PSEN mutations. PiB-PET imaging (23) showed virtually no detectable amyloidosis in all non-carriers and in 4 of the mutation carriers (denoted PIB−), and considerable amyloidosis in 7 mutation carriers (denoted PIB+) (Fig. 1A). A mean cortical binding potential (MCBP) score was calculated from the PET images to obtain a continuous quantitative covariate for statistical analyses.
Fig. 1. PET images and isotopic enrichment time course profiles of CSF Aβ peptides.

(A) Composite PET images showing [11C]Pittsburgh compound B binding in participants who are non-carriers of PSEN mutations (left column), and PSEN mutation carriers who lack (PIB−, middle column) or have (PIB+, right column) evidence of amyloidosis. (B, C) Average Aβ isotopic kinetic time course profiles in CSF showing the Aβ42:40, Aβ38:40, and Aβ42:38 isotopic enrichment ratios (B, middle panel: Aβ38:40, blue circles, Aβ42:38 green squares, and Aβ42:40 red triangles) and as enrichment ratios normalized to plasma leucine plateau enrichments (C, lower panel: Aβ38 blue circles, Aβ40 green squares, Aβ42 red triangles). Aβ38 and Aβ40 present similar labeling profiles in all subject groups, whereas Aβ42 kinetics deviate from Aβ38 and Aβ40 only in mutation carriers, as evident in the Aβ42:40 and Aβ42:38 ratio profiles. Data points represent the mean ± SD for group-averaged values, and the solid lines represent the model fits to the data using the model in Fig. 2.
Differential Aβ isoform kinetics by mutation and amyloid deposition
Plasma leucine enrichment approximated a constant plateau during the 9-h intravenous 13C6-leucine infusion, and then rapidly decreased after the infusion was stopped (Fig. S1). The 13C6-leucine isotopic enrichments of Aβ38, Aβ40, and Aβ42 recovered from CSF were compared between mutation carriers, with or without amyloidosis, and non-mutation carriers to address the relationship between Aβ isoform metabolic kinetics, mutation status, and amyloid deposition.
To compare Aβ isoform kinetics, ratios of labeled Aβ isoform enrichments in the CSF were plotted so that a ratio of one indicates the same isotopic enrichment and kinetics between Aβ isoforms. The Aβ38:Aβ40 labeling ratio was approximately one from 5 to 36 hours in all participant groups (Fig. 1B, no significant group by time interaction), indicating similar kinetics between Aβ38 and Aβ40. Similarly, the Aβ42:40 and Aβ42:38 labeling ratios were nearly constant at one over time in non-carriers. However, in mutation carriers, the Aβ42:40 and Aβ42:38 labeling ratios were elevated during early time points and decreased at later time points (Fig. 1B) in relation to plaque status. Repeated measures ANOVA revealed a significant group by time interaction for both the Aβ42:40 and Aβ42:38 labeling ratios (P<0.0001), with a significant linear contrast (P<0.01) between groups at ≤9 hours and at ≥18 hours showing an increasing magnitude of effect (non-carrier < mutation carrier PIB− < mutation carrier PIB+). The Aβ isoform enrichment mismatch was more pronounced in participants with amyloid deposition (PIB+), caused by an earlier and lower Aβ42 peak with a flatter terminal tail compared to Aβ38 and Aβ40 (Fig. 1C). The time to reach peak 13C-labeling in each Aβ isoform was measured for each participant. The Aβ38:Aβ40 peak time ratio was not different between mutation carrier and non-carrier groups (1.01±0.01 vs. 1.00±0.01, respectively). In contrast, Aβ42 peaked at the same time as Aβ40 in the non-carrier group (Aβ42:Aβ40 peak time ratio = 1.01±0.03), whereas Aβ42 peaked significantly earlier than Aβ40 in the mutation group (peak time ratio = 0.93±0.05, p=0.015 mutation effect, p<0.001 for PIB MCBP score).
A Multi-compartment model to describe Aβ isoform kinetics
A comprehensive compartmental model was developed to quantify steady state Aβ isoform kinetic parameters for each participant dataset. The model incorporated the plasma leucine, Aβ enrichment time course profiles, and the CSF Aβ isoform concentrations for each subject (schematically depicted in Fig. 2). The Supplementary Material shows curve fits for each Aβ isoform time course for each participant (Figs. S3–S5). A reversible exchange compartment was necessary to fit the sigmoidal decay of many labeling curves, especially Aβ42 in PIB+ participants. The model included an irreversible loss of each soluble Aβ isoform that was not recovered in CSF. The rate constants for transfer between compartments in the model were optimized for each participant and mean values for each parameter are summarized in Table S1. The model describes three fundamental processes that affect Aβ kinetics: production, reversible exchange, and irreversible loss, and accounts for the effect of these processes on CSF Aβ concentrations.
Fig. 2. Schematic diagram of compartmental model of Aβ38, Aβ40, and Aβ42 metabolism.

Solid black triangles depict sampling sites for plasma leucine and CSF Aβ peptides. Production of Aβ peptides is signified by yellow arrows, exchange by blue arrows, and irreversible loss by red arrows. The model incorporated the labeling time course of plasma 13C6-leucine, APP production and processing to C99 peptide, and Aβ38, Aβ40, or Aβ42 production from C99. The labeled Aβ38, Aβ40, and Aβ42 that are sampled in CSF are presumed to be soluble within the “brain” compartment. The soluble Aβ peptides may exchange with other unlabeled Aβ structures, may be transported to CSF, or may be lost due to other processes (e.g. transport to blood, plaque deposition, or cellular degradation). Transport through CSF is modeled as a three compartment time delay.
Increased Aβ42 and Aβ42:Aβ40 production with PSEN mutations regardless of amyloid deposition
The mutation carriers had an 18% higher absolute Aβ42 production rate (P<0.05 for mutation effect) and a 24% higher Aβ42:Aβ40 production rate ratio (P<0.0001 for mutation effect) than the non-mutation carriers, whereas amyloid load as measured by PIB-PET did not have a significant effect on Aβ42 production rate (Table 2, Fig. 3A, Fig. S5). There were no differences in Aβ38 or Aβ40 absolute production rates or the Aβ38:Aβ40 production rate ratio between carrier and non-carrier groups (Table 2).
Table 2.
CSF Aβ isoform concentrations and selected model kinetic parameters.
| Non-carriers (n=12) | Mutation carriers (n=11) | P-valuesa | ||
|---|---|---|---|---|
| Production rate, ng/h (e.g. C99 pool size × kAβ42) | Mutation status | PIB MCBP score | ||
| Aβ38 | 106[41] | 111[50] | 0.603 | 0.571 |
| Aβ40 | 418±83 | 452±138 | 0.621 | 0.901 |
| Aβ42 | 57[19] | 67[35] | 0.038 | 0.769 |
| Aβ38:Aβ40 ratio | 0.267[0.021] | 0.252[0.052] | 0.692 | 0.179 |
| Aβ42:Aβ40 ratio | 0.140±0.011 | 0.174±0.020 | 9×10−5 | 0.312 |
| Percentage of flux going to exchange (%)b | Mutation status | PIB status | ||
| Aβ38 | 9.8±16.6 | 0c | 0.190 | 0.376 |
| Aβ40 | 7.8±13.9 | 1.2±4.1 | 0.316 | 0.249 |
| Aβ42 | 5.8±11.5 | 50.8±57.6 | 0.004 | 0.001 |
| Permanent loss of soluble Aβ to all fates (fractional turnover rate, FTR) (pools/h) (e.g. v42+kCSF) | Mutation status | PIB MCBP score | ||
| Aβ38 | 0.144±0.046 | 0.124±0.049 | 0.802 | 0.054 |
| Aβ40 | 0.156[0.055] | 0.109[0.035] | 0.990 | 0.024 |
| Aβ42 | 0.147[0.049] | 0.198[0.086] | 0.065 | 0.548 |
| Aβ38:40 ratio | 0.964±0.038 | 1.013±0.047 | 0.157 | 0.115 |
| Aβ42:40 ratio | 0.942±0.080 | 1.553±0.382 | 0.0016 | 0.0003 |
| CSF concentration by IP-MS (ng/mL) | Mutation status | PIB MCBP score | ||
| Aβ38 | 2.05[0.69] | 1.82[1.00] | 0.296 | 0.105 |
| Aβ40 | 7.15±1.80 | 7.79±1.89 | 0.199 | 0.272 |
| Aβ42 | 1.01[0.39] | 0.80[0.52] | 0.537 | 0.007 |
| Aβ38:Aβ40 ratio | 0.272±0.014 | 0.256±0.053 | 0.803 | 0.068 |
| Aβ42:Aβ40 ratio | 0.149±0.013 | 0.121±0.042 | 0.720 | 0.003 |
Parameters were determined based on the best-fit to a compartmental model of Aβ turnover and production. Outcomes were compared by mutation status using ANOVA after adjusting for fibrillar amyloid deposition by treating PIB MCBP score as a covariate. Kinetic parameters are identified with terminology (Fig. 2). P < 0.05 in bold.
P-values by ANOVA based on mutation status with PIB MCBP score as a covariate.
Analyzed by non-parametric Mann-Whitney U test due to non-normal distribution, based on mutation status and PIB status (rather than PIB MCBP score).
No exchange observed in any subject.
Fig. 3. Aβ concentrations and selected kinetic parameters.

Parameters for Aβ production rate (A), fractional turnover rate (FTR) (B), and baseline CSF concentrations (C) are shown for each group by mutation status and without (PIB−) or with (PIB+) evidence of amyloid deposition. Each parameter was measured for Aβ38 (1st column), Aβ40 (2nd column), Aβ42 (3rd column) and the Aβ42: Aβ40 ratio (4th column), and compared by mutation status and amyloid deposition. Error bars indicate 95% confidence intervals (CI). Mutation status compared when amyloid deposition did not affect magnitude or significance. * P<0.05, ** P<0.01, *** P<0.001, p-values by ANOVA based on mutation status with PIB MCBP score as a covariate. Non-carriers, blue circles; mutation carriers PIB− (MC PIB−), green squares; mutation carriers PIB+ (MC PIB+), red triangles. Each point represents one participant.
Exchange process required to fit Aβ kinetic curves
To optimally fit the shape and peak magnitude of Aβ isoform enrichment time courses, a compartment was required to model reversible exchange of newly synthesized labeled Aβ peptides with a pre-existing pool of unlabeled Aβ (Fig. 2). The exchange process was of minimal magnitude in non-mutation carriers, in which only ~10% of the flux of newly synthesized Aβ38, 40 or 42 underwent exchange (Table 2). The percent of Aβ38 and Aβ40 that underwent exchange was not significantly different between mutation carriers and non-carriers. However, the exchange for Aβ42 was significantly greater in carriers compared to the non-carriers (51±58% vs. 6±12% of flux, respectively, P=0.004 for mutation effect, P=0.001 for PIB status) (Table 2). The exchange process for Aβ42, combined with a faster turnover rate of Aβ42 (see below), provided an excellent fit to the entire shape of the Aβ42 enrichment time course in all groups including mutation carriers with amyloid deposition (mean R2 for all participants of 0.994, 0.995, and 0.987 for Aβ38, Aβ40 and Aβ42, respectively).
Higher irreversible loss of Aβ42 in amyloid deposition
The fractional turnover rate (FTR, pools/h) of soluble Aβ is the rate constant for irreversible loss of soluble Aβ to all metabolic fates; this process is kinetically distinct from reversible exchange. The physiology of the system suggests that the FTR includes irreversible losses to the CSF or bloodstream, degradation, and deposition into amyloid plaques. The Aβ40 FTR was significantly slower in PIB+ compared to PIB− participants (P=0.024 for PIB MCBP score) and trended towards significance for Aβ38 (P=0.054 for PIB MCBP score), but neither was affected by mutation status (Table 2, Fig. 3B). This decreased turnover rate correlated with the extent of amyloidosis measured by PET PIB. In contrast, Aβ42 FTR trended towards an increase in mutation carriers (P=0.065 for mutation effect) independent of amyloid load (Table 2, Fig. 3B). The Aβ38:Aβ40 FTR ratio was not significantly different between non-carrier and mutation carrier groups, but the Aβ42:Aβ40 FTR ratio was 65% higher in mutation carriers (P<0.002 for both mutation status and PIB MCBP score) (Table 2, Fig. 3B).
Decreased CSF Aβ42 concentrations only during amyloid deposition
The measured basal concentration of CSF Aβ isoforms were compared by mutation status and PIB MCBP score (Table 2 and Fig. 3C). The Aβ42 CSF concentration and the Aβ42:Aβ40 CSF concentration ratio was significantly reduced in association with amyloid deposition (P=0.003 for PIB MCBP score; not significant by mutation status), whereas there were no differences between groups for the CSF Aβ38, Aβ40, or Aβ38:Aβ40 concentration ratio (Table 2).
Discussion
Our study of in vivo CNS Aβ isoform kinetics in participants with PSEN mutations confirms the hypothesis that the production rate of Aβ42 is increased relative to Aβ40 in humans with PSEN mutations associated with ADAD, and also revealed increased exchange of soluble Aβ42 and increased irreversible loss of soluble Aβ42 associated with the presence of fibrillar amyloid plaques. The finding of increased Aβ42 production was predicted by some in vitro studies (11, 12) and evidence of altered γ-secretase function in carriers of PSEN1 or PSEN2 ADAD mutations (24). Here, we directly quantify increased Aβ42 production in vivo in humans due to mutations that cause ADAD, and confirm that such mutations increase CNS Aβ42 production rates in vivo. The ratio of Aβ42 to Aβ40 production rates was greater than 15% for almost all mutation carriers (average 17.4%), whereas non-carriers were less than 15% (average 14%), suggesting tight physiological control that is disrupted in the presence of a mutation. There was a ~25% relative increase in Aβ42 to Aβ40 production rate in mutation carriers, which correlates with the 40% decrease in Aβ production due to a mutation that is protective for the development of AD (7). Changes in the Aβ42 to Aβ40 production rate ratio are likely to have a large impact on the rate of Aβ42 deposition into plaques because in vitro studies have demonstrated that even relatively small changes in the Aβ42:Aβ40 ratio significantly impact aggregation kinetics and morphology (15) that could promote plaque formation. The Aβ42:Aβ40 ratio and the total amount of Aβ42 have been hypothesized to affect the age of onset of ADAD(25). Some PSEN mutations in cultured cells in vitro have been reported to decrease Aβ40 production or loss of overall Aβ production (16), however, our in vivo SILK studies did not confirm decreased Aβ40 or total Aβ production.
Our finding of a faster irreversible FTR of soluble Aβ42 relative to Aβ38 or Aβ40 in the presence of PSEN mutations or plaques was not anticipated. This faster relative FTR was a primary factor causing the Aβ42 isotopic enrichment curve to peak earlier than Aβ38 or Aβ40 in the presence of amyloidosis (Fig. 1). We cannot determine the physiological mechanisms that underlie this irreversible FTR, but it includes all losses of soluble Aβ peptides including transport into CSF or plasma, proteolytic degradation, and deposition into amyloid plaques. The rate constant for transport to CSF is likely to be similar for all Aβ isoforms since the transport of soluble Aβ isoforms to CSF is driven by fluid flow (26). Therefore, the faster relative FTR of Aβ42 in the presence of plaques implies a greater loss of Aβ42 from the system to fates other than CSF. Although we cannot identify the specific fates of peptides permanently lost from the system, our observations are consistent with increased deposition of Aβ42 into amyloid plaques (27, 28) relative to Aβ38 and Aβ40, which subsequently causes increased Aβ42 in brain plaques(29) as shown in Video 1, and a reduction in the amount of Aβ42 recovered in CSF (30). Our finding of increased irreversible FTR of Aβ42 in subjects with PSEN mutations but no amyloid load suggests increased Aβ42 deposition may be occurring in these subjects, and SILK studies of Aβ42 turnover kinetics may provide a sensitive window into altered processes before amyloidosis is detectable by PET PIB.
Another unanticipated finding was a reversible exchange process whereby a portion of newly synthesized (i.e. isotopically labeled) peptides exchange with pre-existing unlabeled Aβ. The structure of the pre-existing unlabeled Aβ structures cannot be determined from this tracer study, but they could potentially consist of oligomers or aggregates (31, 32) or may reflect reversible binding of Aβ42 to the surface of amyloid plaques before structural alterations cause permanent assimilation into plaques (33, 34).
We previously reported that late onset Alzheimer’s disease was associated with a slower clearance rate for Aβ40 and Aβ42 based on a monoexponential slope applied to the terminal phase (24–36 h) of the SILK enrichment time course (18). The present study utilized a compartmental model to fit the SILK enrichment time courses over the full experimental period (0–36 h) in a manner consistent with known physiological processes (35). This compartmental model confirms the previous report that the FTR is slower for Aβ40 with increased insoluble amyloid plaque deposition (Table 2). The exchange process described by our compartmental model was minimal for Aβ38 and Aβ40 in the presence or absence of amyloid plaques (Table 2), and thus the terminal tail approximates a monoexponential shape for these isoforms. The observation that the irreversible FTRs of Aβ38 and Aβ40 are negatively correlated with PIB MCBP score (Table 2) suggests that the presence of plaques substantially alters physiological processes (e.g. fluid flow perfusion through brain tissue) that retards the transfer of soluble Aβ peptide from the brain to CSF. An outcome of this decreased transport to the CSF may be loss of the normal physiological diurnal pattern of CSF Aβ concentrations (36, 37).
The present study also confirms that the terminal slope of the Aβ42 enrichment curve is slower in the presence of plaques. We observed that Aβ42 irreversible FTR was increased in the presence of PSEN mutations independent of plaque load. The SILK tracer technique thus reveals abnormalities in Aβ42 before plaques are detectable by PET PIB. Indeed, 3 of the 4 mutation carriers who did not have detectable amyloid plaques by PET PIB had features of Aβ42 kinetics that were more similar to mutation carriers with amyloid plaques than to non-carriers, including Aβ42:Aβ40 FTR ratio > 1.1, and evidence of Aβ42 exchange (Fig. S2). These features strongly suggest that plaque development or some form of Aβ aggregation has been initiated in these individuals, but fibrillar plaque load has not reached the threshold of PET PIB detection. In contrast, only one mutation carrier who was PET PIB negative had no evidence of Aβ42 exchange. This participant also had the highest Aβ42:Aβ40 CSF concentration ratio (0.215; Fig. S4) of all participants in our study (compare to Figs. S2 and S3; this ratio was 27% higher than the next highest concentration ratio observed in individual participants, 0.169), demonstrating that elevated Aβ42:Aβ40 CSF concentration ratios are only observed in ADAD persons in the absence of amyloid plaques. As amyloid deposition in the caudate may be one of the earliest deposition regions, we evaluated caudate PIB as a sensitive measure of amyloid deposition. The caudate PIB and MCBP PIB were highly correlated with each other with a Pearson correlation coefficient of 0.94, p=5×10−11; no kinetic parameters changed significance with amyloidosis as measured by caudate PIB.
Our findings offer a context for the recent report that CSF Aβ42 concentration is elevated in ADAD individuals decades prior to predicted age for onset of clinical symptoms of dementia, and then drops below normal as plaques develop (2, 38) (see Fig. 4 for schematic of proposed process). Our finding of increased Aβ42 production in mutation carriers regardless of the amount of amyloid deposition indicates that increased Aβ42 production precedes amyloid deposition, and likely occurs decades before the onset of dementia and is possibly present throughout life. The long delay in the emergence of plaque deposits of Aβ42 even in the presence of overproduction of Aβ42 suggests the presence of an initial slow process (e.g. initial Aβ nucleation event in which monomeric Aβ forms small aggregates) followed by a growth phase of Aβ polymerization (39). We therefore hypothesize that Aβ42 production rate (solid gold line, Fig. 4) remains above “normal” (solid black line) throughout life due to their PSEN genotype. ADAD individuals have an increased irreversible loss of soluble Aβ42 (FTR solid red line, Fig. 4) that tracks or precedes PET-detectable amyloid deposition, consistent with a faster removal of soluble Aβ42 as it is deposited into plaques. This results in a decreased recovery of Aβ42 in the CSF, accounting for the decrease in CSF Aβ42 concentration (solid blue line, Fig. 4) as plaque development proceeds (solid green line, Fig. 4). The physiological identity of the pool(s) in exchange with newly labeled soluble Aβ42 identified by SILK (dotted black lines, Fig. 4) cannot be determined in this study. If exchange occurs with micelles or oligomers, exchange may precede PET-detectable amyloid deposition (dotted line 1, Fig. 4). If exchange is with plaque surfaces, it may track or even lag behind amyloid deposition (dotted lines 2&3, Fig. 4). Future studies with greater numbers of participants or longitudinal designs will be required to resolve this question.
Fig. 4.
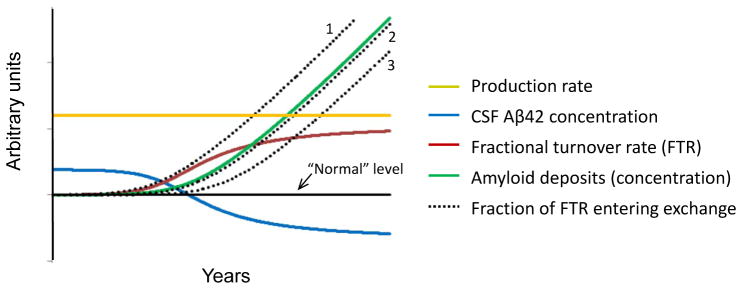
Potential scheme for the time course of plaque deposition in ADAD.
The solid black line signifies “normal” for each measure. The production rate of Aβ42 relative to Aβ40 (gold) remains constant throughout life before and during plaque deposition (production rate was different by mutation status but not by amyloid deposition). CSF concentration of Aβ42 (blue) starts elevated above normal due to overproduction, then decreases to below normal due to an increase in the fractional turnover rate (FTR; red) of Aβ42 relative to Aβ40. As the amount of amyloid plaques increases over time (green), the extent of Aβ42 exchange (dotted) may precede (e.g. if due to oligomer formation), follow closely, or lag behind amyloid deposition (e.g. if due to reversible interaction with amyloid plaques).
Although ADAD represents less than one percent of all AD cases, it has been informative for elucidating the pathophysiology of AD, and can manifest as late-onset AD indistinguishable from the more idiopathic forms (40). The findings of this study further support the amyloid hypothesis and provide quantitative estimates of life-long increases in Aβ42 production that may cause AD in humans. Further, these results indicate profound changes in Aβ kinetics in the presence of amyloid plaques. With increased understanding of the pathogenic causes of AD and the quantitative changes associated with AD pathology, it is hoped that better tests and directed therapeutics can be developed in the future.
Materials and Methods
Study Design
This study took place at the Washington University School of Medicine in St. Louis and was approved by the Human Research Protection Office. All participants completed informed written consent. Two groups of volunteers were enrolled in the study: (1) ADAD mutation carriers either with (n=7) or without (n=4) amyloid deposition; and (2) related family members at risk for carrying a mutation, but who do not carry ADAD mutations (n=12).
Binding of carbon 11-labeled Pittsburgh Compound B-positron emission tomography ([11C] PIB-PET) was used to test for deposition of amyloid plaques in the brain (23). Binding potentials using the cerebellum as a reference region were determined for the prefrontal cortex, precuneus, lateral temporal cortex, and gyrus rectus. The mean cortical PIB binding potential score (PIB MCBP score) was determined by averaging these binding potentials together. A mean PIB MCBP score of 0.18 was considered evidence of amyloid deposition (PIB+), a PIB MCBP score of less than 0.18 was considered to be amyloid deposition negative (PIB−. The highest PIB MCBP score in non-carriers was 0.12.
Participants Demographics
Twenty-three participants from 9 pedigrees (PSEN1 Ala79Val, Leu226Arg, His163Arg, Met146Leu, Met139Ile, Gly217Arg, Ala246Glu, and PSEN2 Asn141Ile) completed Aβ stable isotope labeling kinetics studies. Twelve (52%) of the participants were male. The average age of mutation carriers was 43.2 ± 12.7, and non-carriers was 47.7 ± 14.8 (P > 0.05). Eight PSEN1 mutation carriers and 3 PSEN2 mutation carriers were included in the analysis with sibling controls. Two mutation carriers were rated as having prodromal dementia and one with mild dementia. Eight mutation carriers were cognitively asymptomatic, as were all non-carriers. Seven mutation carriers were PIB+, and 4 of the mutation carriers and all non-carriers were PIB−; the average PIB MCBP score was significantly different between groups, 0.03±0.04 vs. 0.43±0.45 for non-carriers and mutation carriers, respectively (P=0.005). At least one APOE epsilon4 allele was present in 33% (n=4) of the non-carrier group, 25% of the mutation-carrier PIB− group, and 28% of the mutation-carrier PIB+ group (p>0.05).
Sample collection
The SILK clinical study followed the procedure previously described (41). Briefly, intravenous and intrathecal lumbar catheters were placed between 7:30 AM and 9:00 AM, and the collection of samples was started between 8:00 AM and 9:30 AM. After initial CSF and plasma baseline samples were collected, participants were infused with a bolus of 3 mg kg−1 L-[U-13C6] leucine for 10 minutes, followed by 2 mg kg−1 h−1 for the remainder of the first 9 hours. For the 36 hours of the study, 6 mL of CSF were obtained every hour, afterwards the catheters were removed and participants remained on bed rest for 12 hours. Aliquots of CSF were frozen at −80°C immediately in 1-mL polypropylene tubes after being collected. Venous blood samples (12 mL) were obtained at hourly intervals from 1–13 h, then at 17 and 36 h, and plasma stored at −80°C for the determination of plasma leucine enrichment.
Analysis of CSF samples
All procedures relating to clinical studies, sample processing, and sample analysis were performed blinded to mutation, clinical, and amyloid status. From each hour of collection, 1 mL of CSF was thawed, and Aβ was immunopurified and isotopic enrichment of Aβ isoforms were measured using liquid chromatography mass spectrometry similar to prior studies (18), except for dedicated measurement of Aβ38, Aβ40, and Aβ42 C-terminal peptides.
Measurements of Aβ38, Aβ40, and Aβ42 C-terminal peptides
A mid-domain antibody (HJ5.1, anti-Aβ13-28) was covalently bound to CNBr Sepharose beads and stored in a 50% slurry of 0.02% PBS azide at 4°C. The immunopurification mixture was comprised of 800 μL CSF, 20 μL of a solution containing C12N15 Aβ40, Aβ42, and Aβ38 for quantification by internal standard; 12.5 μL 100x protease inhibitor, 110 μL 5M guanidine, and 30 μL antibody-bead slurry. Beads were rotated in an Eppendorf Axygen tube for 2 hours at room temperature, and rinsed with 0.5 M guanidine followed by two rinses of 25 mM ammonium bicarbonate. All liquid was removed by aspiration, and then neat formic acid was added to elute Aβ from the antibody-bead complex. The formic acid supernatant was transferred to a new polypropylene tube and dried in a vacuum dryer (Labconco CentriVap). The sample was reconstituted with 25 μL 25mM ammonium bicarbonate and digested with 2.5 ng Lys-N overnight at 37°C. This solution was dried in the speed-vacuum and resuspended in 10% formic acid, 20% dimethyl sulfoxide. 3 μL of this sample was injected into a NanoAcquity Ultra Performance Liquid Chromatography system, and then analyzed by a XEVO TQ-S mass spectrometer (Waters). MassLynx V4.1 was used to quantify the mass spectrometry data. For determination of plasma 13C6-leucine enrichment, amino acids were recovered from plasma using cation exchange chromatography, converted to N-heptafluorobutyryl n-propyl esters derivatives, and isotopic enrichment (m/z 349 and 355) measured using gas chromatography-negative chemical ionization-mass spectrometry (Agilent 6890N Gas Chromatograph and Agilent 5973N Mass Selective Detector (GC-MS); Agilent, Palo Alto, CA) as described (42). The GC-MS instrument response was calibrated using gravimetric standards of known isotope enrichment.
Compartmental model for Aβ38, Aβ40, and Aβ42 kinetics
A comprehensive steady state compartmental model that accounts for the full time course of Aβ38, Aβ40, and Aβ42 enrichments and CSF concentrations was developed; a schematic diagram of the model is shown in Fig. 2. The model consists of a series of interconnected compartments with first order rate constants that describe the transfer of labeled species between compartments. Modeling was performed using SAAM II (Resource for Kinetic Analysis, University of Washington, Seattle). The model consists of 3 parallel arms, one for each Aβ isoform. The enrichments of plasma leucine and CSF Aβ peptides were measured at frequent time intervals (indicated by solid triangles). Plasma leucine is used as the tracer precursor for CSF Aβ peptides because we have shown that, given a sufficiently long infusion, CSF Aβ enrichments essentially match that of plasma leucine in monkeys (43). A “forcing function” was used to describe the time course of plasma 13C6-leucine enrichment using a linear interpolation between measured samples.
Preliminary modeling revealed that the isotopic enrichment time course for each Aβ isoform could be optimally described by a single compartment coupled with a long time delay that consisted of five sub-compartments. Based on known physiology, two of these delay compartments (representing APP and C99 peptides) were placed in front of the compartments that represent the brain “soluble” Aβ peptides, as in vivo tracer studies in mice show that APP and C99 have relatively long half-lives (~3 h) (44) that should contribute to the overall time delay before labeled Aβ is detected at the lumbar sampling site. The remaining three delay compartments were placed after the “soluble” Aβ compartments, to represent perfusion of labeled peptides through brain tissue and heterogeneous CSF fluid transport processes. Given that preliminary modeling indicated that a single time delay process could be identified within the data, the turnover rates for APP, C99, and each of the 3 CSF delay compartments were set to a single adjustable parameter that affects the overall time delay.
The model takes into consideration that some of the C99 and soluble Aβ peptides are metabolized to fates other than Aβ peptides that appear at the CSF sampling site. The physiological nature of these other losses for soluble Aβ peptides are unknown, but includes all processes that remove soluble peptides irreversibly, e.g. deposition into plaques, cellular uptake, and proteolytic degradation. A compartment in exchange with the “soluble” Aβ peptide was necessary to optimally fit the sigmoid shape of the CSF Aβ enrichment time courses after the peak enrichment, particularly for Aβ42 in the mutation carrier PIB+ group; this exchange process was added for an isoform only if it improved the Akaike Information Criteria (AIC) of the fit as provided by SAAMII. A scaling factor was applied to each of the Aβ isoform enrichments if it improved the AIC, to account for small amounts of isotopic dilution between plasma leucine and the biosynthetic precursor pool (generally < 5%) or to correct for minor calibration errors (generally <10%) in the measurement of isotope enrichments of plasma leucine or Aβ peptides.
Based on the optimized kinetic parameters that describe the shape and magnitude of the CSF Aβ enrichment time course, the model determines the rate constant (pools/h) for production of each Aβ peptide from their common C99 precursor to accurately project the measured baseline CSF Aβ peptide concentrations. The model projects the steady state masses (ng) within and the flux rates (ng/h) between all compartments for each Aβ isoform.
Statistical Analyses
Ratios of isotopic enrichments between peptides at all time points were analyzed by repeated measures ANOVA. When the time by group interaction was significant, enrichment ratios at individual time points were analyzed by ANOVA with a linear contrast to test for significant group effects. ANOVA was performed using a mutation status main affect, adjusting for PIB MCBP score as a covariate. Variables were tested for normal distribution by Shapiro-Wilk criterion; variables that were not normally distributed were log transformed before performing ANOVA. The distribution of rate constants describing the exchange process revealed by the model was analyzed by Mann-Whitney U test as log transformation did not normalize the distribution. Statistics were performed with IBM SPSS, v. 20. A P value < 0.05 was considered to be statistically significant. Unless otherwise noted, results shown are mean ± SD for normally distributed variables, or as median with [interquartile range] for non-normally distributed variables. Graph generations were performed in GraphPad Prism version 5.01 for Windows (GraphPad Software, San Diego, California).
Movie Methods
A video illustrating the isotope labeling model for soluble Aβ42 and Aβ40 was created using Matplotlib (41). Compartments for soluble, exchange, and irreversible loss (plaque) are shown over the MNI152 template image included as part of the FMRIB Software Library (42). The location of the fibrillar plaque compartment in the illustration was determined by thresholding the average PiB image for PiB+ mutation carriers. The growth of the soluble, exchange, and irreversible loss compartments followed the normalized production rates for each peptide. Note that the sizes of the Aβ compartments are greatly exaggerated for visualization purposes.
Supplementary Material
Fig. S1. Time course of plasma leucine isotopic enrichment
Fig. S2. Isotopic enrichment time courses of CSF Aβ peptides in individual non-mutation carrier participants
Fig. S3. Isotopic enrichment time courses of CSF Aβ peptides in mutation carrier participants with amyloid plaques (PIB+)
Fig. S4. Isotopic enrichment time courses of CSF Aβ peptides in mutation carrier participants without amyloid plaques (PIB−)
Fig. S5. Whisker-Box plot and Scatterplot of concentrations and selected kinetic parameters
Video: animated illustration of isotope labeling time course (Video1 FACS.AVI)
Table 1.
Complete list of model parameters.
| Parameter | Non-carriers | Mutation-carrier PIB− | Mutation-carrier PIB+ |
|---|---|---|---|
| kAPP | 1,171 ± 227 | 1,304 ± 602 | 1,291 ± 324 |
| kC99 | 0.666 ± 0.112 | 0.553 ± 0.083 | 0.695 ± 0.096 |
| kAβ38 | 0.062 ± 0.010 | 0.055 ± 0.016 | 0.059 ± 0.008 |
| kAβ40 | 0.238 ± 0.041 | 0.187 ± 0.023 | 0.247 ± 0.037 |
| kAβ42 | 0.033 ± 0.006 | 0.034 ± 0.007 | 0.041 ± 0.006 |
| vC99 | 0.333 ± 0.056 | 0.276 ± 0.041 | 0.347 ± 0.048 |
| v38 | 0.069 ± 0.023 | 0.075 ± 0.027 | 0.054 ± 0.015 |
| v40 | 0.074 ± 0.023 | 0.082 ± 0.037 | 0.050 ± 0.013 |
| v42 | 0.064 ± 0.014 | 0.126 ± 0.072 | 0.120 ± 0.037 |
| kCSF | 0.074 ± 0.023 | 0.082 ± 0.037 | 0.050 ± 0.013 |
| kex38 | 0.020 ± 0.038 | 0.000 ± 0.000 | 0.000 ± 0.000 |
| kex40 | 0.016 ± 0.032 | 0.009 ± 0.018 | 0.000 ± 0.000 |
| kex42 | 0.010 ± 0.021 | 0.041 ± 0.045 | 0.120 ± 0.107 |
| kret | 0.1 | 0.1 | 0.1 |
| kdelay | 0.666 ± 0.112 | 0.553 ± 0.083 | 0.695 ± 0.096 |
| SF38 | 0.937 ± 0.066 | 0.885 ± 0.063 | 0.979 ± 0.092 |
| SF40 | 0.933 ± 0.043 | 0.916 ± 0.078 | 0.977 ± 0.130 |
| SF42 | 0.972 ± 0.102 | 0.879 ± 0.021 | 0.912 ± 0.151 |
aAll units are first order rate constants with units h−1, except for scaling factors (SF), which are unitless, and kAPP, which is a zero order rate constant for APP production (ng/h). Note that kC99 = kdelay; kAβ38+kAβ40+kAβ42 = vC99; and kAβ38+kAβ40+kAβ42+vC99 = kdelay. Values are mean ± SD
Acknowledgments
We thank the participants and their families for their contributions to this study and the Clinical Core of the Adult Children Study for clinical assessments of the participants.
Funding. This work was supported by funding from the National Institute on Aging as a supplement, the Familial Adult Children Study (RJB, 5P01AG026276-S1), to the Antecedent Biomarkers in AD: The Adult Children Study (JCM, 5P01 AG026272). Additional support came from R01-NS065667 (RJB), UL1 RR024992, P41 GM103422, P30 DK056341 (BWP, Nutrition Obesity Research Center), P60 DK020579, and an anonymous foundation.
Footnotes
Author contributions. R.J.B. designed the study and obtained funding. D.M.H., A.G., and J.C.M. assisted in the design of the study. W.S., R.J.B. and R.P. recruited and enrolled participants and performed tracer infusion studies. R.P., R.C. and T.K. performed lab protocol development, sample preparation, and processing. K.M. and V.O. developed mass spectrometry methods and V.O. measured samples by mass spectrometry. B.W.P. and D.L.E. developed and performed kinetic modeling and statistical analyses. R.C. and K.E.Y. analyzed leucine samples. T.B., T.L.S.B., R.J.B., DLE, and BWP developed images and videos. R.J.B. and R.P wrote an initial draft of the paper. The final paper was prepared by BWP, DLE and RJB, which all authors reviewed and approved.
Competing interests. R.J.B. and D.M.H. are co-inventors on U.S. patent 7,892,845 “Methods for measuring the metabolism of neurally derived biomolecules in vivo.” Washington University, with R.J.B. and D.M.H. as co-inventors, has also submitted the U.S. non-provisional patent application “Methods for measuring the metabolism of CNS derived biomolecules in vivo,” serial #12/267,974. R.J.B., D.L.E., and B.W.P. are co-inventors on U.S. Provisional Application 61/728,692 “Methods of Diagnosing Amyloid Pathologies Using Analysis of Amyloid-Beta Enrichment Kinetics”. R.J.B. has consulted for Pfizer, DZNE, Probiodrug AG, Medscape, En Vivo (SAB) and has research grants with AstraZeneca, Merck and Eli Lilly in the past year. Washington University, R.J.B. and D.M.H. have a financial interest in C2N Diagnostics, which uses the SILK methodology in human studies. B.W.P. provides turnover kinetics consultation services for C2N Diagnostics. C2N Diagnostics did not support this work. DMH has consulted for Pfizer, AstraZeneca, and Bristol-Myers Squibb in the last 12 months. His laboratory has received grants from Eli Lilly, AstraZeneca, Pfizer, and C2N Diagnostics that are not related to the content in this manuscript.
TB has served on an advisory board for Eli Lilly and has received research funding from Avid Radiopharmaceuticals. These relationships are not related to the content in the manuscript.
AG has received research funding during the last 12 months from Pfizer, Genentech, AstraZeneca and iPierian and has served as a consultant for Amgen. These relationships are not related to the content in the manuscript.
JM serves on scientific advisory boards for Eisai, Esteve, Janssen Alzheimer Immunotherapy Program, Glaxo-Smith-Kline, Novartis, and Pfizer.
The other authors declare that they have no competing interests.
References
- 1.Bateman RJ, Aisen PS, De Strooper B, Fox NC, Lemere CA, Ringman JM, Salloway S, Sperling RA, Windisch M, Xiong C. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res Ther. 2011:3. doi: 10.1186/alzrt59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N Engl J Med. 2012:367. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002:297. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Haass C, De Strooper B. The presenilins in Alzheimer’s disease--proteolysis holds the key. Science. 1999:286. doi: 10.1126/science.286.5441.916. [DOI] [PubMed] [Google Scholar]
- 5.Wang R, Sweeney D, Gandy SE, Sisodia SS. The profile of soluble amyloid beta protein in cultured cell media. Detection and quantification of amyloid beta protein and variants by immunoprecipitation-mass spectrometry. J Biol Chem. 1996:271. doi: 10.1074/jbc.271.50.31894. [DOI] [PubMed] [Google Scholar]
- 6.Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, Iqbal K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch Biochem Biophy. 1993:301. doi: 10.1006/abbi.1993.1112. [DOI] [PubMed] [Google Scholar]
- 7.Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jonsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer/’s disease and age-related cognitive decline. Nature. 2012:488. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 8.Rovelet-Lecrux A, Hannequin D, Raux G, Meur NL, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nature Genet. 2006:38. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 9.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991:349. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 10.Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, Bird TD, Schellenberg GD. A familial Alzheimer’s disease locus on chromosome 1. Science. 1995:269. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- 11.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature Med. 1996:2. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 12.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer’s Disease-Linked Presenilin 1 Variants Elevate A[beta]1-42/1-40 Ratio In Vitro and In Vivo. Neuron. 1996:17. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 13.Ringman JM, Younkin SG, Pratico D, Seltzer W, Cole GM, Geschwind DH, Rodriguez-Agudelo Y, Schaffer B, Fein J, Sokolow S, Rosario ER, Gylys KH, Varpetian A, Medina LD, Cummings JL. Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology. 2008:71. doi: 10.1212/01.wnl.0000303973.71803.81. [DOI] [PubMed] [Google Scholar]
- 14.Kauwe JS, Jacquart S, Chakraverty S, Wang J, Mayo K, Fagan AM, Holtzman DM, Morris JC, Goate AM. Extreme cerebrospinal fluid amyloid beta levels identify family with late-onset Alzheimer’s disease presenilin 1 mutation. Ann Neurol. 2007:61. doi: 10.1002/ana.21099. [DOI] [PubMed] [Google Scholar]
- 15.Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D’Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B. Neurotoxicity of Alzheimer’s disease A[beta] peptides is induced by small changes in the A[beta]42 to A[beta]40 ratio. EMBO J. 2010:29. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shioi J, Georgakopoulos A, Mehta P, Kouchi Z, Litterst CM, Baki L, Robakis NK. FAD mutants unable to increase neurotoxic Abeta 42 suggest that mutation effects on neurodegeneration may be independent of effects on Abeta. J Neurochem. 2007:101. doi: 10.1111/j.1471-4159.2006.04391.x. [DOI] [PubMed] [Google Scholar]
- 17.Moonis M, Swearer JM, Dayaw MP, St George-Hyslop P, Rogaeva E, Kawarai T, Pollen DA. Familial Alzheimer disease: decreases in CSF Abeta42 levels precede cognitive decline. Neurology. 2005:65. doi: 10.1212/01.wnl.0000171397.32851.bc. [DOI] [PubMed] [Google Scholar]
- 18.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010:330. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreasen N, Hesse C, Davidsson P, Minthon L, Wallin A, Winblad B, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal fluid beta-amyloid(1-42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999:56. doi: 10.1001/archneur.56.6.673. [DOI] [PubMed] [Google Scholar]
- 20.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid A beta(42) in humans. Ann Neurol. 2006:59. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 21.Nikisch G, Hertel A, Kiessling B, Wagner T, Krasz D, Hofmann E, Wiedemann G. Three-year follow-up of a patient with early-onset Alzheimer’s disease with presenilin-2 N141I mutation - case report and review of the literature. European Jjournal of Medical Research. 2008:13. [PubMed] [Google Scholar]
- 22.De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Reports. 2007:8. doi: 10.1038/sj.embor.7400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006:67. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 24.Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of [gamma]-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012:31. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Human Mutat. 2006:27. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- 26.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Science Translational Medicine. 2012:4. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003:23. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong S, Quintero-Monzon O, Ostaszewski BL, Podlisny DR, Cavanaugh WT, Yang T, Holtzman DM, Cirrito JR, Selkoe DJ. Dynamic analysis of amyloid beta-protein in behaving mice reveals opposing changes in ISF versus parenchymal Abeta during age-related plaque formation. J Neurosci. 2011:31. doi: 10.1523/JNEUROSCI.3272-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta. Neuron. 1994;42(43):13. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 30.Grimmer T, Riemenschneider M, Forstl H, Henriksen G, Klunk WE, Mathis CA, Shiga T, Wester HJ, Kurz A, Drzezga A. Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry. 2009:65. doi: 10.1016/j.biopsych.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Nat Acad Sci USA. 1998:95. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science. 2003:300. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 33.Esler WP, Stimson ER, Jennings JM, Vinters HV, Ghilardi JR, Lee JP, Mantyh PW, Maggio JE. Alzheimer’s disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry. 2000:39. doi: 10.1021/bi992933h. [DOI] [PubMed] [Google Scholar]
- 34.Knowles TPJ, Waudby CA, Devlin GL, Cohen SIA, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM. An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science. 2009:326. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 35.Elbert DL, Patterson BW, Ercole L, Ovod V, Kasten T, Mawuenyega K, Yarasheski K, Morris JC, Benzinger T, Holtzman DM, Bateman RJ. Reply to: Fractional synthesis and clearance rates for amyloid [beta] Nature Med. 2011:17. [Google Scholar]
- 36.Huang Y, Potter R, Sigurdson W, Santacruz A, Shih S, Ju YE, Kasten T, Morris JC, Mintun M, Duntley S, Bateman RJ. Effects of Age and Amyloid Deposition on A{beta} Dynamics in the Human Central Nervous System. Arch Neurol. 2011 doi: 10.1001/archneurol.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM. Disruption of the Sleep-Wake Cycle and Diurnal Fluctuation of beta-Amyloid in Mice with Alzheimer’s Disease Pathology. Science Translational Medicine. 2012:4. doi: 10.1126/scitranslmed.3004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, Fagan AM, Shah AR, Alvarez S, Arbelaez A, Giraldo M, Acosta-Baena N, Sperling RA, Dickerson B, Stern CE, Tirado V, Munoz C, Reiman RA, Huentelman MJ, Alexander GE, Langbaum JBS, Kosik KS, Tariot PN, Lopera F. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. The Lancet Neurology. 2012:11. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997:66. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 40.Cruchaga C, Chakraverty S, Mayo K, Vallania FL, Mitra RD, Faber K, Williamson J, Bird T, Diaz-Arrastia R, Foroud TM, Boeve BF, Graff-Radford NR, St Jean P, Lawson M, Ehm MG, Mayeux R, Goate AM N-LNFS Consortium. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PloS one. 2012:7. doi: 10.1371/journal.pone.0031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nature Med. 2006:12. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reeds DN, Cade WT, Patterson BW, Powderly WG, Klein S, Yarasheski KE. Whole-body proteolysis rate is elevated in HIV-associated insulin resistance. Diabetes. 2006:55. doi: 10.2337/db06-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cook JJ, Wildsmith KR, Gilberto DB, Holahan MA, Kinney GG, Mathers PD, Michener MS, Price EA, Shearman MS, Simon AJ, Wang JX, Wu G, Yarasheski KE, Bateman RJ. Acute gamma-secretase inhibition of nonhuman primate CNS shifts amyloid precursor protein (APP) metabolism from amyloid-beta production to alternative APP fragments without amyloid-beta rebound. J Neurosci. 2010:30. doi: 10.1523/JNEUROSCI.1381-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savage MJ, Trusko SP, Howland DS, Pinsker LR, Mistretta S, Reaume AG, Greenberg BD, Siman R, Scott RW. Turnover of amyloid beta-protein in mouse brain and acute reduction of its level by phorbol ester. J Neurosci. 1998:18. doi: 10.1523/JNEUROSCI.18-05-01743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Time course of plasma leucine isotopic enrichment
Fig. S2. Isotopic enrichment time courses of CSF Aβ peptides in individual non-mutation carrier participants
Fig. S3. Isotopic enrichment time courses of CSF Aβ peptides in mutation carrier participants with amyloid plaques (PIB+)
Fig. S4. Isotopic enrichment time courses of CSF Aβ peptides in mutation carrier participants without amyloid plaques (PIB−)
Fig. S5. Whisker-Box plot and Scatterplot of concentrations and selected kinetic parameters
Video: animated illustration of isotope labeling time course (Video1 FACS.AVI)