

Abstract
In neurons, binding of calmodulin (CaM) or calcium-binding protein 1 (CaBP1) to the CaV1 (L-type) voltage-gated calcium channel IQ domain endows the channel with diametrically opposed properties. CaM causes calcium-dependent inactivation (CDI) and limits calcium entry, whereas CaBP1 blocks CDI and allows sustained calcium influx. Here, we combine isothermal titration calorimetry (ITC) with cell-based functional measurements and mathematical modeling to show that these calcium sensors behave in a competitive manner that is explained quantitatively by their apo-state binding affinities for the IQ domain. This competition can be completely blocked by covalent tethering of CaM to the channel. Further, we show that Ca2+/CaM has a sub-picomolar affinity for the IQ domain that is achieved without drastic alteration of calcium binding properties. The observation that the apo-forms of CaM and CaBP1 compete with each other demonstrates a simple mechanism for direct modulation of CaV1 function and suggests a means by which excitable cells may dynamically tune CaV activity.
Keywords: Voltage-gated calcium channel, isothermal titration calorimetry, electrophysiology, calcium sensor proteins, mathematical models
Introduction
High-voltage activated calcium channel (CaV) opening provides the primary source of calcium influx in excitable cells and couples electrical signals to chemical signaling cascades1; 2. A set of calcium-dependent autoregulatory mechanisms shape CaV activity in response to the influx of the permeant ion3; 4 and strongly affect neurotransmitter release, excitation-contraction coupling, and calcium-dependent gene activation4; 5. Crucial among these activity-dependent changes is a process called calcium-dependent inactivation (CDI) that limits calcium entry following channel activation and for which the calcium sensor protein calmodulin is essential3; 4; 6; 7.
CaVs are multi-subunit complexes of four main components8; 9: a pore-forming CaV1 (L-type) or CaV2 (P/Q-, N-, and R-type) CaVα1 subunit1, a cytoplasmic CaVβ subunit10; 11, the membrane anchored subunit CaVα2δ12, and calmodulin (CaM)13. In some neurons members of a family of neuronal calcium sensor proteins similar to CaM, known as CaBPs14 can replace CaM15. This calcium sensor exchange creates CaVs having strikingly different functional properties from those under the control of CaM16; 17; 18; 19; 20; 21; 22. In particular, CaV1.219; 22; 23 and CaV1.316; 17 lack CDI when CaBP1, a CaBP highly expressed in the brain and retina15, is part of the channel complex. CaM and CaBP1 are calcium-binding proteins comprising two lobes that each bear a pair of EF-hands and an interlobe flexible linker15; 23; 24. Structural studies have shown that the calcium-bound forms of the CaM and CaBP1 C-terminal lobes (C-lobes) have similar three-dimensional structures23; 24 and compete for binding to the same channel element, the IQ domain23, whereas the N-terminal lobes (N-lobes) are more divergent23.
Although multiple CaV1 channel segments have been implicated in CaM and CaBP1 function25; 26; 27; 28; 29, the main attachment point for both is the IQ domain of the CaVα1 C-terminal cytoplasmic tail6; 17; 19; 25. Previous studies have shown that CaBP1 and CaM cannot simultaneously bind the CaV1.2 IQ domain17; 19; 23; however, evidence for functional competition in the full-length channel has been lacking. Here, we use thermodynamic measurements to investigate the CaM and CaBP1 competition on the CaV1.2 IQ domain. Our data show that this competition occurs between apo-states of the respective molecules and that although their IQ domain binding sites overlap, the two calcium sensor proteins bind in different ways. Further, we present experiments using Xenopus oocytes that corroborate these biochemical observations and that demonstrate that CaM and CaBP1 can compete directly for control of CaV1.2 CDI in living cells. Importantly, mathematical modeling of the CaM-CaBP1 competition based on the ITC measurements predicts the functional competition measured on full-length channels in the context of a cell membrane. This excellent agreement, which emerges from analysis of measurements made in dramatically different milieus, indicates that the simplified biochemical systems investigated here capture the essence of the mechanism by which CaM and CaBP1 competitively alter channel function.
Results
Ca2+/CaM has sub-picomolar affinity for the CaV1.2 IQ domain
The CaV1.2 IQ domain is the main binding site for both Ca2+/CaM6; 7 and Ca2+/CaBP119. Previous isothermal titration calorimetry (ITC) studies have determined the binding affinities of the individual CaBP1 lobes, N-lobeCaBP1 and Ca2+/C-lobeCaBP1 23, full-length Ca2+/CaBP123, and the individual Ca2+/CaM lobes, Ca2+ /N-lobeCaM and Ca2+/C-lobeCaM 30, for this portion of the CaV1.2 C-terminal tail; however, measurement of the affinity of full-length Ca2+/CaM for the CaV1.2 IQ domain had remained elusive. Hence, we set out to examine this interaction using ITC.
Direct titration of Ca2+/CaM into solution containing the CaV1.2 IQ domain yielded a complex curve (Fig. 1a) that could not be explained by a single binding event. ITC analysis of Ca2+/CaBP1 with the CaV1.2 IQ domain binding posed similar difficulties that were surmounted using a displacement titration strategy coupled with thermodynamic cycle analysis23. Therefore, we tested if a comparable approach would apply here. Indeed, titration of Ca2+/CaM into a preformed Ca2+/C-lobeCaM:CaV1.2 IQ domain complex gave a single transition (Fig. 1b). As the affinity of the Ca2+/C-lobeCaM:CaV1.2 IQ binding reaction is known30, we could use a thermodynamic cycle (Fig. 1c) together with competition ligand binding by displacement analysis31 to determine the affinity of full-length Ca2+/CaM for the CaV1.2 IQ domain. Importantly, there is no detectable interaction between Ca2+/CaM and Ca2+/C-lobeCaM that could interfere with the analysis (Fig. 1d).
Fig. 1. Characterization of Ca2+ 4:CaM-CaV1.2 IQ domain binding.

Exemplar ITC titrations for: a, 75 μM Ca2+ 4:CaM into 7.5 μM CaV1.2 IQ domain and b, 75 μM Ca2+ 4:CaM into 7.5 μM CaV1.2 IQ domain and 50 μM Ca2+ 2:C-lobeCaM. c, Thermodyamic cycle for analysis of the binding of Ca2+ 4:CaM (KA) to the CaV1.2 IQ domain. KB describes Ca2+ 2:C-lobeCaM binding. KC describes the competition of Ca2+ 2:C-lobeCaM by CaM. Exemplar ITC titrations for: d, 75 μM Ca2+ 4:CaM into 50 μM Ca2+ 2:C-lobeCaM and e, 75 μM Ca2+ 2:CaBP1 into 7.5 μM CaV1.2 IQ domain and 11.25 μM Ca2+ 4:CaM. Icons depict ITC components. N-lobes and C-lobes are green and blue, respectively. CaM and CaBP1 are in pastel and dark shades, respectively. White spheres represent Ca2+. CaV1.2 IQ domain is shown in red.
We find that the affinity of Ca2+/CaM for the CaV1.2 IQ domain is exceptionally high, Kd = 850±130 fM, a result that agrees with a previously reported subpicomolar estimate of this interaction32. The affinity of Ca2+/CaM for the CaV1.2 IQ domain is ~3100-fold stronger than that of Ca2+ /C-lobeCaM (Table 1) and indicates that there is a substantial contribution from the Ca2+/N-lobeCaM that agrees with its extensive CaV1.2 IQ domain contacts30; 33. Although extreme, the sub-picomolar affinity of Ca2+ /CaM for the CaV1.2 IQ domain is within the bounds other tight Ca2+/CaM:peptide interactions (cf. a calmodulin kinase II peptide, 70 fM)34 but is many orders of magnitude greater than what Ca2+/CaM displays for the IQ domain of the distantly related voltage-gated sodium channel NaV1.5 (2 μM)35 and the NaV1.5 III–IV loop (3 μM)36.
Table 1.
Titration calorimetry data for interactions between CaM and CaBP1 with the CaV1.2 IQ domain
| n | Setup | N | Kd (M) | ΔH (kcal mol−1) | ΔS (cal mol−1 K−1) | ΔG (kcal mol−1) | KdCaBP1 / KdCaM | Kd(apo) / Kd (Ca2+) | Δ ΔG (kcal mol−1) | |
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| N-lobeBP** | 3 | S | 0.82 ± 0.05 | 1.10 × 10−6 ± 8.0 × 10−8 | 2.09 ± 0.40 | 34.5 ± 1.5 | −7.84 ± 0.04 | - | - | - |
| Ca2+/C-lobeBP** | 4 | S | 0.90 ± 0.07 | 1.05 × 10−8 ± 1.9 × 10−9 | −2.88 ± 0.19 | 25.5 ± 0.6 | −10.52 ± 0.11 | - | - | - |
| Ca2+/CaBP1** | 2 | D | # | 2.9 × 10−10 ± 7.0 × 10−11 | −4.72 ± 0.07 | 26.6 ± 0.7 | −12.38 ± 0.14 | - | - | - |
|
| ||||||||||
| Ca2+ /N-lobeCaM* | 2 | S | 0.85 ± 0.05 | 5.76 × 10−8 ± 3.55 × 10−8 | −1.91 ± 0.14 | 26.7 ± 0.9 | −9.60 ± 0.38 | 19 | - | - |
| Ca2+ /C-lobeCaM* | 2 | S | 0.98 ± 0.18 | 2.63 × 10−9 ± 7.0 × 10−11 | −6.77 ± 0.21 | 14.2 ± 0.9 | −11.31 ± 0.01 | 4.0 | - | - |
| Ca2+/CaM | 2 | D | # | 8.5 × 10−13 ± 1.3 × 10−13 | −5.74 ± 0.35 | 34.5 ± 0.9 | −15.68 ± 0.08 | 340 | - | - |
|
| ||||||||||
| N-lobeBP** | 3 | S | 0.82 ± 0.05 | 1.10 × 10−6 ± 8.0 × 10−8 | 2.09 ± 0.40 | 34.5 ± 1.5 | −7.84 ± 0.04 | - | 1 | 0 |
| C-lobe CaBP1EF34 | 2 | D | # | 3.80 × 10−6 ± 1.30 × 10−6 | 3.77 ± 0.20 | 37.7 ± 1.4 | −7.08 ± 0.21 | − | 360 | −3.44 |
| CaBP1EF34 | 2 | D | # | 9.1 × 10−8 ± 1.3 × 10−8 | −0.82 ± 0.15 | 29.0 ± 0.2 | −9.15 ± 0.08 | − | 310 | −3.24 |
|
| ||||||||||
| N-lobe CaMEF12 | 2 | D | # | > 5 × 10−6 | n/a | n/a | > −6.9 | < 0.2 | > 90 | < −2.78 |
| C-lobeCaMEF34 | 2 | D | # | 2.2 × 10−6 ± 1.0 × 10−8 | −3.79 ± 0.06 | 12.4 ± 0.2 | −7.34 ± 0.02 | 1.7 | 840 | −4.05 |
| CaMEF1234 | 2 | D | # | 5.8 × 10−7 ± 5.0 × 10−8 | −3.36 ± 0.02 | 16.5 ± 0.2 | −8.10 ± 0.06 | 0.14 | 680,000 | −7.58 |
Our previous ITC studies using identical buffer conditions to those used here showed that Ca2+/CaBP1 binds strongly to the CaV1.2 IQ domain (290±70 pM)23. Nevertheless, this value is ~340-fold weaker than the Ca2+/CaM:CaV1.2 IQ domain interaction. In good agreement with these findings, Ca2+/CaBP1 was unable to displace Ca2+/CaM from a preformed Ca2+/CaM:CaV1.2 IQ domain complex (Fig. 1e). This result disagrees with the results reported in pull-down studies of Zhou et al.19; however, the discrepancy is readily explained by the fact that Zhou et al.19 used an CaV1.2 IQ domain construct lacking several key residues that interact with Ca2+/CaM including two that bind Ca2+/N-lobeCaM (cf. 30).
Taken together, these ITC experiments establish that Ca2+/CaM binds the CaV1.2 IQ domain with an exceptionally high affinity that is several hundred-fold stronger than that of Ca2+/CaBP1 (Table 1). As the affinity of the individual Ca2+ /C-lobes differs only three-fold23; 30, the competitive advantage of Ca2+/CaM over Ca2+/CaBP1 for the CaV1.2 IQ domain is almost exclusively due to differences in the contributions from the Ca2+/N-lobe.
apo-CaBP1 binds the CaV1.2 IQ domain more strongly than apo-CaM
The large affinity differences of Ca2+/CaM and Ca2+/CaBP1 for the CaV1.2 IQ domain preclude effective competition by Ca2+/CaBP1. However, because Ca2+/CaM and Ca2+/CaBP1 are likely to be present only for brief periods when local calcium levels rise due to channel activity, we reasoned that the physiologically relevant competition for the CaV1.2 IQ domain might occur in the calcium-free, apo-states of the respective calcium sensors. To look for biochemical evidence that could test this idea, we measured the affinities for the CaV1.2 IQ domain of a set of apo-CaM and apo-CaBP1 mimics. In each, we disabled calcium binding through an established strategy in which alanine replaces the first aspartate of the EF-hand consensus sequence6; 7; 30. ‘EF##’ denotes which EF hands carry this change.
Because of limitations on reaching sufficient apo-lobe mimic concentrations for direct titration due to the weak nature of the interaction (Kds ≥ 100 nM), which would have required concentrations of titrant and target that are not physically well behaved, (~500 μM), we derived the apo-CaM lobe (N-lobeCaMEF12 and C-lobeCaMEF34) and apo-CaBP1 C-lobe (C-lobeCaBP1EF34) affinities for the CaV1.2 IQ domain using displacement ITC (Fig. 2a–c) and an analysis similar to Fig. 1c. These experiments showed that loss of calcium binding capacity in the individual CaM lobes caused a 2–3 order of magnitude affinity reduction relative to the calcium-bound forms: 2,200±10 nM versus 2.63±0.07 nM for C-lobeCaMEF34 and Ca2+/C-lobeCaM and >5000 nM versus 57.6±35.5 nM, for N-lobeCaMEF12 and Ca2+:N-lobeCaM, respectively (Figs. 2a and 2b, Table 1). We measured a similar magnitude change for the only CaBP1 lobe that responds to calcium37, C-lobeCaBP1, (3800±1,300 nM versus 10.5±1.9 nM, for C-lobeCaBP1EF34 and Ca2+/C-lobeCaBP1, respectively) (Fig. 2c, Table 1). Thus, the importance of calcium for elevating CaV1.2 IQ domain binding affinity is shared among the calcium responsive lobes of both CaM and CaBP1.
Fig. 2. Characterization of apo-CaM and apo-CaBP1 CaV1.2 IQ domain binding.

Exemplar ITC titrations for: a, 75 μM Ca2+ 2: C-lobeCaM into 7.5 μM CaV1.2 IQ domain and 50 μM C-lobeCaM EF34. b, 75 μM Ca2+ 2: C-lobeCaM into 7.5 μM CaV1.2 IQ domain and 50 μM N-lobeCaMEF12. c, 75 μM Ca2+ 2: C-lobeCaBP1 into 7.5 μM CaV1.2 IQ domain and 50 μM C-lobeCaBP1EF34. d, 75 μM Ca2+ 2: C-lobeCaM into 7.5 μM CaV1.2 IQ domain and 50 μM CaMEF1234. e, 75 μM Ca2+ 2: C-lobeCaBP1 into 7.5 μM IQ domain and 50 μM CaBP1EF34. Icons depict ITC components. N-lobes and C-lobes are green and blue, respectively. CaM and CaBP1 are in pastel and dark shades, respectively. White spheres represent Ca2+. Black crosses indicate mutated EF-hands. CaV1.2 IQ domain is shown in red.
We also performed competition ITC experiments using apo-mimics of full length CaM and CaBP1 (CaMEF1234 and CaBP1EF34) (Fig. 2d and e, Table 1). These revealed that in the case of CaM, the large change in affinity for the CaV1.2 IQ domain relative to the calcium-bound forms is greatly accentuated in context of the full-length protein, reaching a ~six order of magnitude difference (580±50 nM versus 850±130 fM for CaMEF1234 and Ca2+/CaM, respectively) (Fig. 2d and Table 1). In contrast, the ~300-fold difference for CaBP1 (91±13 nM versus 290±70 pM CaBP1EF34 and Ca2+/CaBP1 Fig. 2e and Table 1) is similar to that of C-lobeCaBP1 alone and is consistent with the inability of N-lobeCaBP1 to undergo a calcium dependent conformational change23. The apo-CaM mimic affinity for the CaV1.2 IQ domain is close to that reported using in cell fluorescence (1035 nM)38 but in poorer agreement with estimates from in vitro fluorescence (~50 nM)39. Strikingly, comparison of the apo-state affinities shows that unlike in the calcium-bound states where Ca2+/CaM has a huge advantage over Ca2+/CaBP1, the CaV1.2 IQ domain favors binding of apo-CaBP1 by ~7-fold over apo-CaM. Thus, the data suggest that under calcium concentrations that favor the apo-states, CaBP1 could compete effectively with CaM for binding to the CaV1.2 IQ domain.
Consequences of CaM-IQ domain affinity for calcium binding
Numerous studies have shown that target engagement can have strong effects on the apparent affinity of CaM for calcium due to thermodynamic linkage32; 40; 41. In this context, the nearly million-fold affinity difference between apo-CaM and Ca2+/CaM for the CaV1.2 IQ domain was surprising. As the average affinity of CaM for calcium has been measured to be ~15 μM42; 43, the large differences associated with IQ domain engagement could be taken to suggest that the calcium affinity of IQ domain bound CaM might be shifted into picomolar range. Such a scenario would cause IQ domain bound CaM to remain in the calcium-bound form even at resting levels of intracellular calcium, which are ~100 nM2; 44. This would put CaM far outside of the range where it could act as a physiologically relevant sensor for calcium-dependent control of channel function.
To try to understand this situation from a quantitative perspective, we constructed a model to evaluate how the measured large changes in protein-protein interactions might lead to changes in the free energy of calcium binding (Fig. 3a and Appendix 1). This analysis shows that there needs to be only a ~30-fold increase (~1.9 kcal mol−1) in the calcium affinity of each CaM EF-hand to account for the ~million-fold (~7.7 kcal mol−1) affinity difference measured for the binding of apo-CaM and Ca2+/CaM to the CaV1.2 IQ domain. Notably, this relative 30-fold change in Ca2+ affinity does not depend on the exact Ca2+ affinities of free CaM. This calcium affinity change is in good agreement with experimental estimates based on CaM:CaV1.2 IQ association39. The rather modest calcium affinity change arises from the fact that the reaction order is higher than a simple single binding event. The affinities of the four EF-hands of CaM have been determined in a number of studies, e.g. 42,45,46. Taking the values that have been used extensively for modeling studies42, e.g.47,48 the affinity change caused by association with the CaV1.2 IQ domain, we calculated the fraction of Ca2+ 4:CaM present in both the free and CaV1.2 IQ domain-associated forms (Fig. 3b). Interestingly, the model indicates that the association of CaM with IQ domain shifts the sensitivity of the CaM calcium response from a range of 10–500 μM to 0.3–10 μM. Such a tuning of calcium sensitivity positions the complex perfectly in range of physiologically relevant intracellular calcium changes2; 43; 44.
Fig. 3. Thermodynamic analysis of CaM, Ca2+, and IQ domain binding.

a, Macroscopic thermodynamic cycle for CaM, Ca2+ and CaV1.2 IQ domain. K1 and K4 describe Ca2+ binding to CaM and the CaM/IQ complex, respectively. K2 and K3 describe IQ domain binding to calcium-free CaM and Ca2+ 4:CaM, respectively. b, Calculated fraction of Ca2+ 4:CaM as a function of Ca2+ concentration for CaM alone (red) or bound to the IQ domain (black curve). a, Macroscopic thermodynamic cycle for CaBP1, Ca2+ and CaV1.2 IQ domain. K1 and K4 describe Ca2+ binding to CaBP1 and the CaBP1/IQ complex, respectively. K2 and K3 describe IQ domain binding to calcium-free CaBP1 and Ca2+ 2:CaBP1, respectively. d, Calculated fraction of Ca2+ 2:CaBP1as a function of Ca2+ concentration for CaBP1 alone (orange) or bound to the IQ domain (grey curve).
Consequences of CaBP1-IQ domain affinity for calcium binding
Functional EF hands are not required for CaBP1 to inhibit CDI23. Nevertheless, given the differences of binding affinity for the CaV1.2 IQ domain between apo-CaBP1, and Ca2+/CaBP1, which are considerably smaller than those for CaM but are still substantial (Table 1), we performed an equivalent mathematical analysis to consequences of IQ domain binding for the EF-hand affinities for CaBP1 (Appendix 2). This analysis shows that association with the CaV1.2 IQ domain alters the affinity of each CaBP1 EF-hand for Ca2+ by 18-fold. This relative change in Ca2+ affinity upon IQ domain is independent of the absolute Ca2+ affinity of free CaBP1. Thus, despite the dramatically smaller change in IQ domain affinity between apo- and Ca2+ -bound forms of CaBP1 relative to CaM (310 versus 680,000), the fact that there are fewer EF-hands in CaBP1 compared to CaM (2 rather than 4) results in affinity changes for each individual EFhand that are similar for CaBP1 and CaM (18 and 29, respectively). Taking the reported dissociation constants of the individual CaBP1 Ca2+ binding sites37, similar to the analysis with CaM, we determined the fraction of the Ca2+ 2:CaBP1 form of CaBP1 as a function of calcium concentration in both the free and CaV1.2 IQ domain-bound forms (Fig. 3d). The reported apparent affinity of free CaBP1 compared for Ca2+ is ~10-fold stronger than that of free CaM. Provided that the affinity changes caused by IQ domain binding are distributed equally among the EF-hands, the analysis indicates that this rank order is maintained upon IQ domain binding and that the CaBP1:CaV1.2 IQ domain complex will bind Ca2+ at lower calcium concentrations than the CaM:CaV1.2 IQ domain complex.
CaBP1 and CaM binding sites comprise different CaV1.2 IQ domain elements
Previous studies have shown that the Ca2+/C-lobes of CaM and CaBP1 have similar structures23; 24 and compete for overlapping binding sites on the CaV1.2 IQ domain23. Given the substantial difference in affinities between the calcium-bound forms of full-length CaM and CaBP1 for the CaV1.2 IQ domain (Table 1), we were interested in defining the structural basis for these differences. Ca2+/CaM engages the CaV1.2 IQ domain using a set of six aromatic anchor residues that are divided into two categories based upon the Ca2+/CaM lobe they contact30: CaV1.2 F1618, F1619 and Y1622 anchor Ca2+/N-lobeCaM; CaV1.2 Y1627, F1628 and F1631 anchor Ca2+/C-lobeCaM. Hence, removal of the Ca2+/N-lobe anchors should reduce the affinity of Ca2+/CaM for the IQ domain while leaving the affinity for the Ca2+/C-lobeCaM unaffected.
To test this idea, we used ITC to determine the affinity of full-length Ca2+/CaM and its individual lobes for a mutant of the CaV1.2 IQ domain having the three Ca2+/N-lobeCaM anchors replaced by alanine (CaV1.2 IQ TripleA). In agreement with the predictions from the structure, ITC shows that Ca2+/C-lobeCaM, binds to CaV1.2 IQ TripleA (Fig. 4a) and has an affinity (1.5±0.2 nM, ΔΔG = 0.14 kcal mol−1) and thermodynamic parameters unchanged from wild-type30 (Tables 1 and 2). In contrast, ITC using Ca2+/N-lobeCaM (Fig. 4b) revealed an affinity for CaV1.2 IQ TripleA that was greatly compromised by the loss of the N-lobe anchors (>5 μM, ΔΔG <−2.7 kcal mol−1) (Tables 1 and 2). In accord with these results, ITC measurements show that the TripleA mutation also reduced the affinity of full-length Ca2+/CaM (170±20 pM, ΔΔG = −3 kcal mol−1) for the CaV1.2 domain (Fig. 4c and Table 2). Together, these data match expectations set by the structure and underscore the importance of the N-lobe anchors to the overall affinity of Ca2+/CaM for the IQ domain.
Fig. 4. Characterization of CaM and CaBP1 binding to the CaV1.2 IQ domain mutant ‘TripleA’.

Exemplar ITC titrations for: a, 75 μM Ca2+ 2:C-lobeCaM into 7.5 μM TripleA. b, 75 μM Ca2+ 2:N-lobeCaM into 7.5 μM TripleA. c, 75 μM Ca2+ 4:CaM into 7.5 μM TripleA and 50 μM Ca2+ 2:C-lobeCaM. d, 75 μM Ca2+ 2:C-lobeCaBP1 into 7.5 μM TripleA. e, 75 μM Ca2+ 2:CaBP1 into 7.5 μM TripleA. f, 75 μM Ca2+ 2:N-lobeCaM into 7.5 μM TripleA. Icons depict ITC components. N-lobes and C-lobes and are green and blue, respectively. CaM and CaBP1 are in pastel and dark shades, respectively. White spheres represent Ca2+. CaV1.2 IQ domain is shown in red. Red star signifies the TripleA mutation.
Table 2.
Titration calorimetry data for interactions between CaM and CaBP1 with CaV1.2 IQ domain TripleA
| n | Setup | N | Kd (M) | ΔH (kcal mol−1) | ΔS (cal mol−1 K−1) | ΔG (kcal mol−1) | KdIQAAA / KdIQ | ΔΔ G (kcal mol−1) | |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| N-lobeCaBP1 | 2 | S | n/a | > 5 × 10−6 | n/a | n/a | > −6.9 | > 4.5 | < −0.9 |
| Ca2+/C-lobeCaBP1 | 2 | S | 0.93 ± 0.01 | 2.21 × 10−7 ± 1.4 × 10−8 | −1.45 ± 0.14 | 25.5 ± 0.6 | −8.65 ± 0.03 | 21 | −1.87 |
| Ca2+/CaBP1 | 2 | S | 0.89 ± 0.19 | 1.09 × 10−8 ± 2.0 × 10−9 | −4.77 ± 1.04 | 19.4 ± 4.0 | −10.35 ± 0.11 | 38 | −2.03 |
|
| |||||||||
| Ca2+/N-lobeCaM | 2 | S | n/a | > 5 × 10−6 | n/a | n/a | > −6.9 | > 90 | < −2.7 |
| Ca2+ /C-lobeCaM | 2 | S | 0.90 ± 0.01 | 1.54 × 10−9 ± 2.1 × 10−10 | −6.44 ± 0.13 | 18.0 ± 0.1 | −11.45 ± 0.08 | 0.6 | 0.14 |
| Ca2+/CaM | 2 | D | # | 1.7 × 10−10 ± 2.0 × 10−11 | −10.6 ± 0.4 | 7.4 ± 1.6 | −12.68 ± 0.07 | 200 | −3.00 |
ΔΔ G = RTln (KdIQAAA / KdIQ), where T = 288K
“#” indicates that N was set to 1.
“S” denotes a simple and “D” a displacement titration
‘n’ indicates the number of independent experiments.
“n/a” means not applicable.
The Ca2+/C-lobe binding sites for CaM and CaBP1 overlap23 but the detailed differences in the binding modes remain unclear. To test whether the strict separation of aromatic residues into N-lobe and C-lobe anchors also held true for CaBP1, we also measured the effect that the TripleA change had on the affinities of full-length Ca2+/CaBP1 and its isolated lobes. In contrast to the results with Ca2+/C-lobeCaM (Fig. 4a), the TripleA change lowered the affinity of Ca2+/C-lobeCaBP1 for the CaV1.2 IQ domain by 1.87 kcal mol−1 (Fig. 4d, Table 2), suggesting that at least some of the N-lobe anchors contribute to Ca2+/C-lobeCaBP1 binding. Similarly, the TripleA change reduced the affinities of both N-lobeCaBP1 (>5 μM, ΔΔG < −0.9 kcal mol−1) (Fig. 4e) and Ca2+/CaBP1 (221±14 nM, ΔΔG = 2.04 kcal mol−1) (Fig. 4f) compared to the wild-type CaV1.2 IQ domain23. The impact of the TripleA change on the affinity of both Ca2+/CaBP1 lobes stands in stark contrast to the results with the Ca2+/CaM lobes where the affinity of Ca2+/N-lobeCaM is reduced but that of Ca2+/C-lobeCaM is spared (Fig. 4a). Further, the strong impact on Ca2+/C-lobeCaBP1, which has a binding site that overlaps with Ca2+/C-lobeCaM 23, indicates that part of the Ca2+/C-lobeCaBP1 binding site includes some of the N-lobe anchors and provides evidence that the binding determinants for Ca2+/CaBP1 and Ca2+/CaM interactions with CaV1.2 IQ domain are not identical.
Functional competition between CaBP1 and CaM on CaV1.2
Given the biochemical evidence that CaM and CaBP1 bind to the CaV1.2 IQ domain in a mutually exclusive way (Fig. 1e)17; 19; 23, we wanted to test whether this biochemical competition could be recapitulated in a functional setting. We used two-electrode voltage clamp experiments in Xenopus oocytes, a system used previously to study CaBP1 function23; 47, to examine whether increasing CaM expression could overcome the functional effects of CaBP1 on CaV1.2. For these studies, we used the CaVβ2a subunit so that there would be a negligible contribution to channel inactivation from voltage-dependent inactivation (VDI)28. Thus, under our experimental conditions measurement of CaV1.2 inactivation when calcium is the permeant ion yields a direct readout of CDI.
As a first attempt to characterize the competition between CaM and CaBP1, we pursued experiments in which CaBP1 synthesis should happen contemporaneously with that of the channel. Due to endogenous CaM, CaV1.2 CDI in oocytes is robust6; 23; 30; 48. We had shown previously that even at a 60:1 excess of CaM:CaV1.2 mRNA, CaM expression did not further affect CDI as measured by the fraction of current decrease at 300 milliseconds (ti300)49. In contrast to these results, changes in CaBP1:CaV1.2 mRNA ratios caused clear alterations in ti300 and loss of CDI (Fig. 5a and b). Based on these experiments, we chose a 1:1 CaBP1:CaV1.2 mRNA ratio as the background for co-injection competition experiments with CaM mRNAs as this ratio achieved nearly full inhibition of CDI (ti300 = 19.3±5.2%) (Fig. 5a and b). Co-injection of 1:1 CaM:CaBP1 mRNA along with the mRNAs for the other channel components resulted in recovery of a small fraction of CDI (ti300 = 28.8±6.4%) (Fig. 5c and d). Increasing the CaM:CaBP1 ratio intensified CDI, which became nearly complete at a ratio of 30:1 CaM:CaBP1 mRNA (ti300 = 52.4±3.2%) (Fig. 5c and d). These experiments support the idea of competitive effects between CaM and CaBP1 for control of CDI.
Fig. 5. CaBP1 and CaM expressed in Xenopus oocytes compete for control of CaV1.2 function.

a, Representative normalized ICa traces at a test potential of +20 mV for Xenopus oocytes co-expressing CaV1.2 and CaBP1 at the indicated ratios. b, Averaged ti300 values from normalized ICa traces at a test potential of +20 mV for CaV1.2 expressed with CaBP1 at the indicated ratios. (n) indicates the number of experiments. c, Representative normalized ICa traces at a test potential of +20 mV for Xenopus oocytes co-expressing CaV1.2 and the indicated ratios of CaBP1 and CaM. d, Averaged ti300 values from normalized ICa traces at a test potential of +20 mV for CaV1.2 expressed with CaBP1 and CaM at the indicated ratio. In all experiments RNA for CaVβ2a and CaVα2δ-1 was injected at concentrations equimolar to CaV1.2. (n) indicates the number of experiments. Trace for 1:1 CaBP1:CaV1.2 in panel a and its corresponding analysis in panel b are reproduced in panels c and d and labeled CaM : CaBP1 0:1.
To test the CaM-CaBP1 competition directly and to exclude factors that might be related to differences in protein synthesis, we examined the effects on CaV1.2 CDI resulting from direct injection of purified CaBP1 and CaM protein. The proteins were injected together with 100 mM BAPTA, and thus, should be in the apo-forms and remain so until the local calcium concentration is raised via voltage-dependent CaV1.2 activation. Injection of CaBP1 at concentrations of 10 μM or higher into Xenopus oocytes expressing CaV1.2α1, CaVβ2a, and CaVα2δ-1 fifteen minutes prior to recording caused unmistakable inhibition of CDI that became nearly complete at 100 μM CaBP1 (ti300 = 6.6±2.2%)(Fig. 6a–c). In contrast, injection of 0.1 μM or 1 μM CaBP1, or buffer alone had no effect (ti300 = 52.9±4.7 %, 56.3±10.7%, and 61.2±5.4%, respectively) (Fig. 6a–c), respectively. This dramatic loss of CDI indicates that the injected CaBP1 competes with endogenous CaM, which is pre-associated with the channel38, for control of CDI.
Fig. 6. Purified CaBP1 and CaM injected into cells expressing CaV1.2 compete for control of channel function.

a, Representative normalized ICa traces at a test potential of +20 mV for Xenopus oocytes expressing CaV1.2 and injected with 50 nl of the indicated concentrations of purified CaBP1 15 minutes prior to recording. b, Averaged ti300 from normalized ICa traces at a test potential of +20 mV from Xenopus oocytes expressing CaV1.2 injected with 50 nl CaBP1 protein at the indicated concentration 15 minutes prior to recording. (n) indicates the number of experiments. c, Voltage-dependence of the averaged normalized current 300 ms after channel activation (100 − ti300) from oocytes expressing CaV1.2 and injected with purified CaBP1 at the following concentrations prior to recording: 0 μM, ■; 1 μM, ◆; 10 μM,
 ; 20 μM, ●; 100 μM, ◆. d, Representative normalized ICa traces at a test potential of +20 mV for Xenopus oocytes expressing CaV1.2 co-injected with the indicated concentrations of purified CaBP1 and CaM. e, Averaged ti300 from normalized ICa traces at a test potential of +20 mV from oocytes expressing CaV1.2 and injected with purified CaM and CaBP1 at the indicated ratios. (n) indicates the number of experiments. f, Voltage-dependence of the averaged normalized current 300 ms after channel activation (100 − ti300) from Xenopus oocytes expressing CaV1.2 and injected with purified CaM and CaBP1 1 at the following concentrations prior to recording: no CaM or CaBP1, ■; 20 μM CaBP1 only, ●; 20 μM CaBP1 and 2 μM CaM, □; 20 μM CaBP1 and 20 μM CaM, ◆; 20 μM CaBP1 and 200 μM CaM,
; 20 μM CaBP1 and 1 mM CaM, ◆. 20 μM CaBP1 trace in panel a and its corresponding analysis in panels in b and c are reproduced in panels d, e, and f and labeled as 0 μM CaM.
; 20 μM, ●; 100 μM, ◆. d, Representative normalized ICa traces at a test potential of +20 mV for Xenopus oocytes expressing CaV1.2 co-injected with the indicated concentrations of purified CaBP1 and CaM. e, Averaged ti300 from normalized ICa traces at a test potential of +20 mV from oocytes expressing CaV1.2 and injected with purified CaM and CaBP1 at the indicated ratios. (n) indicates the number of experiments. f, Voltage-dependence of the averaged normalized current 300 ms after channel activation (100 − ti300) from Xenopus oocytes expressing CaV1.2 and injected with purified CaM and CaBP1 1 at the following concentrations prior to recording: no CaM or CaBP1, ■; 20 μM CaBP1 only, ●; 20 μM CaBP1 and 2 μM CaM, □; 20 μM CaBP1 and 20 μM CaM, ◆; 20 μM CaBP1 and 200 μM CaM,
; 20 μM CaBP1 and 1 mM CaM, ◆. 20 μM CaBP1 trace in panel a and its corresponding analysis in panels in b and c are reproduced in panels d, e, and f and labeled as 0 μM CaM.
We next asked whether co-injection of purified CaM together with CaBP1 could antagonize the CDI loss caused by CaBP1. We chose 20 μM CaBP1 as a basis level for these experiments as this concentration was the minimum required to prevent most of CDI (ti300 = 15.3±3.2%) (Fig. 6). We found, in good agreement with the CaM and CaBP1 mRNA co-injection experiments (Fig. 5), that increasing the CaM:CaBP1 protein ratio caused a substantial recovery of CDI. This recovery became near complete when the CaM:CaBP1 ratio reached 50:1 (ti300 = 50.5±4.9%) (Fig. 6d–f). The reciprocal effects of CaBP1 on native CaM (Fig. 6a–c) and co-injected CaM on CaBP1 (Fig. 6d–f) directly demonstrate that there is competition between CaM and CaBP1 in the context of functional CaV channels in a live cell membrane. Notably, this competition occurs with either the myrisotylated form of CaBP1 (Fig. 5), which causes CaBP1 to localize to membranes50, or in the absence of this membrane anchor (Fig. 6).
Tethering CaM to CaV1.2 blocks the effects of CaBP1 on CDI
As a final test of the CaM:CaBP1 competition, we investigated the response to direct challenge by CaBP1 of a CaV1.2 construct bearing CaM that was covalently tethered to the IQ domain C-terminal end through a glycine-based linker (CaV1.2–CaMT). This unimolecular construct is similar to one described previously51 and should raise the CaM effective concentration to at least millimolar with respect to the IQ domain51. CaV1.2–CaMT showed voltage gating and CDI and properties that were similar to wild-type untethered channels, indicating that the tethering did not greatly alter function (ti300 = 54.6±6.2%) (Fig. 7 and Supplementary Fig. S2). However, one property was completed changed. Unlike wild-type channels for which injection of 100 μM CaBP1 prior to recording caused complete inhibition of CDI (Figs. 6a–c and 7a), CaMT channels were totally resistant to the effects of CaBP1 injection (ti300 = 52.0±8.2%, P=0.56 versus CaV1.2–CaMT without CaBP1 injection) (Fig. 7b). Because CaV1.2 channels bearing a tethered CaM mutant that is incapable of responding to calcium lack CDI51, we did not pursue competition experiments of such channels with CaBP1 as there would be no functional outcome to measure. Taken together with the protein competition results (Fig. 6 and 7a), the resistance of CaMT to the functional effects of CaBP1 provides unequivocal support for direct competition on the IQ domain between CaM and CaBP1 for control of CDI.
Fig. 7. CaV1.2-CaMT channel CDI resists CaBP1 inhibition.

a, Exemplar traces of CaV1.2 with and without saturating amounts of CaBP1. The diagrams indicate Ca2+ channel subunits, CaM and CaBP1. b, Exemplar traces of CaV1.2-CaMT with the same amounts of CaBP1 as in ‘a’.
ITC data predicts functional behavior of the apo-CaM/apo-CaBP1 competition
Although other sites have been implicated in influencing the effects of CaBP1 on channel function25; 26; 27; 28; 29, our studies suggest a simple mechanism in which competition between the apo forms of CaM and CaBP1 on the CaV IQ domain cause channels to have or lack CDI, respectively. In order to test this conceptual model in a quantitative manner, we used the ITC-derived affinities of the apo-state and Ca2+-bound forms of CaM and CaBP1 to derive the relationship between the CaM/CaBP1 ratio and the fraction of channels occupied by either the apo- or Ca2+-bound forms of CaM (Appendix 3) (Fig. 8). We then asked whether either curve predicts the competition measured by CDI levels from full-length functioning CaVs in living cells. Strikingly, the data from the independently measured CDI values observed from our protein competition experiments (Fig. 6d–e) match exceptionally well with the curve defined by the ITC data for apo-state competition.
Fig. 8. Functional outcomes of CaM/CaBP1 competition for CDI match predictions based on ITC measurements.
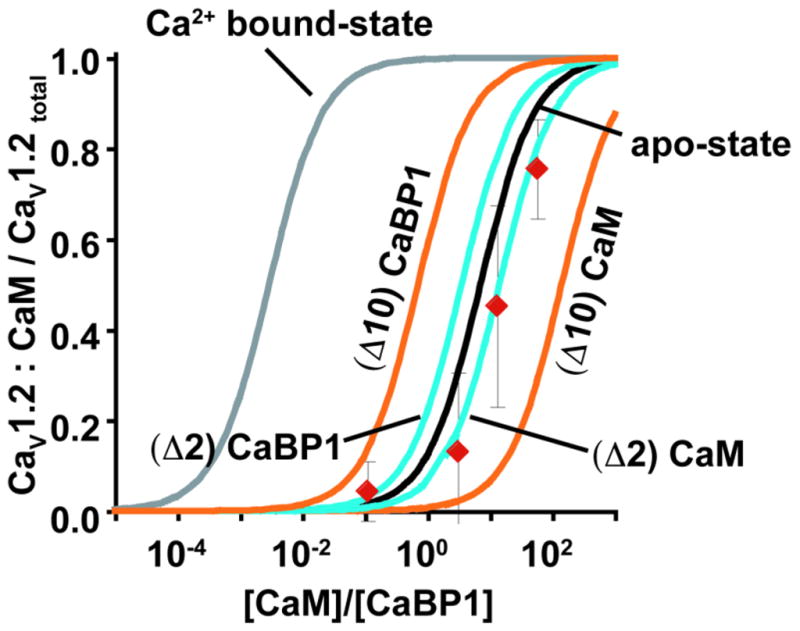
Predicted fraction of CaV1.2 channels bound to CaM as a function of [CaM]/[CaBP1] using values for the apo-state (black) and calcium-bound state (grey) IQ domain affinities measured by ITC (Table 1). Red diamonds show the fraction of CaM:CaV1.2 estimated from CDI measurements at varying ratios of injected CaM and CaBP1 protein. Teal curves show the prediction fraction of CaM-bound channels assuming that the Kd ratios of CaBP1/CaM apo-states differ from the measured values by a factor of 2 in favor of CaBP, (Δ2) CaBP1, or CaM, (Δ2) CaM. Orange curves are calculated assuming that the Kd ratios of CaBP1/CaM apo-states differ from the measured values by a factor of 10 in favor of CaBP, (Δ10) CaBP1, or CaM, (Δ10) CaM.
Even though there is a remarkably good agreement between the predicted CaM occupancy based on the in vitro ITC experiments and the functional competition measured on full-length channels in the membranes of living cells, other factors might affect the binding constants in the cellular setting. These might include CaM and CaBP1 binding sites located elsewhere on the channel, such as the CaV1.2 N-terminal cytoplasmic domain22; 27; 47; 52, or the differences in ionic composition between the in vitro ITC experiments and cell interior. To test the sensitivity of the mathematical model to Kd perturbations, we recalculated the relationship between the CaM/CaBP1 ratio and the fraction of channels occupied by either the apo-forms of CaM or CaBP1 in the case in which the Kds differed from the measured values by a factor of 2 (Fig. 8, teal curves) (i.e. instead of KdapoCaBP1/KdapoCaM = 7.1, KdapoCaBP1/KdapoCaM is either 3.55, a 2-fold advantage for CaBP1 relative to the measured values, or 14.2, a 2-fold advantage for CaM) or 10 (Fig. 8, orange curves) (i.e instead of KdapoCaBP1/KdapoCaM =7.1, KdapoCaBP1/KdapoCaM is either 0.71, a 10-fold advantage for CaBP1, or 71, a 10-fold advantage for CaM). These comparisons clearly show that perturbations as small as ten fold to the Kd in the setting of the cell would greatly disturb the correlation between the measured value and the predicted outcomes. These results strengthen the conclusion that the essence of the functional competition is captured by the reduced system comprising apo-CaM, apo-CaBP1, and the IQ domain and indicate that factors beyond the IQ domain have a minor role, at best, a result that agrees with the recently reported studies from Dascal and collegues53. Thus, a competition between the Ca2+-free forms of CaM and CaBP1 occurring on a single CaV1.2 site, the IQ domain, is sufficient to explain the mechanism by which CaBP1 changes CaV function.
Discussion
Calcium-dependent inactivation (CDI) is a key negative feedback mechanism that determines how much calcium enters excitable cells following CaV voltage-dependent activation3; 4; 9; 54. For CaV1.2 and CaV1.3 channels in some neurons in the brain and retina, this fundamental property can be altered dramatically by association of CaBP1 rather than CaM with the channel16; 17; 19; 22; 55. This change of calcium sensors has profound consequences for the frequency and duration of the CaV generated intracellular calcium signals4; 5, yet the exact mechanism by which this functional alteration occurs has been unclear.
Our studies establish that there is direct competition between the apo-state forms of CaM and CaBP1 for the CaV1.2 IQ domain and that this competition controls the ability of the channel to autoregulate through CDI. This competition can be quantitatively explained based on the in vitro derived ITC parameters for the interactions of the CaV1.2 IQ domain with CaM and CaBP1 (Fig. 8). The exceptionally close agreement between the in vitro and cell-based measurements strongly suggests that the biochemical properties of the CaV1.2 IQ domain in isolation accurately reflect the biologically relevant function of this portion of the channel. Moreover, perturbations to the Kds as small as ten-fold eliminate this agreement between prediction and experiment. This behavioral concordance, observed in the very different contexts of an isolated biochemical system and a full-length channel complex in a living cell, is consistent with the apparent structural independence of the CaV1.2 IQ domain from other parts of the CaV1.2 C-terminal tail26; 56. Interestingly, CaBP4, which is structurally similar to CaBP123 and is able to inhibit CaV1.3 CDI16; 17, has recently been shown to compete with CaM for the CaV1.4 IQ domain and block CDI57. These results to together with the close conservation of CaV1 IQ domain sequences8 strongly suggest that the competitive mechanism we define here is generally used by CaBP-CaV1 pairs.
Although apo-CaM is thought to be pre-associated with the channel complex through the IQ domain25, the details of this interaction have remained unclear. The excellent agreement we find between the in vitro ITC measurements and properties of the apo-CaM:CaV1.2 IQ domain interaction measured in living cells provides strong support for the idea that CaM is associated with the IQ domain under low calcium conditions25; 32. As the IQ domain forms a binding site having an exceptionally high affinity for Ca2+/CaM, the pre-positioning of CaM on this channel element suggests that even though its conformation may change, CaM will remain tethered to the IQ domain during the entire gating cycle.
The well documented ability of substrate interactions to change the affinity of CaM for calcium32; 40; 41; 43 prompted us to investigate the consequences of the sub-picomolar affinity of the Ca2+/CaM:CaV1.2 IQ domain complex. Thermodynamic linkage (Fig. 3a) dictates that the very high ratio of binding constants (680,000) between Ca2+/CaM and CaM for the CaV1.2 IQ domain must increase the calcium affinity of CaM in the IQ domain bound state. Investigation of a thermodynamic model indicates that because of the high order of the reaction, the individual binding constants of each EF hand for calcium do not need to change dramatically (only ~30-fold) in order compensate for large affinity change in the complex. Hence, a large gain in the protein-protein interaction can be achieved without a change of equivalent magnitude in calcium affinity by virtue of the multiple, coupled calcium binding sites on CaM.
The affinity of isolated CaM for calcium is in the micromolar range42, which is outside of the response range of many Ca2+/CaM dependent proteins complexes43. Association of CaM with the CaV1.2 IQ domain tunes the calcium response by ~30-fold into a range, 0.3–10 μM, that allows IQ domain bound CaM to sense calcium concentrations that are much lower than expected based on free CaM and that match well with those expected for intracellular calcium changes2; 43; 44. This situation reinforces a general principle whereby CaM-substrate interactions tune the calcium dependent response of CaM into a physiologically relevant range41; 43. Similar analysis indicates that the reported higher intrinsic affinity of CaBP1 for Ca2+ relative to CaM37 combined with the impact of association with the CaV1.2 IQ domain yields a complex that has a calcium affinity tuned near the lower limit for intracellular calcium levels2; 43; 44. This results suggests that a CaBP1:CaV1.2 IQ domain complex could be responsive to changes in Ca2+ levels; however, as prior studies have demonstrated that CaBP1 does not require functional EF hands to inhibit CaV1.2 CDI23, such a response is not central to the ability of CaBP1 to block CDI.
There is a strong structural similarity between the Ca2+/C-lobes of CaBP1 and CaM23. Although previous studies17; 19; 23 suggest that the Ca2+/CaBP1 and Ca2+/CaM binding sites on the IQ domain overlap, the binding modes are thought to differ23. We show that in contrast to Ca2+/CaM binding, which meets the expectations set by the structure of the Ca2+/CaM:CaV1.2 IQ domain complex30 in which Ca2+/N-lobe and Ca2+/C-lobe interactions are separated into two sets of aromatic anchors, some determinants of the Ca2+/N-lobeCaM site are used by Ca2+/C-lobeCaBP1. These observations further support the notion that despite their common Ca2+/C-lobe architectures23, Ca2+/CaBP1 engages the CaV1.2 IQ domain in a manner that differs from Ca2+/CaM.
Our studies establish that there is direct competition between the apo-state forms of CaM and CaBP1 for the CaV1.2 IQ domain and that this simple mechanism switches the CDI properties of the channel. The demonstration that CaBP1 or CaM can be chased from channels in live cell membranes with the complementary calcium sensor (Figs. 5, 6, and 8) suggests that such competitive mechanisms may be used in vivo for dynamic control of CaV function in cells expressing CaBP1, such as hippocampal CA3 neurons19. In this regard, CaBP1 myristoylation, which localizes CaBP1 to membranes50 but is not involved in CDI inhibition23, could aid the ability of CaBP1 to compete at concentrations lower than those tested here for the non-myristoylated form (Fig. 6). CaV1 channels have a privileged role in excitation-transcription coupling5; 58; hence, changes in the net calcium flux caused by substitution of CaM by CaBP1 could induce long-term effects in neuronal function. Finally, our demonstration that calcium sensors can be exchanged on pre-assembled CaVs suggests that this property may enable the introduction of components bearing novel chemical reactivity to probe the conformational changes that underlie CDI.
Materials and Methods
Expression and Purification
Expression and purification of CaBP1 Δ2–15 and CaM, their individual lobes, and the CaV1.2 IQ domain were as described previously23; 30. The CaV1.2 IQ domain TripleA mutant (CaV1.2 F1618A, F1619A, Y1622A) was purified using the same procedure as for the wild-type CaV1.2 IQ domain23; 30. CaM and CaBP1 lobes with D→A mutations at the first positions of the EF-hand consensus motif indicated were expressed using procedures similar to those used for the wild-type counterparts23; 30. Full-length CaM and CaBP1 with inactivated EF-hands were expressed and purified similarly to the single lobes23.
Isothermal titration calorimetry
Titrations were performed at 15°C using a VP-ITC Microcalorimeter (MicroCal). Samples were dialyzed overnight at 4°C (Slide-A-Lyzer, 2 kDa molecular weight cut-off, Thermo Scientific) against appropriate buffers. Titrations were conducted in 5 mM KCl, 1 mM CaCl2, 10 mM Hepes, pH 7.4. After centrifugation at 40,000 rpm for 30 min at 4 °C, protein concentration was determined by absorbance at 280 nm59. In cases where precipitation at high concentrations of the syringe component precluded direct titration experiments, such as the EF hand mutants, we used a displacement ITC strategy23; 31. All samples were degassed for 5 minutes prior to loading into the calorimeter. Each ITC experiment consisted of one 4 μl injection followed by 29 injections of 10 μl titrant. To correct the baseline either heat of dilution from titrations of injectant into buffer was subtracted or the final titration points were used to estimate the baseline. Data were processed with MicroCal Origin 7.0 using the binding models indicated in the main text.
Electrophysiology
Human CaV1.2 (α1C77, GenBank CAA84346), rat CaVβ2a (GenBank NP 446303), and CaVα2δ -1(GenBank NM_00182276), co-expressed with either Homo sapiens CaM (GenBank NM_006888) or the short isoform of Homo sapiens CaBP1 (GenBank AF169148), which bears a myristoylation site, were used for two-electrode voltage clamp experiments in Xenopus oocytes. Details of constructs and two-electrode voltage clamp have been described previously23. In short, linearized cDNA was translated into capped mRNA using the T7 mMessenger kit (Ambion). 50 nl of CaVα1, CaVβ, CaVα2δ -1, and CaBP1 or CaM mRNA at the molar ratio indicated were injected into stage VI Xenopus oocytes. For experiments that involved protein injections into oocytes, equimolar mRNA of CaVα1, CaVβ, and CaVα2δ;-1 were injected 48 hours prior to recording. 15 minutes before recording, 50 nl of a mixture of 0.1M BAPTA and the test proteins at the indicated concentrations were injected. The CaV1.2 tethered CaM construct (CaV1.2-CaMT) consists of residues 1–1644 of human CaV1.2, followed by a six residue linker (TGGGGG) and residues 1–147 of human CaM. This linker, together with the six disordered residues from the Ca2+ 4:CaM N-terminus and CaV1.2 IQ C-terminus30, provides sufficient length to span the required distance in a manner compatible with the structure.
Two-electrode voltage-clamp experiments were performed 2 to 3 days post-injection. Oocytes were injected with 50 nl of 100 mM BAPTA four minutes before recording unless stated otherwise to minimize calcium-activated chloride currents. Recording solutions contained 40 mM Ca(NO3)2, 50 mM NaOH, 1 mM KOH, and 10 mM HEPES, adjusted to pH 7.4 using HNO3. Electrodes were filled with 3M KCl and had resistances of 0.3–2.0 MΩ. CDI was measured using 450 ms depolarizations from a −90 mV holding potential to test potentials of −50 to +50 mV in 10 mV steps. Consecutive pulses were separated by 15 s. Leak currents were subtracted using a P/4 protocol. Currents were analyzed with Clampfit 8.2 (Axon Instruments). All results are from at least two independent oocyte batches. The ti300 values were calculated from normalized currents at +20 mV and represent the percentage inactivation after 300 milliseconds.
Supplementary Material
Calmodulin (CaM) and CaBP1 differentially control voltage-gated calcium channels
CaM and CaBP1 apo-state competition on the CaV IQ domain directly controls function
In vitro binding constants predict apo-CaM/apo-CaBP1 competition in live cells
Ca2+/CaM has a sub-picomolar affinity for the IQ domain
Exchange of apo-CaM and apo-CaBP1 provides dynamic modulation of channel behavior
Acknowledgments
This work was supported by grants to D.L.M. from NIH R01 HL080050 and the American Heart Association 0740019N. We thank R. Aldrich and M. Grabe for critical comments on the manuscript, and members of the Minor lab for support throughout these studies. D.L.M. is an AHA Established Investigator.
Abbreviations
- CaM
calmodulin
- Ca2+/CaM
Ca2+ 4:CaM form of CaM
- Ca2+/N-lobeCaM
Ca2+ 2:N-lobeCaM form of CaM N-terminal lobe
- Ca2+/C-lobeCaMCa2+2
C-lobeCaM form of CaM C-terminal lobe
- CaBP1
calcium binding protein 1
- Ca2+/CaBP1
Ca2+ 2:CaBP1 form of CaBP1
- Ca2+/C-lobeCaBP1
Ca2+ 2:C-lobeCaBP1 form of CaBP1 C-terminal lobe
- C-lobeCaBP1EF34 and Ca2+/C-lobeCaBP1
CaMEF1234 and CaBP1EF34, apo-mimics of full length CaM and CaBP1, respectively
- N-lobeCaMEF12 and C-lobeCaMEF34
individual apo-CaM lobe mimics
- C-lobeCaBP1EF34
apo-CaBP1 C-lobe mimic
Appendix 1
We modeled the system of CaM, calcium, and the IQ domain using a thermodynamic cycle (Fig. 3a) governed by the following equations:
| (1) |
| (2) |
| (3) |
| (4) |
Conservation of free energy dictates that the free energy difference of Ca2+ binding is as follows:
| (5) |
Consequently, knowing the free energy values of the individual binding reactions for the calcium-bound and calcium-free forms of CaM with the IQ domain, ΔG2 and ΔG3, dictates the difference in calcium affinity (ΔΔGCaM) for reactions ΔG1 and ΔG4.
The values of ΔG2 and ΔG3 are:
| ( Fig. 2 and Table 1) | (6) |
| ( Fig. 1 and Table 1) | (7) |
Thus, combining equations (5) with (6) and (7) yields the free energy difference corresponding to:
| (8) |
ΔΔG comprises the individual Ca2+ affinities of the four EF hands rather than a single binding reaction. Making the simplifying assumption that all CaM EF-hands are affected equally by binding of the IQ domain, results in each EF-hand free energy difference altered by 1.90 kcal mol−1, one fourth of the total free energy difference of 7.58 kcal mol−1, at 288K, an increase of 28.8 fold in the calcium affinity of each individual EF-hand. Although the CaM EF-hands may not be affected equally, in the context of the limits set by the thermodynamic cycle (Fig. 3a) any unequal changes in EF-hand affinity would result in only modest changes in relation shown in Fig. 3b.
Appendix 2
We modeled the system of CaBP1, calcium, and the IQ domain using a thermodynamic cycle (Supplementary Figure S1), using an equivalent analysis to that presented in Appendix 1 for CaM as follows:
| (9) |
| (10) |
| (11) |
| (12) |
For CaBP1, the values of ΔG2 and ΔG3 are:
| ( Table 1 and Figure 2) | (13) |
| ( Table 1 and ref. 23) | (14) |
Thus, combining equations (5) with (18) and (19) yields an difference of free energy corresponding to:
| (15) |
Making the simplifying assumption that both functional CaBP1 EF-hands are affected equally by binding of the IQ domain specifies that each EF-hand free energy difference altered by 1.62 kcal mol−1, half of 3.23 kcal mol−1, equivalent to in increase of 17.5 fold in the calcium affinity of each individual EF-hand.
Appendix 3
The ITC data (Figure 1 and Table 1) dictates the following relationships for the calcium-bound forms of CaM and CaBP1:
| (27) |
and
| (28) |
| (29) |
Rearranging this equation to yield the ratio between the IQ domains occupied by the two Ca2+ sensors yields:
| (30) |
which can be solved for any given ratio of calmodulin and CaBP1.
We assume that competition between CaBP1 and CaM occurs exclusively on the CaV1.2 IQ domain, allowing us to substitute the full-length channel for the IQ domain.
| (31) |
This assumption ignores the possible influence of other sites on CaV1.2 that have been shown to have affinity for both CaM 25; 27 and CaBP1 19; 22. Notably, such sites have a >5-fold weaker affinity than the CaV1.2 IQ domain for CaM 32; 52.
We furthermore assume that there is always one calcium sensor bound to the CaV1.2 IQ domain, i.e.
| (32) |
For (32), given that without CaM, CaV1.2 trafficking to the plasma membrane is heavily compromised 60, we assume that the amount of CaV1.2 the plasma membrane of the test Xenopus oocyte lacking either calcium sensor is negligible.
Similar analysis for the apo state values (Figure 2 and Table 1) yields the following equations:
| (33) |
and
| (34) |
Equations (31) and (34) can be used to derive the relationship between the CaM/CaBP1 ratio and the fraction of the channel occupied by CaM in high (Fig. 8, grey curve) and low Ca2+ (Fig. 8, black curve).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–55. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 2.Clapham DE. Calcium signaling. Cell. 2007;131:1047–58. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 3.Dunlap K. Calcium channels are models of self-control. J Gen Physiol. 2007;129:379–83. doi: 10.1085/jgp.200709786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christel C, Lee A. Ca(2+)-dependent modulation of voltage-gated Ca(2+) channels. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbagen.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW. Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell. 2012;149:1112–24. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–62. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 7.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–58. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 8.Van Petegem F, Minor DL. The structural biology of voltage-gated calcium channel function and regulation. Biochem Soc Trans. 2006;34:887–93. doi: 10.1042/BST0340887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Findeisen F, Minor DL., Jr Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels (Austin) 2010;4:459–74. doi: 10.4161/chan.4.6.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dolphin AC. beta subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003;35:599–620. doi: 10.1023/b:jobb.0000008026.37790.5a. [DOI] [PubMed] [Google Scholar]
- 11.Buraei Z, Yang J. The {beta} Subunit of Voltage-Gated Ca2+ Channels. Physiol Rev. 2010;90:1461–506. doi: 10.1152/physrev.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies A, Hendrich J, Van Minh AT, Wratten J, Douglas L, Dolphin AC. Functional biology of the alpha(2)delta subunits of voltage-gated calcium channels. Trends Pharmacol Sci. 2007;28:220–8. doi: 10.1016/j.tips.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Pitt GS. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc Res. 2007;73:641–7. doi: 10.1016/j.cardiores.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 14.McCue HV, Haynes LP, Burgoyne RD. The diversity of calcium sensor proteins in the regulation of neuronal function. Cold Spring Harb Perspect Biol. 2010;2:a004085. doi: 10.1101/cshperspect.a004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haeseleer F, Sokal I, Verlinde CL, Erdjument-Bromage H, Tempst P, Pronin AN, Benovic JL, Fariss RN, Palczewski K. Five members of a novel Ca(2+)-binding protein (CABP) subfamily with similarity to calmodulin. J Biol Chem. 2000;275:1247–60. doi: 10.1074/jbc.275.2.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui G, Meyer AC, Calin-Jageman I, Neef J, Haeseleer F, Moser T, Lee A. Ca2+-binding proteins tune Ca2+-feedback to Cav1.3 channels in mouse auditory hair cells. J Physiol. 2007;585:791–803. doi: 10.1113/jphysiol.2007.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang PS, Alseikhan BA, Hiel H, Grant L, Mori MX, Yang W, Fuchs PA, Yue DT. Switching of Ca2+-dependent inactivation of Ca(v)1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci. 2006;26:10677–89. doi: 10.1523/JNEUROSCI.3236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee A, Westenbroek RE, Haeseleer F, Palczewski K, Scheuer T, Catterall WA. Differential modulation of Ca(v)2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci. 2002;5:210–7. doi: 10.1038/nn805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou H, Kim SA, Kirk EA, Tippens AL, Sun H, Haeseleer F, Lee A. Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-type) Ca2+ channels. J Neurosci. 2004;24:4698–708. doi: 10.1523/JNEUROSCI.5523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Few AP, Lautermilch NJ, Westenbroek RE, Scheuer T, Catterall WA. Differential regulation of CaV2.1 channels by calcium-binding protein 1 and visinin-like protein-2 requires N-terminal myristoylation. J Neurosci. 2005;25:7071–80. doi: 10.1523/JNEUROSCI.0452-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lautermilch NJ, Few AP, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by the neuronal calcium-binding protein visinin-like protein-2. J Neurosci. 2005;25:7062–70. doi: 10.1523/JNEUROSCI.0447-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J Biol Chem. 2005;280:29612–9. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]
- 23.Findeisen F, Minor DL., Jr Structural basis for the differential effects of CaBP1 and calmodulin on Ca(V)1.2 calcium-dependent inactivation. Structure. 2010;18:1617–31. doi: 10.1016/j.str.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li C, Chan J, Haeseleer F, Mikoshiba K, Palczewski K, Ikura M, Ames JB. Structural insights into Ca2+-dependent regulation of inositol 1,4,5-trisphosphate receptors by CaBP1. J Biol Chem. 2009;284:2472–81. doi: 10.1074/jbc.M806513200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- 26.Kim EY, Rumpf CH, Van Petegem F, Arant RJ, Findeisen F, Cooley ES, Isacoff EY, Minor DL., Jr Multiple C-terminal tail Ca(2+)/CaMs regulate Ca(V)1.2 function but do not mediate channel dimerization. Embo J. 2010;29:3924–38. doi: 10.1038/emboj.2010.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dick IE, Tadross MR, Liang H, Tay LH, Yang W, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–4. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Findeisen F, Minor DL., Jr Disruption of the IS6-AID Linker Affects Voltage-gated Calcium Channel Inactivation and Facilitation. J Gen Physiol. 2009;133:327–43. doi: 10.1085/jgp.200810143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ravindran A, Lao QZ, Harry JB, Abrahimi P, Kobrinsky E, Soldatov NM. Calmodulin-dependent gating of Ca(v)1.2 calcium channels in the absence of Ca(v)beta subunits. Proc Natl Acad Sci U S A. 2008;105:8154–9. doi: 10.1073/pnas.0711624105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Petegem F, Chatelain FC, Minor DL., Jr Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat Struct Mol Biol. 2005;12:1108–15. doi: 10.1038/nsmb1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sigurskjold BW. Exact analysis of competition ligand binding by displacement isothermal titration calorimetry. Anal Biochem. 2000;277:260–6. doi: 10.1006/abio.1999.4402. [DOI] [PubMed] [Google Scholar]
- 32.Evans TI, Hell JW, Shea MA. Thermodynamic linkage between calmodulin domains binding calcium and contiguous sites in the C-terminal tail of Ca(V)1.2. Biophys Chem. 2011;159:172–87. doi: 10.1016/j.bpc.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fallon JL, Halling DB, Hamilton SL, Quiocho FA. Structure of calmodulin bound to the hydrophobic IQ domain of the cardiac Ca(v)1.2 calcium channel. Structure. 2005;13:1881–6. doi: 10.1016/j.str.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 34.Tse JK, Giannetti AM, Bradshaw JM. Thermodynamics of calmodulin trapping by Ca2+/calmodulin-dependent protein kinase II: subpicomolar Kd determined using competition titration calorimetry. Biochemistry. 2007;46:4017–27. doi: 10.1021/bi700013y. [DOI] [PubMed] [Google Scholar]
- 35.Sarhan MF, Tung CC, Van Petegem F, Ahern CA. Crystallographic basis for calcium regulation of sodium channels. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3558–3563. doi: 10.1073/pnas.1114748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarhan MF, Van Petegem F, Ahern CA. A Double Tyrosine Motif in the Cardiac Sodium Channel Domain III–IV Linker Couples Calcium-dependent Calmodulin Binding to Inactivation Gating. Journal of Biological Chemistry. 2009;284:33265–33274. doi: 10.1074/jbc.M109.052910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wingard JN, Chan J, Bosanac I, Haeseleer F, Palczewski K, Ikura M, Ames JB. Structural analysis of Mg2+ and Ca2+ binding to CaBP1, a neuron-specific regulator of calcium channels. J Biol Chem. 2005;280:37461–70. doi: 10.1074/jbc.M508541200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron. 2001;31:973–85. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- 39.Tang W, Halling DB, Black DJ, Pate P, Zhang JZ, Pedersen S, Altschuld RA, Hamilton SL. Apocalmodulin and Ca2+ calmodulin-binding sites on the CaV1.2 channel. Biophys J. 2003;85:1538–47. doi: 10.1016/s0006-3495(03)74586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milos M, Schaer JJ, Comte M, Cox JA. Microcalorimetric investigation of the interactions in the ternary complex calmodulin-calcium-melittin. J Biol Chem. 1987;262:2746–9. [PubMed] [Google Scholar]
- 41.Zhang M, Abrams C, Wang LP, Gizzi A, He LP, Lin RH, Chen Y, Loll PJ, Pascal JM, Zhang JF. Structural Basis for Calmodulin as a Dynamic Calcium Sensor. Structure. 2012;20:911–923. doi: 10.1016/j.str.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linse S, Helmersson A, Forsen S. Calcium binding to calmodulin and its globular domains. J Biol Chem. 1991;266:8050–4. [PubMed] [Google Scholar]
- 43.Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6:267–76. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- 44.Hille B. Ion Channels of Excitable Membranes. 3. Sinauer Associates, Inc; Sunderland, MA: 2001. [Google Scholar]
- 45.Chao LH, Stratton MM, Lee IH, Rosenberg OS, Levitz J, Mandell DJ, Kortemme T, Groves JT, Schulman H, Kuriyan J. A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin- dependent kinase II holoenzyme. Cell. 2011;146:732–45. doi: 10.1016/j.cell.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiba H, Schneider NS, Matsuoka S, Noma A. A simulation study on the activation of cardiac CaMKII delta-isoform and its regulation by phosphatases. Biophys J. 2008;95:2139–49. doi: 10.1529/biophysj.107.118505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oz S, Tsemakhovich V, Christel CJ, Lee A, Dascal N. CaBP1 regulates voltage-dependent inactivation and activation of Ca(V)1.2 (L-type) calcium channels. J Biol Chem. 2011;286:13945–53. doi: 10.1074/jbc.M110.198424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ivanina T, Blumenstein Y, Shistik E, Barzilai R, Dascal N. Modulation of L-type Ca2+ channels by gbeta gamma and calmodulin via interactions with N and C termini of alpha 1C. J Biol Chem. 2000;275:39846–54. doi: 10.1074/jbc.M005881200. [DOI] [PubMed] [Google Scholar]
- 49.Findeisen F, Tolia A, Arant R, Kim EY, Isacoff E, Minor DL., Jr Calmodulin overexpression does not alter Cav1.2 function or oligomerization state. Channels (Austin) 2011;5:320–4. doi: 10.4161/chan.5.4.16821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haynes LP, Tepikin AV, Burgoyne RD. Calcium-binding protein 1 is an inhibitor of agonist-evoked, inositol 1,4,5-trisphosphate-mediated calcium signaling. J Biol Chem. 2004;279:547–55. doi: 10.1074/jbc.M309617200. [DOI] [PubMed] [Google Scholar]
- 51.Mori MX, Erickson MG, Yue DT. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science. 2004;304:432–5. doi: 10.1126/science.1093490. [DOI] [PubMed] [Google Scholar]
- 52.Benmocha A, Almagor L, Oz S, Hirsch JA, Dascal N. Characterization of the calmodulin-binding site in the N terminus of CaV1.2. Channels (Austin) 2009;3:337–42. doi: 10.4161/chan.3.5.9686. [DOI] [PubMed] [Google Scholar]
- 53.Oz S, Benmocha A, Sasson Y, Sachyani D, Almagor L, Lee A, Hirsch JA, Dascal N. Competitive and non-competitive regulation of calcium-dependent inactivation in CaV1.2 L-type Ca2+ channels by calmodulin and Ca2+-binding protein 1. J Biol Chem. 2013 doi: 10.1074/jbc.M113.460949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halling DB, Aracena-Parks P, Hamilton SL. Regulation of voltage-gated Ca2+ channels by calmodulin. Sci STKE. 2006;2006:er1. doi: 10.1126/stke.3182006er1. [DOI] [PubMed] [Google Scholar]
- 55.Haeseleer F, Imanishi Y, Maeda T, Possin DE, Maeda A, Lee A, Rieke F, Palczewski K. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci. 2004;7:1079–87. doi: 10.1038/nn1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fallon JL, Baker MR, Xiong L, Loy RE, Yang G, Dirksen RT, Hamilton SL, Quiocho FA. Crystal structure of dimeric cardiac L-type calcium channel regulatory domains bridged by Ca2+* calmodulins. Proc Natl Acad Sci U S A. 2009;106:5135–40. doi: 10.1073/pnas.0807487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaltiel L, Paparizos C, Fenske S, Hassan S, Gruner C, Rotzer K, Biel M, Wahl-Schott CA. Complex Regulation of Voltage-dependent Activation and Inactivation Properties of Retinal Voltage-gated Cav1.4 L-type Ca2+ Channels by Ca2+-binding Protein 4 (CaBP4) J Biol Chem. 2012;287:36312–21. doi: 10.1074/jbc.M112.392811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murphy TH, Worley PF, Baraban JM. L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron. 1991;7:625–35. doi: 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- 59.Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–54. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 60.Wang HG, George MS, Kim J, Wang C, Pitt GS. Ca2+/calmodulin regulates trafficking of Ca(V)1.2 Ca2+ channels in cultured hippocampal neurons. J Neurosci. 2007;27:9086–93. doi: 10.1523/JNEUROSCI.1720-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.