

Abstract
The S100 protein family consists of small, dimeric proteins that exert their biological functions in response to changing calcium concentrations. S100B is the best studied member and has been shown to interact with over 20 binding partners in a calcium-dependent manner. The TRTK12 peptide, derived from the consensus binding sequence for S100B, has previously been found to interact with S100A1 and has been proposed to be a general binding partner of the S100 family. To test this hypothesis and gain a better understanding of the specificity of binding for the S100 proteins sixteen members of the human S100 family were screened against this peptide and its alanine variants. Novel interactions were only found with two family members: S100P and S100A2, indicating that TRTK12 selectively interacts with a small subset of the S100 proteins. Substantial promiscuity was observed in the binding site of S100B to accommodate variations in the peptide sequence, while S100A1, S100A2, and S100P exhibited larger differences in the binding constants for the TRTK12 alanine variants. This suggests that single-point substitutions can be used to selectively modulate the affinity of TRTK12 peptides for individual S100 proteins. This study has important implications for the rational drug design of inhibitors for the S100 proteins, which are involved in a variety of cancers and neurodegenerative diseases.
The S100 protein family consists of approximately twenty-five members found exclusively in vertebrates, making it the largest subset of the EF-hand, calcium-binding proteins (1). The name derives from the fact that many members are soluble in 100% ammonium sulfate at neutral pH (2). These proteins are expressed in a tissue-specific manner and have been implicated in a variety of intracellular and extracellular functions, including cell growth and differentiation, cytoskeleton dynamics, regulation of calcium homeostasis, inflammation response, and protein phosphorylation (1, 3–6). In addition, the expression levels have been directly correlated to the severity of neurodegenerative disorders (7–11), inflammatory diseases such as irritable bowel syndrome (12–15), cardiomyopathies (16, 17), and cancer (6, 18–24).
S100 proteins are relatively small (9–13kDa) and exist almost exclusively as homo- or heterodimers in solution (25). Each S100 protein monomer consists of four alpha helices, with helices I and IV arranged in an anti-parallel orientation (1, 26). Calcium binding induces a significant structural rearrangement in the S100 proteins that has been well documented by X-ray crystallography and solution NMR (27–32). In this conformational change, helix III reorients itself (> 40°) relative to helices II and IV, to expose a hydrophobic binding pocket that allows interactions with target proteins and peptides (27). Since the S100 proteins appeared to be functionally distinct from related, calcium-binding proteins, such as calmodulin and parvalbumin, early work focused on identifying high-affinity biological targets to better understand the molecular basis of recognition (33–38).
In 1995, Ivankenov et al. screened a phage display library against S100B and provided the first consensus binding sequence for the S100 proteins: (K/R)-(L/I)-X-W-X-X-I-L (35). A search of the available protein sequences showed a highly homologous region in the C-terminus of the actin-binding protein CapZ: T-R-T-K-I-D-W-N-K-I-L-S. This sequence was later termed TRTK12 (35). Subsequent studies have shown that this peptide interacts with the two C-terminal helices of S100B (Figure 1), as well as the hinge region, and was able to compete with binding for other known targets (35, 39–46). Furthermore, it was shown to bind S100A1, leading some to propose that TRTK12 represents a general binding motif for the S100 protein family (32).
Figure 1.
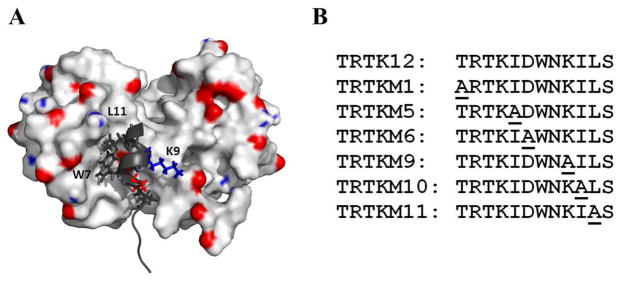
Panel A The NMR solution structure of the wild-type TRTK12 peptide with rat S100B (PDB: 1MWN)(40). For clarity, the binding pocket of a single S100B monomer is shown. Key residues from the TRTK12 peptide are colored and labeled. Panel B: List of TRTK12 peptide sequences. Alanine-substitued residues are underlined.
To gain a better understanding of the specificity of binding within the S100 protein family, we screened the proposed consensus binding sequence (TRTK12) against representative members of the human S100 protein family, using isothermal titration calorimetry (ITC) and steady-state emission fluorescence. Unexpectedly, TRTK12 was found to selectively interact with a small subset of S100 proteins. We confirmed previously reported interactions with S100B and S100A1 proteins. In addition, novel interactions of the wild-type peptide were detected with S100A2 and S100P. These interactions were further characterized in detail using ITC and stopped-flow spectroscopy. The subset of S100 proteins that bound to wild-type TRTK12 was also screened against a variety of TRTK12 alanine variants. Both the thermodynamics and kinetics of binding were then characterized in order to ascertain the individual binding contribution of amino acids within the TRTK12 peptide sequence. For the variants, alanine substitutions were chosen according to the residue-specific interactions in the available structures of the bound S100-TRTK12 complexes (Figure 1)(32, 43, 44). The TRTKM6 (Asp6Ala) and TRTKM9 (Lys9Ala) variants were selected to probe the electrostatic interactions of binding because ionic strength has been reported to significantly affect the binding affinity of the TRTK12 peptide, albeit in a contradictory manner (42, 47). The TRTKM5 (Ile5Ala), TRTKM10 (Ile10Ala), and TRTKM11 (Leu11Ala) variants were selected to query the effects of the hydrophobic residues because the available structures of the S100-TRTK12 complexes suggest that the leucine and isoleucine residues from the peptide contribute most of the buried surface area in the complex with S100B and S100A1 (29, 44). The TRTKM1 (Thr1Ala) variant was used as a control for the N-terminal residues, which are thought to be unstructured and solvent-exposed in the bound complex (32, 40, 43, 44). The TRTK12 peptide variants were found to have unique binding affinities, changes in the heat capacity, and dissociation rates for individual S100 proteins. These dissimilarities suggest that alanine variants of TRTK12 can be used as a starting point for potential peptide-based inhibitors of particular subsets of the S100 proteins.
EXPERIMENTAL PROCEDURES
Protein/Peptide Purification
Representative members of the S100 protein family, including human S100A1, S100A2, S100A3, S100A4, S100A5, S100A6, S100A7, S100A8, S100A9, S100A10, S100A11, S100A12, S100A13, S100P, S100Z, and S100B were overexpressed in Escherichia coli and purified as previously described (48–51). The protein concentrations were determined using a molar extinction coefficient at 280 nm (ε280) of 1,490 M−1cm−1 for S100B, 2,980 M−1cm−1 for S100P, 2,980 M−1cm−1 for S100Z, 8,480 M−1cm−1 for S100A1, 2,980 M−1cm−1 for S100A2, 14,440 M−1cm−1 for S100A3, 2,980 M−1cm−1 for S100A4, 4,470 M−1cm−1 for S100A5, 4,470 M−1cm−1 for S100A6, 4,470 M−1cm−1 for S100A7, 11,460 M−1 cm−1 for S100A8, 6,990 M−1cm−1 for S100A9, 2,980 M−1cm−1 for S100A10, 4,470 M−1cm−1 for S100A11, 2,980 M−1cm−1 for S100A12, and 6,990 M−1cm−1 for S100A13.
All peptides were synthesized at the Penn State College of Medicine Macromolecular Core Facility using standard FMoc chemistry. The N- and C-termini were acetylated and amidated, respectively. The TRTK12 wild-type peptide (Ac-TRTKIDWNKILS-NH2) is derived from the actin-binding protein CapZ and has previously been used in structural studies (3, 5–7). The alanine substituted variants of this sequence, including TRTKM1 (Ac-ARTKIDWNKILS-NH2), TRTKM5 (Ac-TRTKADWNKILS-NH2), TRTKM6 (Ac-TRTKIAWNKILS-NH2), TRTKM9 (Ac-TRTKIDWNAILS-NH2), TRTKM10 (Ac-TRTKIDWNKALS-NH2), and TRTKM11 (Ac-TRTKIDWNKIAS-NH2), were prepared similarly to the wild-type. The peptides were purified using a C18 reverse phase column using a 0–100% methanol gradient in the presence of 0.05–0.065% trifluoroacetic acid (50, 51), and their masses were confirmed via MALDI-TOF mass spectrometry on a Bruker UltraFlex III instrument. The concentration of all peptides was determined using molar extinction coefficients of 5,500 M−1cm−1 at 280 nm (ε280) for TRTK12 (52).
Isothermal titration Calorimetry
ITC measurements were performed using a VP-ITC instrument (MicroCal, Inc., Northhampton, MA) as previously described (50, 51, 53, 54). Prior to all experiments, the protein and peptide were extensively dialyzed into a binding buffer containing 20 mM Tris base, 0.2 mM disodium EDTA, 1 mM TCEP, and 5 mM calcium chloride (pH 7.5). Additional experiments were performed in the same buffer but with 5mM EDTA instead of 5 mM calcium chloride. In general, 0.5–4 mM peptide was injected in 4–10 μL increments into the sample cell (1.5 mL) containing 20–200 μM protein. Due to the low solubility of the TRTKM9 peptide, all titrations performed with this peptide were performed with the protein solution in the syringe and the peptide in the cell. Experiments were performed between 5 and 40 °C, in 5 °C increments. In cases where the enthalpy of binding, ΔH, was close to zero and thus no heat effect was observed, the titration was stopped after three injections. The heat of dilution was measured prior to each experiment by performing at least four peptide injections into buffer, and in all cases was found to be negligible relative to the protein-peptide heats. The resulting binding isotherms were analyzed using the Origin scripts for ITC data analysis provided by MicroCal (Northampton, MA). The following models were used, in order of increasing complexity, and the simplest model that accurately fit the data was accepted.
Single set of identical binding sites model, with one/two peptides per S100 dimer (50, 55):
| (1) |
where Q is the integral heat at each injection, A = 1 + [syringe]t/(n[cell]t) + [syringe]t/[n(1/Kd)[cell]t], n is the stoichiometry of the protein–peptide complex, ΔHcal is the molar heat of peptide binding, Vo is the active cell volume, [syringe]t is the total concentration of the peptide/protein solution in the ITC syringe, [cell]t is the total concentration of the protein/peptide solution in the ITC cell, and Ka is the association constant.
Two-sequential binding sites model, with two peptides per S100 dimer (50, 55):
| (2) |
where Q is the integral heat at each injection, ΔHcal1 and ΔHcal2 are the molar heats of peptide binding, Vo is the active cell volume, Kd1 and Kd2 are the dissociation constants for binding to sites 1 and 2, respectively, and ΔHcal1 and ΔHcal2 are the calorimetric enthalpies for binding to sites 1 and 2, respectively.
Fluorescence Spectroscopy
Steady-state fluorescence titrations were performed using a Fluoromax-4 spectrofluorometer (Horiba Jobin Yvon, Kyoto, Japan) operating FluorEssence v3.5 software. A 10 mm cuvette was used throughout all experiments. All TRTK12 peptides contain a Trp residue. In the available structures of the S100 proteins in complex with the wild-type peptide (S100B, S100A1), this tryptophan becomes buried in a hydrophobic region upon binding. Therefore, changes in Trp emission fluorescence intensity were used to monitor interactions between the TRTK12 peptides and S100 proteins. The experiments were performed in the binding buffer at 25±0.2 °C using a thermostated cell holder. The solution was excited at 295 nm and the emission spectra was monitored from 305 to 420 nm to monitor changes in λmax, while 350 nm was used to specifically monitor changes in intensity. The initial concentration of the TRTK12 peptides in the fluorescence cell was 2–8 μM. Small aliquots of concentrated protein were added until signal saturation, or until the fluorescence signal deviated from unity due to inner filter effects. The experimental emission spectra were corrected by subtracting the emission control spectra of protein added to buffer in the absence of peptide. The fluorescence intensity was also corrected for fluctuations in lamp intensity by dividing the fluorescence signal by the lamp intensity. The data were fit to a single site binding model using Nonlinear Regression Analysis Program (NLREG)(56) and the following equation:
| (3) |
where Q equals the fluorescence intensity at each titration step, Qmax equals the maximum intensity for the fully saturated peptide, [Cell]total equals the molar concentration of peptide at each step, A = Kd + [Cell]total + n[Titrant]total, n equals the number of binding sites, and [Titrant]total equals the molar concentration of the protein at each step.
Anisotropy titrations were performed in the same manner as above, but using Vertical (V) or Horizontal (H) excitation and emission polarizations (IVV, IVH, IHH, and IHV). The G factor was calculated as:
| (4) |
where IHV and IHH represent the corrected intensities for the horizontally polarized light of the peptide in solution. Anisotropy was calculated as:
| (5) |
where 〈r〉 represents the anisotropy at a given titration step and IVV and IVH represent the corrected intensities of the vertically polarized light.
The relative exposure of the Tryptophan residue in the peptide:protein complex was assayed using dynamic quenching with acrylamide. Titrations were performed using 4–8 μM solutions of peptide in the presence or absence of saturating amounts of the S100 proteins and 5 mM CaCl2 or 5mM EDTA. In cases where the affinity for the complex was low, the protein concentration used was limited by the inner filter effect. Titrations were performed until the final concentration of acrylamide was ~0.6 M and the data were then fitted to the modified Stern-Volmer equation (54, 57):
| (6) |
where Io is the initial fluorescence intensity, I is the intensity at a given titration step, KSV is the Stern-Volmer constant, [Q] is the acrylamide concentration at a given titration step, and V[Q] is the constant describing the static component of the quenching reaction. The intensity was corrected for dilution and the average values are reported.
Structure-based Calculations of ΔCp
Using the available three-dimensional structures of S100A1, S100A2, S100B, and S100P in the calcium bound state and/or in complex with a peptide target, ΔASAtot was calculated as previously described (58). In cases where no structure was available, homology models of the peptide-bound state were generated using Modeler (59) and PDB entries 1MQN (40), 1MQ1 (43), 3IQQ (44), and 2KBM (32). Changes in the accessible surface area upon binding, ΔASAtotal, were calculated as previously described (50, 51, 54). Changes in ΔASA were further subdivided into four categories (aliphatic surface area, aromatic surface area, peptide backbone surface area, and polar surface area) and converted into ΔCp using the following empirical relationship (50, 51, 58, 60, 61):
| (7) |
where ΔASAalp, ΔASAarm, and ΔASApol values are the changes in ASA for aliphatic, aromatic, and polar amino acids, respectively, and ΔASAbb values are the changes in ASA for the polypeptide backbone.
Kinetic Stopped Flow koff Measurements
Measurements of protein-peptide dissociation rate constants (koff) were performed using standard stopped-flow methods. Data collection was performed using a JASCO J-815 spectropolarimeter equipped with an SFM 300 mixing module (BioLogic Science Instruments), containing a HDS mixer and a FC-15 observation cuvette. The koff rate constants were evaluated at a minimum of three temperatures between 5°C and 35°C. The temperature of the sample chamber was regulated using a circulating water-bath. After peptide tryptophan excitation at 295 nm from a mercury lamp source, fluorescence emission intensity was collected using a N-WG 320 nm cutoff filter (BioLogic Science Instruments). Slit widths were adjusted between 3 to 5 nm to eliminate or minimize photobleaching. Voltages applied to the photomultiplier tube were set accordingly and ranged from 700–850V. Kinetic traces were collected under an optimal set of push volumes and flow rates for the SFM 300 and kept constant for all experiments. Because peptide binding is calcium-dependent, peptide dissociation was initiated by calcium chelation. For this, preformed protein-peptide complexes (6 μM or 12 μM peptide) in the above binding buffer were mixed in a 1:1 mixing ratio with 40 mM EDTA. Special care was taken to control for inner filter effects by limiting the protein to peptide ratio.
Using the Bio-Kine32 software package (BioLogic Science Instruments), rate constants, koff, were obtained by fitting the exponential change of the emission fluorescence intensity from EDTA-induced peptide dissociation to the following equation:
| (8) |
where I(t) is fluorescence intensity as a function of time, y0 is the initial fluorescence intensity, A is the amplitude of the change between initial and final fluorescence intensity, kapp is the observed kinetic rate constant associated with the fluorescence intensity relaxation, and XT is the sloping baseline correction used in the Bio-Kine32 software, which accounts for the photobleaching effect. Errors were calculated from the standard deviation of five independent traces. A dead time of 4.0 ms was electronically estimated with BioLogic’s Bio-Kine32 software and experimentally confirmed using the dead time assessment procedure of Peterman et. al. (62). Since the experimentally determined rate constants were sufficiently slow, no dead time data correction was made.
RESULTS
The TRTK12 peptide has been shown to bind human S100B and human S100A1 in a calcium-dependent manner (29, 32, 35, 40–44, 46, 47) and has been proposed to be a general S100 protein binding motif (32). To probe the specificity of TRTK12 for these proteins, sixteen representative members of the human S100 protein family were screened for binding using isothermal titration calorimetry (ITC) and emission fluorescence. Unexpectedly, novel interactions were detected with only two S100 proteins: S100A2 and S100P. To gain a better understanding of these interactions, the binding of the wild-type peptide and its alanine variants were further characterized using several biophysical methods.
Interactions of TRTK12 Peptides with S100B
Figure 2A shows the binding isotherm obtained from ITC experiments at 25°C in which the wild-type TRTK12 peptide was titrated into S100B in the presence of 5 mM CaCl2. As reported previously (4), two peptides interact with the S100B dimer through identical binding sites (Equation 1, n=2). The dissociation constant, Kd, obtained from the fit, 1.7±0.1 μM, is in good agreement with previously reported values (44, 47, 50). The binding model is consistent with structures derived from X-ray crystallography and solution NMR involving complexes of TRTK12 and S100B from several orthologs (Homo sapiens, Rattus norveggicus, and Bos Taurus) (5, 17, 18). ITC experiments were performed over the temperature range of 5°C to 40°C. In addition, the thermodynamics of binding were measured for six alanine variants of TRTK12 (see Materials and Methods for abbreviated names and sequences). All peptides, with the exception of TRTKM11, bind with a stoichiometry similar to that of wild-type, i.e. two peptides per S100B dimer (Equation 1, n=2). TRTKM11 appears to interact with a different stoichiometry of one peptide per S100B dimer (Equation 1, n=1). This stoichiometry has previously been observed for several peptides binding to the S100 proteins (50, 63–69). The dissociation constants of wild-type and alanine peptides are listed in Table 1.
Figure 2.

Examples of ITC experiments showing the binding of TRTK12 to S100B, S100A1, S100A2, and S100P in the presence of calcium. Upper plots in each panel represent the raw heat effects as a function of time and lower plots show the cumulative heat effects (●) as a function of the molar ratio of peptide to protein, and the fits to the experimental data using Eq. 1 (—) for binding of TRTK12 to S100B at 25 °C (A), binding of TRTK12 to S100A1 at 25 °C (B), binding of TRTK12 to S100A2 at 25 °C (C), and binding of TRTK12 to S100P at 25 °C (D).
Table 1.
Summary of the dissociation constants, the changes in the heat capacity upon binding, and the kinetic rate constants of the TRTK12 wild-type peptide and alanine variants binding to S100B at 25°C.
| Peptide | Kda μM | ΔCp, expb kJ·mol−1 K−1 | ΔCp, calcc kJ·mol−1 K−1 | koff (s−1)d | kon (1×107 M−1s−1)e |
|---|---|---|---|---|---|
| TRTK12 | 1.7 ± 0.1 (3 ± 1) | −1.2 ± 0.2 | −1.4 ± 0.1 | 53 ± 4 | 3.1 ± 0.3 |
| TRTKM1 | 2.9 ± 1.0 (0.2 ± 0.1) | −1.2 ± 0.1 | −1.3 ± 0.1 | 34 ± 2 | 1.2 ± 0.1 |
| TRTKM5 | 1.2 ± 0.4 (0.6 ± 0.6) | −1.0 ± 0.1 | −1.2 ± 0.1 | 66 ± 7 | 5.5 ± 1.9 |
| TRTKM6 | 1.3 ± 0.6 (5 ± 1) | −1.3 ± 0.1 | −1.5 ± 0.1 | 31 ± 1 | 6.6 ± 1.0 |
| TRTKM9 | 1.7 ± 0.1 (2 ± 1) | −1.5 ± 0.1 | −1.5 ± 0.1 | 53 ± 8 | 3.1 ± 0.5 |
| TRTKM10 | 3.4 ± 1.0 (1 ± 0.1) | −1.0 ± 0.1 | −1.1 ± 0.1 | 36 ± 8 | 1.1 ± 0.4 |
| TRTKM11 | 3.8 ± 1.0 (10 ± 3) | −1.6 ± 0.2 | −1.3 ± 0.1 | n.a. | n.a. |
Dissociation constants obtained using ITC. Values in parentheses were obtained using emission fluorescence assays.
The experimentally determined changes in the heat capacity upon binding (Figure 3).
Structure-based calculations (see Eq. 7) using the average value from homology models based on the human S100B dimer binding to two peptides using PDB: 1MQ1 (43), 1MWN (40), and 3IQQ (44).
koff values obtained using fluorescence stopped-flow spectroscopy (Eq 9).
kon values were calculated using koff rate constants and the dissociation constants obtained from ITC.
Figure 3A shows the temperature dependence of the enthalpy of binding, ΔHcal, for the wild-type peptide and alanine variants to S100B. The slope of temperature dependence corresponds to the heat capacity change upon binding, ΔCp, and these values are summarized in Table 1. It has previously been shown that ΔCp can be predicted using empirical changes in the accessible surface area upon binding, provided that accurate structural models are available (2, 4, 14, 21). The experimentally determined heat capacity change for S100B-TRTK12 (−1.2±0.2 kJ·mol−1·K−1) is in excellent agreement with the structure-based calculated values (Table 1). In addition, the values obtained for all alanine variants but TRTKM11 are within error of the wild-type ΔCp value, suggesting they interact with S100B through a similar structural mode. This is also expected based on the structure-based calculations of ΔCp using homology models of the alanine variants (Table 1). Again, the only notable exception is the TRTKM11 variant, which interacts with the S100B dimer with a novel stoichiometry of one peptide per S100B dimer (Equation 1, n=1), and therefore is inaccurately represented by the homology models.
Figure 3.
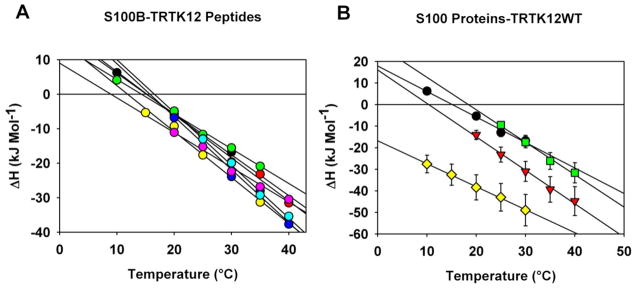
Temperature dependence of the enthalpies of binding, ΔHcal. Panel A: Binding of the TRTK12WT (black circles), TRTK12M1 (red circles), TRTKM5 (green circles), TRTKM6 (yellow circles), TRTKM9 (blue circles), TRTKM10 (pink circles), and TRTKM11 (cyan circles) peptides to the S100B protein in the presence of 5 mM calcium. Solid lines represent linear fits of the ΔHcal temperature dependencies for each peptide-S100B interaction. The slopes of these lines represent the changes in heat capacity, which are summarized in Table 1. Panel B: Temperature dependence of the enthalpies, ΔHcal, of the TRTK12WT peptide binding to S100B (black circles), S100A1 (red inverted triangles), S100A2 (green squares), and S100P (yellow diamonds) in the presence of 5 mM calcium chloride.
Steady-state emission fluorescence and fluorescence anisotropy were used to monitor protein-peptide binding and determine the dissociation constants as a complementary method to ITC. The TRTK12 peptides contain a tryptophan at position 7 (W7), which is buried in a hydrophobic binding pocket in the available structures (29, 32, 43, 44). This burial would be expected to lead to changes in the fluorescence intensity of W7 upon binding because of changes in the polarity of the chemical environment. The S100 proteins, with the exclusion of S100A1, do not contain a tryptophan and therefore contribute minimally to the overall signal intensity. The Kd obtained at 25°C using fluorescence anisotropy for binding of the wild-type peptide to S100B, 3±1 μM, is in good agreement with that obtained from ITC, 1.7±0.1 μM. A similar correspondence in the binding affinities is observed for all alanine variants (Table 1).
Changes in fluorescence intensity in a stopped flow experiment were also used to monitor the koff rates for peptide dissociation (see Materials & Methods). In these experiments, the addition of EDTA to a preformed complex of a S100 protein and the TRTK12 peptides leads to calcium chelation and peptide dissociation. This, in turn, will manifest itself as changes in fluorescence intensity. The emission fluorescence relaxation for peptide dissociation fit well to a single exponential as described by Equation 8. The apparent koff rate obtained for the dissociation of the wild-type TRTK12 peptide from S100B at 35°C, 86±8 s−1, is in good agreement with the previously obtained value from line shape analysis of chemical shifts changes monitored by NMR (50). The kinetic rate constants for the interactions of S100B with all of the TRTK12 peptides are summarized in Table 1. There is only a two-fold difference in the apparent koff rates between the fastest and slowest peptides, consistent with the narrow range of Kd values observed for the alanine variants in ITC experiments.
Interactions of TRTK12 Peptides with S100A1
Figure 2B shows the binding isotherm obtained from ITC experiments at 25°C in which the wild-type TRTK12 peptide was titrated into S100A1 in the presence of 5 mM CaCl2. As with S100B, the stoichiometry of the interaction is two TRTK12 peptides per S100A1 dimer (Equation 1, n=2). The dissociation constant obtained from the fit was found to be 3.4±1.0 μM (Table 3), which is weaker than that of S100B-TRTK12 (Table 1), consistent with a previous report of S100A1 binding this peptide with a two-fold reduced affinity (7). For the TRTK12 alanine variants, the interactions between S100A1 and a given TRTK12 peptide are also characterized by weaker binding affinities, when compared to S100B and the same peptide. This trend is apparent in the dissociation constants measured by both ITC and emission fluorescence (Table 3). In particular, the TRTKM5 peptide binds to S100A1 with ten-fold weaker affinity than to S100B. Overall, there is also a larger variation within the binding affinities of the TRTK12 peptides for S100A1, as compared to S100B.
Table 3.
Summary of the dissociation constants, the changes in the heat capacity upon binding, and the kinetic rate constants of the TRTK12 wild-type peptide and alanine variants binding to S100A1 at 25°C.
| Peptide | Kda μM | ΔCp, expb kJ·mol−1·K−1 | ΔCp, calcc kJ·mol−1·K−1 | koff (s−1)d | kon (1×107 M−1·s−1)e |
|---|---|---|---|---|---|
| TRTK12 | 3.4 ± 1.0 (1 ± 0.5) | −1.6 ± 0.1 | −1.0 ± 0.1 (−1.6 ± 0.1) | 44 ± 8 | 1.2 ± 0.3 |
| TRTKM1 | 1.0 ± 0.2 (0.5 ± 0.1) | −2.0 ± 0.1 | −0.7 ± 0.1 (−1.4 ± 0.1) | 6 ± 2f | 0.6 ± 0.2f |
| TRTKM5 | 11.7 ± 1.0 (6 ± 2) | −1.7 ± 0.2 | −0.7 ± 0.1 (−1.5 ± 0.1) | 77 ± 22 | 0.7 ± 0.2 |
| TRTKM6 | 5.3 ± 3.0 (7 ± 3) | −2.2 ± 0.1 | −0.9 ± 0.1 (−1.7 ± 0.1) | 87 ± 13 | 1.6 ± 1.0 |
| TRTKM9 | 3.1 ± 1.0 (0.6 ± 0.1) | −1.5 ± 0.1 | −1.4 ± 0.1 (−1.7 ± 0.1) | 97 ± 21g | 2.7 ± 0.8g |
| TRTKM10 | 9.9 ± 1.0 (27 ± 15) | −1.7 ± 0.1 | −0.7 ± 0.1 (−1.3 ± 0.1) | 63 ± 6g | 1.7 ± 0.4g |
| TRTKM11 | 4.5 ± 1.9 (2 ± 1) | −1.5 ± 0.1 | −1.3 ± 0.1 (−1.5 ± 0.1) | 71 ± 12 | 1.6 ± 0.7 |
Dissociation constants obtained using ITC. Values in parentheses were obtained using emission fluorescence assays.
The experimentally determined changes in the heat capacity upon binding (Figure 3).
Structure-based calculations (see Eq. 7) homology models based on the human S100A1 dimer binding to two peptides using PDB: 2KBM (32) or (3IQQ) (44).
koff values obtained using fluorescence stopped-flow spectroscopy (Eq 9).
kon values were calculated using koff rate constants and the dissociation constants obtained from ITC.
koff value was measured at 5°C. kon value was calculated for the corresponding temperature.
koff value was measured at 15°C. kon value was calculated for the corresponding temperature.
Figure 3B shows the comparison of the enthalpies of binding of the TRTK12 wild-type peptide to different S100 proteins, including S100A1 and S100B. The absolute values of the enthalpies for S100A1 are significantly lower than those for S100B. The temperature dependence of the enthalpies, which represent ΔCp, is also somewhat different for these proteins (Table 3). For the binding of the alanine variants to S100A1, the ΔCp values also differ from the binding of the wild-type. In particular, binding of the TRTKM6 peptide shows the largest ΔCp value (Table 3). Structure-based calculations of the changes in the heat capacity are listed in Table 3. A comparison shows that, in general, the calculated values are close to the experimentally determined ΔCp values for the binding of the TRTK12 peptides to S100A1. However, the best agreement appears to be with the homology models derived from X-ray structures of S100B-TRTK12 from the Weber group (44), as opposed to those derived from rat S100A1 (32).
Table 3 summarizes the dissociation constants and kinetic rate constants for peptide binding to S100A1. As with S100B, all peptides fit well to a single exponential, consistent with the binding model of two peptides per S100B dimer (Equation 1, n=2). The apparent koff rates are faster than those observed for S100B, suggesting a compensatory effect in the off rate to account for the weaker binding affinities. For both proteins, the kinetics and dissociation constants for all peptides differ by an order of magnitude or less.
Interactions of TRTK12 Peptides with S100A2
The binding of TRTK12 to human S100A2 is a novel interaction, which was identified by us using tryptophan fluorescence and was further confirmed using ITC. Figure 2C shows the binding isotherm obtained from ITC experiments at 25°C in which the wild-type TRTK12 peptide was titrated into S100A2 in the presence of 5 mM CaCl2. Although the binding model for the wild-type peptide is consistent with both S100B and S100A1 (Equation 1, n=2), the dissociation constant is more than an order of magnitude weaker: 48±3 μM. Similarly, the dissociation constants for the binding of the TRTK12 alanine variants to S100A2 are significantly weaker than those for both S100B and S100A1 (Table 4). Notably, the TRTKM5 and TRTKM11 peptides bind to S100A2 with a much weaker affinity than wild-type, three- and ten-fold, respectively. Furthermore, the TRTKM10 peptide, which substitutes a C-terminal isoleucine for an alanine, exhibits no detectable binding by ITC or emission fluorescence (data not shown). As with S100A1, the TRTKM1 peptide appears to bind more tightly to S100A2 than does wild-type. In addition, the experimentally determined ΔCp values of the alanine variants are generally smaller than those of either S100B or S100A1 (Table 4). This possibly indicates that a smaller amount of hydrophobic surface area is buried in the bound state of the S100A2-TRTK12WT complex, although a compensatory effect in the hydrophilic surface area cannot be ruled out until a structure of the bound complex becomes available. There are large deviations in the heat capacity upon binding of the TRTK12 alanine variants to S100A2, relative to both the wild-type and the calculated values based on a variety of structural models (Table 4). This suggests several of the alanine variants may adopt different conformations in the bound complex relative to the wild-type peptide.
Table 4.
Summary of the dissociation constants, the changes in the heat capacity upon binding, and the kinetic rate constants of the TRTK12 wild-type peptide and alanine variants binding to S100A2 at 25°C.
| Peptide | Kda μM | ΔCp, expb kJ·mol−1·K−1 | ΔCp, calcc kJ·mol−1·K−1 |
|---|---|---|---|
| TRTK12 | 48 ± 3 (43 ± 9) | −1.5 ± 0.1 | −1.5 ± 0.1 |
| TRTKM1 | 15.2 ± 1.0 (16 ± 1) | −0.8 ± 0.1 | −1.3 ± 0.1 |
| TRTKM5 | 131.0 ± 6.1 (N/A)c | −0.9 ± 0.1 | −1.3 ± 0.1 |
| TRTKM6 | 21.5 ± 4.0 (23 ± 3) | −0.7 ± 0.1 | −1.5 ± 0.1 |
| TRTKM9 | 45.0 ± 2.0 (17 ± 4) | −0.4 ± 0.1 | −1.5 ± 0.1 |
| TRTKM10d | - | - | - |
| TRTKM11 | 504 ± 9.7 (N/A)c | −1.6 ± 0.1 | −1.5 ± 0.1 |
Dissociation constants obtained using ITC. Values in parentheses were obtained using emission fluorescence assays.
The experimentally determined changes in the heat capacity upon binding (Figure 3).
Structure-based calculations (see Eq. 7) homology models based on the human S100A2 dimer binding to two peptides using PDB:3IQQ (44).
No detectable binding by ITC or emission fluorescence.
We were unable to measure the kinetic rate constants for dissociation of all TRTK12 peptides from S100A2 because there was no detectable change in the emission fluorescence intensity upon the addition of EDTA. Dynamic quenching assays with acrylamide confirmed that W7 of the TRTK12 peptides is buried in the bound complex with S100A2 to a similar extent as with other proteins, such as S100B (Table 2). Furthermore, there are no detectable heats of binding for the TRTK12 peptide and S100A2 in the absence of calcium, which suggests that there is no calcium independent binding. Based on these finding, we conclude that the off rates themselves are too fast to be detectable by the stop flow instrument (instrument dead time: 4 ms). This places a lower bound of koff at 250 s−1 and results in kon rates that are equal or less than those of S100B.
Table 2.
Stern-Volmer (KSV) constants of the TRTK12 peptides in the free and bound state for various complexes with S100B, S100A2, and S100P.
| Peptide Only KSV (M−1) | S100B+Peptide KSV (M−1) | S100A2+Peptide KSV (M−1) | S100P+Peptide KSV (M−1) | |
|---|---|---|---|---|
| TRTK12 | 14.0 ± 0.2 | 2.1 ± 0.1a | 4.9 ± 0.5a | 8.5 ± 0.2 |
| TRTKM1 | 14.9 ± 0.2 | 2.4 ± 0.1 | 4.3 ± 0.2 | n.d. |
| TRTKM5 | 11.9 ± 1.0 | 2.8 ± 0.1 | 8.9 ± 0.2 | 11.2 ± 0.2 |
| TRTKM6 | 11.8 ± 0.2 | 3.7 ± 0.1a | 4.7 ± 0.1a | 11.3 ± 0.3 |
| TRTKM9 | 15.4 ± 0.2 | 7.2 ± 0.2 | 6.1 ± 1.1 | 13.6 ± 0.4 |
| TRTKM10 | 13.6 ± 0.3 | 7.5 ± 0.1 | n.d. | n.d. |
| TRTKM11 | 13.8 ± 0.1 | 7.2 ± 0.1 | 8.7 ± 0.2 | n.d. |
Experiments were performed in the presence of 150mM NaCl.
Interactions of TRTK12 Peptides with S100P
The binding of wild-type TRTK12 with S100P is the only other novel interaction identified for this peptide and the human S100 proteins. Figure 2D shows a typical titration of the TRTK12 peptide into S100P. The dissociation constant, 5.1±1.0 μM, is weaker than that of S100B and S100A1, but still significantly tighter than S100A2 (Table 5). As with S100A2, the TRTKM5 and TRTKM10 peptides exhibit substantially reduced affinities, while the TRTKM11 peptide binds with a similar stoichiometry to S100B (one peptide per S100P dimer, Equation 1, n=1). This stoichiometry has previously been observed for several peptides binding to the S100 proteins (63), most notably to S100B (50, 68), S100A4 (65–67, 69), and to S100A10 (65–67). The results obtained on TRTKM11 peptide suggest significant plasticity in the peptide binding and show an interesting case where a single amino acid substitution can switch between different binding modes. The difference in the affinities does not correlate with the differences in the enthalpies of binding, i.e. there is no enthalpy-entropy compensation (data not shown). In addition, there were no observable changes in the emission fluorescence intensity of the tryptophan upon peptide binding or dissociation to S100P for three of the alanine variants: TRTKM5, TRTKM6, and TRTKM9. This suggests that there may be an alternate mode of binding for these peptides where W7 is solvent exposed in the bound complex. Dynamic quenching with acrylamide was used to probe the relative exposure of W7. Judging by the similarity between the Stern-Volmer constants of the S100P-TRTK12 peptide complexes and the free TRTK12 peptides (Table 2), we conclude that indeed W7 is exposed in the TRTKM5, TRTKM6, and TRTKM9 complexes with S100P.
Table 5.
Summary of the dissociation constants, the changes in the heat capacity upon binding, and the kinetic rate constants of the TRTK12 wild-type peptide and alanine variants binding to S100P at 25°C.
| Peptide | Kda μM | ΔCp, expb kJ·mol−1·K−1 | ΔCp, calcc kJ·mol−1·K−1 | koff (s−1)d | kon (1×107 M−1·s−1)e |
|---|---|---|---|---|---|
| TRTK12 | 5.1 ± 0.3 (11 ± 1) | −1.1 ± 0.1 | −1.7 ± 0.1 | 286 ± 43f | 5.6 ± 0.9f |
| TRTKM1 | 2.0 ± 0.5 (7 ± 3) | −0.9 ± 0.1 | −1.4 ± 0.1 | 135 ± 18 | 6.8 ± 1.9 |
| TRTKM5 | 56.2 ± 4.0 (100 ± 23) | −1.2 ± 0.1 | −1.5 ± 0.1 | - | - |
| TRTKM6 | 7.4 ± 1.0 (16 ± 4) | −0.9 ± 0.1 | −1.6 ± 0.1 | - | - |
| TRTKM9 | 7.1 ± 0.5 (30 ± 10) | −1.0 ± 0.1 | −1.7 ± 0.1 | - | - |
| TRTKM10 | 43.1 ± 3.9 n.d. | −1.3 ± 0.1 | −1.3 ± 0.1 | 32 ± 11g | 0.07 ± 0.03g |
| TRTKM11 | 31.7 ± 0.5 n.d. | −1.8 ± 0.1 | −1.5 ± 0.1 | 151 ± 25h | 0.6 ± 0.1h |
Dissociation constants obtained using ITC. Values in parentheses were obtained using emission fluorescence assays.
The experimentally determined changes in the heat capacity upon binding (Figure 3).
Structure-based calculations (see Eq. 7) using the average value from homology models based on the human S100P dimer binding to two peptides using PDB:3IQQ (44).
koff value obtained using fluorescence stopped-flow spectroscopy (Eq 9).
kon values were calculated using the koff rate constants and the dissociation constants obtained from ITC.
koff value is within error of the dead-time, but is consistent with the temperature-dependent slope of the rate constant.
koff value was measured at 5°C. kon value was calculated for the corresponding temperature.
koff value was measured at 20°C. kon value was calculated for the corresponding temperature.
The absolute enthalpies of binding of the TRTK12 wild-type peptide to S100P are significantly lower than the other S100 proteins, including S100A1 (Figure 3B). However, the changes in the heat capacity upon binding, ΔCp, are very similar to those of S100B (Table 5). As with S100B, the ΔCp values for the binding of the alanine variants to S100P are generally within error of those of wild-type. The only notable exception is TRTKM11, which binds with a different stoichiometry (see above). This suggests that the TRTKM5, TRTKM6, and TRTKM9 variants bind in a manner that compensates for the exposure of W7, possibly by burying other hydrophobic residues in the bound complex.
Because there are no changes in the emission fluorescence intensity for TRTKM5, TRTKM6, and TRKTM10 peptides, the kinetic rate constants of binding to S100P could only be measured for the remaining TRTK12 peptides (Table 5). Although it is a limited data set, the measured koff rates are the fastest observed amongst the other studied S100 proteins.
DISCUSSION
Promiscuity of the S100B Binding Site
Since its discovery in 1965, S100B has become the best studied member of the S100 protein family (70). To date, over 20 binding partners have been identified, including p53, RAGE, tau, NDR, microtubules, intermediate filaments, GFAP, and CapZ (TRTK12) (10, 40–42, 44, 45, 50, 71–74). Despite the promising work of Ivanenkov et al. to elucidate a target binding motif (35, 46), subsequent studies have revealed a large degree of promiscuity within the S100B binding pocket and significant variability within the consensus binding sequence (50, 71, 75). In particular, recent work by Weber et al suggests that no particular residue or position from the original phage display motif, (K/R)-(L/I)-X-W-X-I-L, is a prerequisite for binding (71). Indeed, the only characteristics shared by all available binding partners are the presence of several hydrophobic residues and at least one intervening positive residue. This suggests that the binding affinity of TRTK12 for S100B is probably dominated by non-specific, hydrophobic interactions and only modulated by electrostatic interactions. These minimal requirements for binding S100B may explain why it is able to interact with different targets through novel binding modes, with variations in stoichiometry, affinity and cooperatively (50, 71, 76).
The above assessment is consistent with the results of this study, where no single alanine mutation of the TRTK12 peptide significantly altered the binding affinity or kinetics (observed changed in Kd and koff are less than two-fold, see Table 1). This includes the charge-neutralizing variants, TRTKM6 and TRTKM9, which contain a D6A and K9A substitution, respectively. These results are consistent with the original phage display assay, which found no amino acid preference for position 6 within the TRTK12 sequence, and no preference for a positively charged residue flanking the C-terminal side of the tryptophan (i.e. residue 9) (35). Furthermore, the presence of physiological salt had no observable effect on the dissociation constant of the wild-type peptide and caused only a slight decrease in the apparent koff (data not shown). Since the TRTK12 alanine variants appear to only slightly modulate the peptide binding affinity, our findings suggest that the tryptophan (W7) acts as a bulky hydrophobic anchor for TRTK12 peptide binding to S100B, consistent with previous proposals (43, 71, 77).
This tryptophan 7 of TRTK12 may also play an important role in the kinetics of peptide binding. Previous studies proposed that S100B interacts with several of its binding partners through a “fly-casting mechanism,” where the peptide remains unstructured in a “low affinity” encounter complex, and proceeds to adopt a folded conformation in the final “high affinity” complex (50, 78, 79). This model is increasingly being examined as a rational explanation for the promiscuity of S100B because so many of its binding partners appear to be unstructured in the unbound state (50, 77, 78). Furthermore, computational models suggest that several binding partners of S100B form initial contacts via a large, non-polar residue, such as a tryptophan or phenylalanine (77, 78). The underlying principle is that intrinsically disordered proteins or peptides (IDP), such as TRTK12, have a larger “capture radius” than peptides with a preformed structure (80), and therefore increase the bimolecular association rates (81). A recent review by Kiefhaber et al. quantitatively defined the fly-casting mechanism as a bimolecular association involving an IDP with an apparent kon rate ≥ 1×107 M−1 s−1 (82). This mechanism is consistent with the association rate constants observed for the TRTK12 peptides binding to S100B (Table 1), and the observed changes in the conformational ensemble of the peptide between the free and bound states (40, 41, 44, 50).
TRTK12 Selectively Binds a Subset of S100 Proteins
Previously, it has been suggested that TRTK12 binds to several, distantly related members of the S100 family and therefore represents a general S100 binding motif (32). In contrast, the results of this study indicate that TRTK12 selectively binds S100B and only three other S100 proteins: S100A1, S100P, and S100A2. The available structures of the TRTK12 peptide bound to S100B and S100A1 (29, 32, 43, 44), as well as the sequence alignment of the S100 proteins, suggest a rational basis for the specificity of binding observed in this study. A phylogenetic analysis of the S100 family indicates that two of these proteins, S100A1 and S100P, are the most closely related members to S100B (Figure 5), consistent with the similar affinities and kinetics observed for wild-type TRTK12 binding (Tables 1,3,5). Furthermore, S100A1 exhibits a similar insensitivity to alanine mutagenesis as S100B (Table 3) and also appears to interact with an equally large and diverse set of binding partners (75). These similarities are most likely due not only to the global homology between S100A1 and S100B, but also the sequence conservation within the hinge and C-terminal helix regions (Figure 4). Previous bioinformatics studies have revealed that the hinge and C-terminal helix regions of the S100 proteins contain the largest degree of sequence variability, and a number of crystallographic and solution structures have demonstrated these regions are crucial in S100 target recognition (6, 30, 32, 42, 50, 83–86). The composite binding site for S100B-TRTK12 and S100A1-TRTK12 complexes only differ significantly in terms of their charge distributions. Lys58 of S100A1 has no homologous residue in S100B, and Asp86 of S100B is replaced with an asparagine in the S100A1 sequence. These substitutions may be sufficient to explain the twofold weaker binding affinity of the wild-type peptide for S100A1 in the presence of physiological ionic strength (data not shown).
Figure 5.
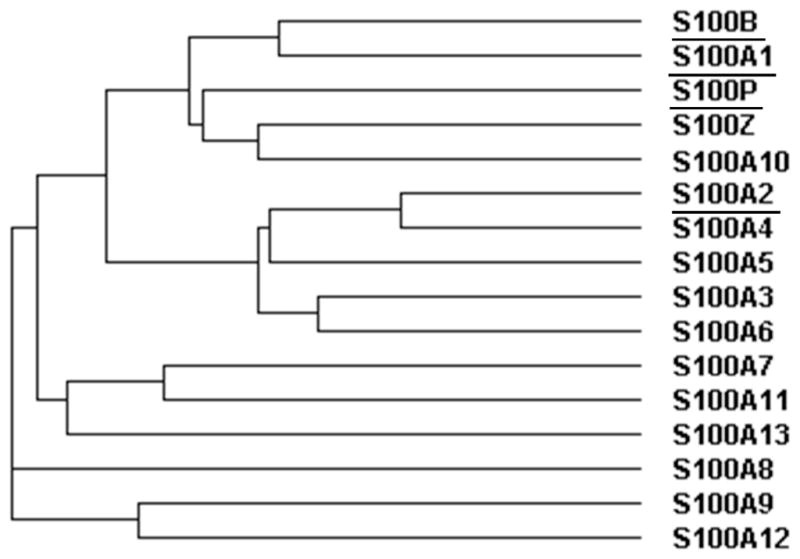
Phylogenetic tree based on the sequence alignment. For clarity, proteins that were found to interact with TRTK12 (S100B, S100A1, S100P, and S100A2) are underlined.
Figure 4.
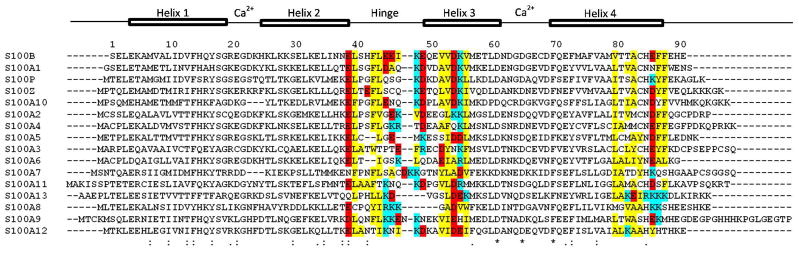
Sequence Comparison of Human S100 Proteins. Sequence alignment of human S100 family members generated using ClustalOmega (106). For clarity, residue numbering in the text is always written in reference to the sequence of S100B.
A hydrophobic pocket for W7 of TRTK12 in the structure of S100B-peptide complex is formed by several residues including I36, L44, V52, and V56. The corresponding positions in the sequence of S100A1 are L36, L44, A52 and V56 (Figure 4). Many of the residues that form the binding pocket in the S100B-TRTK12 and S100A1-TRTK12 complexes are not conserved in other S100 proteins. This sequence difference likely leads to the increased sensitivity of the TRTK12 alanine variants for S100A2 and S100P and the inability of most S100 proteins to bind the TRTK12 peptide. In the absence of structural information for TRTK12 complexes with S100P and S100A2, it is difficult to identify the “hot-spot(s)” that define binding specificity. Therefore, one can only provide a possible rational based on the sequence alignment. For example, within the hinge region of S100P, several substitutions change both the net polarity and charge, relative to S100B. These include S41P, E45Q, E46S, and Q50K. Several residues within helices 3 and 4 have also been replaced, including V56L, M79A, and V80I which modify the shape of the hydrophobic pocket and are likely responsible for the reduced affinity of the TRTKM5, TRTKM10, and TRTKM11 variants to S100P. This may also explain why the TRTKM5, TRTKM6, and TRTKM9 alanine variants are still able to bind S100P, despite the apparent exposure of W7 to solvent (Table 2). Interestingly, S100A10 and S100Z, which are closely related to S100B, S100A1, and S100P, do not appear to interact with the TRTK12WT peptide (Table 1). This may be the result of non-conserved substitutions within the hinge region, including H42E, E45S, and I47Q for S100Z and I36M, I47Q, and V56I for S100A10. In addition, S100A10 and S100Z both have an unusually long, unstructured region following helix 4 that consists of four Lys residues, which may electrostatically repulse the TRTK12 peptide and/or partially occlude the binding site.
S100A2 is the only member of the S100A2/S100A3/S100A4/S100A5/S100A6 phylogenetic branch that is able to bind TRTK12 (Figure 5). Furthermore, it appears to be the most sensitive S100 protein to the TRTK12 motif, as demonstrated by the ability of the TRKTM11 variant, which substitutes an alanine for isoleucine, to completely abolish binding. Similarly, the TRTKM5 and TRTKM11 variants, which contain I5A and L11A substitutions, respectively, bind S100A2 with significantly weaker affinities (Table 3). This is likely the result of key substitutions within the hinge (S41P, E45G, I47K, K48V, and Q50E) and C-terminal helices (V52L, V56L, and A83M), relative to S100B. A sequence comparison demonstrates that the other members of this phylogenetic branch contain significant variations in the number and type of charged residues in their hinge region, relative to S100B (Figure 4). In particular, they lack a number of conserved glutamic acid and aspartic acid residues, which may contribute to the inability of the positively charged TRTK12 peptide to bind to these proteins. Furthermore, S100A3 is believed to have a calcium dissociation constant ≥ 30 mM, meaning that, within this study, the majority of the population would have remained in the apo state, with its hydrophobic binding site secluded (87). S100A3 has been proposed to bind zinc with high affinity, which induces a conformational change similar to that observed for other S100 proteins in the presence of calcium, but the effects of Zn2+-S100A3 were not probed in this study. S100A4 is similar to S100Z in that it has a large, positively charged unstructured region following the C-terminal helix. This region is thought to play an important role in the tetramerization of S100A4 by binding to the canonical binding site of an adjacent dimer (88), but it may also fold back onto the protein surface and interfere with the binding sites within an individual dimer. S100A5 and S100A6 both contain a truncated hinge region, as demonstrated by their three and two residue gaps, respectively, relative to the other S100 proteins (Figure 4). In addition, the residues in this hinge region are not conserved, relative to the S100B. A glycine occupies the position of Glu45 in both S100A5 and S100A6, and neither of the sequences contains His42, Phe43, or Ile47. The increased sequence divergence within the S100A2 subgroup, especially in terms of polar and charged residues, results in a binding pocket that is incompatible with the TRTK12 peptide.
Selective Inhibitions of S100 Proteins
Differential expression of individual S100 proteins has been implicated in a wide variety of aberrant cellular processes, making them prime candidates for therapeutic targeting (1, 4, 5, 23, 75). However, each individual S100 protein has been associated with a variety of diverse, tissue-specific, and sometimes antagonistic, functions. For example, up regulation of S100B has been observed in Alzheimer’s disease (7, 8, 10), Parkinson’s disease (11, 89, 90), Down’s syndrome (9), epilepsy (91), melanoma (23, 92, 93), and glioblastomas (94). In contrast, down regulation of S100A1 is associated with an increased likelihood of cardiovascular disease and cardiomyopathies (16, 17). Up regulation of S100A2 has been shown to suppress tumor growth in certain epithelial tissues (95), but to promote metastasis within the lungs and cervix (96, 97). The interactions of S100 proteins with their cellular targets is further complicated by the ability of these proteins to localize and associate with other family members, such as in the S100B/S100A1 heterodimer (98), and the ability of multiple S100 proteins to interact with the same binding partner, such as p53 (22, 71, 99). Therefore, there is a need to selectively inhibit individual S100 proteins, without detrimentally affecting the biological pathways of closely related homologs.
The results of this study indicate that single point variants of the TRTK12 peptide are sufficient to selectively target S100B, or any subset of TRTK12 binding-competent S100 proteins that include S100B. For example, TRTKM1 exhibits two to four-fold enhanced affinity for S100A1, S100A2, and S100P relative to the wild-type peptide, while binding to S100B remains unchanged. Thus, this peptide can putatively act as a micromolar inhibitor for binding of cellular targets for all four S100 proteins. Similarly, the wild-type or TRTKM9 peptides can be used to selectively inhibit S100B, S100A1 and S100P and not S100A2, because S100A2 binds these peptides an order of magnitude weaker (Table 4). Likewise, the TRTKM11 peptide can be used to preferentially bind S100B and S100A1, as opposed to S100A2 or S100P. Finally, the TRTKM5 peptide can be used to selectively inhibit only S100B as the dissociation constants for S100A1, S100P, and S100A2 for this peptide are one to two orders of magnitude weaker than S100B.
These in vitro results suggest a potential strategy to target S100 proteins with individual, peptide-based inhibitors. However, ideal drug candidates usually require nanomolar efficacy in vivo (100), as opposed to the micromolar dissociation constants observed in this study. Furthermore, peptides have traditionally been avoided as therapeutic candidates because of their low bioavailability, decreased membrane permeability, and high likelihood of degradation and clearance by the cellular machinery (101). Nonetheless, several groups have recently published work demonstrating that these drawbacks can be overcome by stabilizing peptides with “hydrocarbon staples” (102). In several cases, these peptides exhibit enhanced binding affinity, as well as increased cell permeability and resistance to degradation (101–105). In particular, they have shown promise as cancer therapeutics by binding to previously “undruggable” targets, such as members of the pro-survivor BL-2 family or HDMX/MDMX apoptotic proteins (103, 105). The S100 proteins derived from the A1/A11/B/P/Z and A2/A3/A4/A5/A6 subgroups are thought to directly and indirectly regulate these pathways through interactions with the p53, HDM2, and HDM4 proteins (50, 71, 76). In particular, S100B is currently the best biological prognosticator for malignant melanoma, with serum concentrations of the protein inversely proportional to long-term survival rates (93). This suggests that a modified TRTK12 peptide may represents a promising starting point for an in vivo inhibitor of S100B and/or related S100 proteins. In addition, the small size of the TRTK12 peptide and low sensitivity of its binding to ionic strength are advantageous over other binding partners of S100B, such as the C-terminus of p53. The ability to individually inhibit homologous proteins, with similar sequences and tertiary structures, represents an important step towards rational drug design.
Supplementary Material
Acknowledgments
Instrumentation at the Core Facilities at the Center of Biotechnology and Interdisciplinary Studies at RPI were used for some of the experiments reported in this paper. We thank Dr. Werner Streicher for his guidance and insights.
Funding Source Statement: This work was supported by a grant from NIH/NIGMSRO1-GM054537
Abbreviations used
- ITC
isothermal titration calorimetry
- EF-hand calcium-binding
domain consisting of a helix—loop—helix structure
- Fmoc
fluorenylmethyloxycarbonyl chloride
- TCEP
tris(2-carboxyethyl)phosphine
Footnotes
Supporting Information: Additional three figures providing details on stopped-flow, fluorescence anisotropy, and fluorescence quenching. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schafer BW, Heizmann CW. The S100 family of EF-hand calcium-binding proteins: functions and pathology. Trends Biochem Sci. 1996;21:134–140. doi: 10.1016/s0968-0004(96)80167-8. [DOI] [PubMed] [Google Scholar]
- 2.Deloulme JC, Mbele GO, Baudier J. S100 proteins. From purification to functions. Methods Mol Biol. 2002;172:185–198. doi: 10.1385/1-59259-183-3:185. [DOI] [PubMed] [Google Scholar]
- 3.Donato R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol. 2001;33:637–668. doi: 10.1016/s1357-2725(01)00046-2. [DOI] [PubMed] [Google Scholar]
- 4.Donato R. Functional roles of S100 proteins, calcium-binding proteins of the EF-hand type. Biochim Biophys Acta. 1999;1450:191–231. doi: 10.1016/s0167-4889(99)00058-0. [DOI] [PubMed] [Google Scholar]
- 5.Donato R. Intracellular and extracellular roles of S100 proteins. Microsc Res Tech. 2003;60:540–551. doi: 10.1002/jemt.10296. [DOI] [PubMed] [Google Scholar]
- 6.Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature) Biochem Biophys Res Commun. 2004;322:1111–1122. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- 7.Chaves ML, Camozzato AL, Ferreira ED, Piazenski I, Kochhann R, Dall’igna O, Mazzini GS, Souza DO, Portela LV. Serum levels of S100B and NSE proteins in Alzheimer’s disease patients. J Neuroinflammation. 2010;7:6–7. doi: 10.1186/1742-2094-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson PJ, Watts HR, Jen S, Gentleman SM, Moncaster JA, Walsh DT, Jen LS. Differential effects of interleukin-1beta and S100B on amyloid precursor protein in rat retinal neurons. Clin Ophthalmol. 2009;3:235–242. doi: 10.2147/opth.s2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esposito G, Imitola J, Lu J, De Filippis D, Scuderi C, Ganesh VS, Folkerth R, Hecht J, Shin S, Iuvone T, Chesnut J, Steardo L, Sheen V. Genomic and functional profiling of human Down syndrome neural progenitors implicates S100B and aquaporin 4 in cell injury. Hum Mol Genet. 2008;17:440–457. doi: 10.1093/hmg/ddm322. [DOI] [PubMed] [Google Scholar]
- 10.Leclerc E, Sturchler E, Vetter SW. The S100B/RAGE Axis in Alzheimer’s Disease. Cardiovasc Psychiatry Neurol. 2010;2010:539–581. doi: 10.1155/2010/539581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Wang H, Zhang L, Xu Y, Deng W, Zhu H, Qin C. S100B transgenic mice develop features of Parkinson’s disease. Arch Med Res. 2011;42:1–7. doi: 10.1016/j.arcmed.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Goyette J, Geczy CL. Inflammation-associated S100 proteins: new mechanisms that regulate function. Amino Acids. 2011;41:821–842. doi: 10.1007/s00726-010-0528-0. [DOI] [PubMed] [Google Scholar]
- 13.Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81:28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- 14.Foell D, Wittkowski H, Ren Z, Turton J, Pang G, Daebritz J, Ehrchen J, Heidemann J, Borody T, Roth J, Clancy R. Phagocyte-specific S100 proteins are released from affected mucosa and promote immune responses during inflammatory bowel disease. J Pathol. 2008;216:183–192. doi: 10.1002/path.2394. [DOI] [PubMed] [Google Scholar]
- 15.Kaiser T, Langhorst J, Wittkowski H, Becker K, Friedrich AW, Rueffer A, Dobos GJ, Roth J, Foell D. Faecal S100A12 as a non-invasive marker distinguishing inflammatory bowel disease from irritable bowel syndrome. Gut. 2007;56:1706–1713. doi: 10.1136/gut.2006.113431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Remppis A, Greten T, Schafer BW, Hunziker P, Erne P, Katus HA, Heizmann CW. Altered expression of the Ca(2+)-binding protein S100A1 in human cardiomyopathy. Biochim Biophys Acta. 1996;1313:253–257. doi: 10.1016/0167-4889(96)00097-3. [DOI] [PubMed] [Google Scholar]
- 17.Rohde D, Ritterhoff J, Voelkers M, Katus HA, Parker TG, Most P. S100A1: a multifaceted therapeutic target in cardiovascular disease. J Cardiovasc Transl Res. 2010;3:525–537. doi: 10.1007/s12265-010-9211-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarabykina S, Scott DJ, Herzyk P, Hill TJ, Tame JR, Kriajevska M, Lafitte D, Derrick PJ, Dodson GG, Maitland NJ, Lukanidin EM, Bronstein IB. The dimerization interface of the metastasis-associated protein S100A4 (Mts1): in vivo and in vitro studies. J Biol Chem. 2001;276:24212–24222. doi: 10.1074/jbc.M009477200. [DOI] [PubMed] [Google Scholar]
- 19.Gibadulinova A, Tothova V, Pastorek J, Pastorekova S. Transcriptional regulation and functional implication of S100P in cancer. Amino Acids. 2010;41:885–892. doi: 10.1007/s00726-010-0495-5. [DOI] [PubMed] [Google Scholar]
- 20.Takenaga K, Nakamura Y, Endo H, Sakiyama S. Involvement of S100-related calcium-binding protein pEL98 (or mts1) in cell motility and tumor cell invasion. Jpn J Cancer Res. 1994;85:831–839. doi: 10.1111/j.1349-7006.1994.tb02955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakazato Y, Ishizeki J, Takahashi K, Yamaguchi H. Immunohistochemical localization of S-100 protein in granular cell myoblastoma. Vol. 49. 1982. pp. 1624–1628. [DOI] [PubMed] [Google Scholar]
- 22.Mueller A, Schafer BW, Ferrari S, Weibel M, Makek M, Hochli M, Heizmann CW. The calcium-binding protein S100A2 interacts with p53 and modulates its transcriptional activity. J Biol Chem. 2005;280:29186–29193. doi: 10.1074/jbc.M505000200. [DOI] [PubMed] [Google Scholar]
- 23.Salama I, Malone PS, Mihaimeed F, Jones JL. A review of the S100 proteins in cancer. Eur J Surg Oncol. 2008;34:357–364. doi: 10.1016/j.ejso.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Botelho HM, Fritz G, Gomes CM. Analysis of s100 oligomers and amyloids. Methods Mol Biol. 2012;849:373–386. doi: 10.1007/978-1-61779-551-0_25. [DOI] [PubMed] [Google Scholar]
- 25.Streicher WW, Lopez MM, Makhatadze GI. Modulation of quaternary structure of S100 proteins by calcium ions. Biophys Chem. 2010;151:181–186. doi: 10.1016/j.bpc.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee YC, Volk DE, Thiviyanathan V, Kleerekoper Q, Gribenko AV, Zhang S, Gorenstein DG, Makhatadze GI, Luxon BA. NMR structure of the Apo-S100P protein. J Biomol NMR. 2004;29:399–402. doi: 10.1023/B:JNMR.0000032617.88899.4b. [DOI] [PubMed] [Google Scholar]
- 27.Dempsey AC, Walsh MP, Shaw GS. Unmasking the annexin I interaction from the structure of Apo-S100A11. Vol. 11. 2003. pp. 887–897. [DOI] [PubMed] [Google Scholar]
- 28.Wright NT, Inman KG, Levine JA, Cannon BR, Varney KM, Weber DJ. Refinement of the solution structure and dynamic properties of Ca(2+)-bound rat S100B. J Biomol NMR. 2008;42:279–286. doi: 10.1007/s10858-008-9282-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright NT, Varney KM, Ellis KC, Markowitz J, Gitti RK, Zimmer DB, Weber DJ. The three-dimensional solution structure of Ca(2+)-bound S100A1 as determined by NMR spectroscopy. J Mol Biol. 2005;353:410–426. doi: 10.1016/j.jmb.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 30.Lee YT, Dimitrova YN, Schneider G, Ridenour WB, Bhattacharya S, Soss SE, Caprioli RM, Filipek A, Chazin WJ. Structure of the S100A6 complex with a fragment from the C-terminal domain of Siah-1 interacting protein: a novel mode for S100 protein target recognition. Biochemistry. 2008;47:10921–10932. doi: 10.1021/bi801233z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bertini I, Das Gupta S, Hu X, Karavelas T, Luchinat C, Parigi G, Yuan J. Solution structure and dynamics of S100A5 in the apo and Ca2+-bound states. J Biol Inorg Chem. 2009;14:1097–1107. doi: 10.1007/s00775-009-0553-1. [DOI] [PubMed] [Google Scholar]
- 32.Wright NT, Cannon BR, Wilder PT, Morgan MT, Varney KM, Zimmer DB, Weber DJ. Solution structure of S100A1 bound to the CapZ peptide (TRTK12) J Mol Biol. 2009;386:1265–1277. doi: 10.1016/j.jmb.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zimmer DB, Van Eldik LJ. Analysis of the calcium-modulated proteins, S100 and calmodulin, and their target proteins during C6 glioma cell differentiation. J Cell Biol. 1989;108:141–151. doi: 10.1083/jcb.108.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selinfreund RH, Barger SW, Pledger WJ, Van Eldik LJ. Neurotrophic protein S100 beta stimulates glial cell proliferation. Proc Natl Acad Sci U S A. 1991;88:3554–3558. doi: 10.1073/pnas.88.9.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ivanenkov VV, Jamieson GA, Jr, Gruenstein E, Dimlich RV. Characterization of S-100b binding epitopes. Identification of a novel target, the actin capping protein, CapZ. J Biol Chem. 1995;270:14651–14658. doi: 10.1074/jbc.270.24.14651. [DOI] [PubMed] [Google Scholar]
- 36.Henzl MT, Davis ME, Tan A. Leucine 85 is an important determinant of divalent ion affinity in rat beta-parvalbumin (Oncomodulin) Biochemistry. 2008;47:13635–13646. doi: 10.1021/bi8014899. [DOI] [PubMed] [Google Scholar]
- 37.Henzl MT, Tanner JJ. Solution structure of Ca2+-free rat beta-parvalbumin (oncomodulin) Protein Sci. 2007;16:1914–1926. doi: 10.1110/ps.072837307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henzl MT, Tanner JJ. Solution structure of Ca2+-free rat alpha-parvalbumin. Protein Sci. 2008;17:431–438. doi: 10.1110/ps.073318308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rezvanpour A, Phillips JM, Shaw GS. Design of high-affinity S100-target hybrid proteins. Protein Sci. 2009;18:2528–2536. doi: 10.1002/pro.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inman KG, Yang R, Rustandi RR, Miller KE, Baldisseri DM, Weber DJ. Solution NMR structure of S100B bound to the high-affinity target peptide TRTK-12. J Mol Biol. 2002;324:1003–1014. doi: 10.1016/s0022-2836(02)01152-x. [DOI] [PubMed] [Google Scholar]
- 41.McClintock KA, Shaw GS. Assignment of 1H, 13C and 15N resonances of human Ca2+-S100B in complex with the TRTK-12 peptide. J Biomol NMR. 2002;23:255–256. doi: 10.1023/a:1019886414743. [DOI] [PubMed] [Google Scholar]
- 42.McClintock KA, Van Eldik LJ, Shaw GS. The C-terminus and linker region of S100B exert dual control on protein-protein interactions with TRTK-12. Biochemistry. 2002;41:5421–5428. doi: 10.1021/bi011732m. [DOI] [PubMed] [Google Scholar]
- 43.McClintock KA, Shaw GS. A novel S100 target conformation is revealed by the solution structure of the Ca2+-S100B-TRTK-12 complex. J Biol Chem. 2003;278:6251–6257. doi: 10.1074/jbc.M210622200. [DOI] [PubMed] [Google Scholar]
- 44.Charpentier TH, Thompson LE, Liriano MA, Varney KM, Wilder PT, Pozharski E, Toth EA, Weber DJ. The effects of CapZ peptide (TRTK-12) binding to S100B-Ca2+ as examined by NMR and X-ray crystallography. J Mol Biol. 2010;396:1227–1243. doi: 10.1016/j.jmb.2009.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garbuglia M, Verzini M, Rustandi RR, Osterloh D, Weber DJ, Gerke V, Donato R. Role of the C-terminal extension in the interaction of S100A1 with GFAP, tubulin, the S100A1- and S100B-inhibitory peptide, TRTK-12, and a peptide derived from p53, and the S100A1 inhibitory effect on GFAP polymerization. Biochem Biophys Res Commun. 1999;254:36–41. doi: 10.1006/bbrc.1998.9881. [DOI] [PubMed] [Google Scholar]
- 46.Ivanenkov VV, Dimlich RV, Jamieson GA., Jr Interaction of S100a0 protein with the actin capping protein, CapZ: characterization of a putative S100a0 binding site in CapZ alpha-subunit. Biochem Biophys Res Commun. 1996;221:46–50. doi: 10.1006/bbrc.1996.0542. [DOI] [PubMed] [Google Scholar]
- 47.Barber KR, McClintock KA, Jamieson GA, Jr, Dimlich RV, Shaw GS. Specificity and Zn2+ enhancement of the S100B binding epitope TRTK-12. J Biol Chem. 1999;274:1502–1508. doi: 10.1074/jbc.274.3.1502. [DOI] [PubMed] [Google Scholar]
- 48.Gribenko A, Lopez MM, Richardson JM, 3rd, Makhatadze GI. Cloning, overexpression, purification, and spectroscopic characterization of human S100P. Protein Sci. 1998;7:211–215. doi: 10.1002/pro.5560070123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gribenko AV, Hopper JE, Makhatadze GI. Molecular characterization and tissue distribution of a novel member of the S100 family of EF-hand proteins. Biochemistry. 2001;40:15538–15548. doi: 10.1021/bi0114731. [DOI] [PubMed] [Google Scholar]
- 50.Wafer LN, Streicher WW, McCallum SA, Makhatadze GI. Thermodynamic and kinetic analysis of peptides derived from CapZ, NDR, p53, HDM2, and HDM4 binding to human S100B. Biochemistry. 2012;51:7189–7201. doi: 10.1021/bi300865g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Streicher WW, Lopez MM, Makhatadze GI. Annexin I and annexin II N-terminal peptides binding to S100 protein family members: specificity and thermodynamic characterization. Biochemistry. 2009;48:2788–2798. doi: 10.1021/bi8019959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 53.Lopez MM, Makhatadze GI. Isothermal titration calorimetry. Methods Mol Biol. 2002;173:121–126. doi: 10.1385/1-59259-184-1:121. [DOI] [PubMed] [Google Scholar]
- 54.Gribenko AV, Guzman-Casado M, Lopez MM, Makhatadze GI. Conformational and thermodynamic properties of peptide binding to the human S100P protein. Protein Sci. 2002;11:1367–1375. doi: 10.1110/ps.0202202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiseman T, Williston S, Brandts JF, Lin L-N. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Vol. 179. 1989. pp. 131–137. [DOI] [PubMed] [Google Scholar]
- 56.Sherrod P. NLREG Version 6.3. Nashville, TN: 2005. Nonlinear Regression Analysis Program. [Google Scholar]
- 57.Eftink MR, Ghiron CA. Exposure of tryptophanyl residues in proteins. Quantitative determination by fluorescence quenching studies. Biochemistry. 1976;15:672–680. doi: 10.1021/bi00648a035. [DOI] [PubMed] [Google Scholar]
- 58.Makhatadze GI, Privalov PL. Energetics of protein structure. Adv Protein Chem. 1995;47:307–425. doi: 10.1016/s0065-3233(08)60548-3. [DOI] [PubMed] [Google Scholar]
- 59.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci Chapter. 2007;2Unit 2:9. doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 60.Wafer LN, Streicher WW, Makhatadze GI. Thermodynamics of the Trp-cage miniprotein unfolding in urea. Proteins. 2010;78:1376–1381. doi: 10.1002/prot.22681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brokx RD, Lopez MM, Vogel HJ, Makhatadze GI. Energetics of target peptide binding by calmodulin reveals different modes of binding. J Biol Chem. 2001;276:14083–14091. doi: 10.1074/jbc.M011026200. [DOI] [PubMed] [Google Scholar]
- 62.Peterman BF. Measurement of the dead time of a fluorescence stopped-flow instrument. Anal Biochem. 1979;93:442–444. doi: 10.1016/s0003-2697(79)80176-1. [DOI] [PubMed] [Google Scholar]
- 63.Rezvanpour A, Shaw GS. Unique S100 target protein interactions. Gen Physiol Biophys. 2009;28(Spec No Focus):F39–46. [PubMed] [Google Scholar]
- 64.Elliott PR, Irvine AF, Jung HS, Tozawa K, Pastok MW, Picone R, Badyal SK, Basran J, Rudland PS, Barraclough R, Lian LY, Bagshaw CR, Kriajevska M, Barsukov IL. Asymmetric mode of Ca(2)(+)-S100A4 interaction with nonmuscle myosin IIA generates nanomolar affinity required for filament remodeling. Vol. 20. 2012. pp. 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ozorowski G, Milton S, Luecke H. Structure of a C-terminal AHNAK peptide in a 1:2:2 complex with S100A10 and an acetylated N-terminal peptide of annexin A2. Acta Crystallogr D Biol Crystallogr. 2013;69:92–104. doi: 10.1107/S0907444912043429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dempsey BR, Rezvanpour A, Lee TW, Barber KR, Junop MS, Shaw GS. Structure of an asymmetric ternary protein complex provides insight for membrane interaction. Vol. 20. 2012. pp. 1737–1745. [DOI] [PubMed] [Google Scholar]
- 67.Rezvanpour A, Santamaria-Kisiel L, Shaw GS. The S100A10-annexin A2 complex provides a novel asymmetric platform for membrane repair. J Biol Chem. 2011;286:40174–40183. doi: 10.1074/jbc.M111.244038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Dieck J, Brandt T, Teufel DP, Veprintsev DB, Joerger AC, Fersht AR. Molecular basis of S100 proteins interacting with the p53 homologs p63 and p73. Vol. 29. 2010. pp. 2024–2035. [DOI] [PubMed] [Google Scholar]
- 69.Kiss B, Duelli A, Radnai L, Kekesi KA, Katona G, Nyitray L. Crystal structure of the S100A4-nonmuscle myosin IIA tail fragment complex reveals an asymmetric target binding mechanism. Proc Natl Acad Sci U S A. 2012;109:6048–6053. doi: 10.1073/pnas.1114732109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moore B. A soluble protein characteristic of the nervous system. Biochem Biophys Res Commun. 1965:739–744. doi: 10.1016/0006-291x(65)90320-7. [DOI] [PubMed] [Google Scholar]
- 71.Wilder PT, Lin J, Bair CL, Charpentier TH, Yang D, Liriano M, Varney KM, Lee A, Oppenheim AB, Adhya S, Carrier F, Weber DJ. Recognition of the tumor suppressor protein p53 and other protein targets by the calcium-binding protein S100B. Biochim Biophys Acta. 2006;1763:1284–1297. doi: 10.1016/j.bbamcr.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 72.Yu WH, Fraser PE. S100beta interaction with tau is promoted by zinc and inhibited by hyperphosphorylation in Alzheimer’s disease. J Neurosci. 2001;21:2240–2246. doi: 10.1523/JNEUROSCI.21-07-02240.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sorci G, Agneletti AL, Bianchi R, Donato R. Association of S100B with intermediate filaments and microtubules in glial cells. Biochim Biophys Acta. 1998;1448:277–289. doi: 10.1016/s0167-4889(98)00134-7. [DOI] [PubMed] [Google Scholar]
- 74.Garbuglia M, Verzini M, Sorci G, Bianchi R, Giambanco I, Agneletti AL, Donato R. The calcium-modulated proteins, S100A1 and S100B, as potential regulators of the dynamics of type III intermediate filaments. Braz J Med Biol Res. 1999;32:1177–1185. doi: 10.1590/s0100-879x1999001000001. [DOI] [PubMed] [Google Scholar]
- 75.Santamaria-Kisiel L, Rintala-Dempsey AC, Shaw GS. Calcium-dependent and -independent interactions of the S100 protein family. Biochem J. 2006;396:201–214. doi: 10.1042/BJ20060195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Dieck J, Lum JK, Teufel DP, Fersht AR. S100 proteins interact with the N-terminal domain of MDM2. FEBS Lett. 2010;584:3269–3274. doi: 10.1016/j.febslet.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 77.Staneva I, Huang Y, Liu Z, Wallin S. Binding of two intrinsically disordered peptides to a multi-specific protein: a combined Monte Carlo and molecular dynamics study. PLoS Comput Biol. 2012;8:e1002682. doi: 10.1371/journal.pcbi.1002682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen J. Intrinsically disordered p53 extreme C-terminus binds to S100B(betabeta) through “fly-casting”. J Am Chem Soc. 2009;131:2088–2089. doi: 10.1021/ja809547p. [DOI] [PubMed] [Google Scholar]
- 79.Trizac E, Levy Y, Wolynes PG. Capillarity theory for the fly-casting mechanism. Proc Natl Acad Sci U S A. 2010;107:2746–2750. doi: 10.1073/pnas.0914727107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gsponer J, Babu MM. The rules of disorder or why disorder rules. Prog Biophys Mol Biol. 2009;99:94–103. doi: 10.1016/j.pbiomolbio.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 81.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Natl Acad Sci U S A. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kiefhaber T, Bachmann A, Jensen KS. Dynamics and mechanisms of coupled protein folding and binding reactions. Curr Opin Struct Biol. 2011;22:21–29. doi: 10.1016/j.sbi.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 83.Zimmer DB, Eubanks JO, Ramakrishnan D, Criscitiello MF. Evolution of the S100 family of calcium sensor proteins. Cell Calcium. 2013;53:170–179. doi: 10.1016/j.ceca.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 84.Rety S, Osterloh D, Arie JP, Tabaries S, Seeman J, Russo-Marie F, Gerke V, Lewit-Bentley A. Structural basis of the Ca(2+)-dependent association between S100C (S100A11) and its target, the N-terminal part of annexin I. Vol. 8. 2000. pp. 175–184. [DOI] [PubMed] [Google Scholar]
- 85.Bhattacharya S, Large E, Heizmann CW, Hemmings B, Chazin WJ. Structure of the Ca2+/S100B/NDR kinase peptide complex: insights into S100 target specificity and activation of the kinase. Biochemistry. 2003;42:14416–14426. doi: 10.1021/bi035089a. [DOI] [PubMed] [Google Scholar]
- 86.Rustandi RR, Baldisseri DM, Drohat AC, Weber DJ. Structural changes in the C-terminus of Ca2+-bound rat S100B (beta beta) upon binding to a peptide derived from the C-terminal regulatory domain of p53. Protein Sci. 1999;8:1743–1751. doi: 10.1110/ps.8.9.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fohr UG, Heizmann CW, Engelkamp D, Schafer BW, Cox JA. Purification and cation binding properties of the recombinant human S100 calcium-binding protein A3, an EF-hand motif protein with high affinity for zinc. J Biol Chem. 1995;270:21056–21061. doi: 10.1074/jbc.270.36.21056. [DOI] [PubMed] [Google Scholar]
- 88.Gingras AR, Basran J, Prescott A, Kriajevska M, Bagshaw CR, Barsukov IL. Crystal structure of the Ca(2+)-form and Ca(2+)-binding kinetics of metastasis-associated protein, S100A4. FEBS Lett. 2008;582:1651–1656. doi: 10.1016/j.febslet.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 89.Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, Schulte C, Mustafa S, Synofzik M, Vukovic Z, Itohara S, Berg D, Teismann P. S100B is increased in Parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-alpha pathway. Brain. 2012;135:3336–3347. doi: 10.1093/brain/aws250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schaf DV, Tort AB, Fricke D, Schestatsky P, Portela LV, Souza DO, Rieder CR. S100B and NSE serum levels in patients with Parkinson’s disease. Parkinsonism Relat Disord. 2005;11:39–43. doi: 10.1016/j.parkreldis.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 91.Griffin WS, Yeralan O, Sheng JG, Boop FA, Mrak RE, Rovnaghi CR, Burnett BA, Feoktistova A, Van Eldik LJ. Overexpression of the neurotrophic cytokine S100 beta in human temporal lobe epilepsy. J Neurochem. 1995;65:228–233. doi: 10.1046/j.1471-4159.1995.65010228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nonaka D, Chiriboga L, Rubin BP. Differential expression of S100 protein subtypes in malignant melanoma, and benign and malignant peripheral nerve sheath tumors. J Cutan Pathol. 2008;35:1014–1019. doi: 10.1111/j.1600-0560.2007.00953.x. [DOI] [PubMed] [Google Scholar]
- 93.Tarhini AA, Stuckert J, Lee S, Sander C, Kirkwood JM. Prognostic significance of serum S100B protein in high-risk surgically resected melanoma patients participating in Intergroup Trial ECOG 1694. J Clin Oncol. 2009;27:38–44. doi: 10.1200/JCO.2008.17.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Camby I, Nagy N, Lopes MB, Schafer BW, Maurage CA, Ruchoux MM, Murmann P, Pochet R, Heizmann CW, Brotchi J, Salmon I, Kiss R, Decaestecker C. Supratentorial pilocytic astrocytomas, astrocytomas, anaplastic astrocytomas and glioblastomas are characterized by a differential expression of S100 proteins. Brain Pathol. 1999;9:1–19. doi: 10.1111/j.1750-3639.1999.tb00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagy N, Brenner C, Markadieu N, Chaboteaux C, Camby I, Schafer BW, Pochet R, Heizmann CW, Salmon I, Kiss R, Decaestecker C. S100A2, a putative tumor suppressor gene, regulates in vitro squamous cell carcinoma migration. Lab Invest. 2001;81:599–612. doi: 10.1038/labinvest.3780269. [DOI] [PubMed] [Google Scholar]
- 96.Bulk E, Sargin B, Krug U, Hascher A, Jun Y, Knop M, Kerkhoff C, Gerke V, Liersch R, Mesters RM, Hotfilder M, Marra A, Koschmieder S, Dugas M, Berdel WE, Serve H, Muller-Tidow C. S100A2 induces metastasis in non-small cell lung cancer. Clin Cancer Res. 2009;15:22–29. doi: 10.1158/1078-0432.CCR-08-0953. [DOI] [PubMed] [Google Scholar]
- 97.Suzuki F, Oridate N, Homma A, Nakamaru Y, Nagahashi T, Yagi K, Yamaguchi S, Furuta Y, Fukuda S. S100A2 expression as a predictive marker for late cervical metastasis in stage I and II invasive squamous cell carcinoma of the oral cavity. Oncol Rep. 2005;14:1493–1498. [PubMed] [Google Scholar]
- 98.Deloulme JC, Assard N, Mbele GO, Mangin C, Kuwano R, Baudier J. S100A6 and S100A11 are specific targets of the calcium- and zinc-binding S100B protein in vivo. J Biol Chem. 2000;275:35302–35310. doi: 10.1074/jbc.M003943200. [DOI] [PubMed] [Google Scholar]
- 99.Fernandez-Fernandez MR, Rutherford TJ, Fersht AR. Members of the S100 family bind p53 in two distinct ways. Protein Sci. 2008;17:1663–1670. doi: 10.1110/ps.035527.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Andrew Hilton PT, Walkinshaw Malcolm. Biological Ligands. In: Jerry Atwood JS, editor. Encyclopedia of Supramolecular Chemistry. Marcel Dekker; New York, NY: 2004. [Google Scholar]
- 101.Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- 102.Kim YW, Kutchukian PS, Verdine GL. Introduction of all-hydrocarbon i,i+3 staples into alpha-helices via ring-closing olefin metathesis. Org Lett. 2010;12:3046–3049. doi: 10.1021/ol1010449. [DOI] [PubMed] [Google Scholar]
- 103.Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky LD. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell. 2010;18:411–422. doi: 10.1016/j.ccr.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Estieu-Gionnet K, Guichard G. Stabilized helical peptides: overview of the technologies and therapeutic promises. Expert Opin Drug Discov. 2011;6:937–963. doi: 10.1517/17460441.2011.603723. [DOI] [PubMed] [Google Scholar]
- 105.LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, Fisher JK, Godes M, Pitter K, Kung AL, Walensky LD. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122:2018–2031. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.