

Abstract
AKI induces the renoprotective upregulation of survivin expression in kidney epithelial cells, but the underlying mechanisms have not been identified. To determine the role of survivin in renal recovery from AKI, we generated mice with renal proximal tubule-specific deletion of survivin (survivinptKO). Renal survivin expression increased substantially in response to ischemia-reperfusion (I/R) injury in control littermates but remained minimal in survivinptKO mice. Functional and histologic data indicated similar degrees of renal injury in survivinptKO and control mice 24 hours after reperfusion, but recovery was markedly delayed in survivinptKO mice. In MCT cells, a mouse renal proximal tubule cell line, ATP depletion by antimycin A treatment upregulated survivin expression through a phospho-STAT3–dependent pathway. In wild-type mice, inhibition of STAT3 kinase diminished I/R-induced upregulation of STAT3 phosphorylation and survivin expression and delayed recovery. Furthermore, I/R injury activated Notch-2 signaling, and a γ-secretase inhibitor suppressed I/R-induced Notch-2 signaling, STAT3 phosphorylation, and survivin expression and delayed recovery. In MCT cells, inhibition of γ-secretase similarly attenuated antimycin A-induced Notch-2 activation, upregulation of survivin, and phosphorylation of STAT3, but STAT3 kinase inhibition did not prevent Notch-2 activation. Therefore, these data suggest that STAT3 phosphorylation and subsequent upregulation of survivin expression mediated by Notch-2 signaling in renal proximal tubule epithelial cells aid in the functional and structural recovery of the kidney from AKI.
AKI is a common clinical condition encountered in both hospital and outpatient settings, and it is an important cause of morbidity and mortality. Approximately 600,000 cases of AKI are reported each year in the United States.1 Despite significant preclinical and clinical research, supportive therapy remains the only treatment option for AKI patients, and to date, the mortality rates of AKI remain high.2 It has also become increasingly clear that AKI is associated with the development of CKD.3,4 Ischemia-reperfusion (I/R) injury is one of the most common causes of AKI in clinical practice, and the underlying pathogenesis involves injury to nephron segments from both the ischemia itself and the resulting inflammatory response. The S3 segment of proximal tubules is most vulnerable to I/R injury.5 As a result of I/R injury, renal proximal tubule cells exhibit mitochondrial dysfunction, ATP depletion, impaired solute and ion transport, loss of cell polarity, and cytoskeletal disruption,6 and they may ultimately undergo apoptosis or necrosis.
The kidney has a remarkable capacity for regeneration, which is evidenced by the ability to recover renal function after moderate I/R injury. Genetic cell lineage tracing studies have shown that intrinsic surviving epithelial cells are the source of the cells that affect tubule repair in response to I/R injury.7 The Notch pathway is an evolutionarily conserved intercellular signaling pathway responsible for the control of cell fate and tissue morphogenesis during development, and it is also involved in tissue maintenance and repair in adult tissue.8 The Notch family consists of four different single-pass type I transmembrane receptors, which display both redundant and unique functions. Notchs 1, 2, 3, and 4 and their ligands Δ-like 1 (DLL1), DLL3, and DLL4 as well as Jagged-1 and -2 are expressed on the surface of neighboring cells.9 A key event that results in activation of Notch signaling after ligand binding is cleavage of the Notch intracellular domain (NICD; cleaved Notch) by γ-secretase. NICD translocates to the nucleus, where it directly affects gene expression. Generation of cleaved Notch and expression of its target genes, such as Hairy/enhancer of split (HES), have been used as markers to detect activation of the Notch pathway. The Notch pathway-regulated gene expression occurs in both developing and adult kidneys, and Notch signaling pathways are activated in the adult kidney after injury.10 Recent studies have documented that Hes protein induced by activation of Notch receptor signaling pathways may function as a nonreceptor scaffold protein in the nucleus that can recruit and allow Janus kinase 2 (JAK2) to phosphorylate signal transducers and activators of transcription 3 (STAT3), thereby leading to activation of an STAT3 signaling pathway.11
Survivin, which is the smallest member of the inhibitor of apoptosis proteins family, is a 16.5 kDa intracellular protein that has been implicated in regulation of apoptosis, cell division, and cell cycle in cancer cells as well as normal tissues through caspase-dependent and -independent mechanisms.12 Survivin is usually expressed at very low or undetectable levels in normal tissues but highly expressed in most cancer tissues.13 Upregulation of survivin gene expression is closely associated with activation of several oncogenic pathways, including growth factor receptor signaling, STAT activation, and the phosphatidylinositol 3 kinase/Akt signaling pathway.14–16 Of interest, survivin is a component of centrosomes and microtubules of the metaphase and anaphase spindles, and it plays an important role in cell division. Survivin also functions as an apoptosis inhibitor by modulating caspase-9–dependent pathways.17 Survivin intersects with multiple cellular signaling networks related to cancer development and progression, such as regulation of kinetochore attachment and spindle formation, modulation of the p53 checkpoint, regulation of angiogenesis, modulation of cellular stress responses and inhibition of apoptosis. Because of its pleotropic effects, survivin has been identified as a potential cancer drug target.18
Previous studies have indicated that survivin is expressed in normal adult rat, mouse, and human kidney epithelial cells, predominantly in the proximal tubule,19 and it protects the kidney from apoptosis in nephrotoxin-induced kidney injury by suppression of p53 gene expression.20 However, the underlying molecular mechanism of how survivin expression is upregulated in response to AKI has not been previously clarified. Mice with global deletion of survivin die at an early stage of development because of defects in mitosis, indicating that survivin is indispensable for survival.21 Therefore, in the current study, we used renal proximal tubule cell-specific survivin deletion mice (survivinptKO) to examine the role of renal proximal tubule survivin expression in the functional and structural recovery of the kidney from AKI and investigate the mechanisms underlying regulation of survivin expression in this clinically relevant situation.
Results
I/R Markedly Induced Survivin Expression in the Kidney
Previous studies with transgenic mice expressing decreased survivin levels indicated a potential role to inhibit kidney tubule cell apoptosis induced by folic acid and cisplatin.20 To explore the role of survivin expression in kidney regeneration after I/R injury, we performed a time course of survivin expression in renal cortex at different time points after I/R injury and found that survivin expression was markedly increased within 48 hours and remained elevated for at least 6 days after reperfusion, the longest time point examined (Figure 1A). To determine the relationship between the upregulation of survivin expression and kidney recovery from I/R injury, we generated renal proximal tubule-specific survivin knockout mice (survivinptKO) by crossing survivin floxed (survivinf/f) mice22 with γ-glutamyl transpeptidase promoter-driven Cre transgenic mice (γGT-Cre+)23 (Figure 1, B and C). Effective deletion of survivin in renal proximal tubules was verified by immunoblotting of renal cortical lysates (Figure 1D). In survivinptKO mice, upregulation of survivin expression in response to I/R injury was markedly blunted compared with survivinf/f mice (Figure 1E).
Figure 1.

Upregulation of survivin expression was attenuated in renal proximal tubule-specific survivin knockout mice in response to renal I/R injury. (A) Wild-type Balb/c mice were subjected to sham or bilateral renal I/R injury. At different time points after surgery, renal cortex tissue lysates were subjected to immunoblotting with antibodies against survivin. (B) Schematic for the generation of survivinptKO mice by crossing γGT-Cre mice with survivinf/f mice. (C) Representative PCR data using 1.8% agarose gels to analyze the presence of the survivin floxed allele (577-bp product) and γGT-Cre gene (370-bp product). Lanes 1 and 4, Survivinf/w(Surf/w); GGT-Cre−; lane 2, Surw/w; GGT-Cre−; lanes 3 and 6, Surf/w; GGT-Cre+; lane 5, Surf/f; GGT-Cre− (Ctrl); lane 7, Surf/f; GGT-Cre+ (SurptKO). (D) Representative immunoblotting of the renal cortex lysates confirmed deletion of survivin in the renal cortex. (E) Representative immunoblotting of the renal cortex lysates harvested from survivinptKO and survivinf/f mice after 48 hours of either sham or I/R surgery. d, day; h, hour.
Survivin Deletion in Renal Proximal Tubules Delayed the Recovery of Kidney Structure and Function after I/R Injury
When survivinptKO mice and survivinf/f mice were subjected to I/R injury, we observed similarly impaired renal function in both survivinptKO and their littermate control mice 24 hours after I/R injury. BUN levels were 114±30 versus 115±33 mg/dl (n=5–7). However, 10 days after reperfusion, BUN levels remained more elevated in survivinptKO mice compared with their littermate controls (40±10 versus 22±2 mg/dl, P<0.05) (Figure 2A). Histologic evidence of injury was comparable at 24 hours; however, at day 14, there was minimal residual injury in littermate controls, whereas survivinptKO mice still had persistent proximal tubule dilation, epithelial simplification, and cast formation, which is shown in Figure 2B.
Figure 2.

Selective deletion of survivin in renal proximal tubules delayed renal functional and structural recovery from I/R injury. After I/R injury of the survivinptKO mice or their control littermates, (A) BUN was measured at different time points, (B) histologies (200×) at days 1 and 14 after I/R injury were presented, and (C) tubular damage was scored as indicated in Concise Methods. Values are mean ± SEM (n=5–7 for each group). ***P<0.001; **P<0.01; *P<0.05.
Ki-67 immunofluorescence staining indicated that Ki-67–positive cells are primarily lotus tetragonolobus lectin-positive tubular epithelial cells, especially at the later time points after I/R, and survivin deletion in proximal tubular epithelial cells delayed cell proliferation after I/R injury (Figure 3, A and B). Some Ki-67–positive cells seem to be interstitial cells, especially at earlier time points after I/R, but no significant difference was detected between the survivinptKO and survivinf/f mice. We hypothesize that these interstitial Ki-67–positive cells are infiltrating or locally proliferating inflammatory cells, which was shown in our previous study.24 Terminal deoxynucleotidyl transferase-mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL) staining indicated that more tubular epithelial cells underwent apoptosis in response to I/R injury in survivinptKO mice than in survivinf/f mice (Figure 3, C and D), consistent with previous reports indicating that survivin functions as an apoptosis inhibitor.17
Figure 3.
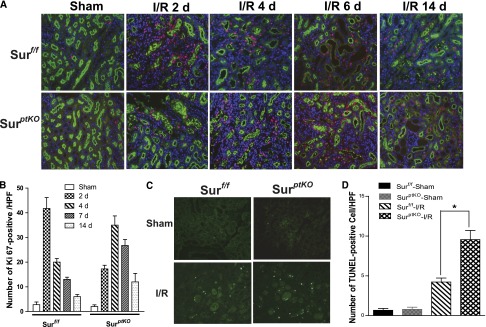
Renal proximal tubule-specific survivin deletion delayed cell proliferation and increased apoptotic cells after I/R injury. (A) Ki-67 staining in survivinptKO or survivinf/f mouse kidneys was performed at days 0, 2, 4, 7, and 14 after I/R injury. (B) The positively stained cells from A were counted in each field. Original magnification, ×400. (C) TUNEL staining in survivinptKO or survivinf/f mouse kidneys was performed at day 2 after I/R injury. (D) The positively stained cells from C were counted in each field. Original magnification, ×200. Values are mean ± SEM (n=5–7 for each group). *P<0.05.
STAT3 Activation Mediated Survivin Expression Upregulation in Response to I/R Injury
Previous studies have shown that the expression of survivin can be upregulated by growth factors.16,25 Because we previously found that epidermal growth factor receptor (EGFR) signaling is activated in response to I/R,26 we subjected renal proximal tubule-specific EGFR knockout mice26 to I/R injury. Contrary to our expectation, in these mice, survivin upregulation in response to I/R injury was not altered compared with the counterpart wild-type control mice (data not shown).
To determine upstream signals mediating survivin upregulation in response to I/R injury, we treated the well characterized mouse renal proximal tubule cell line, MCT, with antimycin A (an inhibitor of oxidative phosphorylation) for 35 minutes to deplete cellular ATP followed by reconstitution of nutrients by changing back to normal cell culture medium to mimic in vivo I/R injury. STAT proteins are a family of latent transcription factors that are produced in many cell types and activated by tyrosine phosphorylation and dimerization in response to a wide variety of extracellular stimuli, many of which have effects on cell growth and survival.27 STAT3 phosphorylation-mediated survivin upregulation has been reported in various cancer cells,15,27 and a study of a brain hypoxia mouse model indicated that hypoxia induced upregulation of survivin expression through a growth factor-independent pathway.28 We found that STAT3 phosphorylation and survivin expression were both increased within 48 hours after nutrient reconstitution (Figure 4A). Downregulation of STAT3 expression by specific small interfering RNAs (siRNAs) attenuated the survivin upregulation (Figure 4B).
Figure 4.

Antimycin A treatment followed by nutrient reconstitution induced phospho-STAT3–dependent upregulation of survivin expression in MCT cells. (A) MCT cells were made quiescent in DMEM medium containing 1% serum for 24 hours and then incubated with 0.5 µM antimycin A for 35 minutes to deplete cellular ATP, after which time the cells were washed one time and cultured with full-culture DMEM medium containing 10% serum for the indicated times. After separation by SDS-PAGE, the cell lysates were subjected to immunoblotting analysis with the indicated antibodies. (B) Control or STAT3-specific siRNAs were transfected in MCT cell for 48 hours before subjecting to antimycin A treatment followed by nutrient reconstitution for 48 hours, and cell lysates were subjected to immunoblotting analysis with the indicated antibodies. Anti-A, antimycin A; min, minutes; Veh, vehicle.
In wild-type Balb/c mice, administration of a selective STAT3 kinase inhibitor (S3I-201)29 did not alter the degree of initial renal functional and histologic injury, but it markedly slowed the functional and structural recovery of the kidney from I/R injury (Figure 5). Immunoblotting of renal cortical lysates revealed markedly increased phospho-STAT3 expression that was localized predominantly to nuclei of injured tubules within 48 hours after I/R injury (Figure 6, A and B). In addition to inhibiting phospho-STAT3 expression, S3I-201 inhibited survivin expression upregulation (Figure 6, C and D) and increased apoptotic cell number in response to I/R injury (Figure 6E).
Figure 5.

Administration of the STAT3 kinase inhibitor, S3I-201, delayed renal functional and structural recovery from I/R injury. After administration with or without 10 mg/kg S3I-201 (ST3I) by gavage starting 1 day before I/R injury in wild-type Balb/c mice, (A) BUN was measured at different time points, (B) histologies (200×) at days 1 and 10 after I/R injury were presented, and (C) tubule damage was scored as indicated in Concise Methods. Values are mean ± SEM (n=5–7 for each group). **P<0.01; *P<0.05.
Figure 6.

The STAT3 kinase inhibitor, S3I-201, decreased STAT3 phosphorylation and survivin expression induced by I/R injury. At different time points after I/R injury in wild-type Balb/c mice, (A) renal cortical lysates were subjected to immunoblotting analysis, and (B) kidney tissues were subjected to immunohistochemistry analysis with antibody against phospho-STAT3. Administration of 10 mg/kg S3I-201 (ST3I) by gavage was begun 1 day before surgery. Two days after sham or I/R injury, (C) renal cortical lysates were subjected to immunoblotting analysis with indicated antibodies, (D) kidney tissues were subjected to immunohistochemistry analysis with antibody against phospho-STAT3, (E) kidney tissues were subjected to TUNEL staining, and (F) the positive staining cells were counted in each field. Original magnification, ×200. Values are mean ± SEM (n=5–8 for each group). HPF, high-powered field. *P<0.05.
Activation of Notch 2 Signaling Initiated STAT3 Kinase Activation and Survivin Upregulation in Response to I/R Injury
Activation of Notch signaling has been reported to play a key role in nephrogenesis,30 and recent studies using conditional knockout mice showed that the differentiation of proximal nephron structures requires the presence of Notch-2 but not Notch-1.31 The DLL1/Notch-2/Hes-1 pathway is strongly upregulated in proximal tubules of rat kidney after ischemic AKI; upregulated Notch signaling induces the proliferation of renal tubular cells in vitro,32 but the underlying mechanisms have not been elucidated.
In the present study, we observed marked increases in cleaved Notch-2 expression by 24 hours after injury, with increased expression for 10 days after I/R (Figure 7A). Expressions of the Notch signaling ligand, Jagged-1, and Notch signaling target gene, Hes-1, were also increased by 48 hours after I/R injury (Figure 7A). Administration of a γ-secretase inhibitor [2,2-dimethyl-N-((S)-6-oxo-6,7-dihydro-5H-dibenzo[b,d]azepin-7-yl)-N′(2,2,3,3,3-pentafluoro-propyl)-malonamide] (RO4929097)33 not only inhibited expression of cleaved Notch-2 and Hes-1 but also inhibited STAT3 phosphorylation and survivin expression upregulation in response to I/R injury (Figure 7B). Furthermore, administration of RO4929097 also significantly delayed the functional and structural recovery of the kidney from I/R injury (Figure 7C).
Figure 7.

Activation of the Notch-2 signaling pathway mediated STAT3 kinase activation and survivin upregulation in response to I/R injury. (A) At different time points after I/R injury, wild-type Balb/c mouse renal cortical lysates were subjected to immunoblotting analysis with indicated antibodies. Starting 1 day before surgery, wild-type Balb/c mice were administered a γ-secretase inhibitor (GSI), RO4929097, 5 mg/kg by intraperitoneal injection. (B) Renal cortical lysates were harvested 2 days after surgery and subjected to immunoblotting analysis with indicated antibodies. (C) Representative histology (200×) at days 1 and 10 after I/R injury treatment with or without RO4929097 are presented.
In MCT cells, we also detected increases in the expression of Jagged-1, cleaved Notch-2, and Hes-1 in response to antimycin A treatment followed by 48 hours of nutrient reconstitution (Figure 8). Pretreatment of the cells with RO4929097 (500 nM) markedly attenuated expression of cleaved Notch-2, Hes-1, phospho-STAT3, and survivin (Figure 8A). When the cells were exposed to S3I-201 (20 μM) before treatment with antimycin A, upregulation of phospho-STAT3 and survivin were inhibited without affecting upregulation of Jagged-1, cleaved Notch-2, or Hes-1 (Figure 8B).
Figure 8.

Antimycin A treatment followed by nutrient reconstitution induced Notch-2 cleavage and phospho-STAT3–dependent upregulation of survivin expression in MCT cells. MCT cells were made quiescent in DMEM medium containing 1% serum for 24 hours. (A) Then, they were incubated with either vehicle or RO4929097 (500 nM) for 30 minutes before the addition of 0.5 µM antimycin A for 35 minutes to deplete cellular ATP, after which time the cells were washed one time and cultured with full-culture DMEM medium containing 10% serum with or without RO4929097 (500 nM) for 48 hours. (B) Alternately, they were incubated with either vehicle or S3I-201 (20 μM) for 30 minutes before the addition of 0.5 µM antimycin A for 35 minutes to deplete cellular ATP, after which time the cells were washed one time and cultured with full-culture DMEM medium containing 10% serum with or without S3I-201 (20 μM) for 48 hours. After separation by SDS-PAGE, the cell lysates were subjected to immunoblotting analysis with the indicated antibodies.
Discussion
Our results indicate that survivin expression was markedly increased in response to I/R injury, but this increased expression was absent in survivinptKO mice, indicating that the increased survivin expression was localized almost entirely to the proximal tubule, the major site of I/R-induced injury. Both the functional and structural recovery of the kidney from I/R injury were delayed in survivinptKO mice compared with their littermate controls. To elucidate the underlying mechanisms of survivin upregulation in response to I/R injury, we determined that STAT3 phosphorylation markedly increased in cultured renal proximal tubule cells in response to ATP depletion and renal cortical tubules in response to I/R injury. S3I-201 not only inhibited STAT3 phosphorylation but also markedly attenuated survivin upregulation in response to I/R injury or in the in vitro model of proximal tubule cell injury. STAT3 phosphorylation-mediated survivin upregulation was initiated through activation of Notch-2 signaling in response to I/R injury, indicating that Notch-2–dependent upregulation of survivin expression in renal proximal tubule epithelial cells plays an important role in the functional and structural recovery of the kidney after AKI.
Expression of survivin is regulated by transcription, differential splicing, and post-translational modifications. Previous studies have linked expression of survivin to growth factor-mediated signaling.16 Because our previous studies indicated an important role for EGFR signaling in recovery from AKI,26,34 we initially hypothesized that increased survivin expression was secondary to EGFR signaling. However, survivin expression in response to I/R injury was comparable in mice with specific renal proximal tubule EGFR deletion and their littermate controls, suggesting the existence of alternative pathway activation. In other systems, STAT3 has been shown to mediate survivin upregulation,15,27 and we found that I/R injury increased STAT3 phosphorylation, which was also not altered in the proximal tubule EGFR deletion mice (data not shown). In vitro studies using cultured proximal tubule cells showed a role for STAT3 to increase survivin expression, and inhibition of STAT3 phosphorylation in vivo not only inhibited I/R-induced survivin upregulation but also delayed recovery.
Furthermore, our data indicate that I/R injury markedly increased expression of the Notch receptor ligand Jagged1, cleaved intracellular Notch-2, and increased expression of the Notch target gene Hes-1. Treatment of the mice with a γ-secretase inhibitor, RO4929097, prevented Notch-2 cleavage and Hes-1 expression upregulation, and also, it inhibited STAT3 phosphorylation and survivin expression and delayed the structural and functional recovery of the kidney from I/R injury.
Therefore, we propose that I/R injury leads to increased expression of the Notch ligand Jagged-1, which binds to and activates Notch receptors on adjacent cells, followed by sequential cleavage by ADAMs (A Disintegrin And Metalloproteinase) to yield membrane-anchored Notch extracellular truncation fragment and γ-secretase to release NICD, which translocates to the nucleus and associates with an Hes-1 gene repressor protein, CSL (CBF1/SUH/LAG-1), which is named after the genes of CBF1(centromere binding factor 1, in human), SUH (suppressor of hairless homolog in Drosophila melanogaster), and LAG-1 (longevity-assurance-gene 1 in Caenorhabditis elegans), thereby activating Hes-1 gene transcription. The upregulated Hes-1 protein may then serve as a scaffold protein to recruit and activate the JAK-STAT3 signaling pathway to mediate upregulation of survivin expression, thereby protecting the injured cell from apoptosis and promoting epithelial cell proliferation, which is shown in Figure 9. Alternatively, Notch signaling could also induce expression of other cytokines or growth factors that then activate JAK-STAT3 signaling in an autocrine fashion. In summary, this study provides genetic and pharmacologic evidence that activation of the Notch receptor signaling pathway mediates STAT3 phosphorylation to upregulate survivin expression, which in turn, plays an important role in the functional and morphologic recovery of the kidney after AKI.
Figure 9.

Proposed mechanism for the role of Notch signaling pathway activation to mediate STAT3 phosphorylation-dependent survivin upregulation during recovery from AKI. ADAM, A Disintegrin And Metalloproteinase; CSL, CBF1/SUH/LAG-1; NEXT, Notch extracellular truncation fragment.
Concise Methods
Reagents and Antibodies
STAT3 phosphorylation inhibitor S3I-201 was purchased from EMD Millipore Chemicals (Billerica, MA), and the γ-secretase inhibitor RO4929097 was purchased from Selleck Chemicals (Houston, TX). Hes-1 antibody was from EMD Millipore Chemicals (Billerica, MA). Antibodies against survivin, phospho-STAT3, STAT3, cleaved Notch-1, and cleaved Notch-2 were from Cell Signaling Technology (Beverly, MA). Jagged-1 antibody was from BD Biosciences (San Jose, CA), Ki-67 antibody was from Vector Laboratories (Burlingame, CA), and β-actin antibody and all secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Generation of SurvivinptKO Null Mice
All experiments were approved by the Vanderbilt University Institutional Animal Use and Care Committee, and all experiments were conducted according to National Institutes of Health guidelines. Survivinf/f mice were generated by flanking exons 1–4 of the survivin gene with two LoxP sites as previously described.22 These survivinf/f mice were crossed with transgenic mice expressing Cre recombinase under the control of γGT-Cre+.23 The γGT promoter has been used to successfully target different genes of interest primarily to the renal proximal tubules.23,35 Age-matched survivinf/f littermates lacking the γGT-Cre transgene were used as wild-type controls for survivinptKO mice. Survivin floxed mice were genotyped by PCR as previously indicated,22 and 5′-AGGTGTAGAGAAGGCACTTAGC-3′ and 5′-CTAATCGCCATCTTCCAGCAGG-3′ were used for PCR verification of the γGT-Cre expression.
I/R Surgery
Nine- to ten-week-old survivinptKO mice and their littermates or wild-type Balb/c mice (Jackson Laboratory, Bar Harbor, ME) were anesthetized by peritoneal injection of 50 mg/kg nembutal sodium. Bilateral renal pedicles were exposed using a midline ventral incision and then clamped for 35 minutes at room temperature followed by reperfusion, which was verified by visual inspection of the kidneys. Sham animals had an incision plus 35 minutes of waiting time without clamping. After ischemia or sham treatment, muscle or skin layers were sutured, respectively; 0.8 ml prewarmed 0.9% saline solution were administered to each mouse immediately before closing the incision to prevent dehydration.
Measurement of Renal Function
Mouse blood samples were collected at 1, 2, 4, 7, 10, and 14 days after surgery, and BUN levels were measured as previously described.26
Histologic Examination
Kidneys embedded in paraffin were sectioned at 5 μm and stained with hematoxylin/eosin by standard methods. Tubular damage was scored by calculation of the percentage of tubules in the corticomedullary junction26 that displayed cell necrosis, loss of the brush border, cast formation, and tubular dilatation: 0, none; 1, ≤10%; 2, 11%–25%; 3, 26%–45%; 4, 46%–75%; –5, >76%. At least 10 high-power fields (magnification, ×200) per section for each sample were examined (n=5–7).
Immunoblotting
Procedures were performed as previously described with minor modifications.35 Briefly, survivinptKO mice and their littermates or wild-type Balb/c mice were euthanized at 1, 2, 4, and 6 or 10 days after surgery, and mouse kidney cortexes were dissected and homogenized in radioimmunoprecipitation (RIPA) buffer. MCT cells were harvested in RIPA buffer after the indicated treatment. After centrifugation at 10,000 × g for 15 minutes at 4°C, equal amounts of protein lysate were loaded directly on 10% or 15% SDS-PAGE, transferred onto Immobilon-P transfer membranes (Millipore, Bedford, MA), and probed with the indicated primary antibodies. The primary antibodies were detected with peroxidase-labeled goat anti-rabbit IgG and exposed on film by using enhanced chemiluminescence (Amersham Biosciences, Little Chalfont, Buckinghamshire, United Kingdom).
TUNEL Staining
TUNEL is a commonly used method for detection of DNA fragmentation that results from apoptotic signaling cascades. The assay relies on the presence of nicks in the DNA that can be identified by terminal deoxynucleotidyl transferase, an enzyme that catalyzes the addition of dUTPs that are secondarily labeled with a marker. Therefore, we used TUNEL staining in the present study to detect apoptotic cells. Specifically, mice were euthanized at day 2 after I/R injury, and kidneys were harvested followed by overnight fixation in 4% paraformaldehyde. The fixed kidneys were dehydrated through a graded series of ethanol, embedded in paraffin, sectioned (5 µm), and mounted on glass slides. The DeadEnd Fluorometric TUNEL System (Promega, Madison, WI) was used, and the staining was performed as per the product protocol. Briefly, after deparaffinization and rehydration, the slides were permeabilized in 0.2% Triton X-100 in PBS for 5 minutes. After being washed in PBS two times for 5 minutes each, the slides were equilibrated with 100 μl Equilibration Buffer at room temperature for 5–10 minutes; then, the tissues were labeled with 50 μl/section terminal deoxynucleotidyl transferase reaction mix at 37°C in a humidified chamber for 60 minutes. The reaction was stopped by Immersion slides in 2× SSC for 15 minutes. Finally the slides were mounted and covered after being washed in PBS three times for 5 minutes each. The images were captured using an Axiocam HRc digital camera (Carl Zeiss Inc.) and processed using Zeiss LSM Image Browser and Photoshop (Adobe Systems Inc., San Jose, CA). Positive cells were counted at magnification of ×200, and at least 10 fields per section for each sample were examined (n=5–7).
Immunohistochemistry
Mice were euthanized at different time points after I/R injury, and kidneys were harvested followed by overnight fixation in 4% paraformaldehyde. The fixed kidneys were dehydrated through a graded series of ethanol, embedded in paraffin, sectioned (5 µm), and mounted on glass slides. After deparaffinization and rehydration, antigen retrievals were performed by using Antigen Unmasking Solution (Vector, Burlingame, CA). The sections were blocked with 2.5% normal goat serum and then incubated with primary antibodies against STAT3 (at 1:100) overnight at 4°C. The sections were washed three times in PBS, and VECTASTAIN ABC Kits (Vector, Burlingame, CA) were used following the manufacturer’s instruction. The sections were counterstained with hematoxylin. Images were captured using an AxioCam HRc digital camera (Carl Zeiss), and the positive-staining cells were counted in each ×400 field (10 fields were randomly chosen for each section). Values are mean ± SEM (n=5–7 for each group).
Immunofluorescence Staining
Ki-67 is a nuclear protein that is associated with and may be necessary for cellular proliferation. The Ki-67 antigen is exclusively detected within the cell nucleus during interphase, and most of the protein is relocated to the surface of the chromosomes in mitosis. Because Ki-67 protein is present during all active phases of the cell cycle (G1, S, G2, and mitosis) but absent from resting cells, Ki-67 staining has become a reliable marker to detect proliferating cells. Accordingly, we performed Ki-67 staining for the detection of cell proliferation in the current study. Briefly, mice were euthanized at different time points after I/R injury, and kidneys were harvested followed by overnight fixation in 4% paraformaldehyde. The fixed kidneys were dehydrated through a graded series of ethanol, embedded in paraffin, sectioned (5 µm), and mounted on glass slides. After deparaffinization and rehydration, antigen retrieval was performed by using Antigen Unmasking Solution (Vector, Burlingame, CA). The sections were blocked with 2.5% BSA in PBS; the sections were incubated with primary antibodies against Ki-67 (1:400) overnight at 4°C and then incubated with Alexa Fluor 594-conjugated secondary antibodies and the fluorescein-labeled lotus tetragonolobus lectin (Vector, Burlingame, CA) for 1 hour. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Images were captured by using an Axioplan2 fluorescence microscope and AxioCam HRc digital camera (Carl Zeiss). The positive staining cells were counted in each ×400 field (10 fields were randomly chosen for each section). Values are mean ± SEM (n=5–7 for each group).
Cell Culture
The mouse proximal tubular epithelial cell line, MCT, was a gift from Eric Neilson36 and maintained in high-glucose (4.5 g/L) DMEM containing 10% FCS and 100 IU/mL penicillin-streptomycin in a humidified incubator at 37°C. Cells were made quiescent in DMEM medium containing 1% serum for another 24 hours, and then, the quiescent MCT cells were incubated with 0.5 µM antimycin A for 35 minutes to deplete cellular ATP followed by restoration of substrate supply in full-culture DMEM medium containing 10% serum for indicated times. For the inhibitor experiments, the cells were incubated with or without RO4929097 (500 nM) or S3I-201 (20 μM) for 30 minutes before treatment with 0.5 µM antimycin A followed by restoration of substrate supply in full-culture DMEM medium containing 10% serum with or without inhibitors for 48 hours. The cells were lysed in RIPA buffer, and lysates were subjected to immunoblotting analysis.
STAT3 siRNA Transfection
Silencer Negative Control 1 siRNA (AM4611; Applied Biosystems/Ambion, Austin, TX), Thermo Scientific Dharmacon SMARTpool: ON-TARGETplus STAT3 siRNAL-040794–01–0005, or SMARTpool: siGENOME STAT3 siRNA M-040794–00–0005 (Thermo Fisher Scientific, Lafayette, CO) was transfected into subconfluent MCT cells by the Lipofectamine method (Invitrogen Corporation, Carlsbad, CA) as we described previously.35 Forty-eight hours after transfection, cells were made quiescent in DMEM medium containing 1% serum for another 24 hours, and then, the quiescent MCT cells were incubated with 0.5 µM antimycin A for 35 minutes to deplete cellular ATP followed by restoration of substrate supply in full-culture DMEM medium containing 10% serum for the indicated time. The cells were lysed in RIPA buffer, and lysates were subjected to immunoblotting analysis.
Statistical Analyses
Data are presented as mean ± SEM from separate experiments. An unpaired t test was used for statistical analysis, and for multiple group comparisons, ANOVA and Bonferroni t tests were used; a P value<0.05 compared with control was considered statistically significant.
Disclosures
None.
Acknowledgments
This work was supported by funds from the Department of Veterans Affairs, National American Heart Association Scientist Development Grant 0630274N, National Institutes of Health R01 Grant DK83575 (to J.-K.C.), and National Institutes of Health Grants DK51265 (to R.C.H.), DK62794 (to R.C.H.), and DK7934 (to R.C.H.). E.M.C. is supported by the Canadian Institutes of Health Research, holds a CSL-Behring Research Chair and a Canada Research Chair in Endothelial Cell Biology, and is an Adjunct Scientist with the Canadian Blood Services.
Some of the data in this paper were presented as an oral presentation during a free communication session at the American Society of Nephrology Meeting October 30–November 4, 2012, in San Diego, CA.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.Rifkin DE, Coca SG, Kalantar-Zadeh K: Does AKI truly lead to CKD? J Am Soc Nephrol 23: 979–984, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lameire N, Van Biesen W, Vanholder R: The changing epidemiology of acute renal failure. Nat Clin Pract Nephrol 2: 364–377, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE: The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int 79: 1361–1369, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishani A, Nelson D, Clothier B, Schult T, Nugent S, Greer N, Slinin Y, Ensrud KE: The magnitude of acute serum creatinine increase after cardiac surgery and the risk of chronic kidney disease, progression of kidney disease, and death. Arch Intern Med 171: 226–233, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Bonventre JV: Mechanisms of ischemic acute renal failure. Kidney Int 43: 1160–1178, 1993 [DOI] [PubMed] [Google Scholar]
- 6.Nony PA, Schnellmann RG: Mechanisms of renal cell repair and regeneration after acute renal failure. J Pharmacol Exp Ther 304: 905–912, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Duffield JS, Humphreys BD: Origin of new cells in the adult kidney: Results from genetic labeling techniques. Kidney Int 79: 494–501, 2011 [DOI] [PubMed] [Google Scholar]
- 8.Wilson A, Radtke F: Multiple functions of Notch signaling in self-renewing organs and cancer. FEBS Lett 580: 2860–2868, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Chiba S: Notch signaling in stem cell systems. Stem Cells 24: 2437–2447, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Gupta S, Li S, Abedin MJ, Wang L, Schneider E, Najafian B, Rosenberg M: Effect of Notch activation on the regenerative response to acute renal failure. Am J Physiol Renal Physiol 298: F209–F215, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Kamakura S, Oishi K, Yoshimatsu T, Nakafuku M, Masuyama N, Gotoh Y: Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat Cell Biol 6: 547–554, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Ambrosini G, Adida C, Altieri DC: A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 3: 917–921, 1997 [DOI] [PubMed] [Google Scholar]
- 13.Mita AC, Mita MM, Nawrocki ST, Giles FJ: Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res 14: 5000–5005, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Asanuma H, Torigoe T, Kamiguchi K, Hirohashi Y, Ohmura T, Hirata K, Sato M, Sato N: Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res 65: 11018–11025, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Aoki Y, Feldman GM, Tosato G: Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 101: 1535–1542, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Dan HC, Jiang K, Coppola D, Hamilton A, Nicosia SV, Sebti SM, Cheng JQ: Phosphatidylinositol-3-OH kinase/AKT and survivin pathways as critical targets for geranylgeranyltransferase I inhibitor-induced apoptosis. Oncogene 23: 706–715, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Altieri DC: Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 22: 8581–8589, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Altieri DC: Targeted therapy by disabling crossroad signaling networks: The survivin paradigm. Mol Cancer Ther 5: 478–482, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Lechler P, Wu X, Bernhardt W, Campean V, Gastiger S, Hackenbeck T, Klanke B, Weidemann A, Warnecke C, Amann K, Engehausen D, Willam C, Eckardt KU, Rödel F, Wiesener MS: The tumor gene survivin is highly expressed in adult renal tubular cells: Implications for a pathophysiological role in the kidney. Am J Pathol 171: 1483–1498, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kindt N, Menzebach A, Van de Wouwer M, Betz I, De Vriese A, Conway EM: Protective role of the inhibitor of apoptosis protein, survivin, in toxin-induced acute renal failure. FASEB J 22: 510–521, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL, Choo KH: Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol 10: 1319–1328, 2000 [DOI] [PubMed] [Google Scholar]
- 22.Xing Z, Conway EM, Kang C, Winoto A: Essential role of survivin, an inhibitor of apoptosis protein, in T cell development, maturation, and homeostasis. J Exp Med 199: 69–80, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG: Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110: 341–350, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, Yin H, Wong K, Miyazawa T, Chen J, Chang I, Singh A, Harris RC: CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest 122: 4519–4532, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaira V, Lee CW, Goel HL, Bosari S, Languino LR, Altieri DC: Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene 26: 2678–2684, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Chen JK, Chen J, Thomas G, Kozma SC, Harris RC: S6 Kinase 1 knockout inhibits Uninephrectomy-or diabetes-induced renal hypertrophy. Am J Physiol, Renal Physiol 297, F585–593, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen Y, Devgan G, Darnell JE, Jr, Bromberg JF: Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc Natl Acad Sci U S A 98: 1543–1548, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conway EM, Zwerts F, Van Eygen V, DeVriese A, Nagai N, Luo W, Collen D: Survivin-dependent angiogenesis in ischemic brain: Molecular mechanisms of hypoxia-induced up-regulation. Am J Pathol 163: 935–946, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunning PT, Katt WP, Glenn M, Siddiquee K, Kim JS, Jove R, Sebti SM, Turkson J, Hamilton AD: Isoform selective inhibition of STAT1 or STAT3 homo-dimerization via peptidomimetic probes: Structural recognition of STAT SH2 domains. Bioorg Med Chem Lett 17: 1875–1878, 2007 [DOI] [PubMed] [Google Scholar]
- 30.McCright B, Gao X, Shen L, Lozier J, Lan Y, Maguire M, Herzlinger D, Weinmaster G, Jiang R, Gridley T: Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development 128: 491–502, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Cheng HT, Kim M, Valerius MT, Surendran K, Schuster-Gossler K, Gossler A, McMahon AP, Kopan R: Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 134: 801–811, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi T, Terada Y, Kuwana H, Tanaka H, Okado T, Kuwahara M, Tohda S, Sakano S, Sasaki S: Expression and function of the Delta-1/Notch-2/Hes-1 pathway during experimental acute kidney injury. Kidney Int 73: 1240–1250, 2008 [DOI] [PubMed] [Google Scholar]
- 33.Luistro L, He W, Smith M, Packman K, Vilenchik M, Carvajal D, Roberts J, Cai J, Berkofsky-Fessler W, Hilton H, Linn M, Flohr A, Jakob-Røtne R, Jacobsen H, Glenn K, Heimbrook D, Boylan JF: Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res 69: 7672–7680, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Chen JK, Wang SW, Moeckel G, Harris RC: Importance of functional EGF receptors in recovery from acute nephrotoxic injury. J Am Soc Nephrol 14: 3147–3154, 2003 [DOI] [PubMed] [Google Scholar]
- 35.Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, Threadgill DW, Neilson EG, Harris RC: EGFR signaling promotes TGFβ-dependent renal fibrosis. J Am Soc Nephrol 23: 215–224, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haverty TP, Kelly CJ, Hines WH, Amenta PS, Watanabe M, Harper RA, Kefalides NA, Neilson EG: Characterization of a renal tubular epithelial cell line which secretes the autologous target antigen of autoimmune experimental interstitial nephritis. J Cell Biol 107: 1359–1368, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]