

Lpd is an essential, evolutionary conserved regulator of the Scar/WAVE complex during cell migration in vivo.
Abstract
Cell migration is essential for development, but its deregulation causes metastasis. The Scar/WAVE complex is absolutely required for lamellipodia and is a key effector in cell migration, but its regulation in vivo is enigmatic. Lamellipodin (Lpd) controls lamellipodium formation through an unknown mechanism. Here, we report that Lpd directly binds active Rac, which regulates a direct interaction between Lpd and the Scar/WAVE complex via Abi. Consequently, Lpd controls lamellipodium size, cell migration speed, and persistence via Scar/WAVE in vitro. Moreover, Lpd knockout mice display defective pigmentation because fewer migrating neural crest-derived melanoblasts reach their target during development. Consistently, Lpd regulates mesenchymal neural crest cell migration cell autonomously in Xenopus laevis via the Scar/WAVE complex. Further, Lpd’s Drosophila melanogaster orthologue Pico binds Scar, and both regulate collective epithelial border cell migration. Pico also controls directed cell protrusions of border cell clusters in a Scar-dependent manner. Taken together, Lpd is an essential, evolutionary conserved regulator of the Scar/WAVE complex during cell migration in vivo.
Introduction
Tightly controlled cell migration is essential for the development of multicellular organisms, and deregulation is a hallmark of diseases such as metastatic cancer (Hanahan and Weinberg, 2011). The force for cell migration is largely provided by actin polymerization at the leading edge of cells, the lamellipodium, and is controlled by actin-binding proteins including Ena/VASP and the Arp2/3 complex. These proteins are recruited to the leading edge by regulators such as Scar/WAVE for the Arp2/3 complex or Lpd for Ena/VASP proteins. The Scar/WAVE complex is composed of five proteins (Sra1/Pir121, Nap1, Scar/WAVE1-3, Abi1-3, and HSPC300) and is activated by Rac to interact with the Arp2/3 complex, thereby nucleating branched actin filament networks. In this way, both Scar/WAVE and Arp2/3 complexes regulate cell migration (Suetsugu et al., 2003; Yan et al., 2003; Insall and Machesky, 2009; Campellone and Welch, 2010; Michael et al., 2010; Suraneni et al., 2012; Wu et al., 2012). However, the regulation of the Scar/WAVE complex in migrating cells is not well understood.
Ena/VASP proteins localize to lamellipodia, tips of filopodia, and focal adhesions, and regulate lamellipodial dynamics and cell migration. Ena/VASP regulate actin filament length at the leading edge of cells by temporarily protecting actin filament ends from capping protein and recruiting polymerization-competent G-actin bound to profilin. Scar/WAVE–Arp2/3–mediated actin filament branching and Ena/VASP-regulated actin filament elongation together control speed and stability of lamellipodial protrusions, but it is not known how these mechanisms are coordinated (Bear et al., 2001, 2002; Krause et al., 2003; Pula and Krause, 2008).
Lpd and its Drosophila melanogaster orthologue Pico interact with Ena/VASP proteins, and harbor a proline-rich region with putative SH3 domain binding sites, a Ras association (RA) domain, and a pleckstrin homology (PH) domain. Lpd localizes to lamellipodia, and both RA and PH domains cooperate in membrane targeting of Lpd upon growth factor stimulation of fibroblasts. Lpd recruits Ena/VASP proteins to lamellipodia and to dorsal ruffles of fibroblasts, thereby controlling lamellipodia protrusion dynamics, dorsal ruffling of fibroblasts, axon elongation, and branching of primary hippocampal neurons, but its role in mesenchymal and epithelial cell migration is unknown. Surprisingly, knockdown of Lpd decreased F-actin content, resulted in the absence of a dense lamellipodial F-actin meshwork, and impaired lamellipodium formation (Krause et al., 2004; Lyulcheva et al., 2008; Michael et al., 2010). These phenotypes were not observed with loss of Ena/VASP, which suggests that Lpd regulates other effectors of the actin cytoskeleton in addition to Ena/VASP. Interestingly, recent reports suggest that the Lpd orthologue in Caenorhabditis elegans, MIG-10, may directly or indirectly bind to Abi-1, and both genetically interact to regulate axon guidance, synaptic vesicle clustering, and excretory canal outgrowth in C. elegans (Stavoe et al., 2012; Xu and Quinn, 2012; McShea et al., 2013).
Here, we show that Lpd is in complex with Scar/WAVE, mediated by a direct binding of the Abi SH3 domain to three sites in Lpd. In addition, Lpd directly interacts with active Rac, which positively regulates the Lpd–Scar/WAVE interaction. Therefore, Lpd functions as a Rac effector and controls lamellipodia formation via the Scar/WAVE complex. Lpd knockout (KO) mouse embryonic fibroblasts (MEFs) are impaired in cell migration, whereas Lpd overexpression dramatically increased cell migration speed in a Scar/WAVE-dependent manner. Most Lpd KO mice die shortly after birth, and the few surviving mice are reduced in body weight and display missing pigmentation on their ventral side because fewer migrating neural crest (NC)–derived melanoblasts reach their target during development. In agreement, Lpd and the Scar/WAVE complex cooperate to regulate NC migration in vivo and in vitro in Xenopus laevis. This cooperation is evolutionary conserved in invertebrates because Lpd’s orthologue Pico also binds the Scar/WAVE complex and regulates epithelial collective border cell migration in the fly ovary in a Scar/WAVE-dependent manner. Taken together, we have identified a novel pathway in which Lpd functions as an important evolutionary conserved regulator of the Scar/WAVE complex during cell migration in vivo.
Results
Lpd colocalizes and interacts with the Scar/WAVE complex
We previously reported that Lpd knockdown impairs lamellipodium formation (Krause et al., 2004). In contrast, loss of the Lpd-binding proteins Ena/VASP only altered lamellipodial dynamics (Bear et al., 2002; Krause et al., 2003). Therefore, we hypothesized that the Scar/WAVE complex, a key regulator of lamellipodia, may mediate Lpd’s function in lamellipodium formation.
Coimmunoprecipitation with GFP-Lpd and Myc-tagged Scar/WAVE complex revealed that Lpd interacts with both Scar/WAVE1 (Fig. 1 A) and Scar/WAVE2 complexes (Fig. 1 B). Endogenous Lpd and Scar/WAVE1 also coimmunoprecipitated from lysates of primary cortical neurons (Fig. 1 C), which suggests that Lpd and Scar/WAVE indeed form a protein complex in cells.
Figure 1.

Lamellipodin interacts with the Scar/WAVE complex. (A and B) Coimmunoprecipitation using Lpd or IgG control antibodies from HEK293 cell lysates expressing GFP-Lpd and the tagged Scar/WAVE complex including FLAG-WAVE1 (A) and Myc-WAVE2 (B). Myc-HSPC300 is not shown. (C) Endogenous Scar/WAVE1 and Lpd coimmunoprecipitate from lysates of primary cortical neurons using Lpd antibodies but not with IgG control. (D) Knockdown of Lpd by siRNA in B16F1 cells does not reduce expression of the Scar/WAVE complex (HSPC300 not shown) or Arp3. Loading control: Tubulin. (E–G) Endogenous Lpd (green) colocalizes with Scar/WAVE1 (E), Abi1 (F), and Sra1 (G; red) at the very edge of lamellipodia in B16F1 mouse melanoma cells. Representative line scan from multiple experimental repeats across the leading edge (location indicated on merged images) shows colocalization of Lpd (green) and Scar/WAVE1 (E), Abi1 (F), and Sra1 (G; red). Bar, 25 µm. See also Fig. S1.
The stability of the Scar/WAVE complex is tightly controlled, such that knocking down one protein of the core complex results in proteasomal degradation of the remaining proteins of the complex (Kunda et al., 2003). The expression of the Scar/WAVE complex was unaltered when Lpd protein levels were reduced using siRNA (Fig. 1 D), which suggests that Lpd is not part of the core complex but may associate with it to control its function.
Both Lpd and the Scar/WAVE complex have been reported to localize to lamellipodia (Hahne et al., 2001; Stradal et al., 2001; Krause et al., 2004). Consistently, immunofluorescence analysis revealed that Lpd colocalizes with Scar/WAVE1 (Figs. 1 E and S1), Abi1 (Figs. 1 F and S1), and Sra1 (Figs. 1 G and S1) at the very edge of lamellipodia in B16F1 mouse melanoma cells (Fig. 1, E–G) or CAD mouse neuronal cells (Fig. S1).
The interaction between Lpd and the Scar/WAVE complex is mediated by the Abi SH3 domain and three sites in Lpd
Abi harbors an SH3 domain, which could bind to putative SH3 binding sites in the C terminus of Lpd. To test this, we purified a GST-Abi1-SH3 domain fusion protein and pulled down Lpd from NIH3T3 cell lysates, revealing an interaction of the Abi SH3 domain with Lpd (Fig. 2 A). In Far-Western blot experiments with fragments of Lpd, we observed that only full-length Abi1 but not a truncated form without the SH3 domain (Abi1ΔSH3) directly interacted with two fragments (GST-Lpd-C4 and GST-Lpd-C6) and weakly with a third (GST-Lpd-C5; Fig. 2, B–D). A Far-Western experiment on a peptide array covering the C terminus of Lpd as 12-mer peptides that overlap each other by three amino acids revealed two strong class II SH3 domain–binding sites: one in each fragment Lpd-C4 and Lpd-C6, and one weak class II binding site in fragment Lpd-C5 (Fig. 2, E and F).
Figure 2.

Lpd directly interacts with the SH3 domain of Abi. (A) Pull-down of Lpd from NIH/3T3 cell lysate using the GST-Abi-SH3 domain or GST as control. (B–D) Far Western overlay on different GST-Lpd truncation mutants (B) or GST control using purified (C) MBP-Abi1full-length or (D) MBP-Abi1ΔSH3 was detected with anti-MBP antibodies. Three independent experiments were performed. (E) Far-Western overlay with MBP-Abi1full-length on a peptide array covering the C terminus of Lpd with 12-mer peptides overlapping each other by three amino acids was detected with anti-MBP antibodies. (F) Table shows Abi SH3 domain–binding motifs in the Lpd sequence. The two GST-Lpd fragments highlighted in red correspond to the most strongly interacting Lpd fragments in the Far-Western experiment in C. The amino acid residues highlighted in yellow correspond to the core residues required for class II SH3 domain binding.
To test whether the SH3 domain of Abi mediates the interaction between Lpd and the Scar/WAVE complex, we attempted to coimmunoprecipitate GFP-Lpd and all Myc-tagged components of the Scar/WAVE complex, including either full-length Abi1 or Abi1ΔSH3. This analysis revealed that expression of Abi1ΔSH3 disrupts the interaction between Lpd and the Scar/WAVE complex (Fig. 3, A and B), which suggests that the SH3 domain of Abi is required for this interaction. To explore whether the three Abi SH3 domain binding sites are sufficient for the Lpd interaction with Abi, we mutated them in full-length Lpd (LpdAbiMut). Comparing coimmunoprecipitations between wild-type (WT) Lpd or LpdAbiMut and Abi1 revealed that LpdAbiMut is defective in Abi binding (Fig. 3, C and D). Because Lpd interacts with Ena/VASP proteins (Krause et al., 2004), we also coimmunoprecipitated Lpd with GFP-Abi and GFP-VASP from HEK cell lysates (Fig. S2 A), which suggests that Lpd forms a complex with both Abi and Ena/VASP proteins.
Figure 3.

The interaction between Lpd and the Scar/WAVE complex is mediated by the Abi SH3 domain and positively regulated by active Rac. (A–D) The Abi SH3 domain and three Abi binding sites in Lpd mediate the interaction between Lpd and the Scar/WAVE complex. Immunoprecipitation using Lpd antibodies or IgG control from HEK293 cell lysates (A) expressing GFP-Lpd and all Myc-tagged components of the Scar/WAVE complex, including Myc-Abi1full-length (A, left), Myc-Abi1ΔSH3 (A, right), or GFP-Abi and GFP-Lpd (C, left) or GFP-LpdAbiMut (C, right), show coimmunoprecipitation between Lpd and all components of the Scar/WAVE complex only when the Abi SH3 domain is present (A; Myc-HSPC300 is not shown) or between Lpd and GFP-Abi only when the Abi binding sites are present (B). Western blot: anti-GFP. (B and D) Comparison of efficiency of coimmunoprecipitation of Lpd with all components of the Scar/WAVE complex (B) or GFP-Lpd or GFP-LpdAbiMut with Abi (D). Quantification of band intensity of chemiluminescence imaged with a charge-coupled device camera. (B) Coimmunoprecipitation is reduced by >90%. Error bars indicate mean ± SEM, n = 3. One-way analysis of variance (ANOVA) and Tukey’s test were used; **, P < 0.01. (D) Coimmunoprecipitation is reduced by >94%. Error bars indicate mean ± SEM, n = 3. An unpaired t test was used; ***, P < 0.001. (E and F) Lpd and the RA-PH domains of Lpd are in complex with active Rac. Purified GTPγS- or GDPβS-loaded GST-Rac, or GST only as control, on Sepharose beads were incubated with lysates from HEK cells expressing GFP-Lpd (E) or GFP-Lpd-RAPH (F) and bound with GFP-Lpd or GFP-Lpd-RAPH. Samples were detected in a Western blot against GFP. (G) The RA-PH domains of Lpd directly interact with active Rac. Purified GTPγS- or GDPβS-loaded GST-Rac or GST only as control Sepharose beads were incubated with MBP-Lpd-RAPH or MBP only as control, and direct interaction was detected in a Western blot against MBP. (H and I) The interaction between Lpd and Abi is positively regulated by active Rac. Immunoprecipitation using Lpd-specific antibodies or IgG control from HEK293 cell lysates expressing GFP-Abi, GFP-Lpd, and dominant-active Rac (DA Rac; H, left) or dominant-negative Rac (DN Rac; H, middle), or empty vector control (H, right) show increased coimmunoprecipitation between Lpd and GFP-Abi only when dominant-active Rac is coexpressed. Western blot: anti-GFP. (I) Comparison of efficiency of coimmunoprecipitation of Lpd with GFP-Abi from blots in H. Quantification of band intensity of chemiluminescence imaged with a charge-coupled device camera. Coimmunoprecipitation is increased by >100% compared with empty vector and 150% compared with DN-Rac. Error bars indicate mean ± SEM, n = 3. One-way ANOVA and Tukey’s test were used. *, P < 0.05; **, P < 0.01.
Collectively, we have shown that the interaction between Lpd and Abi is direct and mediated by the SH3 domain of Abi and three binding sites in Lpd, and that this interaction is sufficient and necessary for the interaction of Lpd with the Scar/WAVE complex.
Lpd directly interacts with active Rac, which positively regulates the interaction between Lpd and the Scar/WAVE complex
It has been shown that MIG-10 is in complex with Rac (Quinn et al., 2008). To explore whether Lpd is also in complex with Rac, we pulled down GFP-Lpd or the GFP-Lpd-RA-PH domains from HEK lysates with purified GST-Rac. This revealed that Lpd (Fig. 3 E) and the RA-PH domains of Lpd are in complex with active Rac (Fig. 3 F). To assess whether this interaction is direct we pulled down purified maltose binding protein (MBP)-tagged RA-PH domains of Lpd with GTPγS- or GDPβS-loaded purified GST-Rac protein and found that it preferentially directly interacts with active Rac (Fig. 3 G).
This prompted us to explore whether active Rac regulates the interaction between Lpd and the Scar/WAVE complex. We expressed GFP-Lpd and GFP-Abi with and without all Myc-tagged components of the Scar/WAVE complex in HEK cells and found that significantly more Abi coimmunoprecipitates with Lpd when dominant-active Rac (DA Rac) was coexpressed compared with coexpression of dominant-negative Rac (DN Rac) or when Rac was not coexpressed (Fig. 3, H and I; and Fig. S2, B and C). This suggests that Lpd functions as a novel Rac effector because the interaction between Lpd and the Scar/WAVE complex is positively regulated by active Rac.
Lpd regulates cell migration via Abi and the Scar/WAVE complex
The Scar/WAVE complex regulates lamellipodium formation by activating the Arp2/3 complex (Machesky and Insall, 1998; Miki et al., 1998), and both complexes regulate cell migration (Suetsugu et al., 2003; Yan et al., 2003; Suraneni et al., 2012; Wu et al., 2012). We identified Lpd as a protein that regulates lamellipodia formation (Krause et al., 2004), but its role in mesenchymal and epithelial cell migration had not been determined.
To explore the role of Lpd in mesenchymal cell migration, we generated conditional KO MEFs (Lpd WT MEFs) from Lpd KO mice (see Fig. 6) and transduced them with retroviruses conferring 4-OH-tamoxifen (4-OHT) inducible CreERT2 (Oskarsson et al., 2006). When treated with 4-OHT, these Lpd WT MEFs lose the Lpd gene and Lpd expression (Lpd KO MEFs; Fig. 4 A). Expression levels of Scar/WAVE1, RIAM, Mena, VASP, or EVL did not change in the Lpd KO MEFs compared with Lpd WT MEFs (Figs. 4 B and S2 D). Lpd KO MEFs were impaired in lamellipodium formation (Fig. 4, C and D), which is consistent with earlier observations that Lpd knockdown cells are devoid of lamellipodia (Krause et al., 2004).
Figure 6.

Lpd functions to regulate melanoblast migration. (A) Conditional Lpd KO mice were generated by flanking exon 4 with loxP sites. Cre-mediated recombination of the loxP sites results in the removal of exon 4, creating a frame shift between exon 3 and 5 and premature termination. (B and C) Conditional Lpd KO mice crossed with β-actin-Cre mice on a mixed genetic background produced mice with a reduced body size (−20.6 ± 3.0% SEM; ****, P ≤ 0.0001, unpaired t test), which also display missing pigmentation on the ventral side (D). (E and F) To visualize melanoblasts, DCT-LacZtg/tg;β-actin-Cretg/+;Lpdflox/flox whole-mount embryos at E14.5 were stained for β-galactosidase expression in the melanoblasts. (E) Areas within three 1 mm × 1.5 mm boxes positioned at the middle of the trunk between the fore and hind limbs were quantified in WT and KO animals. Bar, 1.5 mm. (F) Lpd KO mice show a significant reduction in the number of melanoblasts in all three boxes. Bar, 150 µm. Melanoblast numbers were reduced by ∼60%, ∼40%, and ∼30% for boxes 1, 2, and 3, respectively (20 KO or WT embryos from three litters; unpaired t test; *, P < 0.05; error bars indicate SEM). See also Fig. S4.
Figure 4.

Lpd regulates cell spreading. (A and B) Western blot of cell lysates of Lpd WT and Lpd KO MEFs using anti-Lpd (A) or Scar/WAVE1 (B). Loading control: anti-HSC70. (C and D) F-actin staining (phalloidin) in Lpd WT (C) and Lpd KO MEFs (D). Arrows in C indicate the presence of lamellipodia in Lpd WT MEFs. Arrowheads in D indicate the absence of lamellipodia. (E and F) F-actin staining (phalloidin) determines the area of Lpd WT (E) and Lpd KO MEFs (F) after 60 min of spreading on fibronectin. (G) Quantification of the spreading area of MEFs from E and F. Values are mean ± SEM (error bars) of 131 (KO) or 155 (WT) cells. Unpaired, two-tailed t test: ****, P ≤ 0.0001. Bars, 25 µm.
Cells use lamellipodia for spreading on extracellular matrix and during cell migration. Lpd KO MEFs spread significantly more slowly (spread area reduced by 38% at 60 min) compared with Lpd WT MEFs (Fig. 4, E–G). We then analyzed random cell migration and found that migration speed and persistence were significantly reduced by 45% and 36%, respectively (Fig. 5, A and B). Likewise, directional migration into a scratch wound was impaired in the Lpd KO MEFs compared with the Lpd WT MEFs (reduced by 50%; Fig. 5, C and D). In agreement, we found that directional migration into a scratch wound was also highly reduced when Lpd expression was knocked down in the Rat2 fibroblast cell line (Fig. S3, A and B), which indicates that this defect is not cell line dependent. WT Rat2 fibroblasts migrated with a polarized lamellipodium, whereas Lpd knockdown cells migrated by extending filopodia (Video 2), which was also observed in the Lpd KO MEFs (Video 1), similar to Arp2/3 knockdown and KO cells (Suraneni et al., 2012; Wu et al., 2012). We also overexpressed Lpd in MDA-MB231 breast cancer cells and found that this dramatically increased migration in a scratch wound healing experiment (Fig. 5 E).
Figure 5.

Lpd regulates cell migration via Abi and the Scar/WAVE complex. (A and B) Quantification of velocity (A) and persistence (B) of randomly migrating Lpd WT or KO MEFs. Mean population speed and persistence (dt = 2, TR = 4; see Materials and methods for calculation). Results are mean ± SEM (error bars), with three independent experiments. ****, P ≤ 0.0001, unpaired t test. (C and D) A confluent layer of WT or KO Lpd MEFs was scratched, and the area of the scratch measured at 0 and 24 h. Bar, 500 µm. Area closure is shown as the percentage of WT cells. (D) Results are mean ± SEM, with four independent experiments. ***, P ≤ 0.001, unpaired t test. See also Fig. S3 and Videos 1 and 2. (E and F) Lpd overexpression increases cell migration speed via Abi and Scar/WAVE. MDA-MB231 breast cancer cells, stably expressing Nap1-specific (Nap1 shRNA 1 or 2) or scrambled control shRNA were transiently transfected with GFP-Lpd or GFP as control (E) or GFP-Lpd, GFP-LpdEVMut, GFP-LpdAbiMut, GFP-LpdEV+AbiMut, or GFP as control (F). A confluent cell layer was scratched and the area of the scratch was measured at 0 and 24 h. Area closure is shown as percentage increase over GFP cells. Results are mean ± SEM (error bars), from three independent experiments. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ns, not significant; one-way ANOVA was used. (E) Tukey’s test. (F) Newman-Keuls method.
Because the Scar/WAVE complex regulates lamellipodia formation and cell migration through activation of the Arp2/3 complex (Machesky and Insall, 1998; Suetsugu et al., 2003; Yan et al., 2003), we hypothesized that Lpd may function upstream of the Scar/WAVE complex to control cell migration. To explore this, we stably knocked down Nap1, which leads to proteasomal degradation of the other members of the Scar/WAVE complex (Kunda et al., 2003). Overexpressing GFP-Lpd resulted in increased wound closure in nontargeting shRNA control but not in Nap1 knockdown cell lines (Fig. 5 E), which suggests that Lpd function in cell migration is mediated by the Scar/WAVE complex.
To further assess whether the direct interaction between Lpd and the Scar/WAVE complex is required for Lpd’s role in cell migration, we overexpressed the Lpd mutant in all three Abi binding sites (GFP-LpdAbiMut), which significantly reduced wound closure compared with GFP expression in MDA-MB231 cells, whereas overexpression of GFP-Lpd increased cell migration (Fig. 5 F).
To explore Ena/VASP’s contribution to Lpd’s role in cell migration, we overexpressed an Lpd mutant in all seven Ena/VASP binding sites (GFP-LpdEVMut) and a GFP-Lpd mutant harboring mutations in both Ena/VASP and Abi binding sites (GFP-LpdEV+AbiMut). We observed that the GFP-LpdEVMut increased cell migration into the scratch wound to a similar extent as WT Lpd. In contrast, the double mutant GFP-LpdEV+AbiMut decreased cell migration compared with GFP expression to a similar extent as the Lpd Abi binding mutant (GFP-LpdAbiMut; Fig. 5 F).
Collectively, this suggests that Lpd’s function in cell migration is not mediated by Ena/VASP proteins but is predominantly facilitated by the Scar/WAVE complex.
Lpd KO mice have pigmentation defects
To explore the role of Lpd in cell migration in vivo, we generated conditional Lpd KO mice (Fig. 6 A). We crossed these mice to PGK-Cre (ubiquitous deletion) mice on a pure C57BL/6 genetic background to delete the Lpd gene in all cells. The PGK-Cre;Lpdflox/flox KO mice have a reduced body size, show no ingestion of milk, and tend to die shortly after birth. From crosses of PGK-Cretg/+;Lpdflox/+ × PGK-Cre+/+;Lpdflox/flox we obtained the expected Mendelian ratio directly after birth (postnatal day 0 [P0]), but at P10 (n = 208) only 10.6% of PGK-Cretg/+;Lpdflox/flox compared with the expected 25% survived. Many of these PGK-Cre tg/+;Lpdflox/flox mice died before the age of 6 wk, and those that reached sexual maturity were infertile.
We also used β-actin-Cre (ubiquitous deletion) mice on a mixed genetic background to obtain more viable Lpd KO mice. Western blots of tissue lysates from homozygous β-actin-Cretg/+;Lpdflox/flox mice showed loss of Lpd protein expression as expected (Fig. S4 A). Some homozygous β-actin-Cretg/+;Lpdflox/flox mice died within 4 wk after birth (at P14 only 42% and at P28 only 33% of expected mutants survived). The surviving β-actin-Cretg/+;Lpdflox/flox mice had a reduced body weight (reduced by 20%; Fig. 6, B and C) but were viable, and some were fertile, most likely due to modifier genes differently expressed in the mixed genetic background. Interestingly, the few surviving PGK-Cretg/+;Lpdflox/flox mice and the β-actin-Cretg/+;Lpdflox/flox mice displayed missing pigmentation on their ventral side (“white belly spots”; Fig. 6 D), which suggests NC/melanoblast migration defects. We observed that Lpd is expressed in melanoblasts in agreement with a putative role for Lpd in this cell type (Fig. S4 B).
NC cells that are destined to become melanoblasts emigrate at embryonic day 9.5 (E9.5) from the neural tube after undergoing EMT. Dopachrome tautomerase (DCT) is a melanogenic enzyme required for hair and skin pigmentation. DCT is expressed from E10.5 and serves as a marker for the melanoblast lineage migrating dorsolaterally through the dermis around E11.5. At E13.5, these cells migrate from the dermis into the epidermis, where they distribute evenly at E14.5 (Wehrle-Haller et al., 2001; Lin and Fisher, 2007). To visualize melanoblasts, we used a transgenic mouse line (DCT-lacZ mice) that expresses lacZ from the DCT promoter and allows visualization of melanoblast distribution by β-galactosidase staining in whole mount embryos (Mackenzie et al., 1997). We generated DCT-lacZtg/tg;β-actin-Cretg/+;Lpdflox/flox embryos to compare the number and distribution of melanoblasts at E14.5 with that of WT (DCT-lacZtg/tg;β-actin-Cre+/+;Lpdflox/flox) littermates. We quantified the number of melanoblasts in the trunk region (three 1.5-mm × 1-mm boxes between fore and hind limb; Fig. 6 E) where ventral depigmentation had been observed (Fig. 6, D and E). We observed that Lpd KO embryos had significantly fewer melanoblasts in all three areas (reduced by: box 1, ∼60%; box 2, ∼40%; box 3, ∼30%) along the dorso-ventral path (Fig. 6 F), which suggests that Lpd functions to regulate NC/melanoblast development. However, this analysis does not allow us to distinguish between a problem in NC formation versus migration. To test this, we examined NC development in Xenopus embryos, a system in which NC migration can be directly assessed in vivo (Theveneau and Mayor, 2012).
Lpd and the Scar/WAVE complex cooperate to regulate NC migration in vivo
X. laevis embryos were injected with Lpd mRNA, antisense morpholinos (Lpd MO, Abi MO), and dominant-negative Lpd constructs (Lyulcheva et al., 2008) containing only the N terminus, including the RA-PH domain of Lpd (Lpd N1, Lpd N6), and NC migration was analyzed by examining expression of the NC marker gene Twist (Hopwood et al., 1989). Overexpression of Lpd did not affect NC migration. Strikingly, knockdown of Lpd or Abi expression and expression of Lpd dominant-negative constructs (Lpd N1, Lpd N6) impaired the migration of the NC streams (Fig. 7, A–D), which indicates that Lpd, Abi, and the Scar/WAVE complex regulate NC migration in vivo.
Figure 7.

Lpd regulates NC migration via Abi. (A) In situ hybridization for Twist (Hopwood et al., 1989; migratory NC marker) in control embryos or embryos injected with Lpd mRNA, Lpd, or Abi MOs (Lpd MO or Abi MO) or the dominant-negatives Lpd N1, Lpd N6, and Abi1ΔSH3. Black arrowheads, normally migrating NC streams. Red arrowheads, streams with impaired migration. Blue asterisks, the eye. (B) Summary of phenotypes. Lpd overexpression has no effect on overall NC cell migration. Lpd MO, Abi MO, and the dominant-negatives Lpd N1, Lpd N6, and Abi1ΔSH3 all impair NC cell migration. (C) Percentages of embryos with normal migration along the dorso-ventral axis (nctl = 26, nLpdRNA = 41, nLpdMO = 37, nLpdN1 = 26, nLpdN6 = 49, nAbiMO = 16, and nAbi1ΔSH3 = 24). (D) Mean distance of migration along the dorso-ventral axis as a percentage compared with control embryos (nctl = 12, nLpdRNA = 14, nLpdMO = 12, nLpdN1 = 14, nLpdN6 = 14, nAbiMO = 12, nAbi1ΔSH3 = 12). One-way ANOVA and Dunnett’s test were used. ***, P < 0.001. (E) In situ hybridization for Twist in Lpd or Abi (Lpd MO or Abi MO), Lpd mRNA and Lpd MO, or Abi mRNA and Abi MO or control MO injected embryos. (F) Mean distance of migration along the dorso-ventral axis compared with control MO embryos (nctl MO = 13, nLpdMO = 14, nLpdMO = 26, nLpdMO+LpdmRNA = 20, nAbiMO = 19, nAbiMO+AbimRNA = 26). One-way ANOVA and Dunnett’s test were used. **, P < 0.01; ns, nonsignificant. (G–I) Lpd and Abi function cell-autonomously in NC migration. (G) Schematic diagram of graft experiment in H. (H) NC from MO-treated embryos or WT embryos was grafted into WT or MO-treated embryos, and NC migration was analyzed. (I) Quantification of NC migration phenotypes from H. Numbers (embryos with inhibition of NC migration/total): nctl>ctl = 0/4 (0%), nLpdMO>ctl = 4/6 (66%), nctl>LpdMO = ¼ (25%), nAbiMO>ctl = 4/5 (80%), nctl>AbiMO = 0/5 (0%). Bars, 150 µm.
Coexpression of Lpd or Abi mRNA with the Lpd or Abi MOs rescued NC migration, which suggests that the effect of the Lpd and Abi MO is specific (Fig. 7, E and F). No effect on NC formation was observed under any treatment (Fig. S5), which indicates that Lpd and the Scar/WAVE complex specifically function in NC migration.
To test whether Lpd and Abi act within NC cells or in the surrounding tissue to control NC migration, we performed experiments in which we grafted NC tissue from Lpd or Abi MO–injected embryos into nontreated control hosts. Conversely, we grafted control NC tissue into embryos injected with Lpd or Abi MOs. We observed that Lpd- or Abi-deficient NC grafted into normal hosts exhibited migration defects in 60–80% of embryos, whereas when normal NC was grafted into Lpd or Abi deficient hosts, NC migration was normal (Fig. 7, G–I). This experiment indicates that Lpd and Abi function cell-autonomously to regulate NC migration.
Because Lpd functions in cell migration upstream of the Scar/WAVE complex (Fig. 5, E and F) and interacts with the Scar/WAVE complex via the SH3 domain of Abi (Fig. 2 and Fig. 3, A–D), we hypothesized that the Abi1ΔSH3 construct would block the interaction between Lpd and the Scar/WAVE complex and therefore might have a dominant-negative function. Notably, expression of Abi1ΔSH3 impaired NC migration (Fig. 7, A–D), which suggests that Lpd and the Scar/WAVE complex cooperate to regulate NC migration.
Lpd’s function in regulating lamellipodia formation and cell migration is mediated by the Scar/WAVE complex
As previously described for other cell types (Krause et al., 2004), Lpd was localized to the very edge of lamellipodia in Xenopus NC cells (Fig. 8 A and Video 3). To further analyze the function of Lpd and the Scar/WAVE complex in lamellipodium protrusion and cell migration, the behavior of Xenopus NC explants was analyzed in vitro using time-lapse video microscopy. Overexpression of Lpd or Lpd dominant-negative (Lpd N6) led to a significant increase or decrease in the size of the lamellipodia at the edge of explants, respectively (Fig. 8, B–E; and Video 4). Importantly, the increase in lamellipodia size induced by Lpd could be reversed by coexpression of dominant-negative Abi (Abi1ΔSH3; Fig. 8, B and E; and Video 4), which indicates that Lpd’s function to regulate lamellipodia size is mediated by the Scar/WAVE complex.
Figure 8.
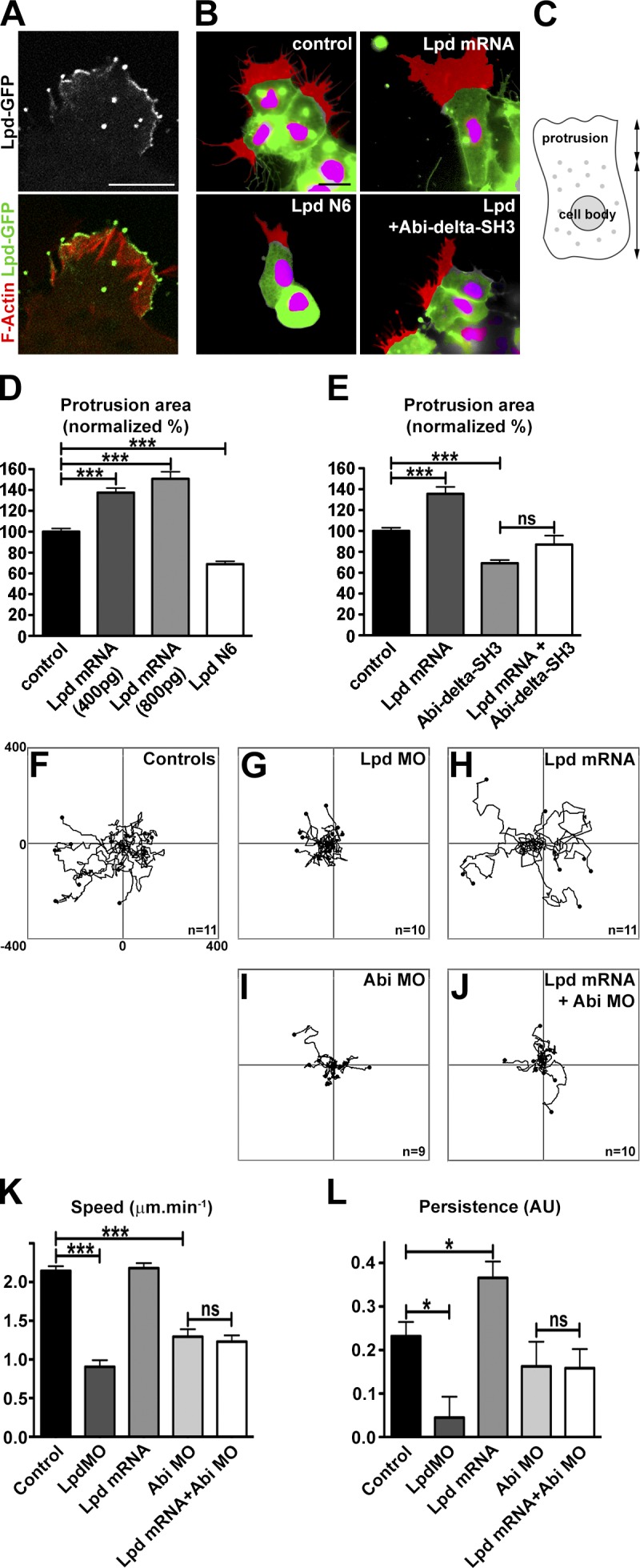
Lpd regulates lamellipodia protrusion and cell migration via Abi in Xenopus NC cells. (A) Localization of Lpd-GFP in Xenopus NC cells cultured on fibronectin. Actin filaments are stained with TRITC-Phalloidin. (B) Cells expressing nuclear mCherry (pseudocolored magenta by thresholding) and membrane GFP (pseudocolored green by thresholding) were used to analyse cell protrusions (pseudocolored red). Cell protrusions were defined as the area of protrusion that extends beyond the cell body. Cell protrusions are shown for control cells and cells injected with Lpd mRNA, Lpd N6 or co-injected with Lpd mRNA and Abi-Δ-SH3. Note that Lpd overexpression leads to enlarged protrusions. This effect is abolished by co-injection with Abi-Δ-SH3.. (C–E) Area of cell protrusions expressed as a proportion of normal protrusions (graph in D: nctl = 58, nLpd800pg = 33, nAbi1ΔSH3 = 24, nLpd+Abi1ΔSH3 = 17 [one-way ANOVA and Dunnett’s test; ***, P < 0.001]; graph in E: nctl = 154, nLpd400pg = 148, nLpd800pg = 157, nLpdN6 = 121 [one-way ANOVA and Tukey’s test; ***, P < 0.001]). (F–J) Tracks of NC cells cultured on fibronectin (a subset of the analyzed tracks are shown). (K and L) Velocity and persistence from tracks performed on all conditions (nctl = 543, nLpdMO = 240, nLpdmRNA = 297, nAbiMO = 180, nLpd+AbiMO = 290). One-way ANOVA and Dunnett’s test were used. *, P < 0.05; ***, P < 0.001. Error bars indicate SEM. See also Fig. S5 and Videos 3–6.
Because Lpd and the Scar/WAVE complex regulate NC migration in vivo, we explored their cooperation in more detail using cells from dissociated explants. Lpd overexpression increased persistence, whereas knockdown reduced migration speeds and persistence of NC cells (Fig. 8, F–L; and Video 5). To test whether the Scar/WAVE complex is required for Lpd’s function in migration of individual NC cells, we overexpressed Lpd and knocked down Abi with a MO. The increase in persistence upon Lpd overexpression was dependent on the presence of Abi (Fig. 8 L and Video 6), which suggests that in NC cells the Scar/WAVE complex functions downstream of Lpd during cell migration.
Collectively, these data suggest that in both mammalian cells and Xenopus NC cells, Lpd’s function in regulating cell migration is mediated by the Scar/WAVE complex.
Lpd and the Scar/WAVE complex regulate epithelial collective cell migration in vivo
So far we have shown that Lpd controls cell migration in mesenchymal cells, such as fibroblasts, melanoblasts, and NC cells; however, epithelial cells are also migratory during collective cell migration. To examine whether Lpd’s function to regulate the Scar/WAVE complex is a general feature of different modes of cell migration and to test whether it is evolutionarily conserved in invertebrates, we turned to a model for collective cell migration in the Drosophila ovary. In this system, a group of epithelial-derived somatic follicle cells are recruited into a migratory cluster of border cells that delaminates from the epithelium and migrates between neighboring nurse cells to the oocyte. First, we examined whether the fly orthologue of Lpd, Pico, forms a complex with Scar/WAVE. GST-Pico but not GST pulled down all five proteins of the tagged Scar/WAVE complex expressed in HEK293 cells (Fig. 9 A), indicating that this interaction is evolutionary conserved. To test the involvement of pico and Scar in border cell migration, we specifically knocked down their expression in the border cell cluster by RNAi. Both pico and Scar RNAi resulted in reduced border cell migration at stage 9 (Fig. 9 B). pico overexpression also impaired border cell migration at stage 9 and 10 (Fig. 9, B and C), which suggests that Lpd/Pico and the Scar/WAVE complex regulate collective cell migration.
Figure 9.

pico regulates Drosophila border cell migration via SCAR. (A) GST-Pico, the fly orthologue of Lpd, pulled down all Myc-tagged components of the Scar/WAVE complex. Myc-HSPC300 is not shown. (B) pico knockdown or overexpression, or SCAR RNAi under the control of slbo-GAL4 abrogate migration at stage 9. (B, top) Representative images of WT and defective egg chambers. Green, GFP-labeled border cells; red, DNA; blue, F-actin. The box and whiskers plot shows mean border cell position: distance of the cluster relative to the most anterior position of the overlying follicle cells (broken lines). Top and bottom box: 75th and 25th quartile; whiskers indicate minimum and maximum. One-way ANOVA and Dunnett’s test were used. ***, P < 0.001; n = 50. (C) pico overexpression abrogates migration at stage 10A, and this is ameliorated by SCAR RNAi. Histogram summarizes migration defects in the indicated genotypes. Migration was calculated as a percentage of the distance traveled to the oocyte/nurse cell boundary (broken lines in top panels). For each genotype, n = 50 egg chambers.
To explore whether pico function in this process is mediated by the Scar/WAVE complex, we tested if pico and Scar genetically interact. The effect of pico overexpression on border cell migration was ameliorated by Scar RNAi, which suggests that pico function is indeed mediated by the Scar/WAVE complex during collective cell migration (Fig. 9 C). To further test this, we quantified the migration speed by live-cell imaging and observed that both pico RNAi and pico overexpression reduced the rate of border cell cluster movement per frame in the first half of migration (Fig. 10 A). Consistent with our earlier observations, Scar RNAi rescued the migration defect induced by pico overexpression.
Figure 10.

pico regulates Drosophila border cell migration via SCAR. (A–D) Analysis of time-lapse images of LifeAct-GFP–labeled border cells, using c306-GAL4 to drive the indicated genotypes (graphs indicate mean ± SEM; WT, n = 9; PVRDN, n = 9; pico RNAi, n = 12; pico OE, n = 12; SCAR RNAi, n = 8; SCAR RNAi, pico OE, n = 7). (A) Graph summarizing migration rate/frame calculated using a custom macro (Poukkula et al., 2011). (B) Graph showing percentage frames from the first half of migration with tumbling border cell clusters (see Materials and methods). (B, right) GFP-labeled clusters display a polarized or tumbling phenotype. Bar, 15 µm. (A and B) Tests used were one-way ANOVA (P < 0.0001) and Tukey’s test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Graph showing number of cellular extensions per frame, irrespective of their direction. One-way ANOVA and Dunnett’s test were used. *, P < 0.05; **, P < 0.01. (D) Graph summarizing percentage extensions/frame at front, back, or sides of the cluster. (D, right) Image of a border cell cluster before and after image segmentation. The body of the cluster is shown in blue, cellular extensions at the front in green, the back in red, and the sides in white. White lines indicate quadrants, representing front, back, and sides for quantification. Bar, 5 µm.
Border cell migration is characterized by polarized cell behavior in the first half of migration, followed by dynamic collective behavior, in which the cluster travels in shuffling and tumbling movements (Bianco et al., 2007). Unlike WT border cells, which normally show little or no tumbling in the first half of migration, we observed a high frequency of premature tumbling in pico-overexpressing clusters, which was dependent on Scar (Fig. 10 B), similar to what has been observed when overexpressing a dominant-negative PDGF receptor (PVRDN) in border cells (Poukkula et al., 2011). To explore the underlying causes of migration defects further, we quantified the number and direction of actin-based cellular protrusions from the border cell clusters. Overexpression of PVRDN resulted in a reduction in the total number of protrusions and a higher proportion of protrusions at the rear of the cluster when compared with WT, as reported previously (Poukkula et al., 2011). In many respects, the effect of pico and Scar RNAi on both the number and directionality of protrusions was similar to, but weaker than, PVRDN. pico overexpression too showed similar effects to PVRDN, with the notable exception that the absolute number of protrusions did not change, but many more rear facing protrusions appeared (Fig. 10, C and D). Importantly, Scar knockdown ameliorated the pico overexpression phenotype and restored the WT distribution of protrusions at the back of the cluster (Fig. 10 D), further demonstrating that pico function is mediated by the Scar/WAVE complex.
Discussion
Here we reveal that Lpd colocalizes with the Scar/WAVE complex at the very edge of lamellipodia and directly interacts with this complex by binding to the Abi-SH3 domain. Active Rac directly binds Lpd, thereby regulating the interaction between Lpd and the Scar/WAVE complex. We therefore postulate that Lpd acts as a platform to link active Rac and the Scar/WAVE complex at the leading edge of cells to regulate Scar/WAVE-Arp2/3 activity and thereby lamellipodium formation and cell migration.
Knockdown of Lpd expression (Krause et al., 2004) or KO of Lpd (Fig. 4) highly impaired lamellipodium formation, phenocopying the effect of Scar/WAVE complex knockdown on lamellipodium formation (Machesky and Insall, 1998; Innocenti et al., 2004; Steffen et al., 2004). Conversely, we observed that overexpression of Lpd increased lamellipodia size in Xenopus NC cells, and this was dependent on the interaction with Abi, linking it to the Scar/WAVE complex. Overexpression of Pico, the Lpd fly orthologue, aberrantly increased the number and frequency of cellular protrusions at the rear of border cell clusters in a Scar-dependent manner, which suggests that the regulation of Scar/WAVE by Lpd is evolutionary conserved. Collectively, these data suggest that Lpd functions to generate lamellipodia via the Scar/WAVE complex.
We found that Lpd or Pico knockdown or Lpd KO impaired cell migration in vitro and in vivo in Drosophila, Xenopus, and mice. Lpd KO or knockdown cells were unable to migrate via lamellipodia but instead migrated very slowly by extending filopodia. The same residual migration mode had been observed for Arp2/3 knockdown cells (Suraneni et al., 2012; Wu et al., 2012). Arp2/3 is activated by the Scar/WAVE complex to regulate cell migration (Insall and Machesky, 2009; Campellone and Welch, 2010). We also observed that both Lpd and Abi knockdown impaired NC migration in vivo. Consistently, we found that Lpd and Abi-Scar/WAVE are in the same pathway regulating cell migration. This is consistent with recent studies suggesting that the Lpd orthologue in C. elegans, mig-10, genetically interacts with abi-1 to regulate axon guidance, synaptic vesicle clustering, and excretory canal outgrowth in C. elegans (Stavoe et al., 2012; Xu and Quinn, 2012; McShea et al., 2013). Collectively, our results suggest that Lpd functions in cell migration via the Scar/WAVE complex in mammalian cells, Xenopus NC cells, and Drosophila border cells.
Lpd not only interacts with the Scar/WAVE complex but also directly binds to Ena/VASP proteins (Krause et al., 2004; Michael et al., 2010). Ena/VASP proteins regulate actin filament length by temporarily preventing capping of barbed ends and by recruiting profilin-actin to the growing end of actin filaments (Bear et al., 2002; Krause et al., 2003). In contrast, the Scar/WAVE–Arp2/3 complexes increase branching of actin filaments. Lamellipodia with a highly branched actin network protrude more slowly but are more persistent, whereas lamellipodia with longer, less branched actin filaments protrude faster but are less stable and quickly turn into ruffles (Bear et al., 2002; Krause et al., 2003). We observed that Lpd overexpression increases cell migration in a Scar/WAVE- and not Ena/VASP-dependent manner (Fig. 5 F). This is consistent with a predominant function of Scar/WAVE downstream of Lpd to regulate a highly branched actin network supporting persistent lamellipodia protrusion and cell migration. Other actin-dependent cell protrusions such as axon extension or dorsal ruffles of fibroblasts require Lpd-Ena/VASP–mediated F-actin structures (Michael et al., 2010).
Collective cell migration describes a group of cells that moves together and affect each other (Rørth, 2012), and various types of collective cell migration exists during development and cancer invasion (Friedl and Gilmour, 2009; Friedl et al., 2012). Xenopus NC cells migrate as loose streams, whereas Drosophila border cells migrate as a cluster of cells with close cell–cell contacts (Rørth, 2012; Theveneau and Mayor, 2012). We found that Rac regulates Lpd and Scar/WAVE interaction and that both are required for Xenopus NC migration, which is consistent with our previous work in which Rac activity mediates this type of migration (Carmona-Fontaine et al., 2008; Matthews et al., 2008; Theveneau et al., 2010). Similarly, NC–derived melanoblast migration in the mouse depends on Rac–Scar/WAVE–Arp2/3 (Li et al., 2011), and we found that Lpd functions in this process as well.
Drosophila border cell clusters migrate through the fly egg chamber in two phases: an early part characterized by large and persistent front extensions, which are regulated predominantly by PVR (the fly PDGF receptor); and a late part characterized by dynamic collective “tumbling” behavior (Bianco et al., 2007; Poukkula et al., 2011). Surprisingly, Pico overexpression resulted in the appearance of a higher proportion of rear facing extensions, a phenotype previously observed with dominant-negative PVR (Prasad and Montell, 2007), causing premature tumbling of the border cell cluster. This suggests that Pico function is normally tightly controlled to stabilize specific extensions and functions also in guidance of collective cell migration. Because Lpd-Scar/WAVE control single cell migration as well as collective cell migration, this suggests that they function as general regulators of cell migration.
Collectively, we have identified a novel pathway in which Lpd functions as an essential, evolutionary conserved regulator of the Scar/WAVE complex during cell migration in vivo.
Materials and methods
Molecular biology, plasmids, and reagents
The following materials were used. GFP-Lpd (Krause et al., 2004). Lpd-N1 (aa 1–592) and Lpd-N6 (aa 242–592) was amplified from human Lpd (AY494951), cloned into KpnI-EcoRI of pENTR3C (Invitrogen), and transferred into pDEST-EGFP (which was generated by subcloning the destination cassette [Invitrogen] into pEGFP-N1 [Takara Bio Inc.]). Pico-L (Lyulcheva et al., 2008) in pDEST27 (Invitrogen). Sra1 (CYFIP1; HsCD00042136), PIR121 (CYFIP2; HsCD00045545), Nap1 (NCKAP1; HsCD00045562), Abi-2 (HsCD00042752), and HSPC300 (C3orf10; HsCD00045008) in pDONR221 (Harvard Institute of Proteomics). pDONR221-WAVE-2 (Deutsches Ressourcenzentrum für Genomforschung). hsAbi1d (BC024254; Geneservice) full-length and Abi1d-Δ-SH3 (aa 1–417) cloned into pENTR11 and transferred to pRK5-Myc-DEST, pV16-gateway (MBP). Abi1d SH3 (aa 410–472) in pGEX-6P1. pCDNA3-2×FLAG-hsScar/WAVE1 was a gift from J. Scott (University of Washington, Seattle, WA). Nap1-shRNA-1 (5′-GCTCACCATCCTCAACGAC-3′) and Nap1-shRNA-2 (5′-CCAGATTGCTGCAGCTTTG-3′) in pLL3.7puro. Xenopus laevis Lpd (5′-TCTTCATCCGATAATTGCTCCATCT-3′) and Abi (5′-AACATCTGTAGCTCAGCCATCTTCC-3′) MOs (Gene Tools LLC).
Antibodies
The following antibodies were used: Lpd pab 3917 (Krause et al., 2004), RIAM 4613 (Lafuente et al., 2004), VASP pab 2010 (Bear et al., 2000; a gift from F. Gertler, Massachusetts Institute of Technology, Cambridge, MA), EVL mAb 84H1 (Lanier et al., 1999), Mena mAb A351F7D9 (Lebrand et al., 2004), Hsc70 mAb (Santa Cruz Biotechnology, Inc.), GST pab (GE Healthcare), Myc mAb 9E10, FLAG mAb M2 (Sigma-Aldrich), GFP mAb (Roche), Scar1 (BD), Sra1 mAb 30A4 (Synaptic Systems), Nap1 pab 5151 (peptide HAVYKQSVTSSA; Eurogentec), Abi mAb (Abcam), Arp3 mAb (BD), Tubulin, β-actin mAbs (Sigma-Aldrich), MBP mAb (New England Biolabs, Inc.), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mAb (EMD Millipore). Secondary antibodies used were: HRP-goat anti–rabbit, goat anti–mouse, and rabbit anti–goat (Dako).
Cell culture
MEFs isolated from E13 embryos from conditional Lpd KO mice were immortalized using the 3T3 method (Todaro and Green, 1963) and transduced with retroviruses conferring 4-OHT inducible CreERT2 (Oskarsson et al., 2006; a gift from A. Trumpp, Deutsches Krebsforschungszentrum, Heidelberg, Germany). The melanoblast stem cell line melb-a was grown using conditions described in Sviderskaya et al. (1995; provided by E. Sviderskaya, Wellcome Trust Functional Genomics Cell Bank at St. George’s, University of London, London, England, UK). B16F1, HEK293FT, NIH/3T3, MDA-MB231, or Lpd WT and KO MEFs were grown in DMEM (high glucose 4,500 mg/liter). CAD cells were grown in DMEM-Hepes/F12-Ham’s (1:1), 10% FCS, or 12.5% FCS for MEFs, or 10% calf serum for NIH/3T3, l-Glutamine, and penicillin/streptomycin. Cells were maintained at 37°C in 5% CO2 and transfected with Lipofectamine 2000 (HEK293FT) or LTX (MDA-MB231; Invitrogen). Primary cortical neurons were prepared from E16 mice as described for E18 rats (Goslin and Banker, 1989). In brief: the cortices of E16 mouse embryos were dissected and incubated for 20 min in 0.25% Trypsin, HBSS-Hepes, washed 3× with 5 ml plating media, triturated 25× with a P1000 tip, and plated on poly-d-lysine–coated dishes in plating media (Neurobasal [Invitrogen], 5% FCS, 2% B27 [Invitrogen], l-glutamine, and penicillin/streptomycin) and maintained without FCS at 37°C in 5% CO2 and lysed after 36 h.
Scratch, random migration, and cell spreading assays
Confluent control- or tamoxifen-treated Lpd MEFs or MDA-MB-231 cells were scratched with a P200 pipette tip and treated with mitomycin C, and the scratch area was measured at 0 and 24 h with ImageJ. Control- or tamoxifen-treated Lpd MEFs were plated onto fibronectin (10 µg/ml)-coated coverslips for 60 min (spreading) or in 12-well dishes for 12 h and imaged for 24 h every 5 min.
Quantification of cell migration speed and persistence
Cell track coordinates were reformatted into CEL format and imported into Mathematica for analysis using the Chemotaxis Analysis Notebook v1.5β by G. Dunn (King’s College London, England, UK). Sample frequency at which speed measurements were taken and used to calculate mean track speed (MTS) was defined as the lowest time interval (dt) at which the persistence of the population of cells was stable. Because different populations and groups have different persistence profiles, it was necessary to select the lowest common dt that was applicable to all groups being compared. This interval was also used to calculate persistence, which is the total sum displacement over the straight-line distance for a fixed time ratio (TR). e.g., TR = 4 means that the straight-line distance from start of dt1 to the end point of dt4 was divided by the sum displacement of four consecutive dt intervals (dt1+ dt2 + dt3 + dt4). Tracks are generally long enough for multiple Sum[dt]/TR persistence measurements. The persistences for each track were therefore averaged to obtain mean track persistence (MTP), which is an accurate estimate for the persistence of a cell throughout the length of movies, and overcomes traditional persistence measurement obstacles of track length, sample frequency, and positional tracking error. MTS and MTP were averaged across each population of cells to obtain mean population speed (MPS) and mean population persistence (MPP) at designated dt and TR. The persistence profile of each population at increasing dt and fixed TR of four was plotted and used to determine the point at which each population’s persistence profile plateaued, indicating, therefore, that the population persistence was stable. For MEF migration, the lowest common dt was 30 min. For the Xenopus MO migration experiments the lowest dt was 21 min. Persistences were compared at TR = 4 and TR = 2 for MEFs and Xenopus, respectively.
Immunofluorescence analysis and imaging
For immunofluorescence analysis, cells were plated on nitric acid–washed coverslips (Hecht Assistant) and fixed with 4% paraformaldehyde-PHEM (60 mM Pipes, 25 mM Hepes, 10 mM EGTA, 2 mM MgCl2, and 0.12 M sucrose). Goat anti–rabbit or anti–mouse Alexa Fluor 488 or 568 (Invitrogen) secondary antibodies were used, and cells were mounted (Prolong Gold; Invitrogen). A microscope (IX 81 [Olympus], with filter wheels [Sutter], an ASI X-Y stage, CascadeII 512B camera [Photometrics], and 4× UPlanFL, 10× UPlanFL, 60× Plan-Apochromat NA 1.45, or 100× UPlan-Apochromat S NA 1.4 objective lenses) was used, and line scans were analyzed with MetaMorph software.
Immunoprecipitation, GST pull-downs, and Western blotting
Cells were harvested in lysis buffer (50 mM Tris HCL, 200 mM NaCl, 1% NP-40, 2 mM MgCl2, 10% glycerol, pH 7.4, 1 mM Na3VO4, 10 mM NaF, and protease inhibitors [complete mini without EDTA]; Roche). Lysates were incubated on ice for 30 min and centrifuged at 17,000 g at 4°C for 15 min. Protein concentration was then determined (Pierce BCA protein assay kit; Thermo Fisher Scientific). Lysates were precleared with protein A beads (Thermo Fisher Scientific) and incubated with glutathione beads for GST pull-downs or with antibody or control IgG, followed by 1% BSA blocked protein A beads. Beads were washed with lysis buffer, separated on SDS-PAGE gels, transferred onto Immobilon-P membranes (EMD Millipore), blocked in 5% BSA, and probed with the indicated antibodies, followed by HRP secondary antibodies (Dako). Blots were developed with the ECL kit (Thermo Fischer Scientific) and x-ray film, or the Immun-Star WesternC ECL kit (Bio-Rad Laboratories) using the Bio-Rad Imager and ImageLab software.
Far-Western blot and peptide array
GST and MBP fusion proteins were purified from BL21 E. coli using glutathione (GE Healthcare) or amylose (New England Biolabs, Inc.) beads. Western blots of purified GST-Lpd fragments or custom-made peptide arrays (Cancer Research UK services) were overlayed as described previously (Niebuhr et al., 1997) with purified MBP-Abi full-length or MBP-Abi-Δ-SH3, and MBP was detected with anti-MBP antibodies (New England Biolabs, Inc.).
Generation of Lpd conditional KO mice
All experiments were performed according to UK Home Office regulations. Lpd conditional KO mice were generated using C57BL6 ES cells (TaconicArtemis GmbH) by flanking exon 4 with loxP sites (Fig. 5 A), and the selection cassette was removed by crossing to Flp mice and crossed to PGK-Cre (Lallemand et al., 1998; general deleter) C57BL6 (provided by A. Behrens, London Research Institute, Cancer Research UK, London, England, UK), β-actin-Cre (Lewandoski and Martin, 1997), or DCT-lacZtg/tg (Mackenzie et al., 1997; provided by I. Jackson, Medical Research Council, Edinburgh, Scotland, UK) mice on a mixed genetic background.
Whole-mount β-galactosidase staining of embryos
DCT-lacZtg/tg;β-actin-Cretg/+;Lpdflox/flox and DCT-lacZtg/tg;β-actin-Cre+/+;Lpdflox/flox embryos were used at E14, with time of gestation calculated using the day of detection of a vaginal plug as E1. Embryos were dissected in ice-cold PBS and fixed in 0.25% glutaraldehyde at 4°C for 40 min on a rolling platform. Fixed embryos were washed in ice-cold PBS for 15 min at 4°C on a rolling platform before incubating in permeabilization buffer (2 mM MgCl2, 0.02% NP-40, and 0.01% sodium deoxycholate in PBS, without Ca2+ or Mg2+) for 30 min and two further washes for 15 min at room temperature before staining with β-galactosidase substrate solution (2 mM MgCl2, 0.02% NP-40, 0.01% sodium deoxycholate, 5 mM K3Fe, 5 mM K4Fe, and 0.4 mg/ml 5-bromo-4-chloro-3-indolyl β-D-galactosidase) for 24–48 h in darkness, at 4°C on a rolling platform. Embryos not fully stained after this time were transferred to 37°C or left at 4°C in stain solution until staining was completed. Embryos were post-fixed in 4% PFA for 2 h at 4°C and transferred into PBS.
Melanoblast quantification
Both left and right side trunk regions were imaged using a microscope (SMZ1500; Nikon) and camera (Exi Aqua; QImaging). Melanoblast numbers from whole-mount embryos were quantified using MetaMorph Integrated Morphometric Analysis software within three 1 mm × 1.5 mm boxes positioned at the middle of the trunk between the fore and hind limbs. The three boxes were aligned next to each other with the most distal box (from the somites) placed closest to the umbilical cord. Melanoblast numbers were quantified from the boxes by running an automated object count from the MetaMorph Integrated Morphometric Analysis software using automated thresholding of the images. Melanoblasts were identified as objects by screening for objects that are within 10–88 pixels to filter nonspecific staining of veins and other small artifacts.
Xenopus methods
NC culture.
Vitelline membranes were peeled off from embryos at stage 15 using tweezers, and the embryos were left to recover. Around stage 18, the superficial pigmented layer was removed and NC cells were taken out using a hair mounted on a glass pipette. The NC explants were then cut is small pieces and transferred into a fibronectin-coated dish and cultured in modified Danilchick’s medium at room temperature.
In situ hybridization.
Embryos were fixed in MEMFA for 1 h at room temperature and washed several times in methanol. Embryos were then rehydrated by a series of methanol solutions of decreasing concentration, washed in PBS plus 0.1% Tween 20, and bleached in 5% H2O2 solution for a few minutes. A brief post-fixation in 3.7% in formaldehyde was performed, then embryos were washed in PBS and passed into a formamide-based hybridization buffer. Embryos were incubated overnight at 67°C in hybridization buffer containing a digoxigenin-labeled antisense probe (1 µg/ml). Probes are washed using formamide-based solutions, and PBS with 0.1% Tween 20. Embryos were passed into a blocking solution (TBS, 10% serum) and incubated overnight with an alkaline-phosphatase–coupled anti-digoxigenin antibody (1/3,000). Antibody was washed in TBS serum for one day and one night. Staining is performed in alkaline-phosphatase buffer containing NBT and BCIP (3 µl/ml). The mRNA probes used were Slug (Mayor et al., 1995) and Twist (Hopwood et al., 1989).
Time-lapse analysis.
For cell tracking, NC cells were dissociated in low-calcium/magnesium Danilchik’s medium and were cultured as single cells on fibronectin-coated glass coverslip monitored using a 10× objective lens on an inverted microscope (Axiovert; Carl Zeiss) equipped with a camera (Hamamatsu Photonics). Pictures were acquired every 3 min. Cell tracking was performed using ImageJ Manual Tracking plug-in and Imaris spot tracker software.
Cell protrusion analysis.
NC cells were plated on plastic Petri dishes and monitored using a 63× water-immersion lens on an upright microscope (Leica). Protrusions were defined as the region devoid of vitelline platelets.
Drosophila methods
Analysis of fixed Drosophila egg chambers.
Ovaries of 3-d-old females fed on fresh yeast were dissected in PBS and fixed with 4% paraformaldehyde, and stained with Alexa Fluor 633 Phalloidin (Invitrogen). At stage 9, the distance between the center of the border cell cluster and the anterior columnar follicle cells (“border cell position”) was measured in ImageJ (http://rsb.info.nih.gov/ij/). For stage 10 egg chambers, the percentage of the total migration was calculated using the distance from the anterior edge of the egg chamber to the center of the cluster as a percentage of the total migration distance (to the anterior columnar follicle cells). Transgenic lines for RNAi knockdown of pico and SCAR were as described previously (Jonchere and Bennett, 2013). UAS-SCAR RNAi (VDRC 21908) was provided by Vienna Drosophila RNAi Center. Genotypes of fixed samples were as follows:
slbo-GAL4, UAS-GFP, his2A-RFP/+.
slbo-GAL4, UAS-GFP, his2A-RFP/ UAS-pico RNAi (line 9); UAS-pico RNAi (line 4)/+.
slbo-GAL4, UAS-GFP, his2A-RFP/+; UAS-pico/+.
slbo-GAL4, UAS-GFP, his2A-RFP/UAS-SCAR RNAi (VDRC 21908).
slbo-GAL4, UAS-GFP, his2A-RFP/UAS-SCAR RNAi (VDRC 21908); UAS-pico/+.
Live imaging of Drosophila egg chambers.
Egg chambers from fattened ovaries (see “Drosophila methods“) were dissected and cultured at 25°C in imaging media (Schneider’s medium, 15% FBS, 1 µg/ml insulin, and 1 µg/ml streptomycin and penicillin) as described by Prasad et al. (2007), with minor modifications. In brief, ovaries were removed from the abdomen of fattened flies and separated into individual fresh droplets of media before dissection of single ovarioles. Egg chambers were placed between two coverslips in the center of a gas-permeable Lumox dish (Sigma-Aldrich). A third coverslip was placed on top to prevent movement while imaging and surrounded by halocarbon oil 27 (Sigma-Aldrich). Confocal stacks were taken at multiple positions using a confocal microscope (LSM 710; Carl Zeiss) equipped with a 488-nm argon ion laser, fitted to an inverted microscope with a 20× 0.75 NA Fluor objective. 2.5-µm-thick z sections (∼10–15 images/stack) were taken at each position at 2-min intervals. If necessary, imaging was paused briefly during the time lapse to adjust the focus. The time lapses were cropped at 50% (early stage) for analysis. Maximum intensity projections were generated, and subsequent analysis was performed using a custom macro for ImageJ to analyze border cell migration behavior and protrusion dynamics, as described in Poukkula et al. (2011; provided by P. Rorth [National University of Singapore, Singapore] for custom macros). In brief, time-lapse movies were split into the relevant stages of migration, and processed into binary images after individual thresholding of each movie. Images of border cell clusters were segmented into cluster cell body and cellular protrusions. Protrusions were then categorized by position with respect to the leading cell: front, 315–45°; back, 135–225°; side, 45–135° or 225–315°. In addition to logging the protrusion behavior, the macro automatically tracked the movement of the cluster in xy, enabling information such as migration speed and directed movement to be calculated. Rate per frame was calculated based on the mean distance the cluster center had moved from one frame to the next. Early tumbling was calculated as the mean percentage of frames per time lapse movie that showed rounded clusters, exhibiting changes in the position of individual cells within the cluster for two or more consecutive frames in the first half of migration. Genotypes of time-lapse images were as follows (some of the fly strains were provided by B. Stramer [King’s College London, London, UK], Denise Montell [Johns Hopkins University, Baltimore, MA], Vienna Drosophila RNAi Center, and Bloomington Drosophila Stock Center):
c306-GAL4/+; UAS-LifeAct-GFP/+.
c306-GAL4/+; UAS-LifeAct-GFP/UAS-pico RNAi (line 9); UAS-pico RNAi (line 4)/+.
c306-GAL4/+; UAS-LifeAct-GFP/+; UAS-pico/+.
c306-GAL4/+; UAS-LifeAct-GFP/UAS-SCAR RNAi (VDRC 21908).
c306-GAL4/+; UAS-LifeAct-GFP/+; UAS-PvrDN/+.
c306-GAL4/+; UAS-LifeAct-GFP/UAS-SCAR RNAi (VDRC 21908); UAS-pico/+.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software (see figure legends).
Online supplemental material
Fig. S1 shows the colocalization of Lpd with Scar/WAVE1, Abi1, and Sra1 at the leading edge of the CAD neuronal cell line. Fig. S2 shows Western blots that indicate that Lpd can be in complex with both Abi1 and VASP, that the interaction between Lpd and the Scar/WAVE complex is positively regulated by active Rac, and that protein levels of RIAM, Mena, EVL, and VASP are not altered in Lpd KO MEFs. Fig. S3 shows that knocking down Lpd expression reduces cell migration in scratch wound healing experiments in Rat2 fibroblasts. Fig. S4 demonstrates the absence of Lpd expression in Lpd KO mice and the presence of Lpd expression in mouse melanoblasts in Western blots. Fig. S5 shows that overexpression or knockdown of Lpd has no effect on NC induction in Xenopus. Videos 1 and 2 show one of the Lpd KO MEF (1) and one of the Rat2 fibroblasts scratch wound healing assays (2) used for quantification in Fig. 5 (C and D) and Fig. S3. Video 3 demonstrates that GFP-Lpd localizes to the leading edge of Xenopus NC cells. Video 4 shows that Lpd overexpression increases lamellipodia size. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201304051/DC1.
Supplementary Material
Acknowledgments
We thank Pernille Rorth for custom macros; Brian Stramer, Denise Montell, Vienna Drosophila RNAi Center, and Bloomington Drosophila Stock Center for Drosophila strains; Frank Gertler, John Scott, and Andreas Trumpp for sharing plasmids and antibodies; and Ian Jackson and Axel Behrens for sharing mice. We thank Elena Sviderskaya for the melb-a melanoblast stem cell line. We also thank Ang Li (Beatson Institute, Glasgow, Scotland, UK) for help with the β-galactosidase protocol.
L. Dodgson was supported by a Medical Research Council Capacity Building studentship. D. Soong is supported by a British Heart Foundation grant (RE/08/003). M. Kotini is supported by a PhD scholarship from the Latsis Public Benefit Foundation. E. Theveneau is supported by a Wellcome Trust Value in People award. M. Michael was supported by a Medical Research Council studentship. C. Navarro was supported by a European Molecular Biology Organization long-term fellowship. D. Bennett was supported by grants from Cancer Research UK (C20691/A11834; C20691/A6678), North West Cancer Research (CR847), and the Wellcome Trust (084659/Z/08/Z). R. Mayor is supported by grants from Medical Research Council UK (MR/J000655/1), Biotechnology and the Biological Sciences Research Council, and the Wellcome Trust. M. Krause is supported by grants from the Biotechnology and Biological Sciences Research Council (BB/G00319X/1; BB/F011431/1; BB/J000590/1) and the Wellcome Trust (077429/Z/05/Z; 082907/Z/07/Z).
Author contributions: A.-L. Law, C. Bodo, C. Navarro, and M. Krause performed the biochemistry; A. Vehlow, A.-L. Law, U. Perera, and C. Navarro performed the cell biology; and A.-L. Law, C. Bodo, and M. Krause performed the mouse experiments. D. Soong and A.-L. Law quantified cell migration and spreading. G.A. Dunn conceived and wrote the Mathematica notebook for analysis of cell migration. L. Dodgson and E. Taylor performed experiments on fixed Drosophila samples; L. Dodgson performed all Drosophila live imaging experiments and analyzed the data. D. Bennett conceived, designed, and analyzed the Drosophila data. M. Kotini and E. Theveneau performed all the Xenopus experiments; R. Mayor designed and analyzed the Xenopus data. M. Krause conceived, designed, and analyzed the mammalian experiments and wrote the manuscript. All authors contributed to the writing of the manuscript.
Footnotes
Abbreviations used in this paper:
- 4-OHT
- 4-OH-tamoxifen
- ANOVA
- analysis of variance
- DCT
- dopachrome tautomerase
- E
- embryonic day
- KO
- knockout
- MBP
- maltose binding protein
- MEF
- mouse embryonic fibroblast
- MO
- morpholino
- NC
- neural crest
- P
- postnatal day
- PH
- pleckstrin homology
- RA
- Ras association
- TR
- time ratio
- WT
- wild type
References
- Bear J.E., Loureiro J.J., Libova I., Fässler R., Wehland J., Gertler F.B. 2000. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 101:717–728 10.1016/S0092-8674(00)80884-3 [DOI] [PubMed] [Google Scholar]
- Bear J.E., Krause M., Gertler F.B. 2001. Regulating cellular actin assembly. Curr. Opin. Cell Biol. 13:158–166 10.1016/S0955-0674(00)00193-9 [DOI] [PubMed] [Google Scholar]
- Bear J.E., Svitkina T.M., Krause M., Schafer D.A., Loureiro J.J., Strasser G.A., Maly I.V., Chaga O.Y., Cooper J.A., Borisy G.G., Gertler F.B. 2002. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 109:509–521 10.1016/S0092-8674(02)00731-6 [DOI] [PubMed] [Google Scholar]
- Bianco A., Poukkula M., Cliffe A., Mathieu J., Luque C.M., Fulga T.A., Rørth P. 2007. Two distinct modes of guidance signalling during collective migration of border cells. Nature. 448:362–365 10.1038/nature05965 [DOI] [PubMed] [Google Scholar]
- Campellone K.G., Welch M.D. 2010. A nucleator arms race: cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 11:237–251 10.1038/nrm2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Fontaine C., Matthews H.K., Kuriyama S., Moreno M., Dunn G.A., Parsons M., Stern C.D., Mayor R. 2008. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature. 456:957–961 10.1038/nature07441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P., Gilmour D. 2009. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 10:445–457 10.1038/nrm2720 [DOI] [PubMed] [Google Scholar]
- Friedl P., Locker J., Sahai E., Segall J.E. 2012. Classifying collective cancer cell invasion. Nat. Cell Biol. 14:777–783 10.1038/ncb2548 [DOI] [PubMed] [Google Scholar]
- Goslin K., Banker G. 1989. Experimental observations on the development of polarity by hippocampal neurons in culture. J. Cell Biol. 108:1507–1516 10.1083/jcb.108.4.1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne P., Sechi A., Benesch S., Small J.V. 2001. Scar/WAVE is localised at the tips of protruding lamellipodia in living cells. FEBS Lett. 492:215–220 10.1016/S0014-5793(01)02239-6 [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. 2011. Hallmarks of cancer: the next generation. Cell. 144:646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hopwood N.D., Pluck A., Gurdon J.B. 1989. A Xenopus mRNA related to Drosophila twist is expressed in response to induction in the mesoderm and the neural crest. Cell. 59:893–903 10.1016/0092-8674(89)90612-0 [DOI] [PubMed] [Google Scholar]
- Innocenti M., Zucconi A., Disanza A., Frittoli E., Areces L.B., Steffen A., Stradal T.E., Di Fiore P.P., Carlier M.F., Scita G. 2004. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat. Cell Biol. 6:319–327 10.1038/ncb1105 [DOI] [PubMed] [Google Scholar]
- Insall R.H., Machesky L.M. 2009. Actin dynamics at the leading edge: from simple machinery to complex networks. Dev. Cell. 17:310–322 10.1016/j.devcel.2009.08.012 [DOI] [PubMed] [Google Scholar]
- Jonchere V., Bennett D. 2013. Validating RNAi phenotypes in Drosophila using a synthetic RNAi-resistant transgene. PLoS ONE. 8:e70489 10.1371/journal.pone.0070489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M., Dent E.W., Bear J.E., Loureiro J.J., Gertler F.B. 2003. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu. Rev. Cell Dev. Biol. 19:541–564 10.1146/annurev.cellbio.19.050103.103356 [DOI] [PubMed] [Google Scholar]
- Krause M., Leslie J.D., Stewart M., Lafuente E.M., Valderrama F., Jagannathan R., Strasser G.A., Rubinson D.A., Liu H., Way M., et al. 2004. Lamellipodin, an Ena/VASP ligand, is implicated in the regulation of lamellipodial dynamics. Dev. Cell. 7:571–583 10.1016/j.devcel.2004.07.024 [DOI] [PubMed] [Google Scholar]
- Kunda P., Craig G., Dominguez V., Baum B. 2003. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr. Biol. 13:1867–1875 10.1016/j.cub.2003.10.005 [DOI] [PubMed] [Google Scholar]
- Lafuente E.M., van Puijenbroek A.A., Krause M., Carman C.V., Freeman G.J., Berezovskaya A., Constantine E., Springer T.A., Gertler F.B., Boussiotis V.A. 2004. RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Dev. Cell. 7:585–595 10.1016/j.devcel.2004.07.021 [DOI] [PubMed] [Google Scholar]
- Lallemand Y., Luria V., Haffner-Krausz R., Lonai P. 1998. Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res. 7:105–112 10.1023/A:1008868325009 [DOI] [PubMed] [Google Scholar]
- Lanier L.M., Gates M.A., Witke W., Menzies A.S., Wehman A.M., Macklis J.D., Kwiatkowski D., Soriano P., Gertler F.B. 1999. Mena is required for neurulation and commissure formation. Neuron. 22:313–325 10.1016/S0896-6273(00)81092-2 [DOI] [PubMed] [Google Scholar]
- Lebrand C., Dent E.W., Strasser G.A., Lanier L.M., Krause M., Svitkina T.M., Borisy G.G., Gertler F.B. 2004. Critical role of Ena/VASP proteins for filopodia formation in neurons and in function downstream of netrin-1. Neuron. 42:37–49 10.1016/S0896-6273(04)00108-4 [DOI] [PubMed] [Google Scholar]
- Lewandoski M., Martin G.R. 1997. Cre-mediated chromosome loss in mice. Nat. Genet. 17:223–225 10.1038/ng1097-223 [DOI] [PubMed] [Google Scholar]
- Li A., Ma Y., Yu X., Mort R.L., Lindsay C.R., Stevenson D., Strathdee D., Insall R.H., Chernoff J., Snapper S.B., et al. 2011. Rac1 drives melanoblast organization during mouse development by orchestrating pseudopod- driven motility and cell-cycle progression. Dev. Cell. 21:722–734 10.1016/j.devcel.2011.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.Y., Fisher D.E. 2007. Melanocyte biology and skin pigmentation. Nature. 445:843–850 10.1038/nature05660 [DOI] [PubMed] [Google Scholar]
- Lyulcheva E., Taylor E., Michael M., Vehlow A., Tan S., Fletcher A., Krause M., Bennett D. 2008. Drosophila pico and its mammalian ortholog lamellipodin activate serum response factor and promote cell proliferation. Dev. Cell. 15:680–690 10.1016/j.devcel.2008.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky L.M., Insall R.H. 1998. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 8:1347–1356 10.1016/S0960-9822(98)00015-3 [DOI] [PubMed] [Google Scholar]
- Mackenzie M.A., Jordan S.A., Budd P.S., Jackson I.J. 1997. Activation of the receptor tyrosine kinase kit is required for the proliferation of melanoblasts in the mouse embryo. Dev. Biol. 192:99–107 10.1006/dbio.1997.8738 [DOI] [PubMed] [Google Scholar]
- Matthews H.K., Marchant L., Carmona-Fontaine C., Kuriyama S., Larraín J., Holt M.R., Parsons M., Mayor R. 2008. Directional migration of neural crest cells in vivo is regulated by Syndecan-4/Rac1 and non-canonical Wnt signaling/RhoA. Development. 135:1771–1780 10.1242/dev.017350 [DOI] [PubMed] [Google Scholar]
- Mayor R., Morgan R., Sargent M.G. 1995. Induction of the prospective neural crest of Xenopus. Development. 121:767–777 [DOI] [PubMed] [Google Scholar]
- McShea M.A., Schmidt K.L., Dubuke M.L., Baldiga C.E., Sullender M.E., Reis A.L., Zhang S., O’Toole S.M., Jeffers M.C., Warden R.M., et al. 2013. Abelson interactor-1 (ABI-1) interacts with MRL adaptor protein MIG-10 and is required in guided cell migrations and process outgrowth in C. elegans. Dev. Biol. 373:1–13 10.1016/j.ydbio.2012.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael M., Vehlow A., Navarro C., Krause M. 2010. c-Abl, Lamellipodin, and Ena/VASP proteins cooperate in dorsal ruffling of fibroblasts and axonal morphogenesis. Curr. Biol. 20:783–791 10.1016/j.cub.2010.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H., Suetsugu S., Takenawa T. 1998. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 17:6932–6941 10.1093/emboj/17.23.6932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niebuhr K., Ebel F., Frank R., Reinhard M., Domann E., Carl U.D., Walter U., Gertler F.B., Wehland J., Chakraborty T. 1997. A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena/VASP family. EMBO J. 16:5433–5444 10.1093/emboj/16.17.5433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T., Essers M.A., Dubois N., Offner S., Dubey C., Roger C., Metzger D., Chambon P., Hummler E., Beard P., Trumpp A. 2006. Skin epidermis lacking the c-Myc gene is resistant to Ras-driven tumorigenesis but can reacquire sensitivity upon additional loss of the p21Cip1 gene. Genes Dev. 20:2024–2029 10.1101/gad.381206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poukkula M., Cliffe A., Changede R., Rørth P. 2011. Cell behaviors regulated by guidance cues in collective migration of border cells. J. Cell Biol. 192:513–524 10.1083/jcb.201010003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pula G., Krause M. 2008. Role of Ena/VASP proteins in homeostasis and disease. Handbook Exp. Pharmacol. 186:39–65 10.1007/978-3-540-72843-6_3 [DOI] [PubMed] [Google Scholar]
- Prasad M., Montell D.J. 2007. Cellular and molecular mechanisms of border cell migration analyzed using time-lapse live-cell imaging. Dev. Cell. 12:997–1005 10.1016/j.devcel.2007.03.021 [DOI] [PubMed] [Google Scholar]
- Prasad M., Jang A.C., Starz-Gaiano M., Melani M., Montell D.J. 2007. A protocol for culturing Drosophila melanogaster stage 9 egg chambers for live imaging. Nat. Protoc. 2:2467–2473 10.1038/nprot.2007.363 [DOI] [PubMed] [Google Scholar]
- Quinn C.C., Pfeil D.S., Wadsworth W.G. 2008. CED-10/Rac1 mediates axon guidance by regulating the asymmetric distribution of MIG-10/lamellipodin. Curr. Biol. 18:808–813 10.1016/j.cub.2008.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rørth P. 2012. Fellow travellers: emergent properties of collective cell migration. EMBO Rep. 13:984–991 10.1038/embor.2012.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavoe A.K., Nelson J.C., Martínez-Velázquez L.A., Klein M., Samuel A.D., Colón-Ramos D.A. 2012. Synaptic vesicle clustering requires a distinct MIG-10/Lamellipodin isoform and ABI-1 downstream from Netrin. Genes Dev. 26:2206–2221 10.1101/gad.193409.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen A., Rottner K., Ehinger J., Innocenti M., Scita G., Wehland J., Stradal T.E. 2004. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. 23:749–759 10.1038/sj.emboj.7600084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stradal T., Courtney K.D., Rottner K., Hahne P., Small J.V., Pendergast A.M. 2001. The Abl interactor proteins localize to sites of actin polymerization at the tips of lamellipodia and filopodia. Curr. Biol. 11:891–895 10.1016/S0960-9822(01)00239-1 [DOI] [PubMed] [Google Scholar]
- Suetsugu S., Yamazaki D., Kurisu S., Takenawa T. 2003. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev. Cell. 5:595–609 10.1016/S1534-5807(03)00297-1 [DOI] [PubMed] [Google Scholar]
- Suraneni P., Rubinstein B., Unruh J.R., Durnin M., Hanein D., Li R. 2012. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol. 197:239–251 10.1083/jcb.201112113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sviderskaya E.V., Wakeling W.F., Bennett D.C. 1995. A cloned, immortal line of murine melanoblasts inducible to differentiate to melanocytes. Development. 121:1547–1557 [DOI] [PubMed] [Google Scholar]
- Theveneau E., Mayor R. 2012. Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 366:34–54 10.1016/j.ydbio.2011.12.041 [DOI] [PubMed] [Google Scholar]
- Theveneau E., Marchant L., Kuriyama S., Gull M., Moepps B., Parsons M., Mayor R. 2010. Collective chemotaxis requires contact-dependent cell polarity. Dev. Cell. 19:39–53 10.1016/j.devcel.2010.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro G.J., Green H. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17:299–313 10.1083/jcb.17.2.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrle-Haller B., Meller M., Weston J.A. 2001. Analysis of melanocyte precursors in Nf1 mutants reveals that MGF/KIT signaling promotes directed cell migration independent of its function in cell survival. Dev. Biol. 232:471–483 10.1006/dbio.2001.0167 [DOI] [PubMed] [Google Scholar]
- Wu C., Asokan S.B., Berginski M.E., Haynes E.M., Sharpless N.E., Griffith J.D., Gomez S.M., Bear J.E. 2012. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 148:973–987 10.1016/j.cell.2011.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Quinn C.C. 2012. MIG-10 functions with ABI-1 to mediate the UNC-6 and SLT-1 axon guidance signaling pathways. PLoS Genet. 8:e1003054 10.1371/journal.pgen.1003054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C., Martinez-Quiles N., Eden S., Shibata T., Takeshima F., Shinkura R., Fujiwara Y., Bronson R., Snapper S.B., Kirschner M.W., et al. 2003. WAVE2 deficiency reveals distinct roles in embryogenesis and Rac-mediated actin-based motility. EMBO J. 22:3602–3612 10.1093/emboj/cdg350 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.