

Abstract
ATP-sensitive K+ (KATP) channels are abundant membrane proteins in cardiac myocytes that are directly gated by intracellular ATP and form a signaling complex with metabolic enzymes, such as creatine kinase. KATP channels are known to be essential for adaption to cardiac stress, such as ischemia; however, how all the molecular components of the stress response interact is not fully understood. We examined the effects of decreasing the KATP current density on Ca2+ and mitochondrial homeostasis and ischemic preconditioning. Acute knockdown of the pore-forming subunit, Kir6.2, was achieved using adenoviral delivery of short hairpin RNA targeted to Kir6.2. The acute nature of the knockdown of Kir6.2 accurately shows the effects of Kir6.2 depletion without any compensatory effects that may arise in transgenic studies. We also investigated the effect of reducing the KATP current while maintaining KATP channel protein in the sarcolemmal membrane using a nonconducting Kir6.2 construct. Only 50% KATP current remained after Kir6.2 knockdown, yet there were profound effects on myocyte responses to metabolic stress. Kir6.2 was essential for cardiac myocyte Ca2+ homeostasis under both baseline conditions before any metabolic stress and after metabolic stress. Expression of nonconducting Kir6.2 also resulted in increased Ca2+ overload, showing the importance of K+ conductance in the protective response. Both ischemic preconditioning and protection during ischemia were lost when Kir6.2 was knocked down. KATP current density was also important for the mitochondrial membrane potential at rest and prevented mitochondrial membrane potential oscillations during oxidative stress. KATP channel density is important for adaption to metabolic stress.
Keywords: ATP-sensitive K+ channels, calcium, cardioprotection, ischemia-reperfusion injury, mitochondrial permeability transition
the cardiovascular system operates under a broad range of demands, varying between periods of rest to bursts of intense exercise. During ischemic heart disease, the cardiac muscle experiences an extreme and pathological metabolic challenge. There is exquisite control of mitochondrial metabolism so that the ATP supply is balanced for the wide range of cardiac workloads. Indeed, any mismatch in ATP supply and demand can have fatal consequences (4). The maintenance of cardiac energetic homeostasis depends on the cellular responses to metabolic stress. ATP-sensitive K+ (KATP) channels have a critical role in maintaining cellular energetic homeostasis in physiological conditions, and their role is highlighted in the pathological setting of ischemic heart disease (53). KATP channels are the most abundantly expressed membrane proteins in cardiac muscle (13). In ventricular myocytes, where they were first discovered (36), octomeric sarcolemmal KATP channels predominantly comprise four pore-forming Kir6.2 subunits and four sulphonylurea receptor (SUR)2A subunits (40).
The damage to cardiac muscle by ischemia-reperfusion injury is caused, in part, by Ca2+ overload (38). KATP channels play an adaptive role during metabolic stress by opening when cellular ATP levels are low, as in ischemia (34). This hyperpolarizes the sarcolemmal membrane potential in an attempt to prevent Ca2+ overload (3, 30). High intracellular Ca2+ is deleterious to ventricular myocytes because in addition to preventing correct contractile function, it can trigger opening of the mitochondrial permeability transition pore (mPTP), which is an irreversible step toward myocyte death particularly during ischemia-reperfusion injury (16, 23).
Ischemic preconditioning (IPC) is a cardioprotective phenomenon in which short periods of ischemia reduce the impact of a prolonged ischemic event (33). KATP channels have also been proposed to play a key role in this intrinsic protective mechanism of the heart. KATP channel openers have been shown to mimic the protective effects of IPC (51), whereas KATP channel blockers ablated the effects of preconditioning (21).
The importance of KATP channels in adaptation to stress has also been highlighted in a number of transgenic studies. IPC is lost in mice lacking functional KATP channels (22). Furthermore, Kir6.2 knockout mice develop ventricular arrhythmia and sudden death because they have lost their ability to adapt to exercise or sympathetic challenge (54). Although the ATP sensitivity of KATP channels underlies their protective function during ischemia, stressed hearts of Kir6.2 knockout and control mice do not show a decrease of total cellular ATP concentration (54), suggesting that there may be other mechanisms that account for the role of KATP channels in cardioprotection. The full extent of the role of KATP channels may not be revealed by transgenic studies due to compensatory effects that are known to occur since the Kir6.2 knockout adult mouse shows only mild impairment of glucose homeostasis despite being predicted to be diabetic (37).
Sarcolemmal KATP channels are sensitive to mitochondrial ATP production through signaling complexes with metabolic enzymes important for cellular energy homeostasis, including adenylate kinase and creatine kinase (1, 27, 41). This creates a phosphotransfer bridge from the sarcolemmal membrane to the mitochondria, which allows metabolic fine tuning of excitability for the wide range of physiological workloads. How these signaling complexes influence KATP channel opening during ischemia is not fully understood. The aim of this study was to investigate the contribution of K+ flux and the signaling complex on Ca2+ and mitochondrial homeostasis.
Gene knockout and transgenic approaches are powerful tools but have some significant limitations. In addition to genetic background-dependent variations in phenotype, developmental compensation has the potential to obscure or distort “true” phenotypes. This is illustrated by the remodeling of excitation-contraction coupling seen in mice expressing an ATP-insensitive KATP channel (12). The function of KATP channels in ischemia remains to be unequivocally explained. It is estimated that opening of as few as 1% of KATP channels would be sufficient to shorten action potential duration and thus be protective, suggesting considerable redundancy, yet increased channel expression has been reported to be cardioprotective (9).
In this study, we used we used RNA interference to probe the effects of acute reductions in channel expression and investigate the role of the KATP channel subunit Kir6.2 in IPC and Ca2+ homeostasis. In addition, dominant negative, nonconducting Kir6.2 was expressed to distinguish between the roles of KATP channels in ion conduction or as part of a metabolic signaling complex. The effect of decreasing KATP channel density on mitochondrial function, particularly mPTP opening, was also investigated.
MATERIALS AND METHODS
Adenovirus construction.
Recombinant adenovirus encoding two separate rat RNA interfering short hairpin sequences [short hairpin (sh)RNA] for knockdown of Kir6.2 were generated using pAdEasy (25).
Cassettes expressing shRNAs were created by ligating the following oligonucleotides into pSilencer adeno 1.0 CMV (Ambion): 6.2 shRNA-A, 5′-TCGAGGAAAGGCAACTGCAACGTCTTCAAGAGAGACGTTGCAGTTGCCTTTCTTA-3′ and 5′-CTAGTAAGAAAGGCAACTGCAACGTCTCTCTTGAAGACGTTGCAGTTGCCTTTCC-3′; and 6.2 shRNA-B, 5′-TCGAGTTCGGGAACACCGTTAAAGTTCAAGAGACTTTAACGGTGTTCCCGAATTA-3′ and 5′-CTAGTAATTCGGGAACACCGTTAAAGTCTCTTGAACTTTAACGGTGTTCCCGAAC-3′.
The control shRNA sequence (Ambion) was a nontargeting shRNA with limited sequence similarity to known genes in the rat. For the nonconducting Kir6.2 construct, pore loop mutated (GFG to AFA) dominant negative Kir6.2 (29) cDNA was generated by overlap PCR and cloned into pIRES2-DsRed2 (Clontech). An insert including the mutant Kir6.2, IRES, and DsRed2 sequences was subcloned into pENTR-1A (Invitrogen) and recombined with pAd-CMV-DEST (Invitrogen) to produce recombinant adenovirus.
These cassettes included a modified cytomegalovirus promoter, shRNA insert, and polyA terminator and were subcloned into the pAdTrack shuttle vector and verified by sequencing. Viral genomes were assembled by transformation of electrocompetent Esherichia coli BJ5183 carrying pAd-easy1 with Pme1 linearized shuttle vector constructs. Appropriately recombined isolates were propagated in DH5α. Human embryonic kidney-293 cells were used for adenoviral amplification. Ventricular myocytes were infected with the adenovirus in supplement-free medium 199 immediately after isolation. After 3 h of incubation with adenovirus, media were replaced with supplemented medium 199, and myocytes were cultured for 24 h. This resulted in >90% efficiency gene transfer as detected by green fluorescent protein (GFP) fluorescence.
Isolation and culture of single ventricular myocytes.
Adult male Wistar rats (250–300 g) were killed by cervical dislocation, and single ventricular myocytes were isolated by enzymatic dissociation as previously described (38). All experiments had the required UK Home Office approval as designated by the Animals (Scientific Procedures) Act of 1986. This investigation also conformed with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85–23, Revised 1996). After isolation, myocytes were kept in medium 199 supplemented with ITS+3 [Sigma, which contains insulin (0.02 mg/ml), transferrin (0.01 mg/ml), sodium selenite (0.01 μg/ml), linoleic acid (9.4 μg/ml), oleic acid (9.4 μg/ml), and BSA (1 mg/ml)] as well as sodium pyruvate (1 mg/ml), triiodo-l-thyronine (10 nM), l-carnitine (2 mM), creatine (5 mM), and taurine (5 mM) with 1% penicillin and streptomycin for 24 h in 5% CO2 at 37°C.
Electrophysiology.
A conventional whole cell patch-clamp technique was used to record membrane currents from isolated cardiac myocytes with an Axopatch 200 amplifier. Signals were digitized using a Digidata 1322A interface, and records were acquired and analyzed using pCLAMP 9.2 software (Molecular Devices, Sunnyvale, CA). Patch pipettes were made from thick-walled borosilicate glass, filled with a solution containing (in mM) 140 KCl, 2 ATP, 0.1 ADP, 0.1 GTP, 1 MgCl2, 10 HEPES, and 5 BAPTA titrated to pH 7.2 with NaOH, and had resistances of 5–6 MΩ. Cells were superfused with normal Tyrode solution containing (in mM) 135 NaCl, 5 KCl, 0.33 NaH2PO4, 5 Na-pyruvate, 10 glucose, 1 MgCl2, 2 CaCl2, and 10 HEPES titrated to pH 7.4 with NaOH. P1075 and glibenclamide were from Tocris Bioscience (Abingdon, UK), and all other reagents were from Sigma-Aldrich (Dorset, UK).
Measurement of contractile activity and intracellular Ca2+ concentration.
Myocytes were placed in a 500-μl chamber on the stage of a Nikon inverted microscope, continuously superfused at 5 ml/min with Tyrode solution or metabolic inhibition Tyrode solution [which contained NaCN (2 mM) and iodoacetic acid (1 mM) without glucose or pyruvate], and stimulated at 1 Hz by electrical field stimulation. Contractile activity was determined from observation of fields containing 8–15 cells using a charge-coupled device camera (Orca2, Hamamatsu, Cairn Research, Kent, UK). To measure intracellular Ca2+ concentration ([Ca2+]i), myocytes were loaded with 5 μM indo-1 AM or fura-2 AM (Invitrogen, Paisley, UK). Indo-1 was excited at 340 nm, the emitted light was split with a 440-nm dichroic filter, and the resulting images passed through 485- and 405-nm bandpass filters, respectively. The indo-1 ratio (405-to-485-nm fluorescence ratio), corresponding to Ca2+ bound/Ca2+ free, was calculated using AQM Advance 6 software (Kinetic Imaging, Cairn Research). For fura-2 experiments, single myocytes were excited alternately with 340- and 380-nm light from a monochromator (deltaRAM, Photon Technology, West Sussex, UK). Fluorescence intensity was collected at 510 nm with a photomultiplier tube and analyzed with a video imaging system (Photon Technology). Free Ca2+ values were estimated using in vitro calibration for all treatment groups. The KD may differ in the cell, so the values given are intended to be an estimated indication of [Ca2+]i rather than the precise intracellular concentration.
Measurement of mitochondrial membrane potential.
Ventricular myocytes in Tyrode solution were loaded with the fluorescent dye tetramethylrhodamine methyl ester (TMRM; 5 μM, Invitrogen). In this assay, TMRM sequesters into the mitochondria, where fluorescence is autoquenched. TMRM-loaded myocytes were then continuously illuminated at 546 nm, generating ROS, and emission was measured using a long-pass filter of >560 nm at a rate of 0.5 Hz (Orca2, Hamamatsu, Cairn Research). Accumulation of ROS induced mPTP opening. This allows TMRM to leave the mitochondria and dequench in the cytosol, which is observed as an increase in fluorescence. An increase in TMRM fluorescence is used as an index of mitochondrial membrane potential, which, in these circumstances, reflects the opening of the mPTP (24). Regions of interest were placed around all viable myocytes in the field of view, and fluorescence changes were plotted against time (Kinetic Imaging, Cairn Research).
Ischemic pelleting.
Freshly isolated myocytes were infected with adenovirus and cultured for 24 h as described above. Myocytes were placed in 1.8-ml tubes and allowed to settle under gravity into a pellet. To simulate ischemia, excess supernatant was removed, and mineral oil was layered on top to exclude gaseous exchange. Samples were incubated at 37°C in a 5% CO2 incubator. To simulate reperfusion, the oil was removed, fresh medium 199 was added, and myocytes were incubated for a further 60 min. After reperfusion, myocytes were resuspended in Tyrode solution containing sodium amytal (3 mM), trypan blue (0.4%), and paraformaldyde (0.5%). Myocytes were counted, and trypan blue exclusion was used as an index of survival.
Western blot analysis.
A sample of cardiac myocytes infected with control or Kir6.2 shRNA adenovirus was taken and lysed. Equal amounts of protein, measured by the Bradford assay, from each treatment group were loaded in each well. Samples were subjected to SDS-PAGE and transferred to nitrocellulose using a wet transfer cell. The Kir6.2 antibody (a kind gift from Dr Norman, University of Leicester), which has been previously characterized (43), was used to detect Kir6.2 protein levels and was normalised to total ERK protein (Cell Signaling Technology, Hitchin, UK). Densitometry was determined using Felix software (Photon Technology).
Statistics.
Data are presented as means ± SE from at least three independent experiments. Statistical significance was calculated using Student's t-test and one-way ANOVA with Tukey's post hoc test where appropriate. P values of <0.05 were accepted as significant.
RESULTS
Decrease of KATP channel protein and current by silencing shRNA targeted to Kir6.2.
The cellular consequences of reduced expression of Kir6.2 in cardiac myocytes were investigated by RNA interference with shRNA. An adenoviral system for delivering shRNA sequences to adult rat cardiac myocytes was developed. Two independent Kir6.2 subunit-specific sequences, designated shRNA-A and shRNA-B, were used to knock down Kir6.2. In addition, a control shRNA containing an RNA interfering sequence with limited sequence similarity to known genes in the rat was used. Whole cell sarcolemmal KATP current was evoked by the KATP channel opener P1075 (10 μM) from myocytes voltage clamped to 0 mV, and the current was blocked by the specific KATP channel inhibitor glibenclamide (10 μM; Fig. 1A). Currents were normalized for cell size to show KATP current density. In contrast to the control, expression of Kir6.2 shRNA-A or Kir6.2 shRNA-B sequences for 24 h resulted in a significant decrease in KATP current density (Fig. 1, A and C).
Fig. 1.

Short hairpin (sh)RNA knockdown of ATP-sensitive K+ (KATP) currents in cardiac myocytes. A: whole cell recordings of KATP currents from isolated ventricular myocytes after 24-h infection with adenovirus expressing either control shRNA, Kir6.2 shRNA-A, or Kir6.2 shRNA-B sequences. Currents were recorded at 0 mV, and P1075 (10 μM) and glibenclamide (10 μM) were bath applied, as indicated by the horizontal bars. B: whole cell voltage-clamp recordings of voltage-gated Ca2+ currents from single ventricular myocytes after 24-h infection with either control shRNA, Kir6.2 shRNA-A, or Kir6.2 shRNA-B adenoviruses. Ca2+ currents were evoked by a 240-ms voltage step from −40 to 0 mV. C: peak KATP currents normalized for cell size (in pA/pF) of myocytes infected with control shRNA (n = 17), Kir6.2 shRNA-A (n = 12), or Kir6.2 shRNA-B (n = 5). **P < 0.01 vs. control. D: mean peak Ca2+ currents normalized for myocyte size (in pA/pF) recorded from cardiomyocytes infected with either control shRNA, Kir6.2 shRNA-A, or Kir6.2 shRNA-B adenoviruses (n = 3 for each). E: representative Western blots showing Kir6.2 protein from isolated ventricular myocytes after 24-h infection expressing control shRNA, Kir6.2 shRNA-A, or Kir6.2 shRNA-B. Total ERK was used as a loading control. F: average densitometry showing the effects of Kir6.2 shRNA on Kir6.2 protein levels. n = 3 Western blots from 3 hearts. **P < 0.01. Stats were done on raw densitometry values.
To exclude the possibility of a general reduction of ion channel function, L-type Ca2+ channel currents were evoked by a voltage step from −40 to 0 mV and recorded from cardiac myocytes infected with Kir6.2 shRNA virus (Fig. 1B). Mean normalized Ca2+ current amplitudes showed no significant differences between control, Kir6.2 shRNA-A, or Kir6.2 shRNA-B virus-infected myocytes (Fig. 1D).
In addition to the specific and significant decrease in KATP channel current density, Western blot analysis confirmed that KATP channel protein levels were significantly decreased in cardiac myocytes expressing either Kir6.2 shRNA-A or shRNA-B (Fig. 1, E and F).
Protection by IPC is abolished after knockdown of Kir6.2.
To investigate the effect of acute Kir6.2 knockdown on the protective effect of IPC, the ischemic pelleting technique was used (8). Myocytes were incubated under a layer of mineral oil to create an ischemic environment for 60 min. When the oil layer was removed, myocytes were resuspended in fresh medium to simulate reperfusion for 60 min. IPC was induced by incubating a pellet of myocytes for 10 min under oil and 10 min of reperfusion before the prolonged ischemia. Control myocytes were maintained in medium 199 throughout. The protocols used are shown in Fig. 2A. The population of live and dead myocytes was assessed by the ability to exclude trypan blue at the end of reperfusion (Fig. 2B). In contrast to the control virus-infected population of myocytes, the survival of those expressing Kir6.2 shRNA-A was not increased by IPC (Fig. 2C). Furthermore, knockdown of Kir6.2 resulted in a significant decrease in the survival of myocytes subjected to ischemia (32 ± 4%, n = 21 experiments) compared with myocytes infected with the control virus (42 ± 4%, n = 25 experiments, P < 0.05). In summary, control virus-infected myocytes showed significant protection by IPC; however, the protection was completely lost in Kir6.2 knockdown myocytes. Also, after ischemia, the percentage of dead myocytes was greater in Kir6.2 knockdown myocytes compared with control myocytes.
Fig. 2.

Kir6.2 knockdown prevents ischemic preconditioning (IPC). A: ischemic pelleting protocols for control, ischemia (Isch), and IPC. The solid horizontal bars show the incubation times under mineral oil, and the open horizontal bars show incubation times in medium 199. B: representative images of myocytes at the end of the ischemic pelleting protocols. Control, ischemia, and IPC results are shown for control virus (top) or Kir6.2 shRNA-A virus (bottom). Scale bars = 200 μm. C: mean percentages of control or Kir6.2 shRNA-A virus-infected myocytes surviving at the end of the ischemic pelleting protocols (4 experiments from 4 hearts). The percentage of untreated myocytes surviving was 68 ± 6% (n = 5 experiments). Numbers of myocytes were as follows: control virus protocol (control: 542, ischemia: 768, and IPC: 717) and Kir6.2 shRNA-A virus protocol (control: 676, ischemia: 825, and IPC: 770). *P < 0.05 and **P < 0.01 vs. ischemia (by one-way ANOVA with Tukey's post hoc test). NS, not significant.
Effect of Kir6.2 knockdown on Ca2+ homeostasis.
The ability of cardiac myocytes to maintain Ca2+ homeostasis during ischemia-reperfusion is central to the recovery of contractile function and myocyte survival. To investigate the effect of Kir6.2 knockdown on Ca2+ homeostasis, the Ca2+ indicator indo-1 was used. To simulate the decrease in cellular ATP that occurs in ischemia, myocytes were challenged with metabolic inhibition solution, which inhibits glycolysis and oxidative phosphorylation and thus reduces cellular ATP production. Myocytes were perfused with metabolic inhibition solution (4 min) and reperfused with Tyrode solution for 10 min in a contracting myocyte model of ischemia-reperfusion injury. Ventricular myocytes infected for 24 h with either control, shRNA-A, or shRNA-B adenovirus were stimulated to contract synchronously with electrical field stimulation at 1 Hz. Initially, control myocytes responded to field stimulation; shortly after metabolic inhibition solution, myocytes stopped responding to field stimulation, and at the end of reperfusion, the majority of myocytes regained their contractile function, although with elevated intracellular Ca2+ (Fig. 3, A and C). Initially, all Kir6.2 knockdown myocytes responded to field stimulation, but in contrast to control myocytes, they developed much higher intracellular Ca2+ at the end of reperfusion, and many did not regain their contractile function (Fig. 3, B and C). Interestingly, the baseline estimated [Ca2+] from Kir6.2 knockdown myocytes measured before the addition of metabolic inhibition solution was significantly elevated compared with control myocytes. Figure 3D shows the percentage of cells that did not recover Ca2+ homeostasis (indicated by estimated intracellular Ca2+ > 350 nM) after metabolic inhibition solution followed by 10 min of reperfusion. After metabolic inhibition solution and reperfusion, 76% of myocytes infected with control shRNA recovered contractile activity; in contrast, only 33% myocytes infected with Kir6.2 shRNA-A recovered contractile function (Fig. 3E). The results from experiments using shRNA-B to knock down Kir6.2 were very similar to those for shRNA-A virus-infected myocytes (data not shown).
Fig. 3.

Effect of Kir6.2 knockdown on Ca2+ homeostasis and contractile recovery after metabolic stress/reperfusion. A: simultaneous recordings of the indo-1 ratio from six control virus-infected myocytes within a field of view during 5-min perfusion with normal Tyrode solution followed by 4 min of metabolic inhibition (MI) solution and 10 min of reperfusion in normal Tyrode solution. Each trace is the ratio recorded from a different myocyte. Fields of myocytes were stimulated at 1 Hz, and images were sampled at 0.1 Hz. These Ca2+ transients ceased when the stimulator was turned off at the end of 10 min of reperfusion. B: recordings of the indo-1 ratio from six Kir6.2 shRNA-A virus-infected myocytes, as in A. C: averaged estimated Ca2+ concentrations for the protocol described in A. For the myocyte populations infected with control (n = 41, 7 experiments) or Kir6.2 shRNA-A virus (n = 77, 11 experiments). D: percentage of myocytes with an estimated diastolic intracellular Ca2+ concentration ([Ca2+]i) of >350 nM before the bath application of MI solution (baseline) and after 10 min of reperfusion with Tyrode solution. ***Statistical significance at P < 0.001 between control and Kir6.2 RNA-A virus-infected myocytes measured at the end of reperfusion. E: percentage of myocytes recovering a contractile response to electrical field stimulation (1 Hz) after 4 min of metabolic inhibition followed by 10 min of reperfusion. Cardiac myocytes were infected for 24 h with either control virus (n = 228 cells, 14 experiments), Kir6.2 shRNA-A virus (n = 79 cells, 10 experiments), or Kir6.2 shRNA-B virus (n = 126, 17 experiments). **P < 0.01 vs. control.
In summary, acute knockdown of Kir6.2 resulted in a significant increase in estimated cellular Ca2+ levels at the beginning of the experiment (baseline) and at the end of reperfusion (after 10 min of reperfusion). The proportion of myocytes that could regain contractile function was significantly decreased by Kir6.2 knockdown.
Effect of dominant negative Kir6.2 on Ca2+ homeostasis.
The metabolic enzymes that couple with KATP channels are known to influence the cardiac myocytes response to stress and the pathophysiology of ischemic heart disease (2). The role of the metabolic signaling complex was assessed by expressing a nonconducting Kir6.2 (dominant negative) subunit in adult ventricular myocytes using an adenoviral vector. In this way, the effect of prevention of the ion conduction properties of the KATP channels while leaving the scaffolding properties of the ion channel intact was tested.
Untreated and control virus-infected myocytes were held at 0 mV. Current was observed upon application of the KATP channel opener P1075, and the current was blocked by application of the specific KATP inhibitor glibenclamide. Expression of nonconducting Kir6.2 for 24 h resulted in a decrease of ∼50% of the KATP current compared with GFP-expressing control virus-infected myocytes (Fig. 4, A and B). Expression of either dominant negative Kir6.2 or Kir6.2-targetting shRNA resulted in comparable patterns of intracellular Ca2+ changes both before and after metabolic inhibition solution and reperfusion challenge; intracellular Ca2+ levels were significantly higher both at baseline and after reperfusion compared with control virus-infected myocytes.
Fig. 4.
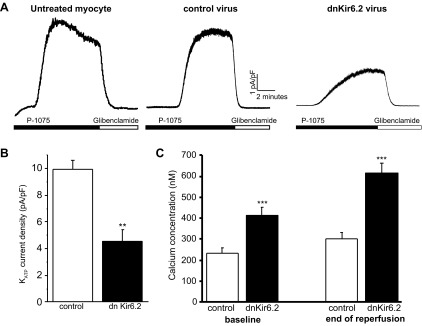
Effect of expression of dominant negative (dn)Kir6.2 on KATP current density and Ca2+ homeostasis. A: whole cell recordings of KATP currents from isolated ventricular myocytes after 24 h either untreated or infected with adenovirus expressing control green fluorescent protein (GFP) or dnKir6.2. Currents were recorded at 0 mV, and P1075 (10 μM) and glibenclamide (10 μM) were bath applied, as indicated by the horizontal bars. B: difference in peak KATP currents normalized for cell size (in pA/pF) of myocytes infected with control GFP adenovirus (n = 5) or dnKir6.2 adenovirus (n = 6). **P < 0.01 vs. control. C: mean estimated [Ca2+]i recorded in normal Tyrode solution before MI solution (baseline) and after 10 min of reperfusion (end of reperfusion) for myocytes infected with control GFP adenovirus (n = 23, 4 experiments) or dnKir6.2 adenovirus (n = 33, 4 experiments). ***P < 0.001 vs. control.
Effect of Kir6.2 knockdown on mitochondrial membrane potential.
The metabolic signaling complex that couples to the KATP channel forms a phsophotransfer bridge to the mitochondria, allowing for high-fidelity coupling of mitochondrial ATP to the excitability of ventricular myocytes. To test the effect of knockdown of Kir6.2 on mitochondrial function, adenovirus-infected cardiac myocytes were loaded with a high concentration of TMRM (5 μM) to access mitochondrial membrane potential. For myocytes infected with the control virus, the fluorescence recorded was initially low because the TMRM fluorescence was quenched in the mitochondria; however, after a period of continuous illumination, ROS accumulation caused a depolarization of the mitochondrial membrane potential, which was observed as an increase in fluorescence. Typical TMRM images from this assay are shown in Fig. 5A. The warmer colors (yellow and red) illustrate high-intensity fluorescence, which occurs on mitochondrial membrane potential depolarization. In contrast, knockdown of Kir6.2 resulted in oscillations in the fluorescence intensity before the final increase in fluorescence intensity. Figure 5B shows typical fluorescence traces of TMRM-loaded myocytes under continuous illumination over the time. Knockdown of Kir6.2 resulted in a number of changes in mitochondrial function. First, the baseline TMRM fluorescence of Kir6.2 shRNA virus-infected myocytes was significantly higher compared with control virus-infected myocytes (Fig. 5, B and C), indicating a reduced mitochondrial membrane potential. Second, knockdown of Kir6.2 significantly increased the number of myocytes that had mitochondrial membrane potential oscillations before the final and irreversible mitochondrial depolarization (Fig. 5, B and D).
Fig. 5.

Changes in mitochondrial membrane potential of myocytes infected with control or Kir6.2 shRNA-A adenovirus. A: example time series showing fluorescence images of representative ventricular myocytes loaded with 5 μM tertramethylrhodamine methyl ester (TMRM) subjected to laser-induced oxidative stress. Time 0 is before oxidative stress. Mitochondrial depolarization is indicated by the increase in the intensity of fluorescence (increase in warm colors, e.g., red indicating the highest and blue the lowest fluorescence intensity). B: typical TMRM fluorescence intensity traces (in arbitrary units) recorded from myocytes infected with control (solid line) or Kir6.2 shRNA-A (shaded line) adenovirus for 24 h. C: baseline TMRM fluorescence (in arbitrary units) recorded at the beginning of the experiment from myocytes infected with control (n = 157) and Kir6.2 shRNA-A (n = 174) for 24 h. ***P < 0.001. D: percentage of myocytes that displayed mitochondrial membrane potential oscillations before the final depolarization for myocytes infected with control adenovirus (n = 77 myocytes, 20 experiments) compared with myocytes infected with Kir6.2 shRNA-A (n = 84 myocytes, 19 experiments) for 24 h. **P < 0.01.
DISCUSSION
The KATP channel pore-forming subunit Kir6.2 was knocked down in isolated rat ventricular myocytes using adenoviral transfer of silencing RNA targeted to Kir6.2. This resulted in a decrease in total Kir6.2 protein compared with control after 24 h and a significant decrease (∼50%) in sarcolemmal KATP current. Ca2+ currents were unaffected by Kir6.2 shRNA adenoviral infection, clarifying specificity to KATP.
To investigate whether knockdown of Kir6.2 affected IPC in ventricular myocytes, the ischemic pelleting technique was used as this has been suggested to provide a realistic model of IPC in isolated myocytes (8). The IPC response was abolished when Kir6.2 was knocked down in cardiac myocytes. While this is consistent with previous knockout studies (22, 44), it shows that even a modest reduction in channel expression is sufficient to block this effect. In the present study, the knockdown of Kir6.2 was acute, allowing little possibility for compensation, which may occur in transgenic mice. Interestingly, knockdown of Kir6.2 markedly reduced the proportion of myocytes surviving ischemia. This, again, is consistent with the inability of Kir6.2 knockout mice to adapt to stress situations such as ischemia, exercise exertion, or β-adrenergic challenge (54), but in this study with acute rather than chronic and partial rather than total ablation of KATP channels. However, it is in contrast to the observation that whole hearts from Kir6.2 knockout mice yielded the same recovery of function and had the same infarct size in response to global ischemia as littermate controls (48). This suggests there is more to understand about the role of KATP channels in response to physiological and pathophysiological stresses.
During metabolic stress, such as ischemia-reperfusion injury, myocytes become overloaded with Ca2+ and maintaining Ca2+ homeostasis becomes increasingly energy consuming. The opening of the sarcolemmal KATP channels has been proposed to oppose this Ca2+ overload by hyperpolarization of the membrane potential (13, 36). KATP channels are an abundant protein in cardiac myocytes, and <1% of KATP channels may need to open to hyperpolarize the membrane potential and thus prevent Ca2+ loading (35, 46). This would suggest that only very extreme changes in KATP channel expression would result in altered myocyte responses to stress. However, despite the Kir6.2 silencing RNA only reducing the KATP current by ∼50% in this study, Ca2+ overload was increased in response to metabolic inhibition solution and reperfusion. This may be because the reduction in KATP channel number decreases the channel density in caveolin-rich microdomains, which could affect responses to local changes in ATP concentration (17).
It is interesting to note that there was a significant increase in the basal estimated [Ca2+]i before any metabolic challenge in the Kir6.2 knockdown myocyte population. This suggests that basal Ca2+ homeostasis is perturbed by a decrease in KATP channel density and that, in addition to their pronounced effects during metabolic stress, sarcolemmal KATP channels may play a role in Ca2+ homeostasis under more normal physiological conditions. This increased basal [Ca2+]i may be a result of increased action potential duration, as both Kir6.2 knockout mouse hearts (20) and hearts from normal mice treated with the KATP blocker tolbutamide (10) show increased action potential duration. Elevated [Ca2+]i may also account for the upregulation of calcineurin- and Ca2+-dependent transcription factors in Kir6.2 knockout mice (49). Indeed, it has been suggested that transgenic mice expressing an ATP-insensitive Kir6.2 mutant that is more active in vivo than wild type compensate for the change in KATP activity by increasing Ca2+ current density (12). However, in this study, we found no change in the Ca2+ current density.
Despite increased baseline estimated [Ca2+]i, all myocytes showed contractile function at the beginning of the experiments, which is consistent with observations in other Kir6.2 knockout studies (44, 52). This might be because the increase in [Ca2+]i was not sufficient to inhibit contraction. During metabolic inhibition solution, the Kir6.2 knockdown myocyte population continued contracting for ∼2 min longer than the control population due to the loss of KATP channel-dependent loss of excitability, which is also consistent with the prolonged contractility reported for Kir6.2 knockout hearts (39).
In addition to conducting K+ efflux, KATP channels form a signaling complex with creatine kinase and adenylate kinase, which forms a phosphotransfer bridge connecting the mitochondria to the sarcolemmal membrane (1, 41). It is possible that a decrease in KATP channel density might disrupt this phosphotransfer bridge (6, 45). Indeed, knockout of creatine kinase from cardiac myocytes causes uncoupling of metabolic signals such that KATP channels open significantly earlier in response to metabolic stress (41). This may also occur in heart failure, when the ability of KATP channels to respond to metabolic challenge is lost due to deficits in the creatine kinase network (26). However, it is unclear whether K+ current is necessary for the adaptive response to stress or whether the KATP channel and surrounding metabolic signaling complex is sufficient for protection (6). To address this issue, a nonconducting dominant negative mutant of Kir6.2 was delivered by adenovirus to adult rat cardiac myocytes. Expression of the nonconducting dominant negative mutant of Kir6.2 resulted in a ∼50% reduction of KATP current density. The response to metabolic inhibition solution and reperfusion was like that of shRNA knockdown myocytes in that there was a significant increase in baseline intracellular Ca2+ in addition to increased Ca2+ overload after metabolic inhibition solution and reperfusion. This suggests that K+ flux is important in the prevention of Ca2+ overload, presumably by hyperpolarizing the membrane potential. Another study (52) with cardiac myocytes expressing dominant negative Kir6.2 showed KATP current reduction by ∼85%, which resulted in a slowed rate of action potential duration shortening, again highlighting a role for KATP channels in adaption to metabolic stress.
The opening of the mPTP is the point of no return in myocyte death after ischemia-reperfusion injury (24). To investigate whether mitochondrial responses to stress were altered by Kir6.2 knockdown, a well-established assay using a high concentration of TMRM and oxidative stress to induce mPTP opening was used (11, 24). First, this experiment showed that TMRM fluorescence was significantly higher in Kir6.2-downregulated myocytes. This suggests a more depolarized mitochondrial membrane potential compared with control myocytes and shows a link between KATP channels and mitochondrial membrane potential under baseline conditions. Second, this experiment showed that knockdown of Kir6.2 resulted in a significant increase in the number of myocytes that showed mitochondrial membrane potential oscillations before the final and irreversible mitochondrial depolarization compared with control myocytes. This has also recently been shown to occur in intact hearts in response to ischemia-reperfusion injury (31). The oscillations occur in metabolic stress because to maintain a hyperpolarized mitochondrial membrane potential, F1/F0-ATPase reverses and consumes ATP (28), but when all the ATP is consumed, the mitochondrial membrane potential depolarizes (5, 47, 50). The glycolytic enzyme phosphofructokinase is sensitive to ATP and exaggerates the oscillations in the cellular ATP-to-ADP ratio until all the glucose is exhausted (50). This has been shown in cardiac myocytes near anoxia when the mitochondrial membrane potential oscillates in synchrony with glycolysis (15). The downregulation of KATP channels could lead to a loss in the finely balanced connection between energy homeostasis and excitability afforded by the metabolic signaling complex. However, it is also possible that the increase in basal [Ca2+]i observed in Kir6.2 knockdown myocytes could increase mitochondrial Ca2+ loading and depolarizes the mitochondrial membrane potential by activation of Ca2+-sensitive respiratory enzymes (7, 19, 28). Inhibition of KATP channels has also been shown to cause mitochondrial Ca2+ loading (32) and enhance mPTP opening (42).
In addition to sarcolemmal KATP channels, a role for mitochondrial KATP channels has been proposed in preconditioning through K+ influx into the matrix of the mitochondria (18). However, the present study was unable to distinguish whether the effects are due to loss of one or both the mitochondrial and sarcolemmal KATP channels. Recently, the mitochondrial KATP channel has been found to be composed of ROMK1 as the pore-forming subunit rather than Kir6.1 or Kir6.2, which lends support to the notion that the effects observed here are due to a decrease in sarcolemmal rather than mitochondrial KATP channel density (14).
In summary, this study shows that KATP channel density is important for Ca2+ homeostasis both before and after metabolic stress. Kir6.2 plays a role in the protective effect of IPC and protection during ischemia, as both were lost when Kir6.2 was knocked down. Kir6.2 influences mitochondrial membrane potential at rest and prevents mitochondrial membrane potential oscillations during oxidative stress. The KATP channel plays a central role in IPC and Ca2+ and mitochondrial homeostasis during stress, and the impact of decreasing channel number is deleterious to myocyte survival.
GRANTS
This work was supported by British Heart Foundation Project Grants PG0404216942 and PG0711224007.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: N.M.S., N.B.S., and D.L. conception and design of research; N.M.S., R.C.S., and D.L. performed experiments; N.M.S. and R.C.S. analyzed data; N.M.S. interpreted results of experiments; N.M.S. prepared figures; N.M.S. drafted manuscript; N.M.S., R.D.R., and D.L. edited and revised manuscript; N.M.S., R.C.S., R.D.R., N.B.S., and D.L. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Robert Norman for the kind gift of the Kir6.2 antibody. The authors are grateful to Diane Everitt and Sonja Khemiri for technical assistance.
REFERENCES
- 1.Alekseev AE, Hodgson DM, Karger AB, Park S, Zingman LV, Terzic A. ATP-sensitive K+ channel channel/enzyme multimer: metabolic gating in the heart. J Mol Cell Cardiol 38: 895–905, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arrell DK, Zlatkovic Lindor J, Yamada S, Terzic A. KATP channel-dependent metaboproteome decoded: systems approaches to heart failure prediction, diagnosis, and therapy. Cardiovasc Res 90: 258–266, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baczko I, Jones L, McGuigan CF, Fox JE, Gandhi M, Giles WR, Clanachan AS, Light PE. Plasma-membrane KATP channel-mediated cardioprotection involves posthypoxic reductions in calcium overload and contractile dysfunction: mechanistic insights into cardioplegia. FASEB J 19: 980–902, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Balaban RS. Metabolic homeostasis of the heart. J Gen Physiol 139: 407–414, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campanella M, Parker N, Tan CH, Hall AM, Duchen MR. IF1: setting the pace of the F1Fo-ATP synthase. Trends Biochem Sci 34: 343–350, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Carrasco AJ, Dzeja PP, Alekseev AE, Pucar D, Zingman LV, Abraham MR, Hodgson D, Bienengraeber M, Puceat M, Janssen E, Wieringa B, Terzic A. Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc Natl Acad Sci USA 98: 7623–7628, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalmers S, McCarron JG. The mitochondrial membrane potential and Ca2+ oscillations in smooth muscle. J Cell Sci 121: 75–85, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Diaz RJ, Wilson GJ. Studying ischemic preconditioning in isolated cardiomyocyte models. Cardiovasc Res 70: 286–296, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Du QY, Jovanovic S, Clelland A, Sukhodub A, Budas G, Phelan K, Murray-Tait V, Malone L, Jovanovic A. Overexpression of SUR2A generates a cardiac phenotype resistant to ischemia. FASEB J 20: 1131–1141, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faivre JF, Findlay I. Effects of tolbutamide, glibenclamide and diazoxide upon action-potentials recorded from rat ventricular muscle. Biochim Biophys Acta 984: 1–5, 1989 [DOI] [PubMed] [Google Scholar]
- 11.Falchi AM, I R, Diana A, Putzolu M, Diaz G. Characterization of depolarization and repolarization phases of mitochondrial membrane potential fluctuations induced by tetramethylrhodamine methyl ester photoactivation. FEBS J 272: 1649–1659, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Flagg TP, Charpentier F, Manning-Fox J, Remedi MS, Enkvetchakul D, Lopatin A, Koster J, Nichols C. Remodeling of excitation-contraction coupling in transgenic mice expressing ATP-insensitive sarcolemmal KATP channels. Am J Physiol Heart Circ Physiol 286: H1361–H1369, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev 90: 799–829, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foster DB, Ho AS, Rucker J, Garlid AO, Chen L, Sidor A, Garlid KD, O'Rourke B. Mitochondrial ROMK channel is a molecular component of mitoKATP. Circ Res 111: 446–454, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganitkevich V, Mattea V, Benndorf K. Glycolytic oscillations in single ischemic cardiomyocytes at near anoxia. J Gen Physiol 135: 307–319, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao WD, Liu YG, Mellgren R, Marban E. Intrinsic myofilament alterations underlying the decreased contractility of stunned myocardium–a consequence of Ca2+-dependent proteolysis? Circ Res 78: 455–465, 1996 [DOI] [PubMed] [Google Scholar]
- 17.Garg V, Jiao JD, Hu KL. Regulation of ATP-sensitive K+ channels by caveolin-enriched microdomains in cardiac myocytes. Cardiovasc Res 82: 51–58, 2009 [DOI] [PubMed] [Google Scholar]
- 18.Garlid KD, Halestrap AP. The mitochondrial KATP channel–fact or fiction? J Mol Cell Cardiol 52: 578–583, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51: 2959–2973, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glukhov AV, Flagg TP, Fedorov VV, Efimov IR, Nichols CG. Differential KATP channel pharmacology in intact mouse heart. J Mol Cell Cardiol 48: 152–160, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross GJ, Auchampach JA. Blockade of ATP-sensitive potassium channels prevents myocardial preconditioning in dogs. Circ Res 70: 223–233, 1992 [DOI] [PubMed] [Google Scholar]
- 22.Gumina RJ, Pucar D, Bast P, Hodgson DM, Kurtz CE, Dzeja PP, Miki T, Seino S, Terzic A. Knockout of Kir6.2 negates ischemic preconditioning-induced protection of myocardial energetics. Am J Physiol Heart Circ Physiol 284: H2106–H2113, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion–a target for cardioprotection. Cardiovasc Res 61: 372–385, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 287: H841–H849, 2004 [DOI] [PubMed] [Google Scholar]
- 25.He TC, Zhou SB, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509–2514, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hodgson DM, Zingman LV, Kane GC, Perez-Terzic C, Bienengraeber M, Ozcan C, Gumina RJ, Pucar D, O'Coclain F, Mann DL, Alekseev AE, Terzic A. Cellular remodeling in heart failure disrupts KATP channel-dependent stress tolerance. EMBO J 22: 1732–1742, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong MY, Kefalogianni E, Bao L, Malester B, Delaroche D, Neubert TA, Coetzee WA. The cardiac KATP channel associates with the glycolytic enzyme complex. FASEB J 25: 2456–2467, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jafri MS, Kotulska M. Modeling the mechanisms of metabolic oscillations in ischemic cardiac myocytes. J Theor Biol 242: 801–817, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Lalli MJ, Johns DC, Janecki M, Liu YG, O'Rourke B, Marban E. Suppression of KATP currents by gene transfer of a dominant negative Kir6.2 construct. Pflügers Arch 436: 957–961, 1998 [DOI] [PubMed] [Google Scholar]
- 30.Lederer WJ, Nichols CG, Smith GL. The mechanism of early contractile failure of isolated rat ventricular myocytes subjected to complete metabolic inhibition. J Physiol 413: 329–349, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyon AR, Joudrey PJ, Jin DZ, Nass RD, Aon MA, O'Rourke B, Akar FG. Optical imaging of mitochondrial function uncovers actively propagating waves of mitochondrial membrane potential collapse across intact heart. J Mol Cell Cardiol 49: 565–575, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marinovic J, Ljubkovic M, Stadnicka A, Bosnjak ZJ, Bienengraeber M. Role of sarcolemmal ATP-sensitive potassium channel in oxidative stress-induced apoptosis: mitochondrial connection. Am J Physiol Heart Circ Physiol 294: H1317–H1325, 2008 [DOI] [PubMed] [Google Scholar]
- 33.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia–a delay of lethal cell injury in ischemic myocardium. Circulation 74: 1124–1136, 1986 [DOI] [PubMed] [Google Scholar]
- 34.Nichols CG, Lederer WJ. The regulation of ATP-sensitive K+ channel activity in intact and permeabilized rat ventricular myocytes. J Physiol 423: 91–110, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nichols CG, Ripoll C, Lederer WJ. ATP-sensitive potassium channel modulation of the guinea-pig ventricular action-potential and contraction. Circ Res 68: 280–287, 1991 [DOI] [PubMed] [Google Scholar]
- 36.Noma A. ATP-regulated K+ channels in cardiac-muscle. Nature 305: 147–148, 1983 [DOI] [PubMed] [Google Scholar]
- 37.Ravier MA, Nenquin M, Miki T, Seino S, Henquin JC. Glucose controls cytosolic Ca2+ and insulin secretion in mouse islets lacking adenosine triphosphate-sensitive K+ channels owing to a knockout of the pore-forming subunit Kir6.2. Endocrinology 150: 33–45, 2009 [DOI] [PubMed] [Google Scholar]
- 38.Rodrigo GC, Lawrence CL, Standen NB. Dinitrophenol pretreatment of rat ventricular myocytes protects against damage by metabolic inhibition and reperfusion. J Mol Cell Cardiol 34: 555–569, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Saito T, Sato T, Miki T, Seino S, Nakaya H. Role of ATP-sensitive K+ channels in electrophysiological alterations during myocardial ischemia: a study using Kir6.2-null mice. Am J Physiol Heart Circ Physiol 288: H352–H357, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Seino S. Physiology and pathophysiology of KATP channels in the pancreas and cardiovascular system–a review. J Diabetes Complications 17: 2–5, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Selivanov VA, Alekseev AE, Hodgson DM, Dzeja PP, Terzic A. Nucleotide-gated KATP channels integrated with creatine and adenylate kinases: amplification, tuning and sensing of energetic signals in the compartmentalized cellular environment. Mol Cell Biochem 256–257: 243–256, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shintani-Ishida K, Inui M, Yoshida K. Ischemia-reperfusion induces myocardial infarction through mitochondrial Ca2+ overload. J Mol Cell Cardiol 53: 233–239, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Singh H, Hudman D, Lawrence CL, Rainbow RD, Lodwick D, Norman RI. Distribution of Kir6.0 and SUR2 ATP-sensitive potassium channel subunits in isolated ventricular myocytes. J Mol Cell Cardiol 35: 445–459, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Suzuki M, Sasaki N, Miki T, Sakamoto N, Ohmoto-Sekine Y, Tamagawa M, Seino S, Marban E, Nakaya H. Role of sarcolemmal KATP channels in cardioprotection against ischemia/reperfusion injury in mice. J Clin Invest 109: 509–516, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Beek JH. Adenine nucleotide-creatine-phosphate module in myocardial metabolic system explains fast phase of dynamic regulation of oxidative phosphorylation. Am J Physiol Cell Physiol 293: C815–C829, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Weiss JN, Venkatesh N, Lamp ST. ATP-sensitive K channels and cellular K loss in hypoxic and ischaemic mammalian ventricle. J Physiol 447: 649–673, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weiss JN, Yang JH. Oscillations at odds in the heart. J Gen Physiol 135: 303–305, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wojtovich AP, Urciuoli WR, Chatterjee S, Fisher AB, Nehrke K, Brookes PS. Kir6.2 is not the mitochondrial KATP channel, but is required for cardioprotection by ischemic preconditioning. Am J Physiol Heart Circ Physiol 304: H1439–H1445, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamada S, Kane GC, Behfar A, Liu XK, Dyer RB, Faustino RS, Miki T, Seino S, Terzic A. Protection conferred by myocardial ATP-sensitive K+ channels in pressure overload-induced congestive heart failure revealed in KCNJ11 Kir6.2-null mutant. J Physiol 577: 1053–1065, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang JH, Yang L, Qu Z, Weiss JN. Glycolytic oscillations in isolated rabbit ventricular myocytes. J Biol Chem 283: 36321–36327, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev 83: 1113–1151, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Zhu ZY, Burnett CML, Maksymov G, Stepniak E, Sierra A, Subbotina E, Anderson ME, Coetzee WA, Hodgson-Zingman DM, Zingman LV. Reduction in number of sarcolemmal KATP channels slows cardiac action potential duration shortening under hypoxia. Biochem Biophys Res Commun 415: 637–641, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zingman LV, Alekseev AE, Hodgson-Zingman DM, Terzic A. ATP-sensitive potassium channels: metabolic sensing and cardioprotection. J Appl Physiol 103: 1888–1893, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, Pucar D, Bienengraeber M, Dzeja PP, Miki T, Seino S, Alekseev AE, Terzic A. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA 99: 13278–13283, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]