

Abstract
We have previously reported that seventy percent ethanol extract of Chrysanthemum indicum Linne (CIE) strongly reduces Epstein-Barr virus (EBV)-transformed lymphoblastoid cell line (LCL) survival by inhibiting virus-encoded latent infection membrane protein 1 (LMP1)-induced NF-κB activation. To identify an active compound(s) in CIE that inhibits LMP1-induced NF-κB activation, activity-guided fractionation was employed. The CH2Cl2 fraction of CIE strongly reduced LMP1-induced NF-κB activation and LCL viability with relatively low cytotoxic effects on primary human foreskin fibroblast (HFF), HeLa or Burkitt’s lymphoma (BL41) cells. Furthermore, lupeol, a pentacyclic triterpene, was identified in the CH2Cl2 fraction of CIE to attenuate LMP1-induced NF-κB activation and LCL viability. This study demonstrates that lupeol is one of active compounds in the CH2Cl2 fraction of CIE that inhibits LMP1-induced NF-κB activation and reduces NF-κB-dependent LCL viability.
Introduction
The NF-κB family of transcription factors plays an important role in tumoirgenesis, and aberrant NF-κB activation is a hallmark of many epithelial and lymphoid-derived cancers [1,2]. NF-κB promotes tumorigenesis by inducing expression of genes involved in cell proliferation, survival, tumor promotion, immortalization, angiogenesis and metastasis [1–3]. In addition, NF-κB is a critical regulator of inflammation and promotes inflammation-associated cancers [4].
The mammalian NF-κB family consists of RelA (p65), RelB, c-Rel, p105/p50 (NF-κB1) and p100/p52 (NF-κB2) [5,6]. In response to extracellular or intracellular stimuli including proinflammatory cytokines, tumor promoters, viral and bacterial infection or DNA damage, homo- or hetero-dimers of NF-κB family members are activated to transactivate various genes [3]. The inhibitor of κB (IκB) kinase (IKK) complex which is composed of two catalytic subunits, IKKα and β, and a regulatory subunit, IKKγ (or NEMO) is a key regulator of NF-κB activation. There are two major signaling pathways leading to NF-κB activation: the IKKβ- and IKKγ-dependent canonical NF-κB pathway and the IKKα-dependent non-canonical (or alternative) NF-κB pathway [5,6].
A canonical NF-κB pathway involves the heterodimeric p65/p50 complexes that are sequestered in the cytoplasm by IκB proteins. In response to various stimuli including pro-inflammatory cytokines, TNF-α and IL-1, and lipopolysaccharide (LPS), IκB proteins are phosphorylated by IKKβ and degraded by the ubiquitin-proteasome pathway allowing nuclear translocation of the p65/p50 complexes. The non-canonical NF-κB pathway which is utilized by lymphotoxin β (LT- β), B cell-activating factor of the TNF family (BAFF) and CD40 involves NF-κB inducing kinase (NIK)- and IKKα-mediated proteolytic processing of p100 into p52 and nuclear translocation of the RelB/p52 complexes [5,6].
Epstein-Barr virus (EBV) belongs to the human γ-herpesvirus family, and latent infection of EBV is causally associated with human lymphoid and epithelial malignancies including Burkitt’s lymphoma, T-cell lymphoma, Hodgkin’s disease, lymphoproliferative disease and nasopharyngeal carcinoma (NPC) [7,8]. In vitro, EBV latently infects primary B lymphocytes and transforms these cells into proliferating lymphoblastoid cell lines (LCLs). The EBV encoded Latent infection Membrane Protein 1 (LMP1) which is expressed in most EBV-associated cancers is essential for EBV-infected primary B lymphocytes conversion to LCLs [7,8]. LMP1 constitutively activate both the non-canonical and the canonical NF-κB pathways through two C-terminal cytoplasmic domains referred to as C-terminal Activation Regions 1 and 2 (CTAR1 and 2), respectively, and LMP1-induced NF-κB activation is critical for EBV-transformed LCL survival [9–18].
We have previously reported that Chrysanthemum indicum Linne extract (CIE) inhibits LMP1-induced NF-κB activation and LCL viability without exhibiting any adverse effect on the viability of cells whose survival is independent of NF-κB activation [19]. Therefore, in this study, we have expanded our investigation to identify an active compound(s) in CIE that inhibits LMP1-induced NF-κB activation and LCL viability by using activity-guided fractionation.
Results
The effect of CIE fractions on LMP1-induced NF-κB activation
To determine active constituents that inhibit LMP1-induced NF-κB activation, CIE was fractionated by sequential solvent extractions (Figure 1). Inhibitory activity of each fraction against LMP1-induced NF-κB activation was determined by using NF-κB-dependent luciferase reporter assays. Among the five tested fractions, the CH2Cl2 fraction reduced LMP1-induced NF-κB activation by 62% (Figure 2A, compare lane 8 with lane 2). The n-Hexane, EtOAc or n-BuOH fraction also reduced LMP1-induced NF-κB activation by 18%, 35% or 31%, respectively (Figure 2A, compare lanes 6, 10, 12 with lane 2).
Figure 1. Fractionation scheme for the CIE.
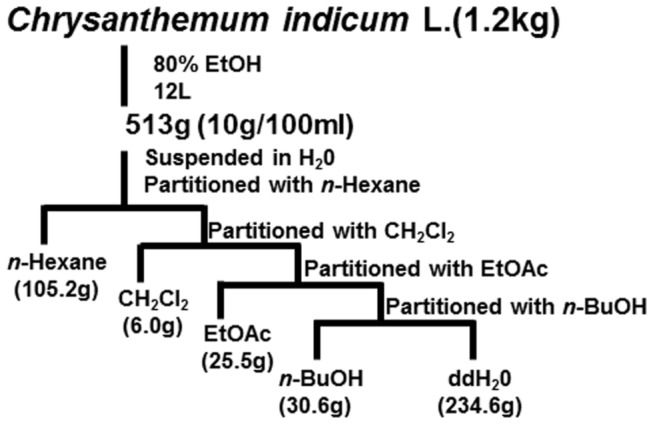
Figure 2. The CH2Cl2 fraction of CIE inhibits LMP1-induced NF-κB activation.
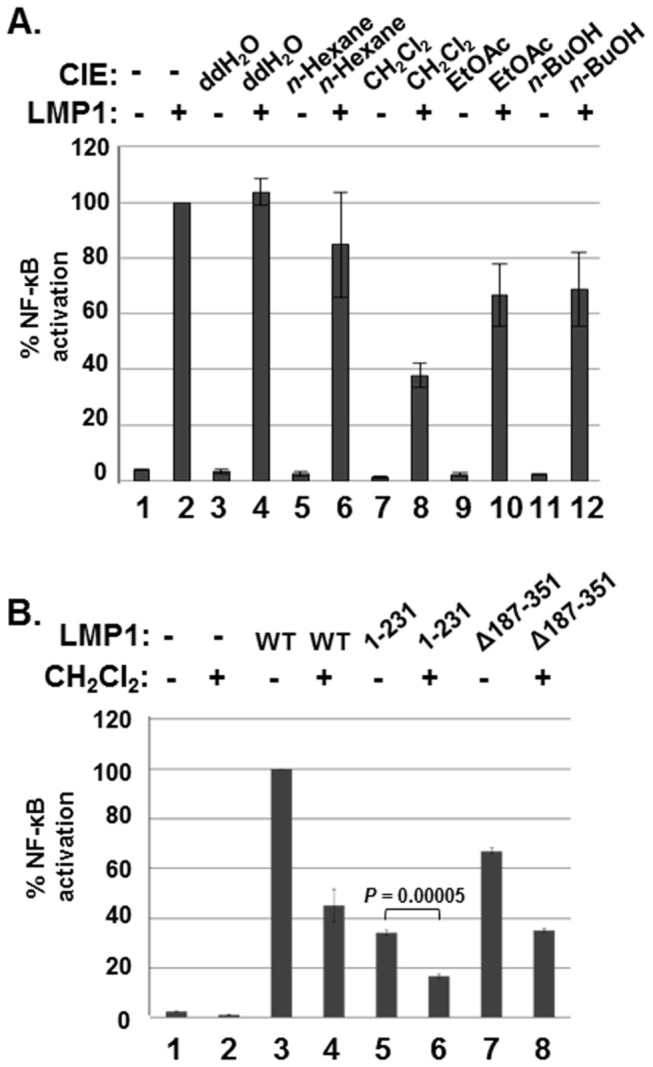
(A) HEK293 cells were co-transfected with pSG5 (lanes 1, 3, 5, 7, 9, and 11) or pSG5-FLAG-LMP1 (lanes 2, 4, 6, 8, 10 and 12) plus NF-κB dependent firefly luciferase and control Renilla luciferase plasmids. Cells were treated with DMSO (lanes 1 and 2) or ddH2O (lanes 3 and 4), n-Hexane (lanes 5 and 6), CH2Cl2 (lanes 7 and 8), EtOAc (lanes 9 and 10) or n-BuOH (lanes 11 and 12) fraction of CIE at 100µg/ml, and luciferase activity was measured using a dual luciferase assay system. (B) HEK293 cells were co-transfected with pSG5 (lanes 1 and 2), pSG5-FLAG-LMP1 WT (lanes 3 and 4), pSG5-FLAG-LMP1 1-231 (lanes 5 and 6) or pSG5-FLAG-LMP1 Δ187-351 (lanes 7 and 8) plus NF-κB dependent firefly luciferase and control Renilla luciferase plasmids. Cells were treated with either DMSO (lanes 1, 3, 5 and 7) or the CH2Cl2 fraction of CIE (lanes 2, 4, 6 and 8) at 100µg/ml, and luciferase activity was measured using a dual luciferase assay system. NF-κB dependent luciferase activity was expressed in RLU by normalizing firefly luciferase activity with constitutive Renilla luciferase activity. To calculate relative luciferase activity, LMP1-induced luciferase activities in the presence of DMSO was set 100%. Significant difference between lanes 5 and 6 was determined by the P value of a two-sample t test (P < 0.0001). Luciferase data shown here represent three independent experiments. (RLU, relative luciferase unit).
The effect of the CH2Cl2 fraction of CIE on LMP1 CTAR1- or CTAR2-induced NF-κB activation was further determined by using LMP1 mutants deleted for CTAR2 (aa 1-231) or CTAR1 (Δ187-351) (Figure 2B). The CH2Cl2 fraction of CIE reduced LMP1 CTAR1- or CTAR2-induced NF-κB activation by 51 or 54%, respectively (Figure 2B, compare lanes 6 and 8 with lanes 5 and 7). Taken together, comparing to other fractions, the CH2Cl2 fraction significantly reduced LMP1-induced NF-κB activation. Thus, the CH2Cl2 fraction of CIE may contain an active compound(s) that inhibits LMP1-induced NF-κB activation.
The effect of the CH2Cl2 fraction of CIE on IKK activation
Since LMP1 activates both non-canonical and canonical NF-κB pathways through CTAR1 and CTAR2, respectively, the effect of the CH2Cl2 fraction of CIE on LMP1-induced IKKα or IKKβ activation was further investigated (Figure 3A). BL41 cells or their LMP1 expressing counterparts were treated with either DMSO or the CH2Cl2 fraction for 24hr, and IKKα or IKKβ activity was determined by Western blot analysis with anti-p100/p52 or anti-phospho-IκBα antibody, respectively. Since the promoters of IκBα and p100 contain κB-binding sites and are regulated by the canonical NF-κB pathway [20–22], the protein levels of p100 and IκBα were induced in BL41 cells expressing LMP1 as previously reported (Figure 3A, compare lane 2 with lane 1) [6,9,23]. In DMSO treated cells, LMP1 induced p100 processing to p52 and IκBα phosphorylation (Figure 3A, compare lane 2 with lane 1). On the other hand, the CH2Cl2 fraction blocked LMP1-induced p100 processing and IκBα phosphorylation (Figure 3A, compare lane 4 with lane 2). The protein levels of p100 and IκBα in cells treated with the CH2Cl2 fraction were decreased due to inactivation of the canonical NF-κB pathway (Figure 3A, compare lanes 3 and 4 with lanes 1 and 2). Furthermore, the CH2Cl2 fraction significantly attenuated both LPS- and IL-1β-induced IKKβ activation (Figure 3B and C, compare lanes 6 to 10 with lanes 1 to 5). Thus, these data suggest that the CH2Cl2 fraction of CIE inhibits NF-κB activation possibly by interfering with IKK activation.
Figure 3. The CH2Cl2 fraction of CIE inhibits IKK activation.
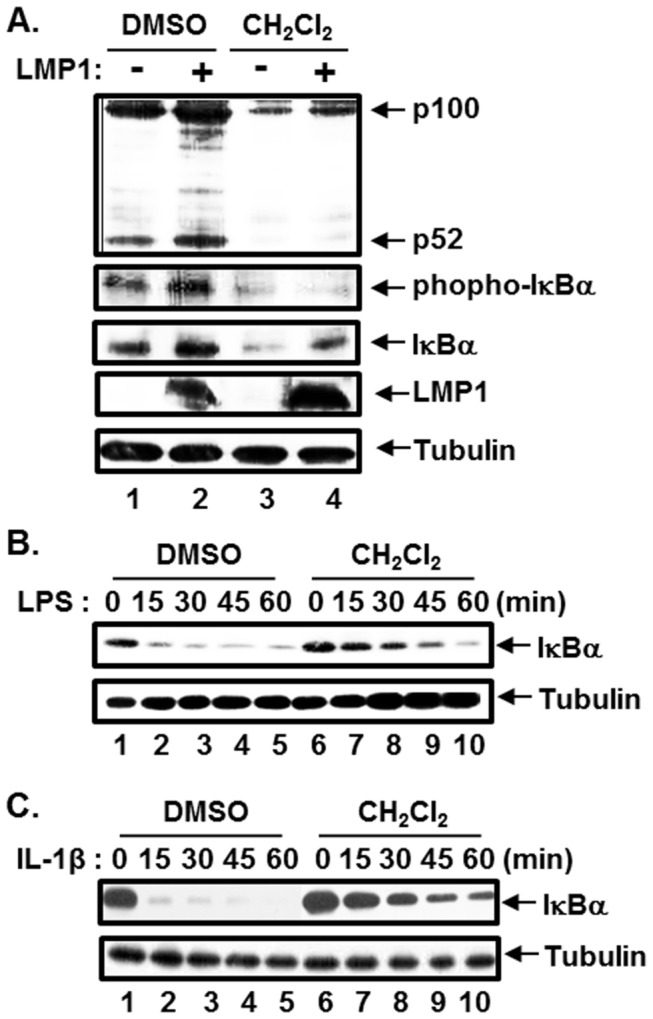
(A) Parental BL41 cells and their counterparts expressing LMP1 were treated with either DMSO or CH2Cl2 fraction of CIE at 100µg/ml for 24hr, and equal amounts of cell extracts were subjected to Western blot analysis with anti-p100/p52, anti-phopho-IκBα, anti-IκBα, anti-LMP1 or anti-tubulin antibody. (B) Raw 264.7 cells were pre-treated with either DMSO or CH2Cl2 fraction of CIE at 100µg/ml for 3hr and stimulated with LPS at 1µg/ml. At 0, 15, 30, 45 and 60 min after LPS treatment, equal amounts of cell extracts were subjected to Western blot analysis with anti-IκBα or anti-tubulin antibody. (C) HeLa cells were pre-treated with either DMSO or CH2Cl2 fraction of CIE at 100µg/ml for 3hr and stimulated with IL-1β at 20ng/ml. At 0, 15, 30, 45 and 60 min after IL-1β treatment, equal amounts of cell extracts were subjected to Western blot analysis with anti-IκBα or anti-tubulin antibody.
The effect of the CH2Cl2 fraction of CIE on LCL viability
Since LMP1-induced NF-κB activation is essential for LCL survival, the effect of the CH2Cl2 fraction of CIE on the viability of LCLs was investigated. LCLs were treated with 100µg/ml of CIE fractions, and the cell viability was measured using the CellTiter-Glo assay at 0, 3, 6, 9, 12 or 24hr after treatment (Figure 4). Consistent with the NF-κB-dependent luciferase reporter data, the CH2Cl2 fraction strongly reduced LCL viability (Figure 4). After treatment with the CH2Cl2 fraction, LCL viability was reduced by 65% at 9hr and by 94% at 24hr (Figure 4). Interestingly, other fractions had almost no effect on LCL viability (Figure 4).
Figure 4. The CH2Cl2 fraction of CIE reduces LCL viability.

LCLs were treated with either DMSO or n-Hexane, CH2Cl2, EtOAc or n-BuOH fraction of CIE at 100µg/ml, and cell viability was determined at 0, 3, 6, 9, 12 or 24hr after treatment using CellTiter-Glo Luminescent Cell Viability Assay. A score of 1.0 indicates that there is no difference in viability between DMSO or CIE fraction treated cells. Luciferase data shown here represent three independent experiments.
To further assess the relative toxicity of the CH2Cl2 fraction in other cell types whose survival is independent of NF-κB activation, LCLs, HFF, HeLa or BL41 cells were treated with 6.25, 12.5, 25, 50 or 100µg/ml of the CH2Cl2 fraction, and the cell viability was measured by using the CellTiter-Glo assay at 24, 48 or 72hr after treatment (Figure 5). The CH2Cl2 fraction reduced LCL viability in a dose- and time-dependent manner, and the half maximal inhibitory concentration (IC50) values for the cytotoxicity of the CH2Cl2 fraction on LCLs at 24, 48 and 72hr were 97.3, 55.8 and 45.2mM, respectively (Figure 5A and Table 1). Interestingly, the CH2Cl2 fraction had very little cytotoxic effect on HFF or HeLa cells. Within the first 24hr after treatment, the CH2Cl2 fraction had no adverse effect on the viability of HFF or HeLa cells (Figure 5B and C). After 72hr treatment with the CH2Cl2 fraction at 100µg/ml, HFF or HeLa cell viability was reduced by 91% and 80%, respectively (Figure 5B and C). BL41 cells were slightly more sensitive to the CH2Cl2 fraction than HFF or HeLa cells. After 24hr treatment with the CH2Cl2 fraction at 100µg/ml, BL41 cell viability was reduced by 72% and 98% (Figure 5D). Nonetheless, the CH2Cl2 fraction was evidently less cytotoxic to HFF, HeLa or BL41 cells with IC50 values of 145.5, 109.7 and 91.4mM, respectively, at 72hr after treatment (Table 1).
Figure 5. The CH2Cl2 fraction of CIE is more cytotoxic toward LCLs than HFF, HeLa or BL41 cells.

(A) LCLs, (B) HFF, (C) HeLa or (D) BL41 cells were treated with either DMSO or CH2Cl2 fraction of CIE at 6.25, 12.5, 25, 50 or 100µg/ml, and cell viability was determined at 24, 48 or 72hr after treatment using CellTiter-Glo Luminescent Cell Viability Assay. A score of 1.0 indicates that there is no difference in viability between DMSO or CH2Cl2 fraction of CIE treated cells. Luciferase data shown here represent three independent experiments.
Table 1. The IC50 value for the cytotoxicity of the CH2Cl2 fraction of CIE.
| Cell | IC50(mM) |
||
|---|---|---|---|
| 24hr | 48hr | 72hr | |
| LCL | 97.3 | 55.8 | 45.2 |
| HFF | ND * | ND * | 145.5 |
| HeLa | ND * | 84.3 | 109.7 |
| BL41 | 150.9 | 93.7 | 91.4 |
not determined
Since NF-κB inhibition induces apoptosis in LCLs [9,10], the apoptotic effect of the CH2Cl2 fraction of CIE in LCL was further assessed. LCLs were treated with 100µg/ml of either DMSO or the CH2Cl2 fraction, and poly (ADP-ribose) polymerase (PARP) cleavage was determined by Western blot analysis at 0, 1, 3, 6, 9, 12 or 24hr after treatment (Figure 6). At 6hr after treatment, the CH2Cl2 fraction induced the PARP cleavage (Figure 6, lane 11). At later time points, the PARP cleavage was further induced in cells treated with the CH2Cl2 fraction (Figure 6, compare lanes 11 to 14 with lanes 4 to 7). Taken together, the CH2Cl2 fraction of CIE is more cytotoxic toward NF-κB-dependent LCLs than NF-κB-independent HFF, HeLa or BL41 cells. The CH2Cl2 fraction of CIE may reduce LCL viability by inducing apoptosis.
Figure 6. The CH2Cl2 fraction of CIE induces apoptosis in LCLs.

LCLs were treated with 100µg/ml of either DMSO or the CH2Cl2 fraction of CIE, and PARP cleavage was determined by Western blot analysis at 0, 1, 3, 6, 9, 12 or 24hr after treatment.
The effect of lupeol isolated from CIE on LMP1-induced NF-κB activation
To identify active compounds, the CH2Cl2 fraction of CIE was further fractionated as described in the Materials and Methods (Figure 7). By performing NF-κB inhibitory activity-guided fractionation, lupeol, a pentacyclic triterpene, was isolated from the CH2Cl2 fraction of CIE (Figure 8A). To further determine the effect of lupeol on LMP1-induced NF-κB activation, HEK293 cells were co-transfected with pSG5 or pSG5-FLAG-LMP1 plus NF-κB dependent firefly luciferase and control Renilla luciferase plasmids and treated with lupeol at 0, 3.125, 6.25, 12.5, 25 or 50µg/ml (Figure 8B). At 50µg/ml, lupeol reduced LMP1-induced NF-κB activation by 34% (Figure 8B, compare lane 12 with lane 1). Thus, lupeol possesses inhibitory activity against LMP1-induced NF-κB activation. Compared to the CH2Cl2 fraction of CIE which reduced LMP1-induced NF-κB activation by 62%, lupeol was less effective to inhibit LMP1-induced NF-κB activation.
Figure 7. Sub-fractionation and isolation scheme for lupeol from the CH2Cl2 fraction of CIE.

Figure 8. Lupeol inhibits LMP1-induced NF-κB activation.
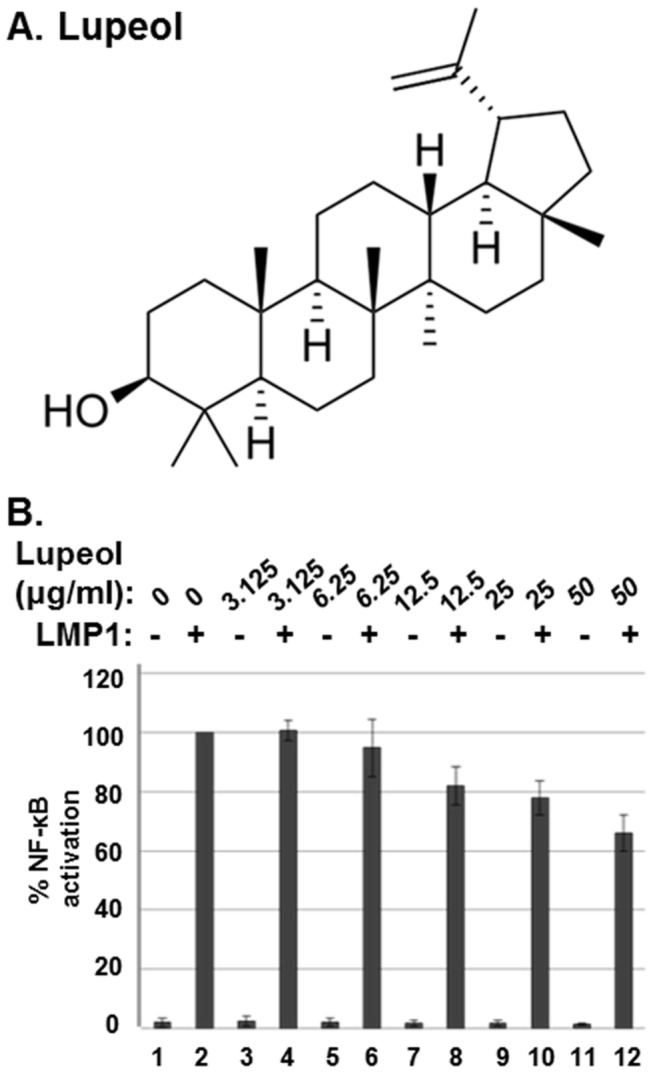
(A) The lupeol structure. (B) HEK293 cells were co-transfected with pSG5 (lanes 1, 3, 5, 7, 9, and 11) or pSG5-FLAG-LMP1 (lanes 2, 4, 6, 8, 10 and 12) plus NF-κB dependent firefly luciferase and control Renilla luciferase plasmids. Cells were treated with lupeol at 0, 3.125, 6.25, 12.5, 25 or 50µg/ml, and luciferase activity was measured using a dual luciferase assay system. NF-κB dependent luciferase activity was expressed in RLU by normalizing firefly luciferase activity with constitutive Renilla luciferase activity. To calculate relative luciferase activity, LMP1-induced luciferase activities in the presence of lupeol at 0µg/ml was set 100%. Luciferase data shown here represent three independent experiments.
The effect of lupeol isolated from CIE on LCL viability
To assess the relative toxicity of lupeol in different cell types, LCLs, HFF, HeLa or BL41 cells were treated with either DMSO or lupeol at 3.125, 6.25, 12.5, 25 or 50µg/ml, and the cell viability was measured by using the CellTiter-Glo assay at 0, 3, 6, 9, 12, 24, 48 or 72hr after treatment (Figure 9). Although lupeol reduced the viability of these cells in a dose- and time-dependent manner at latter time points, it was more cytotoxic toward LCLs than HFF, HeLa or BL41 cells (Figure 9). Within the first 24hr after treatment at 50µg/ml, lupeol reduced LCL viability by 54% (Figure 9A). The IC50 values for the cytotoxicity of lupeol on LCLs at 24, 48 and 72hr were 109.9, 57.6 and 51.8mM, respectively (Table 2). On the other hand, lupeol had very little adverse effect on the viability of HFF or HeLa cells within the first 24hr after treatment (Figure 9B and C). At 50µg/ml, lupeol reduced the viability of HFF cells by 48% and 93% at 48 and 72hr after treatment, respectively (Figure 9B). At the same concentration, lupeol reduced the viability of HeLa cells by 78% and 96% at 48 and 72hr after treatment (Figure 9C). BL41 cells were more sensitive to lupeol than HFF and HeLa cells. Within the first 24hr after treatment at 50µg/ml, lupeol reduced the viability of BL41 cells by 48% (Figure 9D). At 48 and 72hr after treatment, 50µg/ml of lupeol further reduced the viability of BL41 cells by 61% and 92%, respectively (Figure 9D). Nonetheless, lupeol was slightly more effective to reduce the viability of LCLs than BL41 cells. The IC50 value for the cytotoxicity of lupeol on HFF, HeLa and BL41 cells at 72hr after treatment was 79.7, 63.3 and 56.9mM, respectively (Table 2).
Figure 9. Lupeol is more cytotoxic toward LCLs than HFF, HeLa or BL41 cells.

(A) LCLs, (B) HFF, (C) HeLa or (D) BL41 cells were treated with either DMSO or lupeol at 3.125, 6.25, 12.5, 25 or 50µg/ml, and cell viability was determined at 0, 3, 6, 9, 12, 24, 48 or 72hr after treatment using CellTiter-Glo Luminescent Cell Viability Assay. A score of 1.0 indicates that there is no difference in viability between DMSO or lupeol treated cells.
Table 2. The IC50 value for the cytotoxicity of lupeol.
| Cell | IC50(mM) |
||
|---|---|---|---|
| 24hr | 48hr | 72hr | |
| LCL | 109.9 | 57.6 | 51.8 |
| HFF | ND * | ND * | 79.7 |
| HeLa | ND * | 88.3 | 63.3 |
| BL41 | ND * | 68.4 | 56.9 |
not determined
Furthermore, the apoptotic effect of lupeol in LCL was determined. LCLs were treated with 50µg/ml of lupeol, and PARP cleavage was determined by Western blot analysis at 0, 1, 3, 6, 9, 12 or 24hr after treatment (Figure 10). At 9hr after treatment, lupeol induced the PARP cleavage (Figure 9, lane 12). These data indicate that lupeol induces apoptosis in LCLs and is more cytotoxic to LCLs than HFF, HeLa or BL41 cells.
Figure 10. Lupeol induces apoptosis in LCLs.
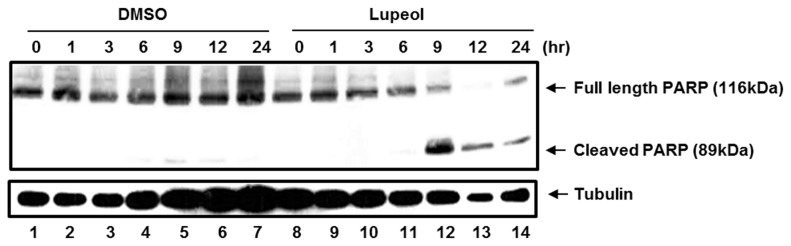
LCLs were treated with 50µg/ml of either DMSO or lupeol, and PARP cleavage was determined by Western blot analysis at 0, 1, 3, 6, 9, 12 or 24hr after treatment.
Discussion
The NF-κB family of transcription factors plays an important role in inflammation-associated tumorigenesis [4]. Constitutive activation of NF-κB induced by either mutation in components of the NF-κB pathway or proinflammatory stimuli in the microenvironment has been proposed to promote tumorigenesis [2,24]. Anti-apoptotic function of NF-κB is a major contributor to the survival of numerous cancer cells [24]. NF-κB induces expression of anti-apoptotic genes including caspase-8/FAS-associated death domain (FADD)-like IL-1β-converting enzyme (FLICE) inhibitory protein (c-FLIP), cellular inhibitor of apoptosis (cIAPs) and Bcl2 family proteins such as A1/BFL1 and Bcl-xL [2,25]. Thus, cancer cells in which NF-κB is constitutively activated are resistance to chemo- and radiation therapies [26,27]. In addition, these genotoxic anti-cancer therapies may be ineffective against cancer cells because genotoxic stress induces NF-κB activation [28].
NF-κB is also activated in lymphoid cancers associated with tumor viruses such as EBV, Kaposi Sarcoma-associated herpesvirus (KSHV) and human T-cell lymphoma virus (HTLV). These tumor viruses encode proteins that induce NF-κB activation which may contribute to lymphomagenesis [24,29]. Indeed, EBV LMP1 changes the growth phenotype of human B lymphocytes and induces B cell lymphoma when expressed in transgenic mice [30–32]. Since LMP1-induced NF-κB activation is critical for EBV-transformed LCL survival [9,10], LMP1-induced NF-κB activation pathway may be an ideal target for the development of novel therapeutic strategies to treat EBV-associated malignancies.
We have previously reported that CIE strongly reduces EBV-transformed LCL viability by inhibiting LMP1-induced NF-κB activation [19]. CIE had almost no adverse effects on the viability of cells in which NF-κB is not activated [19]. C. indicum has been used to treat inflammatory disease in traditional Korean and Chinese medicine [33–40]. CIE inhibits LPS-induced production of inflammatory cytokines possibly by down-regulating NF-κB and MAPK in RAW264.7 cells macrophages [41]. In addition, CIE inhibits LMP1- and IL-1β-induced NF-κB activation by blocking IKK activity [19]. How CIE inhibits IKK is unclear and is the subject of future studies. CIE may inhibit IKK activation directly by targeting IKK and/or indirectly by blocking the function of components upstream of IKK.
By performing NF-κB inhibitory activity-guided fractionation, we found that the CH2Cl2 fraction of CIE strongly reduces LMP1-induced NF-κB activation and LCL survival without adversely affecting the viability of HFF, HeLa or BL41 cells. The CH2Cl2 fraction inhibited LMP1-, IL-1β- and LPS-induced IKK activation. Furthermore, lupeol, a pentacyclic triterpene, was identified in the CH2Cl2 fraction of CIE to inhibit LMP1-induced NF-κB activation. Lupeol is relatively more cytotoxic toward LCLs in which NF-κB is constitutively activated than HFF, HeLa or BL41 cells. Indeed, lupeol has been reported to inhibit NF-κB activation and induces apoptosis in human cancer cells [42–48].
In addition to NF-κB, lupeol affects various cellular signal transduction pathways including Wnt/β-catenin and Akt/protein kinase B (PKB) [44,48,49]. Since the NF-κB, Akt/PKB and Wnt/β-catenin pathways are functionally inter-connected [50–54], lupeol may target a protein(s) commonly utilized by these pathways. At 72hr after treatment, 50µg/ml of lupeol may induce NF-κB-independent cell death in HFF, HeLa or BL41 cells possibly by down-regulating Akt/PKB and/or Wnt/β-catenin pathways. Lupeol may induce both NF-κB-dependent and -independent cell death pathways in LCLs. Thus, the viability of LCLs was relatively more susceptible to lupeol than other cells.
The CH2Cl2 fraction of CIE was more effective to inhibit LMP1-induced NF-κB activation and selectively reduce LCL viability than lupeol. Lupeol may interact synergistically with unknown compounds in the CH2Cl2 fraction of CIE to reduce LMP1-induced NF-κB activation and LCL viability. In addition to the CH2Cl2 fraction of CIE, the EtOAc or n-BuOH fraction also reduced LMP1-induced NF-κB activation by 35% or 31%, respectively. Thus, these fractions may contain additional active compounds that contribute NF-κB inhibitory activity of CIE.
Materials and Methods
Plant material and fractionation
The plants materials and 70% ethanol extracts used in this study were collected from Jeju island in Korea through Korea National Research Resource Center (KNRRC, Medicinal Plants Resources Bank No. 2011-0000538) supported by the Korea Research Foundation, where resources were provided by the Ministry of Education, Science and Technology in 2011. The voucher specimens for the samples (specimen number MPRB-KR-04-00034) were deposited at the herbarium of Department of Life Science, Gachon University (GCU).
Fractionation and isolation of active compounds in CIE
The dried Chrysanthemum indicum Linne (1.2kg) was exhaustively extracted by 70% EtOH. The solvent was then evaporated under reduced pressure, at a temperature not exceeding 40°C, to yield 513 g of a semisolid dark yellow residue. The extract was re-suspended in distilled water and successively fractionated with n-Hexane, dichloromethane (CH2Cl2), ethyl acetate (EtOAc), n-butanol (n-BuOH) and the ddH2O. (Figure 1)
The CH2Cl2 layer (3g) was chromatographed on a column of Silica (2.5 x 20 cm). Elution was carried out with ddH2O followed by 10% stepwise addition of methanol till 100% to give 9 sub-fractions. The 3rd sub-fraction was re-chromatographed on a column of sephadex LH-20. Elution was carried out with CH2Cl2:ddH2O:MeOH = 1:1:3, 1:1:2 and 1:1:1 and then an active compound was isolated by recycling HPLC with solvent, CH3Cl-MeOH (Figure 6).
Identification of lupeol
1H-NMR (CDCl3, 500 MHz) δ: 0.76 (3H, s, H-28), 0.78 (3H, s, H-23), 0.83 (3H, s, H-25), 0.94 (3H, s, H-24), 0.96 (3H, s, H-27), 1.03 (3H, s, H-26), 1.68 (3H, s, H-30), ), 2.37 (1H, dd, J = 11.3, 5.1 Hz, H-19), 3.18 (1H, dd, J = 11.3, 5.1 Hz, H-3), ), 4.57 (1H, br s, H-29α), , 4.69 (1H, br s, H-29β); 13C-NMR (CDCl3, 125 MHz) δ: 14.5 (C-27), 15.3 (C-24), 15.9 (C-26), 16.1 (C-25), 18.0 (C-28), 18.3 (C-6), 19.3 (C-30), 20.9 (C-11), 25.1 (C-12), 27.4 (C-15), 27.4 (C-2), 28.0 (C-23), 29.8 (C-21), 34.3 (C-7), 35.6 (C-16), 37.1 (C-10), 38.0 (C-13), 38.7 (C-1), 38.8 (C-4), 40.0 (C-22), 40.8 (C-8), 42.8 (C-14), 43.0 (C-17), 47.9 (C-18), 48.3 (C-19), 50.4 (C-9), 55.3 (C-5), 79.0 (C-3), 109.3 (C-29), 150.9 (C-20) (Figure 7A).
Cells, plasmids, reagents and transfections
Primary human foreskin fibroblast (HFF), HEK293, HeLa and mouse leukemic monocyte-macrophage Raw 264.7 cells were maintained in Dulbecco’s modified Eagle’s medium (Biowest, Nuaille, France) supplemented with 10% fetal bovine serum (Biowest) and penicillin (100U/ml) and streptomycin (100µg/ml). Burkitt’s lymphoma BL41 cells were maintained in RPMI 1640 (Life Technologies, Carlsbad, CA) medium supplemented with 10% fetal bovine serum (Biowest) and penicillin (100U/ml) and streptomycin (100µg/ml). IB4, an EBV-infected cord blood derived LCLs and BL41 cells in which LMP1 expression can be negatively controlled by Doxycycline (Dox) were kindly provided by Dr. Elliott Kieff (Harvard Medical School) [9,55]. The pcDNA3FLAG-LMP1 was previously described [56]. Recombinant human IL-1β and LPS from E. Coli Serogroup 0111:B4 were purchased from R&D Systems (Minneapolis, MN) and InvivoGen (San Diego, CA), respectively. Effectene for transient transfection was used according to the manufacturer’s directions (Qiagen, Valencia, CA).
Western blot analysis
Cells were collected, fractionated, and transferred to nitrocellulose membranes as described previously [57]. Polyclonal rabbit antibody to p100/p52 was a kind gift from Dr. Ulrich Siebenlist (NIH). Antibodies to phospho-IκBα, IκBα and PARP were purchased from Cell Signaling Technology (Beverly, MA). An antibody to alpha-tubulin was purchased from Sigma Aldrich (St. Louis, MO). Enhanced chemiluminescence detection reagents (Pierce, Rockford, IL) and secondary peroxidase-labeled anti-mouse or anti-rabbit immunoglobulin G antibody (Amersham Biosciences, Piscataway, NJ.) were used according to the manufacturer’s directions.
NF-κB luciferase reporter and cell viability assays
NF-κB luciferase reporter assay was performed as previously described [58]. Cell viability was determined using CellTiter-Glo luminescent cell viability assay (Promega, WI) according to the manufacturer’s directions.
Acknowledgments
The authors are grateful to Jung-Eun Kim and Jung Eun Kwon for technical support and discussion.
Funding Statement
This research was supported by Bio-industry Technology Development Program, Ministry of Agriculture, Food and Rural Affairs (No. 311063-5) and by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education, Science and Technology (MEST) (No. 2010-0003301). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Baud V, Karin M (2009) Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov 8: 33-40. doi: 10.1038/nrd2781. PubMed: 19116625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Karin M, Cao Y, Greten FR, Li ZW (2002) NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2: 301-310. doi: 10.1038/nri807. PubMed: 12001991. [DOI] [PubMed] [Google Scholar]
- 3. Aggarwal BB (2004) Nuclear factor-kappaB: the enemy within. Cancer Cell 6: 203-208. doi: 10.1016/j.ccr.2004.09.003. PubMed: 15380510. [DOI] [PubMed] [Google Scholar]
- 4. Ben-Neriah Y, Karin M (2011) Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol 12: 715-723. doi: 10.1038/ni.2060. PubMed: 21772280. [DOI] [PubMed] [Google Scholar]
- 5. Ghosh S, Karin M (2002) Missing pieces in the NF-kappaB puzzle. Cell 109 Suppl: S81-S96. doi: 10.1016/S0092-8674(02)00703-1. PubMed: 11983155. [DOI] [PubMed] [Google Scholar]
- 6. Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18: 2195-2224. doi: 10.1101/gad.1228704. PubMed: 15371334. [DOI] [PubMed] [Google Scholar]
- 7. Kieff ED, Rickinson AB (2007) Epstein-Barr Virus and Its Replication. In: Knipe DM, Howley PM. Fields Virology. 2007 ed. Philadelphia: Lippincott, Williams, and Wilkins; pp. 2603-2655. [Google Scholar]
- 8. Rickinson AB, Kieff ED (2007) Epstein-Barr Virus. In: Knipe DM, Howley PM. Fields Virology. fifth ed. Philadelphia, Pa: Lippincott, Williams, and Wilkins; pp. 2655-2700. [Google Scholar]
- 9. Cahir-McFarland ED, Carter K, Rosenwald A, Giltnane JM, Henrickson SE et al. (2004) Role of NF-kappa B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells. J Virol 78: 4108-4119. doi: 10.1128/JVI.78.8.4108-4119.2004. PubMed: 15047827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cahir-McFarland ED, Davidson DM, Schauer SL, Duong J, Kieff E (2000) NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proc Natl Acad Sci U S A 97: 6055-6060. doi: 10.1073/pnas.100119497. PubMed: 10811897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitchell T, Sugden B (1995) Stimulation of NF-kappa B-mediated transcription by mutant derivatives of the latent membrane protein of Epstein-Barr virus. J Virol 69: 2968-2976. PubMed: 7707523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huen DS, Henderson SA, Croom-Carter D, Rowe M (1995) The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 10: 549-560. PubMed: 7845680. [PubMed] [Google Scholar]
- 13. Devergne O, McFarland Cahir Ed; Mosialos G, Izumi KM, Ware CF et al. (1998) Role of the TRAF binding site and NF-kappaB activation in Epstein-Barr virus latent membrane protein 1-induced cell gene expression. J Virol 72: 7900-7908. PubMed: 9733827. Available online at: PubMed: 9733827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Devergne O, Hatzivassiliou E, Izumi KM, Kaye KM, Kleijnen MF et al. (1996) Association of TRAF1, TRAF2, and TRAF3 with an Epstein-Barr virus LMP1 domain important for B-lymphocyte transformation: role in NF-kappaB activation. Mol Cell Biol 16: 7098-7108. PubMed: 8943365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C et al. (1995) The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell 80: 389-399. doi: 10.1016/0092-8674(95)90489-1. PubMed: 7859281. [DOI] [PubMed] [Google Scholar]
- 16. Kaye KM, Devergne O, Harada JN, Izumi KM, Yalamanchili R et al. (1996) Tumor necrosis factor receptor associated factor 2 is a mediator of NF-kappa B activation by latent infection membrane protein 1, the Epstein-Barr virus transforming protein. Proc Natl Acad Sci U S A 93: 11085-11090. doi: 10.1073/pnas.93.20.11085. PubMed: 8855313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaye KM, Izumi KM, Mosialos G, Kieff E (1995) The Epstein-Barr virus LMP1 cytoplasmic carboxy terminus is essential for B-lymphocyte transformation; fibroblast cocultivation complements a critical function within the terminal 155 residues. J Virol 69: 675-683. PubMed: 7815530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Izumi KM, McFarland Cahir ED, Riley: EA, Rizzo D, Chen Y et al. (1999) The residues between the two transformation effector sites of Epstein-Barr virus latent membrane protein 1 are not critical for B-lymphocyte growth transformation. J Virol 73: 9908-9916. PubMed: 10559303. Available online at: PubMed: 10559303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim JE, Jun S, Song M, Kim JH, Song YJ (2012) The extract of Chrysanthemum indicum Linne inhibits EBV LMP1-induced NF-kappaB activation and the viability of EBV-transformed lymphoblastoid cell lines. Food Chem Toxicol 50: 1524-1528. doi: 10.1016/j.fct.2012.02.034. PubMed: 22387267. [DOI] [PubMed] [Google Scholar]
- 20. Liptay S, Schmid RM, Nabel EG, Nabel GJ (1994) Transcriptional regulation of NF-kappa B2: evidence for kappa B-mediated positive and negative autoregulation. Mol Cell Biol 14: 7695-7703. PubMed: 7969113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Müller JR, Siebenlist U (2003) Lymphotoxin beta receptor induces sequential activation of distinct NF-kappa B factors via separate signaling pathways. J Biol Chem 278: 12006-12012. doi: 10.1074/jbc.M210768200. PubMed: 12556537. [DOI] [PubMed] [Google Scholar]
- 22. Shih VF, Tsui R, Caldwell A, Hoffmann A (2011) A single NFkappaB system for both canonical and non-canonical signaling. Cell Res 21: 86-102. doi: 10.1038/cr.2010.161. PubMed: 21102550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saito N, Courtois G, Chiba A, Yamamoto N, Nitta T et al. (2003) Two carboxyl-terminal activation regions of Epstein-Barr virus latent membrane protein 1 activate NF-kappaB through distinct signaling pathways in fibroblast cell lines. J Biol Chem 278: 46565-46575. doi: 10.1074/jbc.M302549200. PubMed: 12968033. [DOI] [PubMed] [Google Scholar]
- 24. Karin M (2009) NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol 1: a000141 PubMed: 20066113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sen R (2006) Control of B lymphocyte apoptosis by the transcription factor NF-kappaB. Immunity 25: 871-883. doi: 10.1016/j.immuni.2006.12.003. PubMed: 17174931. [DOI] [PubMed] [Google Scholar]
- 26. Wang CY, Cusack JC Jr., Liu R, Baldwin AS Jr. (1999) Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med 5: 412-417. doi: 10.1038/7410. PubMed: 10202930. [DOI] [PubMed] [Google Scholar]
- 27. Wang CY, Mayo MW, Baldwin AS Jr. (1996) TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science 274: 784-787. doi: 10.1126/science.274.5288.784. PubMed: 8864119. [DOI] [PubMed] [Google Scholar]
- 28. Nakanishi C, Toi M (2005) Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer 5: 297-309. doi: 10.1038/nrc1588. PubMed: 15803156. [DOI] [PubMed] [Google Scholar]
- 29. de Oliveira DE, Ballon G, Cesarman E (2010) NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol 18: 248-257. doi: 10.1016/j.tim.2010.04.001. PubMed: 20452220. [DOI] [PubMed] [Google Scholar]
- 30. Henderson S, Rowe M, Gregory C, Croom-Carter D, Wang F et al. (1991) Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell 65: 1107-1115. doi: 10.1016/0092-8674(91)90007-L. PubMed: 1648447. [DOI] [PubMed] [Google Scholar]
- 31. Kulwichit W, Edwards RH, Davenport EM, Baskar JF, Godfrey V et al. (1998) Expression of the Epstein-Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc Natl Acad Sci U S A 95: 11963-11968. doi: 10.1073/pnas.95.20.11963. PubMed: 9751773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang D, Liebowitz D, Wang F, Gregory C, Rickinson A et al. (1988) Epstein-Barr virus latent infection membrane protein alters the human B-lymphocyte phenotype: deletion of the amino terminus abolishes activity. J Virol 62: 4173-4184. PubMed: 2845129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng W, Li J, You T, Hu C (2005) Anti-inflammatory and immunomodulatory activities of the extracts from the inflorescence of Chrysanthemum indicum Linne. J Ethnopharmacol 101: 334-337. doi: 10.1016/j.jep.2005.04.035. PubMed: 16029939. [DOI] [PubMed] [Google Scholar]
- 34. Chun HS, Kim JM, Choi EH, Chang N (2008) Neuroprotective effects of several korean medicinal plants traditionally used for stroke remedy. J Med Food 11: 246-251. doi: 10.1089/jmf.2007.542. PubMed: 18598165. [DOI] [PubMed] [Google Scholar]
- 35. Kato T, Noguchi K, Miyamoto Y, Suekawa M, Aburada M et al. (1987) Effects of Chrysanthemum indicum Linn. on coronary, vertebral, renal and aortic blood flows of the anesthetized dog. Arch Int Pharmacodyn Ther 285: 288-300. PubMed: 3579429. [PubMed] [Google Scholar]
- 36. Kim IS, Ko HM, Koppula S, Kim BW, Choi DK (2011) Protective effect of Chrysanthemum indicum Linne against 1-methyl-4-phenylpridinium ion and lipopolysaccharide-induced cytotoxicity in cellular model of Parkinson's disease. Food Chem Toxicol 49: 963-973. doi: 10.1016/j.fct.2011.01.002. PubMed: 21219959. [DOI] [PubMed] [Google Scholar]
- 37. Lee do Y, Choi G, Yoon T, Cheon MS, Choo BK et al. (2009) Anti-inflammatory activity of Chrysanthemum indicum extract in acute and chronic cutaneous inflammation. J Ethnopharmacol 123: 149-154. doi: 10.1016/j.jep.2009.02.009. PubMed: 19429354. [DOI] [PubMed] [Google Scholar]
- 38. Pongjit K, Ninsontia C, Chaotham C, Chanvorachote P (2011) Protective effect of glycine max and chrysanthemum indicum extracts against cisplatin-induced renal epithelial cell death. Hum Exp Toxicol, 30: 1931–44. PubMed: 21406484. [DOI] [PubMed] [Google Scholar]
- 39. Su JY, Tan LR, Lai P, Liang HC, Qin Z et al. (2011) Experimental study on anti-inflammatory activity of a TCM recipe consisting of the supercritical fluid CO(2) extract of Chrysanthemum indicum, Patchouli Oil and Zedoary Turmeric Oil in vivo. J Ethnopharmacol. [DOI] [PubMed] [Google Scholar]
- 40. Wang ZD, Huang C, Li ZF, Yang J, Li BH et al. (2010) Chrysanthemum indicum ethanolic extract inhibits invasion of hepatocellular carcinoma via regulation of MMP/TIMP balance as therapeutic target. Oncol Rep 23: 413-421. PubMed: 20043102. [PubMed] [Google Scholar]
- 41. Cheon MS, Yoon T, Lee do Y, Choi G, Moon BC et al. (2009) Chrysanthemum indicum Linne extract inhibits the inflammatory response by suppressing NF-kappaB and MAPKs activation in lipopolysaccharide-induced RAW 264.7 macrophages. J Ethnopharmacol 122: 473-477. doi: 10.1016/j.jep.2009.01.034. PubMed: 19429315. [DOI] [PubMed] [Google Scholar]
- 42. Lee TK, Poon RT, Wo JY, Ma S, Guan XY et al. (2007) Lupeol suppresses cisplatin-induced nuclear factor-kappaB activation in head and neck squamous cell carcinoma and inhibits local invasion and nodal metastasis in an orthotopic nude mouse model. Cancer Res 67: 8800-8809. doi: 10.1158/0008-5472.CAN-07-0801. PubMed: 17875721. [DOI] [PubMed] [Google Scholar]
- 43. Li W, Hao J, Xiao Y (2013) Synthesis and in vitro antitumor activities of lupeol dicarboxylic acid monoester derivatives. Arch Pharm Res: ([MedlinePgn:]) PubMed: 23700293. [DOI] [PubMed] [Google Scholar]
- 44. Prasad S, Madan E, Nigam N, Roy P, George J et al. (2009) Induction of apoptosis by lupeol in human epidermoid carcinoma A431 cells through regulation of mitochondrial, Akt/PKB and NFkappaB signaling pathways. Cancer Biol Ther 8: 1632-1639. doi: 10.4161/cbt.8.17.9204. PubMed: 19625778. [DOI] [PubMed] [Google Scholar]
- 45. Saleem M, Afaq F, Adhami VM, Mukhtar H (2004) Lupeol modulates NF-kappaB and PI3K/Akt pathways and inhibits skin cancer in CD-1 mice. Oncogene 23: 5203-5214. doi: 10.1038/sj.onc.1207641. PubMed: 15122342. [DOI] [PubMed] [Google Scholar]
- 46. Saleem M, Kaur S, Kweon MH, Adhami VM, Afaq F et al. (2005) Lupeol, a fruit and vegetable based triterpene, induces apoptotic death of human pancreatic adenocarcinoma cells via inhibition of Ras signaling pathway. Carcinogenesis 26: 1956-1964. doi: 10.1093/carcin/bgi157. PubMed: 15958516. [DOI] [PubMed] [Google Scholar]
- 47. Saleem M, Kweon MH, Yun JM, Adhami VM, Khan N et al. (2005) A novel dietary triterpene Lupeol induces fas-mediated apoptotic death of androgen-sensitive prostate cancer cells and inhibits tumor growth in a xenograft model. Cancer Res 65: 11203-11213. doi: 10.1158/0008-5472.CAN-05-1965. PubMed: 16322271. [DOI] [PubMed] [Google Scholar]
- 48. Tarapore RS, Siddiqui IA, Adhami VM, Spiegelman VS, Mukhtar H (2013) The dietary terpene lupeol targets colorectal cancer cells with constitutively active Wnt/beta-catenin signaling. Mol Nutr Food Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tarapore RS, Siddiqui IA, Saleem M, Adhami VM, Spiegelman VS et al. (2010) Specific targeting of Wnt/beta-catenin signaling in human melanoma cells by a dietary triterpene lupeol. Carcinogenesis 31: 1844-1853. doi: 10.1093/carcin/bgq169. PubMed: 20732907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen L, Huang K, Han L, Shi Z, Zhang K et al. (2011) beta-catenin/Tcf-4 complex transcriptionally regulates AKT1 in glioma. Int J Oncol 39: 883-890. PubMed: 21720709. [DOI] [PubMed] [Google Scholar]
- 51. Coant N, Ben Mkaddem S, Pedruzzi E, Guichard C, Tréton X et al. (2010) NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate of proliferative progenitor cells in the colon. Mol Cell Biol 30: 2636-2650. doi: 10.1128/MCB.01194-09. PubMed: 20351171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Misra UK, Deedwania R, Pizzo SV (2006) Activation and cross-talk between Akt, NF-kappaB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J Biol Chem 281: 13694-13707. doi: 10.1074/jbc.M511694200. PubMed: 16543232. [DOI] [PubMed] [Google Scholar]
- 53. Guo C, Gasparian AV, Zhuang Z, Bosykh DA, Komar AA et al. (2009) 9-Aminoacridine-based anticancer drugs target the PI3K/AKT/mTOR, NF-kappaB and p53 pathways. Oncogene 28: 1151-1161. doi: 10.1038/onc.2008.460. PubMed: 19137016. [DOI] [PubMed] [Google Scholar]
- 54. Freyberg Z, Ferrando SJ, Javitch JA (2010) Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am J Psychiatry 167: 388-396. doi: 10.1176/appi.ajp.2009.08121873. PubMed: 19917593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hurley EA, Klaman LD, Agger S, Lawrence JB, Thorley-Lawson DA (1991) The prototypical Epstein-Barr virus-transformed lymphoblastoid cell line IB4 is an unusual variant containing integrated but no episomal viral. DNA - J Virol 65: 3958-3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song YJ, Izumi KM, Shinners NP, Gewurz BE, Kieff E (2008) IRF7 activation by Epstein-Barr virus latent membrane protein 1 requires localization at activation sites and TRAF6, but not TRAF2 or TRAF3. Proc Natl Acad Sci U S A 105: 18448-18453. doi: 10.1073/pnas.0809933105. PubMed: 19017798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Song YJ, Stinski MF (2005) Inhibition of cell division by the human cytomegalovirus IE86 protein: role of the p53 pathway or cyclin-dependent kinase 1/cyclin B1. J Virol 79: 2597-2603. doi: 10.1128/JVI.79.4.2597-2603.2005. PubMed: 15681459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bari W, Song YJ, Yoon SS (2011) Suppressed induction of proinflammatory cytokines by a unique metabolite produced by Vibrio cholerae O1 El Tor biotype in cultured host cells. Infect Immun 79: 3149-3158. doi: 10.1128/IAI.01237-10. PubMed: 21576340. [DOI] [PMC free article] [PubMed] [Google Scholar]