

Abstract
This review presents the roles of cardiac sodium channel NaV1.5 late current (late INa) in generation of arrhythmic activity. The assumption of the authors is that proper Na+ channel function is necessary to the maintenance of the transmembrane electrochemical gradient of Na+ and regulation of cardiac electrical activity. Myocyte Na+ channels’ openings during the brief action potential upstroke contribute to peak INa and initiate excitation–contraction coupling. Openings of Na+ channels outside the upstroke contribute to late INa, a depolarizing current that persists throughout the action potential plateau. The small, physiological late INa does not appear to be critical for normal electrical or contractile function in the heart. Late INa does, however, reduce the net repolarizing current, prolongs action potential duration, and increases cellular Na+ loading. An increase of late INa, due to acquired conditions (e.g. heart failure) or inherited Na+ channelopathies, facilitates the formation of early and delayed afterpolarizations and triggered arrhythmias, spontaneous diastolic depolarization, and cellular Ca2+ loading. These in turn increase the spatial and temporal dispersion of repolarization time and may lead to reentrant arrhythmias.
Keywords: Heart, Late INa, Afterpotential, Arrhythmia, Reentry
1. Origins of cardiomyocyte late INa
Although the presence and potential importance of so-called non-inactivating Na+ current in myocytes was recognized as early as 1979,1,2 the roles of this seemingly minor current in arrhythmogenesis were not identified until the demonstration that ‘gain of function’ mutations in the gene SCN5A enhance NaV1.5 late INa and cause the congenital long-QT syndrome type 3 (LQT3).3,4 Pathological roles of late INa in the heart have been reviewed previously.5–18
After opening briefly (about 50 µs at 35°C)19 during the upstroke of the cardiac action potential (AP), individual Na+ channels usually inactivate and remain inactivated until repolarization of the cell membrane. Sodium channel openings after the upstroke create a small ‘late’ current that persists throughout the plateau of the AP. The amplitude of late INa is reported to be ≤0.1% of peak INa in isolated left-ventricular myocytes from the rat,20 guinea pig,21 and human heart.22 Many Na+ channel mutations, pathological conditions, pharmacological agents and toxins delay or destabilize Na+ channel inactivation and increase late INa. The magnitude of late INa in cardiac myocytes may be increased by either acquired conditions such as heart failure,12,23–26 hypoxia/ischaemia,8,27–29 inflammation,30 oxidative stress,31 and thyroid hormone,32 (Table 1) or congenital (inherited) mutations in SCN5A and channel-interacting proteins that cause long-QT syndrome.3,4,15,33,34 Several forms of cardiac Na+ channel dysfunction are direct causes of late INa: (i) delayed or failed inactivation of open channels (i.e. long openings); (ii) transient bursts of re-openings and scattered single late openings of channels that were in an unstable inactivated state; and (iii) fast recovery of channels from inactivation during non-equilibrium conditions, as during repolarization of the AP.19,20,22,35–38 In addition, not all Na+ channels open during the AP upstroke, and those that do not open during peak INa are potentially available to open late. Lastly, within a ‘window’ of voltages that is sufficiently depolarized to cause activation of some Na+ channels but not so depolarized as to cause inactivation of all channels from the closed state, a small equilibrium Na+ current is theoretically present.1 This Na+ window current is not typically referred to as late INa, and its significance is poorly understood. However, it is a potential cause of Na+ loading, especially in depolarized ischaemic myocardium.39 Interestingly, the range of voltages for steady-state window current was shifted by tens of millivolts in a hyperpolarizing direction by membrane stretch.40 This may be partly responsible for a background Na+ current that occurs close to the threshold voltage for Na+ channel activation in myocytes.41
Table 1.
Conditions and agents that have been demonstrated to increase cardiac late INa
| Conditions/endogenous agents | Drugs and toxins |
|---|---|
| Activation of CaMKII | Aconitine |
| Activation of Fyn tyrosine kinase | ATX-II |
| Activation of PKC | Batrachotoxin |
| Angiotensin II | DPI 201–106 and analogues |
| Carbon monoxide | KB130015 |
| 2,3-Diphosphoglycerate | Ouabain (indirectly) |
| Hydrogen peroxide (H2O2) | Pyrethroids (e.g. tefluthrin) |
| Hypoxia, ischaemia | Veratridine |
| Lysophosphatidylcholine | |
| Nitric oxide (NO) | Diseases |
| Palmitoyl-l-carnitine | Heart failure |
| Thyroid hormone T3 | Hypertrophic cardiomyopathy |
2. Myocyte late INa, Na+ and Ca2+ homeostasis, and contractile function
The Na+/Ca2+ exchanger (NCX) and voltage-gated Na+ channels are major routes of Na+ entry into cardiac myocytes.42 Late INa constitutes perhaps one-half of Na+ channel-mediated Na+ entry in ventricular myocytes.43,44 In normal ventricular myocardium at a heart rate of 60/min, late INa-mediated Na+ influx during phase 2 of the AP plateau is estimated to be about 30% of total Na+ influx through Na+ channels.44 Na+ influx during phase 2 can be increased several-fold when late INa is enhanced by, for example, lysophosphatidylcholine and palmitoyl-l-carnitine (lipid metabolites that accumulate during ischaemia), or by H2O2, veratridine, or SCN5A mutations. Enhancement of late INa by five-fold during the AP plateau may double the total Na+ influx into a myocyte during a cardiac cycle.44 In this situation, Na+ influx in phase 2 exceeds that during all other phases of the AP combined.44
The effect of an increase of late INa to raise the intracellular Na+ concentration in cardiac myocytes is well documented. The late INa enhancer veratridine (0.1 µM) increased the intracellular Na+ concentration in a sheep Purkinje fibre by 2.2 mM, accompanied by a 140% increase in twitch tension.45 ATX-II (3 nM) enhanced late INa by four-fold in rat ventricular myocytes and increased the intracellular Na+ concentration by 30%.46 In rabbit myocytes exposed to ATX-II, a two-fold increase of late INa was associated with a four-fold increase of the intracellular Na+ concentration.47 The effect of an increase of late INa to increase the intracellular Na+ concentration appears to be greater in rabbit than rat, as the resting Na+ concentration and Na+/K+ ATPase activity are lower in the rabbit.48,49 Simulated-demand ischaemia (i.e. metabolic inhibition and pacing) of rabbit cardiac myocytes in the absence and presence of the late INa inhibitor ranolazine led to increases of the intracellular Na+ concentration by 13 and 5 mM, respectively.50 In myocytes from failing hearts, the intracellular Na+ concentration is increased by 2–6 mM above normal.51–55 This increase has been attributed to greater Na+ influx due to an enhanced late INa.12,23–26,47,51,56 Increases of Na+ window and/or background current potentially contribute to Na+ influx more in the failing than in the normal heart, and are increased by veratridine and ATX-II as well, apparently due to effects of the latter toxins on the voltage dependence of Na+ channel gating. To our knowledge, the effect of an LQT3 mutation in SCN5A on the intracellular Na+ concentration has yet to be reported.
A late INa-induced increase of the intracellular Na+ concentration alters contractile function. Studies of Purkinje fibres and papillary muscles demonstrate that elevation of the intracellular Na+ concentration by 1–2 mM may cause twitch tension to increase acutely as much as 2.5-fold.43,45,55,57–59 An increase of Na+ concentration (generated by late INa) in the t-tubule subsarcolemmal fuzzy space has been proposed to drive Ca2+ entry via reverse mode NCX.56,60,61 The direction of NCX-mediated ion fluxes is regulated by the electrochemical gradients of Na+ and Ca2+ and by the membrane potential. When intracellular Na+ rises, forward mode (3 Na+ in and 1 Ca2+ out) NCX is reduced, whereas reverse mode (3 Na+ out and 1 Ca2+ in) NCX is increased.51,62 Cohen et al.57 calculated that an increase of the intracellular Na+ concentration from 8 to 10 mM reduces the electrochemical driving force for NCX-mediated Ca2+ efflux by half. An increase of late INa is associated with an increased diastolic Ca2+ concentration in myocytes46,47,63 and isolated hearts.64 An increase of the intracellular Na+ concentration in the hypertrophied/failing heart supports systolic function at low heart rates by increasing Ca2+ influx via NCX.51 However, it is associated with increases of diastolic Ca2+, diastolic contractile tension, and arrhythmias at higher heart rates.47,53–56,65 Increases of late INa and intracellular Na+ have been shown to raise tonic contractile force and myocardial wall stress in the intact heart.66–68 The effects of an increase of late INa on AP duration and ion homeostasis in the guinea-pig ventricular myocyte have been modelled.69 An increase of late INa from 0 to 0.2% of peak INa at a pacing rate of 1 Hz increased AP duration by nearly 2.2-fold and Na+ and Ca2+ concentrations in diastole by 34 and 52%, respectively, and was associated with spontaneous erratic releases of Ca2+ from the sarcoplasmic reticulum.69 An increase of late INa in myocytes isolated from failing human and dog hearts is also associated with spontaneous releases of sarcoplasmic reticular Ca2+ during diastole.56 Drug-induced inhibition of late INa has been shown to reduce Na+-dependent Ca2+ loading and contractile dysfunction of cardiac myocytes from both normal and failing hearts,39,47,56,63–65 and contractile dysfunction in the ischaemic heart.70,71 One may conclude that an enhanced late INa can cause changes of Na+ entry and the transmembrane Na+ gradient that alter cardiac function.
The effects of inhibiting the normal, small, endogenous late INa have not been unequivocally demonstrated due to the lack of a selective blocker of the current. Lidocaine (20 µM) acutely reduced AP duration, twitch tension, and intracellular Na+ concentration in sheep Purkinje fibres.43 The reduction of twitch tension by lidocaine was due in roughly equal parts to the decrease in AP duration and the reduction of intracellular Na+.43 Lidocaine reduces both peak and late INa, and reductions by the drug of contractile force and intracellular Na+ concentration may be due to both actions. Because Na+ channels become more inactivated as heart rate increases, a decrease in late INa at higher rates contributes to rate-dependent shortening of AP duration.72,73 Results of a recent study of the selective late INa inhibitor GS967 indicate that reduction of endogenous late INa in rabbit hearts and isolated ventricular myocytes is associated with a decrease of AP duration, a small but non-significant decrease in intracellular Na+, and no change in Ca2+.74
3. Types and mechanisms of late INa-induced arrhythmic activity
The detrimental electrical effects of an enhanced, pathological late INa are depicted in Figure 1, and include the following: (i) diastolic depolarization during phase 4 of the AP that may lead to spontaneous AP firing and abnormal automaticity, especially of myocytes that are relatively depolarized and have low resting K+ conductance (e.g. low IK1); (ii) an increase of AP duration, due to the depolarizing effect of an increased inward Na+ current during the AP plateau, and which may lead to early after-depolarizations (EADs) and triggered activity, as well as increased spatiotemporal differences of repolarization time, which promote reentrant electrical activity; and (iii) the indirect effects of a late INa-induced increase of Na+ entry to alter Ca2+ homeostasis in myocytes, which may lead to Ca2+ alternans and DADs. Acquired conditions and drugs that enhance late INa (Table 1) are associated with atrial tachyarrhythmias,75–78 ventricular tachyarrhythmias including torsades de pointes (TdP),3,4,79,80 afterpotentials (EADs, DADs), and triggered activity.23,26,76,81 Patients with LQT3 are at a high risk for both ventricular arrhythmias and atrial fibrillation.3,4,15,82,83 All three of the common mechanisms for tachyarrhythmias—abnormal automaticity, afterpotentials, and reentry—can occur as the result of an enhanced late INa.
Figure 1.
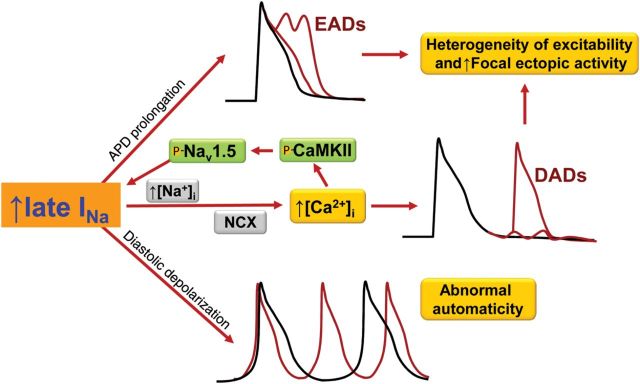
Mechanisms of late INa-induced arrhythmia: EADs, DADs, and spontaneous diastolic depolarization. Not shown, late INa increases spatiotemporal dispersion of repolarization and facilitates reentrant arrhythmic activity. NCX, Na+/Ca2+ exchange; CaMKII, Ca2+/calmodulin-dependent protein kinase II.
4. Diastolic depolarization and abnormal automaticity
Spontaneous diastolic depolarization of a myocyte during phase 4 of the AP occurs normally in pacemaking cells of the central sinoatrial and compact atrioventricular nodes, but is rare in normal intact atrial and ventricular tissues. However, spontaneous diastolic depolarizations are often observed in isolated Purkinje fibres84 and atrial tissue excised from diseased human85–87 and animal88–90 hearts, and are a cause of lethal arrhythmias in the infarcted heart.91 The cause of diastolic depolarization in these cells—which are not normally involved in pacemaking—is unclear. Non-inactivating Na+ current (window or background Na+ current) is observed in the threshold region for Na+ channel activation in Purkinje fibres.92 This finding is consistent with reports that a slowly inactivating, lidocaine and tetrodotoxin (TTX)-sensitive Na+ current contributes to diastolic depolarization of cardiac Purkinje fibres.92–94 In ventricular myocytes, late INa was shown to be present at voltages as negative as −70 mV,95,96 but spontaneous activity of ventricular myocytes in the intact heart appears to be rare in the absence of K+ channelopathies that decrease the resting potential. Spontaneous diastolic depolarization and the rate of AP firing of atrial myocytes can be increased and decreased by late INa enhancers and inhibitors, respectively.77 Late INa was found to be present in atrial myocytes that undergo spontaneous diastolic depolarization, and ATX-II accelerated diastolic depolarization and induced rapid firing of APs in these cells.77 The reactive oxygen species H2O2 increases late INa and causes diastolic depolarization and rapid AP firing of isolated atrial myocytes (Figure 2).77 Atrial myocyte diastolic depolarization and AP firing in the absence and presence of H2O2 were reduced when late INa was inhibited using ranolazine or TTX.77 Voltage-clamp studies of atrial myocytes demonstrated that an inward current is activated by a depolarizing ramp pulse and that the ramp-induced current is blocked by TTX and enhanced by ATX-II, consistent with its identification as late INa.77 These findings suggest that late INa is a cause of spontaneous diastolic depolarization and abnormal automaticity that may contribute to arrhythmogenesis in atrial myocytes and Purkinje fibres.
Figure 2.
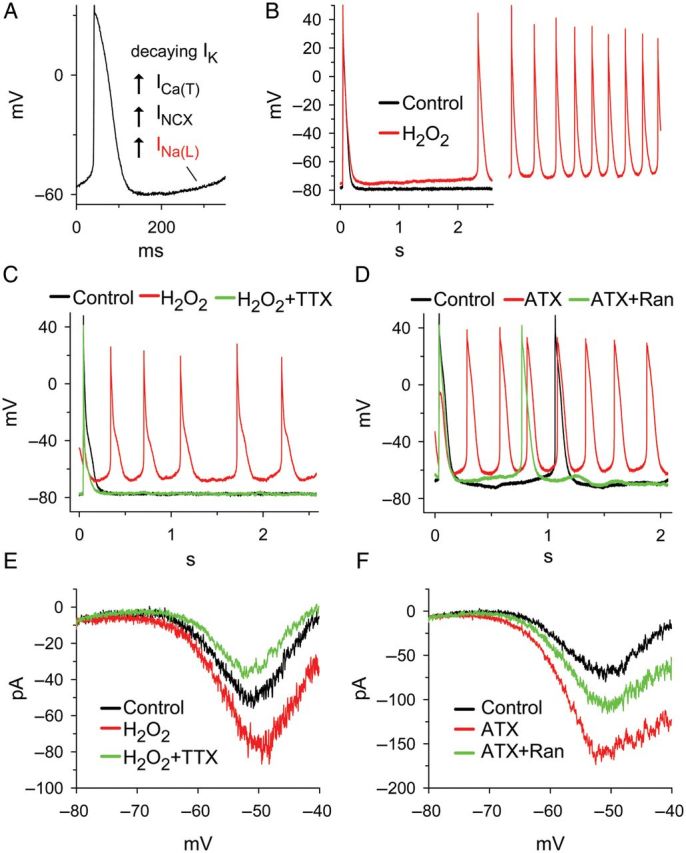
Hydrogen peroxide (H2O2, 50 µM) and anemone toxin-II (ATX, 5 nM) increase late INa and induce diastolic depolarization in guinea pig atrial myocytes. (A) Four proposed mechanisms for diastolic depolarization: decay of the delayed rectifier current, IK; an increase of T-type Ca2+ current, ICa(T); an increase of forward mode Na+/Ca2+ exchange current, INCX; and an increase of late INa. (B) Induction by H2O2 of diastolic depolarization and rapid spontaneous firing in a quiescent atrial myocyte. (C) H2O2-induced spontaneous firing was terminated by tetrodotoxin (TTX, 1 µM). (D) Spontaneous action potential firing of an atrial myocyte was accelerated by ATX. The effect of ATX was attenuated by ranolazine (Ran, 10 µM). (E) Inward late INa in an atrial myocyte was activated by simulating diastolic depolarization using a ramp voltage clamp. H2O2 increased whereas TTX decreased the amplitude of late INa. (F) ATX enhanced whereas Ran inhibited inward late INa activated by ramp voltage clamp pulses. pA, picoamperes; mV, millivolts.
5. AP prolongation and EADs
During the AP plateau, membrane resistance is high (i.e. ionic conductance is low). A modest increase of an inward current such as late INa, or reduction of an outward current such as IKr, can cause marked AP prolongation.81 Late INa is documented to prolong the duration of the AP.21,81,97–99 Tetrodotoxin and lidocaine inhibit late INa and reduce the duration of the AP in Purkinje fibres and ventricular myocytes.2,23,43,92,97,99These findings are consistent with the interpretation that late INa during the AP plateau reduces net repolarizing current (i.e. repolarization reserve100).
EAD are a primary mechanism of arrhythmic activity and there is considerable evidence that their occurrence is facilitated when late INa is enhanced and the AP is prolonged. AP prolongation provides time for L-type Ca2+ channels to recover from inactivation and re-activate.101–104 The resulting Ca2+ ‘window’ current may increase progressively over a range of voltages from −30 to 0 mV to form the upstroke of an EAD.104,105 Calcium influx leads to increases of subsarcolemmal Ca2+, Ca2+-induced Ca2+ release, and Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity.104 An increase of the subsarcolemmal Ca2+ concentration drives forward mode NCX. Forward mode NCX generates inward, depolarizing current that further contributes to AP prolongation.106–108 Both forward mode NCX and late INa (which is increased by CaMKII-mediated Na+ channel phosphorylation) contribute inward current that may be sufficient to overcome outward repolarizing K+ currents and enable an EAD. Single and burst openings of Na+ channels are reported to occur at take-off voltages for EADs.24 Indeed, modelling of Purkinje fibre electrophysiology indicates that late INa is the major inward current responsible for generation of the EAD in that cell.109 Consistent with this hypothesis, the late INa enhancer anthopleurin-A increases the dispersion of repolarization and induces spontaneous tachyarrhythmias that are triggered by subendocardial Purkinje tissue in the dog heart.110 The increase of late INa that is observed in myocytes isolated from failing human and dog hearts is associated with a prolonged AP duration, increased beat-to-beat variability of AP duration, and EADs.23,26 Enhancers of late INa such as ATX-II (Figure 3) and H2O2 cause EADs and TdP.31,65,79,81,111 EADs are common in mice expressing the LQT3 mutant ‘gain-of-function’ Na+ channel ΔKPQ.112 In contrast, inhibitors of late INa reduce occurrences of EADs and TdP in the presence of H2O231,63,113 and IKr blockers.80,114,115 Reentrant and multifocal ventricular fibrillation in aged rat isolated hearts can be induced by rapid pacing or treatment with H2O2; the late INa inhibitor ranolazine suppressed EADs and the number of foci, and terminated ventricular fibrillation in these hearts.113 Inhibition of late INa was recently reported to markedly shorten AP duration and halve the occurrence of EADs in myocytes isolated from patients with hypertrophic cardiomyopathy.116
Figure 3.
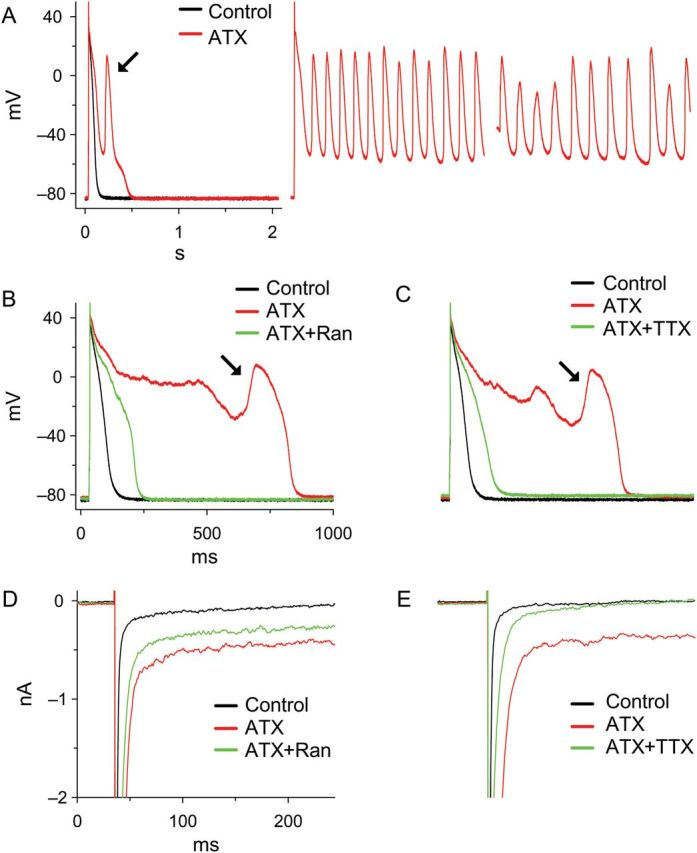
Anemone toxin-II (ATX, 10 nM) induced EADs and enhanced late INa in guinea pig atrial myocytes; these effects were attenuated by either ranolazine (Ran) or tetrodotoxin (TTX). (A) ATX induced EADs and sustained triggered activity. (B and C) ATX-induced EADs were suppressed by either 10 µM Ran or 2 µM TTX. (D and E) ATX increased the late INa activated by square voltage clamps from −90 to −20 mV in a ventricular myocyte. The effect of ATX was attenuated by either 10 µM Ran or 10 µM TTX. nA, nanoamperes; mV, millivolts.
The amplitude of late INa and its contribution to EAD formation depend on heart rate. Late INa is greater at slow than at fast heart rates56,72,117 because an increased rate of Na+ channel opening increases channel inactivation and reduces late INa.56,72 Reduction of late INa with increased rate contributes to the normal reverse-rate dependence of AP duration in Purkinje fibres,109 rabbit hearts,72 and myocytes isolated from failing human hearts.56 Slow pacing rates facilitate long APs and increase late INa and occurrences of EADs and TdP.37,72,103 The role of late INa to increase EAD formation and dispersion of repolarization is increased by heart rate slowing, and the pro-arrhythmic risk associated with an increased late INa is high when heart rate is low.37,118,119
A small increase of late INa that does not cause arrhythmic activity in the normal heart may do so in the heart with reduced repolarization reserve. Low concentrations of the late INa enhancer ATX-II increased the duration of the monophasic AP, but did not cause arrhythmias in rabbit isolated hearts.79,120 However, late INa facilitated the induction of EADs by blockers of the rapid (IKr) or slowly (IKs) activating components of the delayed rectifier K+ current (Figure 4). When low concentrations of E-4031, amiodarone, cisapride, quinidine, moxifloxacin, or ziprasidone alone caused little or no arrhythmic activity in the isolated rabbit heart, combinations of these IKr blockers with ATX-II greatly increased AP duration and caused ventricular tachyarrhythmias.79,120–122 Similarly, in guinea pig isolated ventricular myocytes, low concentrations of ATX-II, the IKr blocker E-4031, and the IKs blocker chromanol 293B individually caused small increases of AP duration.81 However, combinations of ATX-II with either E-4031 or chromanol 293B markedly prolonged AP duration and induced EADs (Figure 4).81 In patients, drugs that block IK prolong the QT interval and may induce EADs.123 However, not all patients exposed to these drugs develop arrhythmias. Genetic analysis revealed that susceptibility to drug-induced long QT syndromes is linked to SCN5A mutations (e.g. L1825P or Y1102) that enhance late INa.124,125 Patients with ‘silent’ Na+ channel gene mutations had normal QT intervals, but developed long QT syndrome and TdP when given an IKr blocker such as cisapride or amiodarone.124,125 An enhanced late INa is therefore a risk factor predisposing to EADs under both acquired (disease and drug-induced) and inherited (LQT1 and LQT2) pathological conditions. An ideal substrate for generation of EADs and TdP in the failing and/or hypertrophic heart is present when late INa is enhanced,26,56 the inward-rectifier K+ current, IK1 is reduced,126,127 NCX, diastolic Ca2+, and sarcoplasmic reticular Ca2+ sparks are increased,128–131 repolarizing K+ currents are reduced,123 and spatial and temporal lability of repolarization is prominent.128,132 Inhibition of late INa reduces EADs in ventricular myocytes isolated from failing and hypertrophic hearts22,26,116 and in left atrial myocytes from hearts of rabbits with left-ventricular hypertrophy.78 Interestingly, stem cell-derived cardiomyocytes generated from an LQT3 mouse model carrying the human ΔKPQ Nav1.5 mutation recapitulate the typical pathophysiological ΔKPQ phenotype, including APD prolongation and EAD development.133
Figure 4.

Synergistic effects of the late INa enhancer ATX-II (3 nM) with either the IKr blocker E4031 (1 µM) or the IKs blocker chromanol 293B (30 µM) to prolong the action potential duration (APD) and induce early afterdepolarizations. The combined effects of ATX-II and E4031 or 293B were attenuated by ranolazine (10 µM) (C and G). Data summarized in (D) and (H). Each bar represents the value (mean ± SEM) of a percentage increase of APD50. *P < 0.05 vs. ATX-II, E4031, or 293B alone; †P < 0.05 vs. ATX-II plus E4031 or ATX-II plus 293B. The duration of drug treatment was 3 min. C, control (absence of drug); A, ATX-II; E4, E-4031; C293B or CB, chromanol 293B; Ran, ranolazine.
The contribution of late INa to EAD formation in phase 3 of the ventricular AP is unclear. In phase 3, L-type Ca2+ channel activation and Ca2+ window current are negligible.62 The more negative membrane potential during phase 3 relative to phase 2 favours Na+ influx. Therefore, increases of both late INa and inward Na+/Ca2+ exchange current134–136 may contribute to the upstroke of EADs during phase 3. Rapid recovery from inactivation and reactivation of Na+ channels is a potential cause of phase 3 EADs and triggered activity.38,137 However, because repolarizing K+ currents during phase 3 are normally robust unless the extracellular [K+]o is reduced and IK1 is inhibited, it would appear that depolarizing currents must be large to elicit an EAD at this time. Depolarizing current flowing electrotonically from myocytes with long APs to those with shorter APs may contribute to initiation of phase 3 EADs in the intact heart.138 Exacerbation of the large repolarization gradients that favour current flow between Purkinje fibres and M cells, on the one hand, and adjacent cells with shorter AP durations, on the other hand, would favour EAD formation110,139–141 and reentrant arrhythmias141 by this ‘extrinsic’ electrotonic mechanism.105 Late INa is inherently greater in Purkinje fibres and M cells139,142 than in other cells in the heart and contributes to AP prolongation and EAD formation in these cells. Enhancement of late INa enables reentrant AP propagation from these endocardial cells with long APs to repolarized myocardium.137
6. Intracellular Na+ and Ca2+ loading and DADs
Transient depolarizations of the cell membrane that follow repolarization of a previous AP are referred to as delayed after-depolarizations. DADs of Purkinje fibres have been recognized for >40 years as a mechanism of digitalis glycoside-induced arrhythmogenesis and non-reentrant triggered activity.143,144 A transient inward current, ITi, was found to be responsible for the DAD,144–146 and inward, forward mode NCX (i.e. entry of 3Na+ with exit of 1 Ca2+) was identified as the source of this current.145–148 ITi and/or DADs have been observed in Purkinje,144 ventricular,145–147 atrial,76,149 pulmonary vein sleeve,150,151 superior vena cava,152 and sinoatrial node153 tissues. DADs are observed under conditions in which myocytes are relatively overloaded with Ca2+, causing Ca2+ to be released from multiple sarcoplasmic reticulum sites into the cytoplasm during diastole;154 this increase of cytoplasmic Ca2+ leads to aftercontractions and forward mode NCX that generates transient inward current and a DAD.101,144,147,148,155–157 Events that promote a combination of an increase of the intracellular Na+ concentration, increased Ca2+ influx (e.g. rapid pacing, catecholamines, block of IKs), decreased Ca2+ efflux, opening of sarcoplasmic reticulum Ca2+ channels (i.e. ryanodine receptors), and reduced outward K+ current (e.g. IK1) during diastole act to facilitate DADs.
The role of late INa in DAD generation is not as a source of inward current, as that is provided by forward mode NCX, but rather to ‘set the stage’ by increasing cellular Ca2+ loading via reverse mode NCX (Figure 1). An increase of late INa can increase the intracellular, subsarcolemmal Na+ concentration, thereby increasing Ca2+ entry via reverse-mode NCX (3 Na+ out, 1 Ca2+ in) during the AP plateau.45,47,48,56,158 The contribution of late INa to Na+ and Ca2+ loading has been referred to as an intrinsic digitalis-like effect.12,26,159 Like digitalis, late INa-mediated Na+ loading (i) may increase Ca2+ entry into the cell, and Ca2+ uptake by sarcoplasmic reticulum, (ii)increase diastolic Ca2+ and reduce the rate and extent of diastolic relaxation, and (iii) give rise to Ca2+ release from the sarcoplasmic reticulum during diastole, and DAD formation (Figure 1).12,47,56,63,65 An increase of late INa prolonged the Ca2+ transient and induced spontaneous Ca2+ waves during rapid pacing of rat isolated hearts.160 Exposure of myocytes to late INa enhancers provokes DADs.27,63,76,161,162 The transient inward current ITI and both DADs and DAD-dependent triggered activity can be induced by ATX-II in guinea pig atrial myocytes.76 DADs induced by cardiac glycosides or other interventions are suppressed by inhibitors of Na+ channels and late INa, including TTX, lidocaine, mexiletine, R56865, and ranolazine.63,65,76,162–164 Inhibition of late INa has also been shown to decrease the incidence of DADs in studies of pulmonary vein and superior vena cava sleeves,152 and in myocytes from hearts of patients with hypertrophic cardiomyopathy.116 These findings implicate increased Na+ entry into myocytes via Na+ channel late INa as a cause of DADs. Inhibition of late INa is a means of reducing occurrences of DADs.
A positive feedback loop between the amplitude of late INa and the activity of CaMKII appears to contribute to DAD formation and arrhythmogenesis. An increase of late INa can lead to myocyte Ca2+ loading and activation of CaMKII.46 CaMKII phosphorylates sodium channel sites in the intracellular linker between domains 1 and 2, and this increases late INa.165–169 CaMKII also phosphorylates cardiomyocyte ryanodine receptor II (RyR2), which increases RyR2 sensitivity to SR Ca2+-induced opening.170 This facilitates more frequent and larger releases of Ca2+ from the SR (i.e. sparks) during diastole,170 and leads to Ca2+ waves and DAD generation.171 Thus increases of late INa and activity of CaMKII increase SR Ca2+ loading and SR Ca2+ release, respectively; together they create a substrate for DAD-induced arrhythmias.
7. Late INa, dispersion of repolarization, and reentry
The mechanism of reentrant arrhythmia involves unidirectional block and conduction around a circuit long enough to enable recovery of excitability at each point in the circuit before the wave of excitation returns.172 Acquired or congenital conditions that exacerbate normal regional differences in duration of the AP and the time sequence of repolarization in the heart create a substrate for reentry.137,173,174 Electrical and/or structural heterogeneity of repolarization, excitability, and conduction among adjacent regions of myocardium are associated with EADs, TdP, and reentrant arrhythmic events.174–178 Results of computational modelling studies indicate that increased spatial and temporal dispersion of AP duration and repolarization time act to increase susceptibility to reentrant arrhythmic activity.179,180
Late INa contributes to the regional differences of AP duration and repolarization in myocardium. Differences in the density of late INa in various cell types (i.e. Purkinje fibre, M cell > endo > epicardium)2,7,142 contribute to transmural differences in AP duration (Figure 5). An increase of late INa can prolong APD more in some cells than others, thereby increasing dispersion of repolarization and providing a potential substrate for reentry. An ATX-II induced increase of late INa increases AP duration more in M and Purkinje cells than in epicardium, and increases transmural dispersion of repolarization time; these effects are attenuated by inhibition of late INa with mexiletine or tetrodotoxin.7,80,181,182 Augmentation of late INa with ATX-II in canine left-ventricular wedge preparations leads to reentrant arrhythmias.80,182 The late INa enhancers anthopleurin-A and veratridine also cause EADs and reentrant arrhythmias in intact isolated guinea pig and rabbit hearts, respectively.183,184 Reduction of late INa decreased transmural dispersion of repolarization and suppressed TdP in canine and rabbit experimental models of LQT1, LQT2, and LQT3 syndromes.16,79,80,115,185
Figure 5.

Spatial (A) and temporal (B) differences in the duration of the action potential (APD) create a substrate for reentrant arrhythmias. Reprinted from Belardinelli et al.140 and Undrovinas et al.65
An increase of late INa also increases the beat-to-beat (temporal) variability of AP duration and repolarization (Figure 5) in isolated myocytes23,81 and intact hearts.115 Late INa and repolarization variability are especially enhanced in myocytes isolated from failing hearts, and reduction of late INa reduces the beat-to-beat variability of AP duration in these cells.26,65 Reduction of late INa by ranolazine decreased the beat-to-beat variability of repolarization caused by treatment of rabbit isolated hearts with the IKr blocker E-4031,115 and the rate dependence of pacing-induced alternans of the beat-to-beat Ca2+ transient amplitude in rat isolated hearts treated with ATX-II.160 The magnitude of T-wave alternans, which is believed to reflect the spatio-temporal heterogeneity of ventricular repolarization, was suppressed by ranolazine in intact pigs subjected to acute coronary stenosis.186 During ventricular fibrillation, dynamic beat-to-beat heterogeneity of AP duration (i.e. alternans) is a major contributor to wave break, a process in which new waves of reentrant excitation are continually formed, thus sustaining fibrillation.187 The effect of ranolazine to reduce beat-to-beat variability of AP duration may reduce wave break and reentrant activity,113 and explain the many clinical and experimental findings that the drug reduces the incidence and duration of arrhythmias.7,9,13,14,16
8. Models of late INa-induced arrhythmogenesis
Experimental animal models for study of the arrhythmogenic effects of enhancing late INa include rabbit,120–122,184 guinea pig,79,81 and rodent isolated hearts,64,71,160 intact pigs,186 and dog isolated left-ventricular wedge preparations.80 Late INa is enhanced in myocytes of dogs and humans with heart failure,23–26 by toxins such as ATX-II,81,111 veratridine,71 and aconitine,159,161 by ischaemia8,27–29,70 and oxidative stress,31,50,63 and by activation of various kinases including CaMKII165–169 (Table 1). Among the inherited gain-of-function NaV1.5 channelopathies that are causes of LQT3, the best-studied is ΔKPQ, a deletion of 3 amino acids in the putative inactivation gate of the Na+ channel.3,4,118 Mice expressing heterozygous knock-in ΔKPQ Na+ channels experience cardiac arrhythmias including TdP and have been used to investigate underlying mechanisms of late INa-associated arrhythmogenesis, including EADs, pause-induced DADs, AP prolongation with increased dispersion, and APD alternans.112,119,188–190 Major limitations of rodent models of LQT3 as guides to an understanding of the consequences of increasing/decreasing late INa in human myocardium include the higher intracellular Na+ concentration in the rodent cardiomyocyte and therefore a reduced role of Na+ entry in the regulation of Ca2+ handling,48,49 and the difficulty of assessing drug effects and channel function in hearts whose APs are so different in shape and duration as those in mouse and man. Both Na+ channel function and drugs actions are heart rate- and voltage-dependent, and Na+ channel drugs have non-specific actions on other ion currents whose amplitude and roles differ in human and rodent hearts.
9. Conclusion and perspectives
Late INa is a small inward current that reduces repolarization reserve and prolongs the duration of the AP in cardiac myocytes. Physiological roles for late INa-mediated Na+ loading to contribute to the normal inotropic state and for late INa-induced AP prolongation to increase the effective refractory period and decrease reentry are theoretically possible but have not been adequately investigated. The amplitude of late INa is increased in many pathological conditions, where it contributes to atrial and ventricular arrhythmogenesis. An increase of late INa due to acquired or inherited Na+ channelopathies abnormally prolongs repolarization and increases the influx of Na+, and via NCX, Ca2+ into the cell. Late INa and NCX-mediated Ca2+ loading increase diastolic force production. AP prolongation and Na+/Ca2+ loading cause CaMKII activation and electrical instability. Enhancement of late INa may lead to automaticity, early and delayed afterdepolarizations, and Ca2+ and AP alternans that facilitate arrhythmias by triggered and reentrant mechanisms. Drugs that reduce late INa have been shown to reduce EADs, DADs, Ca2+ handling defects, and arrhythmias. Interactions between late INa, CaMKII, RyR2, and oxidative stress have been demonstrated, and their potential pathological roles in ischaemic heart disease, heart failure, and arrhythmias are subjects of current and future investigation.
Conflict of interest: L.B. and S.R. are employees of Gilead Sciences. Y.S., C.A., and J.C.S. receive support from Gilead Sciences. Gilead Sciences owns ranolazine and GS967.
Funding
Support was provided by Gilead Sciences. C.A. is supported by grant HL47678 from National Heart Lung and Blood Institute, New York State Stem Cell Science grant C026424, and the Masons of New York State, Florida, Massachusetts, and Connecticut.
References
- 1.Attwell D, Cohen I, Eisner D, Ohba M, Ojeda C. The steady state TTX-sensitive (‘window’) sodium current in cardiac Purkinje fibres. Pflugers Arch. 1979;379:137–142. doi: 10.1007/BF00586939. doi:10.1007/BF00586939. [DOI] [PubMed] [Google Scholar]
- 2.Coraboeuf E, Deroubaix E, Coulombe A. Effect of tetrodotoxin on action potentials of the conducting system of the dog heart. Am J Physiol. 1979;236:H561–H567. doi: 10.1152/ajpheart.1979.236.4.H561. [DOI] [PubMed] [Google Scholar]
- 3.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. doi:10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 4.Bennett PB, Yazawa K, Makita N, George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. doi:10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 5.Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92(Suppl. iv):1–5. doi: 10.1136/hrt.2005.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92(Suppl. iv):6–14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antzelevitch C, Belardinelli L. The role of sodium channel current in modulating transmural dispersion of repolarization and arrhythmogenesis. J Cardiovasc Electrophysiol. 2006;17(Suppl. 1):S79–S85. doi: 10.1111/j.1540-8167.2006.00388.x. doi:10.1111/j.1540-8167.2006.00388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saint DA. The role of the persistent Na+ current during cardiac ischemia and hypoxia. J Cardiovasc Electrophysiol. 2006;17:S96–S103. doi: 10.1111/j.1540-8167.2006.00390.x. doi:10.1111/j.1540-8167.2006.00390.x. [DOI] [PubMed] [Google Scholar]
- 9.Shryock JC, Belardinelli L. Inhibition of late sodium current to reduce electrical and mechanical dysfunction of ischaemic myocardium. Br J Pharmacol. 2008;153:1128–1132. doi: 10.1038/sj.bjp.0707522. doi:10.1038/sj.bjp.0707522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saint DA. The cardiac persistent sodium current: an appealing therapeutic target? Br J Pharmacol. 2008;153:1133–1142. doi: 10.1038/sj.bjp.0707492. doi:10.1038/sj.bjp.0707492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac ‘late sodium current. Pharmacol Ther. 2008;119:326–339. doi: 10.1016/j.pharmthera.2008.06.001. doi:10.1016/j.pharmthera.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Undrovinas A, Maltsev VA. Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovasc Hematol Agents Med Chem. 2008;6:348–359. doi: 10.2174/187152508785909447. doi:10.2174/187152508785909447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hale SL, Shryock JC, Belardinelli L, Sweeney M, Kloner RA. Late sodium current inhibition as a new cardioprotective approach. J Mol Cell Cardiol. 2008;44:954–967. doi: 10.1016/j.yjmcc.2008.03.019. doi:10.1016/j.yjmcc.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 14.Maier LS. A novel mechanism for the treat of angina, arrhythmias, and diastolic dysfunction: inhibition of late INa using ranolazine. J Cardiovasc Pharmacol. 2009;54:279–286. doi: 10.1097/FJC.0b013e3181a1b9e7. doi:10.1097/FJC.0b013e3181a1b9e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337–348. doi: 10.1038/nrcardio.2009.44. doi:10.1038/nrcardio.2009.44. [DOI] [PubMed] [Google Scholar]
- 16.Antzelevitch C, Burashnikov A, Sicouri S, Belardinelli L. Electrophysiological basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. 2011;8:1281–1290. doi: 10.1016/j.hrthm.2011.03.045. doi:10.1016/j.hrthm.2011.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moreno JD, Clancy CE. Pathophysiology of the cardiac late Na current and its potential as a drug target. J Mol Cell Cardiol. 2012;52:608–619. doi: 10.1016/j.yjmcc.2011.12.003. doi:10.1016/j.yjmcc.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trenor B, Cardona K, Gomez JF, Rajamani S, Ferrero JM, Jr, Belardinelli L, et al. Simulation and mechanistic investigation of the arrhythmogenic role of the late sodium current in human heart failure. PLoS One. 2012;7:e32659. doi: 10.1371/journal.pone.0032659. doi:10.1371/journal.pone.0032659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benndorf K. Properties of single cardiac Na channels at 35°C. J Gen Physiol. 1994;104:801–820. doi: 10.1085/jgp.104.5.801. doi:10.1085/jgp.104.5.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patlak JB, Ortiz M. Slow currents through single sodium channels of the adult rat heart. J Gen Physiol. 1985;86:89–104. doi: 10.1085/jgp.86.1.89. doi:10.1085/jgp.86.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiyosue T, Arita M. Late sodium current and its contribution to action potential configuration in guinea pig ventricular myocytes. Circ Res. 1989;64:389–397. doi: 10.1161/01.res.64.2.389. doi:10.1161/01.RES.64.2.389. [DOI] [PubMed] [Google Scholar]
- 22.Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res. 2006;69:116–127. doi: 10.1016/j.cardiores.2005.08.015. doi:10.1016/j.cardiores.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. doi:10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Undrovinas AI, Maltsev VA, Kyle JW, Silverman N, Sabbah HN. Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–1489. doi: 10.1006/jmcc.2002.2100. doi:10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- 25.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. doi:10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 26.Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implication for repolarization variability. Eur J Heart Fail. 2007;9:219–227. doi: 10.1016/j.ejheart.2006.08.007. doi:10.1016/j.ejheart.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J, Corr PB. Palmitoylcarnitine increases [Na+]i and initiates transient inward current in adult ventricular myocytes. Am J Physiol. 1995;268:H2405–H2417. doi: 10.1152/ajpheart.1995.268.6.H2405. [DOI] [PubMed] [Google Scholar]
- 28.Ju Y-K, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammarström AKM, Gage PW. Hypoxia and persistent sodium current. Eur Biophys J. 2002;31:323–330. doi: 10.1007/s00249-002-0218-2. doi:10.1007/s00249-002-0218-2. [DOI] [PubMed] [Google Scholar]
- 30.Ward CA, Bazzazi H, Clark RB, Nygren A, Giles WR. Actions of emigrated neutrophils on Na+ and K+ currents in rat ventricular myocytes. Prog Biophys Mol Biol. 2006;90:249–269. doi: 10.1016/j.pbiomolbio.2005.07.003. doi:10.1016/j.pbiomolbio.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J Physiol. 1997;500:631–642. doi: 10.1113/jphysiol.1997.sp022048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang YG, Dedkova EN, Fiening JP, Ojamaa K, Blatter LA, Lipsius SL. Acute exposure to thyroid hormone increases Na+ current and intracellular Ca2+ in cat atrial myocytes. J Physiol. 2003;546:491–499. doi: 10.1113/jphysiol.2002.032847. doi:10.1113/jphysiol.2002.032847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.George AL., Jr Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. doi:10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abriel H. Cardiac sodium channel NaV1.5 and interacting proteins: physiology and pathophysiology. J Mol Cell Cardiol. 2010;48:2–11. doi: 10.1016/j.yjmcc.2009.08.025. doi:10.1016/j.yjmcc.2009.08.025. [DOI] [PubMed] [Google Scholar]
- 35.Kunze DL, Lacerda AE, Wilson DL, Brown AM. Cardiac Na currents and the inactivating, reopening, and waiting properties of single cardiac Na channels. J Gen Physiol. 1985;86:691–719. doi: 10.1085/jgp.86.5.691. doi:10.1085/jgp.86.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li CZ, Wang XD, Wang HW, Bian YT, Liu YM. Four types of late Na channel current in isolated ventricular myocytes with reference to their contribution to the lastingness of action potential plateau. Acta Physiol Sinica. 1997;49:241–248. [PubMed] [Google Scholar]
- 37.Clancy CE, Tateyama M, Kass RS. Insights into the molecular mechanisms of bradycardia-triggered arrhythmias in long QT-3 syndrome. J Clin Invest. 2002;110:1251–1262. doi: 10.1172/JCI15928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clancy CE, Tateyama M, Liu H, Wehrens XHT, Kass RS. Non-equilibrium gating in cardiac Na+ channels. An original mechanism of arrhythmia. Circulation. 2003;107:2233–2237. doi: 10.1161/01.CIR.0000069273.51375.BD. doi:10.1161/01.CIR.0000069273.51375.BD. [DOI] [PubMed] [Google Scholar]
- 39.Haigney MCP, Lakatta EG, Stern MD, Silverman HS. Sodium channel blockade reduces hypoxic sodium loading and sodium-dependent calcium loading. Circulation. 1994;90:391–399. doi: 10.1161/01.cir.90.1.391. doi:10.1161/01.CIR.90.1.391. [DOI] [PubMed] [Google Scholar]
- 40.Beyder A, Rae JL, Bernard C, Strege PR, Sachs F, Farrugia G. Mechanosensitivity of NaV1.5, a voltage-sensitive sodium channel. J Physiol. 2010;588:4969–4985. doi: 10.1113/jphysiol.2010.199034. doi:10.1113/jphysiol.2010.199034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zilberter Y, Starmer CF, Starobin J, Grant AO. Late Na channels in cardiac cells: the physiological role of background Na channels. Biophys J. 1994;67:153–160. doi: 10.1016/S0006-3495(94)80464-3. doi:10.1016/S0006-3495(94)80464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bers DM, Barry WH, Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res. 2003;57:897–912. doi: 10.1016/s0008-6363(02)00656-9. doi:10.1016/S0008-6363(02)00656-9. [DOI] [PubMed] [Google Scholar]
- 43.Sheu S-S, Lederer WJ. Lidocaine's negative inotropic and antiarrhythmic actions. Dependence on shortening of action potential duration and reduction of intracellular Na+ activity. Circ Res. 1985;57:578–590. doi: 10.1161/01.res.57.4.578. doi:10.1161/01.RES.57.4.578. [DOI] [PubMed] [Google Scholar]
- 44.Makielski JC, Farley AL. Na+ current in human ventricle: implications for sodium loading and homeostasis. J Cardiovasc Electrophysiol. 2006;17:S15–S20. doi: 10.1111/j.1540-8167.2006.00380.x. doi:10.1111/j.1540-8167.2006.00380.x. [DOI] [PubMed] [Google Scholar]
- 45.Brill DM, Wasserstrom JA. Intracellular sodium and the positive inotropic effect of veratridine and cardiac glycoside in sheep Purkinje fibers. Circ Res. 1986;58:109–119. doi: 10.1161/01.res.58.1.109. doi:10.1161/01.RES.58.1.109. [DOI] [PubMed] [Google Scholar]
- 46.Yao L, Fan P, Jiang Z, Viatchenko-Karpinski S, Wu Y, Kornyeyev D, et al. NaV1.5-dependent persistent Na+ influx activates CaMKII in rat ventricular myocytes and N1325S mice. Am J Physiol Cell Physiol. 2011;301:C577–C586. doi: 10.1152/ajpcell.00125.2011. doi:10.1152/ajpcell.00125.2011. [DOI] [PubMed] [Google Scholar]
- 47.Sossalla S, Wagner S, Rasenack ECL, Ruff H, Weber SL, Schöndube FA, et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts: role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;45:32–43. doi: 10.1016/j.yjmcc.2008.03.006. doi:10.1016/j.yjmcc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 48.Shattock MJ, Bers DM. Rat vs. rabbit ventricle: Ca flux and intracellular Na assessed by ion-selective microelectrodes. Am J Physiol Cell Physiol. 1989;256:C813–C822. doi: 10.1152/ajpcell.1989.256.4.C813. [DOI] [PubMed] [Google Scholar]
- 49.Despa S, Islam MA, Pogwizd SM, Bers DM. Intracellular [Na+] and Na+ pump rate in rat and rabbit ventricular myocytes. J Physiol. 2002;539:133–143. doi: 10.1113/jphysiol.2001.012940. doi:10.1113/jphysiol.2001.012940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang XQ, Yamada S, Barry WH. Ranolazine inhibits an oxidative stress-induced increase in myocyte sodium and calcium loading during simulated-demand ischemia. J Cardiovasc Pharmacol. 2008;51:443–449. doi: 10.1097/FJC.0b013e318168e711. doi:10.1097/FJC.0b013e318168e711. [DOI] [PubMed] [Google Scholar]
- 51.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. doi:10.1161/01.CIR.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 52.Pieske B, Maier LS, Piacentino V, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–453. doi: 10.1161/01.cir.0000023042.50192.f4. doi:10.1161/01.CIR.0000023042.50192.F4. [DOI] [PubMed] [Google Scholar]
- 53.Pogwizd SM, Sipido KR, Verdonck F, Bers DM. Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc Res. 2003;57:887–896. doi: 10.1016/s0008-6363(02)00735-6. doi:10.1016/S0008-6363(02)00735-6. [DOI] [PubMed] [Google Scholar]
- 54.Verdonck F, Volders PG, Vos MA, Sipido KR. Increased Na+ concentration and altered Na/K pump activity in hypertrophied canine ventricular cells. Cardiovasc Res. 2003;57:1035–1043. doi: 10.1016/s0008-6363(02)00734-4. doi:10.1016/S0008-6363(02)00734-4. [DOI] [PubMed] [Google Scholar]
- 55.Mills GD, Harris DM, Chen X, Houser SR. Intracellular sodium determines frequency-dependent alterations in contractility in hypertrophied feline ventricular myocytes. Am J Physiol Heart Circ Physiol. 2007;292:H1129–H1138. doi: 10.1152/ajpheart.00375.2006. doi:10.1152/ajpheart.00375.2006. [DOI] [PubMed] [Google Scholar]
- 56.Undrovinas NA, Maltsev VA, Belardinelli L, Sabbah HN, Undrovinas A. Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J Physiol Sci. 2010;60:245–257. doi: 10.1007/s12576-010-0092-0. doi:10.1007/s12576-010-0092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen CJ, Fozzard HA, Sheu SS. Increase in intracellular sodium ion activity during stimulation in mammalian cardiac muscle. Circ Res. 1982;50:651–662. doi: 10.1161/01.res.50.5.651. doi:10.1161/01.RES.50.5.651. [DOI] [PubMed] [Google Scholar]
- 58.Lee CO, Dagostino M. Effect of strophanthidin on intracellular Na ion activity and twitch tension in constantly driven canine cardiac Purkinje fibers. Biophys J. 1982;40:185–198. doi: 10.1016/S0006-3495(82)84474-3. doi:10.1016/S0006-3495(82)84474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eisner DA, Lederer WJ, Vaughan-Jones RD. The quantitative relationship between twitch tension and intracellular sodium activity in sheep cardiac Purkinje fibres. J Physiol. 1984;355:251–266. doi: 10.1113/jphysiol.1984.sp015417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leblanc N, Hume JR. Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum. Science. 1990;248:372–376. doi: 10.1126/science.2158146. doi:10.1126/science.2158146. [DOI] [PubMed] [Google Scholar]
- 61.Barry WH. Na+ ‘fuzzy space’: does it exist, and is it important in ischemic injury? J Cardiovasc Electrophysiol. 2006;17(Suppl. 1):S43–S46. doi: 10.1111/j.1540-8167.2005.00396.x. doi:10.1111/j.1540-8167.2005.00396.x. [DOI] [PubMed] [Google Scholar]
- 62.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd ed. Norwell, MA: Kluwer Academic Publishers; 2001. [Google Scholar]
- 63.Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222. doi: 10.1124/jpet.106.101832. doi:10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 64.Fraser H, Belardinelli L, Wang L, Light PE, McVeigh JJ, Clanachan AS. Ranolazine decreases diastolic calcium accumulation caused by ATX-II or ischemia in rat hearts. J Mol Cell Cardiol. 2006;41:1031–1038. doi: 10.1016/j.yjmcc.2006.08.012. doi:10.1016/j.yjmcc.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 65.Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17(Suppl. 1):S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x. doi:10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoyer K, Song Y, Wang D, Phan D, Balschi J, Ingwall JS, et al. Reducing the late sodium current improves cardiac function during sodium pump inhibition by ouabain. J Pharmacol Exp Ther. 2011;337:513–523. doi: 10.1124/jpet.110.176776. doi:10.1124/jpet.110.176776. [DOI] [PubMed] [Google Scholar]
- 67.Levi AJ, Dalton GR, Hancox JC, Mitcheson JS, Issberner J, Bates JA, et al. Role of intracellular sodium overload in the genesis of cardiac arrhythmias. J Cardiovasc Electrophysiol. 1997;8:700–721. doi: 10.1111/j.1540-8167.1997.tb01834.x. doi:10.1111/j.1540-8167.1997.tb01834.x. [DOI] [PubMed] [Google Scholar]
- 68.Wu Y, Song Y, Belardinelli L, Shryock JC. The late Na+ current (INa) inhibitor ranolazine attenuates effects of palmitoyl-L-carnitine to increase late INa and cause ventricular diastolic dysfunction. J Pharmacol Exp Ther. 2009;330:550–557. doi: 10.1124/jpet.109.151936. doi:10.1124/jpet.109.151936. [DOI] [PubMed] [Google Scholar]
- 69.Christé G, Chahine M, Chevalier P, Pasek M. Changes in action potentials and intracellular ionic homeostasis in a ventricular cell model related to a persistent sodium current in SCN5A mutations underlying LQT3. Prog Biophys Mol Biol. 2008;96:281–293. doi: 10.1016/j.pbiomolbio.2007.07.023. doi:10.1016/j.pbiomolbio.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 70.Le Grand B, Vie B, Talmant JM, Coraboeuf E, John GW. Alleviation of contractile dysfunction in ischemic hearts by slowly inactivating Na+ current blockers. Am J Physiol Heart Circ Physiol. 1995;269:H533–H540. doi: 10.1152/ajpheart.1995.269.2.H533. [DOI] [PubMed] [Google Scholar]
- 71.Le Grand B, Pignier C, Letienne R, Cuisiat F, Rolland F, Mas A, et al. Sodium late current blockers in ischemia reperfusion: Is the bullet magic? J Med Chem. 2008;51:3856–3866. doi: 10.1021/jm800100z. doi:10.1021/jm800100z. [DOI] [PubMed] [Google Scholar]
- 72.Wu L, Ma J, Li H, Wang C, Grandi E, Zhang P, et al. Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation. 2011;123:1713–1720. doi: 10.1161/CIRCULATIONAHA.110.000661. doi:10.1161/CIRCULATIONAHA.110.000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O'Hara T, Virag L, Varro A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol. 2011;7:e1002061. doi: 10.1371/journal.pcbi.1002061. doi:10.1371/journal.pcbi.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Belardinelli L, Liu G, Smith-Maxwell C, Wang WQ, El-Bizri N, Hirakawa R, et al. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J Pharmacol Exp Ther. 2013;344:23–32. doi: 10.1124/jpet.112.198887. doi:10.1124/jpet.112.198887. [DOI] [PubMed] [Google Scholar]
- 75.Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, et al. Altered Na+ currents in atrial fibrillation. J Am Coll Cardiol. 2010;55:2330–2342. doi: 10.1016/j.jacc.2009.12.055. doi:10.1016/j.jacc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 76.Song Y, Shryock JC, Belardinelli L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H2031–H2039. doi: 10.1152/ajpheart.01357.2007. doi:10.1152/ajpheart.01357.2007. [DOI] [PubMed] [Google Scholar]
- 77.Song Y, Shryock JC, Belardinelli L. A slowly inactivating sodium current contributes to spontaneous diastolic depolarization of atrial myocytes. Am J Physiol Heart Circ Physiol. 2009;297:H1254–H1262. doi: 10.1152/ajpheart.00444.2009. doi:10.1152/ajpheart.00444.2009. [DOI] [PubMed] [Google Scholar]
- 78.Guo D, Young L, Wu Y, Belardinelli L, Kowey PR, Yan GX. Increased late sodium current in left atrial myocytes of rabbits with left ventricular hypertrophy: its role in the genesis of atrial arrhythmias. Am J Physiol Heart Circ Physiol. 2010;298:H1375–H1381. doi: 10.1152/ajpheart.01145.2009. doi:10.1152/ajpheart.01145.2009. [DOI] [PubMed] [Google Scholar]
- 79.Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L. Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther. 2004;310:599–605. doi: 10.1124/jpet.104.066100. doi:10.1124/jpet.104.066100. [DOI] [PubMed] [Google Scholar]
- 80.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsades de pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–2047. doi: 10.1161/01.cir.96.6.2038. doi:10.1161/01.CIR.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 81.Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44:192–199. doi: 10.1097/00005344-200408000-00008. doi:10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- 82.Benito B, Brugada R, Perich RM, Lizotte E, Cinca J, Mont L, et al. A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm. 2008;5:1434–1440. doi: 10.1016/j.hrthm.2008.07.013. doi:10.1016/j.hrthm.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 83.Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, et al. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117:1927–1935. doi: 10.1161/CIRCULATIONAHA.107.757955. doi:10.1161/CIRCULATIONAHA.107.757955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Noble D. The Initiation of the Heartbeat. 2nd ed. Oxford: Clarendon Press; 1979. [Google Scholar]
- 85.Trautwein W, Kassebaum DG, Nelson RM, Hecht HH. Electrophysiological study of human heart muscle. Circ Res. 1962;10:306–312. doi: 10.1161/01.res.10.3.306. doi:10.1161/01.RES.10.3.306. [DOI] [PubMed] [Google Scholar]
- 86.Gelband H, Bush HL, Rosen MR, Myerburg RJ, Hoffman BF. Electrophysiologic properties of isolated preparations of human atrial myocardium. Circ Res. 1972;30:293–300. doi: 10.1161/01.res.30.3.293. doi:10.1161/01.RES.30.3.293. [DOI] [PubMed] [Google Scholar]
- 87.Mary-Rabine L, Hordof AJ, Danilo P, Jr, Malm JR, Rosen MR. Mechanisms for impulse initiation in isolated human atrial fibers. Circ Res. 1980;47:267–277. doi: 10.1161/01.res.47.2.267. doi:10.1161/01.RES.47.2.267. [DOI] [PubMed] [Google Scholar]
- 88.Hogan PM, Davis LD. Evidence for specialized fibers in the canine right atrium. Circ Res. 1968;23:387–396. doi: 10.1161/01.res.23.3.387. doi:10.1161/01.RES.23.3.387. [DOI] [PubMed] [Google Scholar]
- 89.Wit AL, Cranefield PF. Triggered and automatic activity in the canine coronary sinus. Circ Res. 1977;41:434–445. doi: 10.1161/01.res.41.4.434. doi:10.1161/01.RES.41.4.434. [DOI] [PubMed] [Google Scholar]
- 90.Cheung DW. Pulmonary vein as an ectopic focus in digitalis-induced arrhythmia. Nature. 1981;294:582–584. doi: 10.1038/294582a0. doi:10.1038/294582a0. [DOI] [PubMed] [Google Scholar]
- 91.Wit AL, Bigger JT., Jr Possible electrophysiological mechanisms for lethal arrhythmias accompanying myocardial ischemia and infarction. Circulation. 1975;52(Suppl. 6):III96–III115. [PubMed] [Google Scholar]
- 92.Carmeliet E. Slow inactivation of the sodium current in rabbit cardiac Purkinje fibres. Pflügers Arch. 1987;408:18–26. doi: 10.1007/BF00581835. doi:10.1007/BF00581835. [DOI] [PubMed] [Google Scholar]
- 93.Carmeliet E, Saikawa T. Shortening of the action potential and reduction of pacemaker activity by lidocaine, quinidine, and procainamide in sheep cardiac Purkinje fibers. Circ Res. 1982;50:257–272. doi: 10.1161/01.res.50.2.257. doi:10.1161/01.RES.50.2.257. [DOI] [PubMed] [Google Scholar]
- 94.Rota M, Vassalle M. Patch-clamp analysis in canine cardiac Purkinje cells of a novel sodium component in the pacemaker range. J Physiol. 2003;548:147–165. doi: 10.1113/jphysiol.2003.039263. doi:10.1113/jphysiol.2003.039263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Saint DA, Ju Y-K, Gage PW. A persistent sodium current in rat ventricular myocytes. J Physiol. 1992;453:219–231. doi: 10.1113/jphysiol.1992.sp019225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sakmann BFAS, Spindler AJ, Bryant SM, Linz KW, Noble D. Distribution of a persistent sodium current across the ventricular wall in guinea pigs. Circ Res. 2000;87:910–914. doi: 10.1161/01.res.87.10.910. doi:10.1161/01.RES.87.10.910. [DOI] [PubMed] [Google Scholar]
- 97.Colatsky TJ. Mechanisms of action of lidocaine and quinidine on action potential duration in rabbit cardiac Purkinje fibers. Circ Res. 1982;50:17–27. doi: 10.1161/01.res.50.1.17. doi:10.1161/01.RES.50.1.17. [DOI] [PubMed] [Google Scholar]
- 98.Liu Y-M, DeFelice LJ, Mazzanti M. Na channels that remain open throughout the cardiac action potential plateau. Biophys J. 1992;63:654–662. doi: 10.1016/S0006-3495(92)81635-1. doi:10.1016/S0006-3495(92)81635-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maltsev VA, Sabbah HN, Higgins RSD, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. doi:10.1161/01.CIR.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 100.Roden DM. Taking the ‘idio’ out of ‘idiosyncratic’: predicting torsades de pointes. PACE. 1998;21:1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. doi:10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 101.Marban E, Robinson SW, Wier WG. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J Clin Invest. 1986;78:1185–1192. doi: 10.1172/JCI112701. doi:10.1172/JCI112701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. doi:10.1161/01.RES.64.5.977. [DOI] [PubMed] [Google Scholar]
- 103.Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J. 1995;68:949–964. doi: 10.1016/S0006-3495(95)80271-7. doi:10.1016/S0006-3495(95)80271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xie L-H, Chen F, Karagueuzian HS, Weiss JN. Oxidative stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. doi:10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weiss JN, Garfinkel A, Karagueuzian HS, Chen P-S, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010;7:1891–1899. doi: 10.1016/j.hrthm.2010.09.017. doi:10.1016/j.hrthm.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Janvier NC, Harrison SM, Boyett MR. The role of inward Na+-Ca2+ exchange current in the ferret ventricular action potential. J Physiol. 1997;498:611–625. doi: 10.1113/jphysiol.1997.sp021887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O'Rourke B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. 2003;93:46–53. doi: 10.1161/01.RES.0000080932.98903.D8. doi:10.1161/01.RES.0000080932.98903.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Spencer CI, Sham JSK. Effects of Na+/Ca2+ exchange induced by SR Ca2+ release on action potentials and afterdepolarizations in guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol. 2003;285:H2552–H2562. doi: 10.1152/ajpheart.00274.2003. [DOI] [PubMed] [Google Scholar]
- 109.Li P, Rudy Y. A model of canine Purkinje cell electrophysiology and Ca2+ cycling. Rate dependence, triggered activity, and comparison to ventricular myocytes. Circ Res. 2011;109:71–79. doi: 10.1161/CIRCRESAHA.111.246512. doi:10.1161/CIRCRESAHA.111.246512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ben Caref E, Boutjdir M, Himel HD, El-Sherif N. Role of subendocardial Purkinje network in triggering torsade de pointes arrhythmia in experimental long QT syndrome. Europace. 2008;10:1218–1223. doi: 10.1093/europace/eun248. doi:10.1093/europace/eun248. [DOI] [PubMed] [Google Scholar]
- 111.Boutjdir M, El-Sherif N. Pharmacological evaluation of early afterdepolarisations induced by sea anemone toxin (ATXII) in dog heart. Cardiovasc Res. 1991;25:815–819. doi: 10.1093/cvr/25.10.815. doi:10.1093/cvr/25.10.815. [DOI] [PubMed] [Google Scholar]
- 112.Thomas G, Killeen MJ, Grace AA, Huang CL. Pharmacological separation of early afterdepolarizations from arrhythmogenic substrate in deltaKPQ SCN5A murine hearts modelling human long QT 3 syndrome. Acta Physiol. 2008;192:505–517. doi: 10.1111/j.1748-1716.2007.01770.x. doi:10.1111/j.1748-1716.2007.01770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Morita N, Lee JH, Xie Y, Sovari A, Qu Z, Weiss JN, et al. Suppression of re-entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J Am Coll Cardiol. 2011;57:366–375. doi: 10.1016/j.jacc.2010.07.045. doi:10.1016/j.jacc.2010.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Orth PM, Hesketh JC, Mak CK, Yang Y, Lin S, Beatch GN, et al. RSD1235 blocks late INa and suppresses early afterdepolarizations and torsades de pointes induced by class III agents. Cardiovasc Res. 2006;70:486–496. doi: 10.1016/j.cardiores.2006.01.026. doi:10.1016/j.cardiores.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 115.Wu L, Rajamani S, Li H, January CT, Shryock JC, Belardinelli L. Reduction of repolarization reserve unmasks the proarrhythmic role of endogenous late Na+ current in the heart. Am J Physiol Heart Circ Physiol. 2009;297:H1048–H1057. doi: 10.1152/ajpheart.00467.2009. doi:10.1152/ajpheart.00467.2009. [DOI] [PubMed] [Google Scholar]
- 116.Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013;127:575–584. doi: 10.1161/CIRCULATIONAHA.112.134932. doi:10.1161/CIRCULATIONAHA.112.134932. [DOI] [PubMed] [Google Scholar]
- 117.Jia S, Lian J, Guo D, Xue X, Patel C, Yang L, et al. Modulation of the late sodium current by ATX-II and ranolazine affects the reverse use-dependence and proarrhythmic liability of IKr blockade. Br J Pharmacol. 2011;164:308–316. doi: 10.1111/j.1476-5381.2010.01181.x. doi:10.1111/j.1476-5381.2010.01181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ blockade and to increases in heat rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. doi:10.1161/01.CIR.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 119.Nagatomo T, January CT, Ye B, Abe H, Nakashima Y, Makielski JC. Rate-dependent QT shortening mechanism for the LQT3 deltaKPQ mutant. Cardiovasc Res. 2002;54:624–629. doi: 10.1016/s0008-6363(02)00265-1. doi:10.1016/S0008-6363(02)00265-1. [DOI] [PubMed] [Google Scholar]
- 120.Wu L, Shryock JC, Song Y, Belardinelli L. An increase in late sodium current potentiates the proarrhythmic activities of low-risk QT-prolonging drugs in female rabbit hearts. J Pharmacol Exp Ther. 2006;316:718–726. doi: 10.1124/jpet.105.094862. doi:10.1124/jpet.105.094862. [DOI] [PubMed] [Google Scholar]
- 121.Wu L, Guo D, Li H, Hackett J, Yan GX, Jiao Z, et al. Role of late sodium current in modulating the proarrhythmic and antiarrhythmic effects of quinidine. Heart Rhythm. 2008;5:1726–1734. doi: 10.1016/j.hrthm.2008.09.008. doi:10.1016/j.hrthm.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wu L, Rajamani S, Shryock JC, Li H, Ruskin J, Antzelevitch C, et al. Augmentation of late sodium current unmasks the proarrhythmic effects of amiodarone. Cardiovasc Res. 2008;77:481–488. doi: 10.1093/cvr/cvm069. doi:10.1093/cvr/cvm069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. doi:10.1016/S0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- 124.Makita N, Horie M, Nakamura T, Ai T, Sasaki K, Yokoi H, et al. Drug-induced long-QT syndrome associated with a subclinical SCN5A mutation. Circulation. 2002;106:1269–1274. doi: 10.1161/01.cir.0000027139.42087.b6. doi:10.1161/01.CIR.0000027139.42087.B6. [DOI] [PubMed] [Google Scholar]
- 125.Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, Beggs AH, et al. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297:1333–1336. doi: 10.1126/science.1073569. doi:10.1126/science.1073569. [DOI] [PubMed] [Google Scholar]
- 126.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure. Roles of sodium-calcium exchange, inward rectifier potassium current, and residual β-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. doi:10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 127.Fauconnier J, Lacampagne A, Rauzier J-M, Vassort G, Richard S. Ca2+-dependent reduction of IK1 in rat ventricular cells: a novel paradigm for arrhythmia in heart failure? Cardiovasc Res. 2005;68:204–212. doi: 10.1016/j.cardiores.2005.05.024. doi:10.1016/j.cardiores.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 128.Lakatta EG, Talo A, Capogrossi MC, Spurgeon HA, Stern MD. Spontaneous sarcoplasmic reticulum Ca2+ release leads to heterogeneity of contractile and electrical properties of the heart. Basic Res Cardiol. 1992;87(Suppl. 2):93–104. doi: 10.1007/978-3-642-72477-0_9. [DOI] [PubMed] [Google Scholar]
- 129.Bers DM, Pogwizd SM, Schlotthauer K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol. 2002;97(Suppl. 1):36–42. doi: 10.1007/s003950200027. [DOI] [PubMed] [Google Scholar]
- 130.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. doi:10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 131.Hoeker GS, Katra RP, Wilson LD, Plummer BN, Laurita KR. Spontaneous calcium release in tissue from the failing canine heart. Am J Physiol Heart Circ Physiol. 2009;297:H1235–H1242. doi: 10.1152/ajpheart.01320.2008. doi:10.1152/ajpheart.01320.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–2529. doi: 10.1161/CIRCULATIONAHA.104.494476. doi:10.1161/CIRCULATIONAHA.104.494476. [DOI] [PubMed] [Google Scholar]
- 133.Malan D, Friedrichs S, Fleischmann BK, Sasse P. Cardiomyocytes obtained from induced pluripotent stem cells with long-QT syndrome 3 recapitulate typical disease-specific features in vitro. Circ Res. 2011;109:841–847. doi: 10.1161/CIRCRESAHA.111.243139. doi:10.1161/CIRCRESAHA.111.243139. [DOI] [PubMed] [Google Scholar]
- 134.Milberg P, Pott C, Fink M, Frommeyer G, Matsuda T, Baba A, et al. Inhibition of the Na+/Ca2+ exchanger suppresses torsades de pointes in an intact heart model of long QT syndrome-2 and long QT syndrome-3. Heart Rhythm. 2008;5:1444–1452. doi: 10.1016/j.hrthm.2008.06.017. doi:10.1016/j.hrthm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 135.Volders PGA, Vos MA, Szabo B, Sipido KR, de Groot SHM, Gorgels APM, et al. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc Res. 2000;46:376–392. doi: 10.1016/s0008-6363(00)00022-5. doi:10.1016/S0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- 136.Choi BR, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and torsade de pointes in rabbit hearts with type 2 long QT syndrome. J Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. doi:10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Flaim SN, Giles WR, McCulloch AD. Arrhythmogenic consequences of Na+ channel mutations in the transmurally heterogeneous mammalian left ventricle: analysis of the I1768 V SCN5A mutation. Heart Rhythm. 2007;4:768–778. doi: 10.1016/j.hrthm.2007.02.009. doi:10.1016/j.hrthm.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 138.Maruyama M, Lin S-F, Xie Y, Chua S-K, Joung B, Han S, et al. Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-QT syndrome. Circ Arrhythm Electrophysiol. 2011;4:103–111. doi: 10.1161/CIRCEP.110.959064. doi:10.1161/CIRCEP.110.959064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Antzelevitch C, Shimizu W, Yan G-X, Sicouri S, Weissenburger J, Nesterenko VV, et al. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;10:1124–1152. doi: 10.1111/j.1540-8167.1999.tb00287.x. doi:10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 140.Belardinelli L, Antzelevitch C, Vos MA. Assessing predictors of drug-induced torsade de pointes. TIPS. 2003;24:619–625. doi: 10.1016/j.tips.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 141.Poelzing S, Rosenbaum DS. Cellular mechanisms of torsade de pointes. Novartis Found Symp. 2005;266:204–217. [PubMed] [Google Scholar]
- 142.Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch CA. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol Heart Circ Physiol. 2001;281:H689–H697. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]
- 143.Ferrier GR, Saunders JH, Mendez C. A cellular mechanism for the generation of ventricular arrhythmias by acetylstrophanthidin. Circ Res. 1973;32:600–609. doi: 10.1161/01.res.32.5.600. doi:10.1161/01.RES.32.5.600. [DOI] [PubMed] [Google Scholar]
- 144.Kass RS, Lederer WJ, Tsien RW, Weingart R. Role of calcium ions in transient inward currents and aftercontractions induced by strophanthidin in cardiac Purkinje fibres. J Physiol. 1978;281:187–208. doi: 10.1113/jphysiol.1978.sp012416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Karagueuzian HR, Katzung BG. Voltage-clamp studies of transient inward current and mechanical oscillations induced by ouabain in ferret papillary muscle. J Physiol. 1982;327:255–271. doi: 10.1113/jphysiol.1982.sp014230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fedida D, Noble D, Rankin AC, Spindler AJ. The arrhythmogenic transient inward current iTi and related contraction in isolated guinea-pig ventricular myocytes. J Physiol. 1987;392:523–542. doi: 10.1113/jphysiol.1987.sp016795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Capogrossi MC, Houser SR, Bahinski A, Lakatta EG. Synchronous occurrence of spontaneous localized calcium release from the sarcoplasmic reticulum generates action potentials in rat cardiac ventricular myocytes at normal resting membrane potential. Circ Res. 1987;61:498–503. doi: 10.1161/01.res.61.4.498. doi:10.1161/01.RES.61.4.498. [DOI] [PubMed] [Google Scholar]
- 148.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. doi:10.1161/01.RES.87.9.774. [DOI] [PubMed] [Google Scholar]
- 149.Henning B, Wit AL. The time course of action potential repolarization affects delayed afterdepolarization amplitude in atrial fibers of the canine coronary sinus. Circ Res. 1984;55:110–115. doi: 10.1161/01.res.55.1.110. doi:10.1161/01.RES.55.1.110. [DOI] [PubMed] [Google Scholar]
- 150.Wangcharoen W, Chen YC, Chen YJ, Chen SY, Yeh HI, Lin CI, et al. Aging increases pulmonary veins arrhythmogenesis and susceptibility to calcium regulatory agents. Heart Rhythm. 2007;4:1338–1349. doi: 10.1016/j.hrthm.2007.06.023. doi:10.1016/j.hrthm.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 151.Chang SL, Chen YC, Yeh YH, Lin YK, Wu TJ, Lin CI, et al. Heart failure enhanced pulmonary vein arrhythmogenesis and dysregulated sodium and calcium homeostasis with increased calcium sparks. J Cardiovasc Electrophysiol. 2011;22:1378–1386. doi: 10.1111/j.1540-8167.2011.02126.x. doi:10.1111/j.1540-8167.2011.02126.x. [DOI] [PubMed] [Google Scholar]
- 152.Sicouri S, Blazek J, Belardinelli L, Antzelevitch C. Electrophysiological characteristics of canine superior vena cava sleeve preparations: effect of ranolazine. Circ Arrhythm Electrophysiol. 2012;5:371–379. doi: 10.1161/CIRCEP.111.969493. doi:10.1161/CIRCEP.111.969493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Joung B, Zhang H, Shinohara T, Maruyama M, Han S, Kim D, et al. Delayed afterdepolarization in intact canine sinoatrial node as a novel mechanism for atrial arrhythmia. J Cardiovasc Electrophysiol. 2011;22:448–454. doi: 10.1111/j.1540-8167.2010.01905.x. doi:10.1111/j.1540-8167.2010.01905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Katra RP, Laurita KR. Cellular mechanism of calcium-mediated triggered activity in the heart. Circ Res. 2005;96:535–542. doi: 10.1161/01.RES.0000159387.00749.3c. doi:10.1161/01.RES.0000159387.00749.3c. [DOI] [PubMed] [Google Scholar]
- 155.Kort AA, Capogrossi MC, Lakatta EG. Frequency, amplitude, and propagation velocity of spontaneous Ca++-dependent contractile waves in intact adult rat cardiac muscle and isolated myocytes. Circ Res. 1985;57:844–855. doi: 10.1161/01.res.57.6.844. doi:10.1161/01.RES.57.6.844. [DOI] [PubMed] [Google Scholar]
- 156.Tweedie D, Harding SE, MacLeod KT. Sarcoplasmic reticulum Ca content, sarcolemmal Ca influx and the genesis of arrhythmias in isolated guinea-pig cardiomyocytes. J Mol Cell Cardiol. 2000;32:261–272. doi: 10.1006/jmcc.1999.1070. doi:10.1006/jmcc.1999.1070. [DOI] [PubMed] [Google Scholar]
- 157.Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+-Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ Res. 2008;103:509–518. doi: 10.1161/CIRCRESAHA.108.176677. doi:10.1161/CIRCRESAHA.108.176677. [DOI] [PubMed] [Google Scholar]
- 158.Faber GM, Rudy Y. Action potential and contractility changes in [Na+]i overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78:2392–2404. doi: 10.1016/S0006-3495(00)76783-X. doi:10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Honerjager P. Cardioactive substances that prolong the open state of sodium channels. Rev Physiol Biochem Pharmacol. 1982;92:1–74. doi: 10.1007/BFb0030502. [DOI] [PubMed] [Google Scholar]
- 160.Wasserstrom JA, Sharma R, O'Toole MJ, Zheng J, Kelly JE, Shryock J, et al. Ranolazine antagonizes the effects of increased late sodium current on intracellular calcium cycling in rat isolated heart. J Pharmacol Exp Ther. 2009;331:382–391. doi: 10.1124/jpet.109.156471. doi:10.1124/jpet.109.156471. [DOI] [PubMed] [Google Scholar]
- 161.Jurevichus JA, Rosenshtraukh LV. Mechanism of delayed afterdepolarization caused by aconitine. Adv Myocardiol. 1982;3:159–176. doi: 10.1007/978-1-4899-5561-6_17. [DOI] [PubMed] [Google Scholar]
- 162.Rosen MR, Danilo P. Effects of tetrodotoxin, lidocaine, verapamil, and AHR-2666 on ouabain-induced delayed afterdepolarizations in canine Purkinje fibers. Circ Res. 1980;46:117–124. doi: 10.1161/01.res.46.1.117. doi:10.1161/01.RES.46.1.117. [DOI] [PubMed] [Google Scholar]
- 163.Uchida H, Ozawa E, Watanabe Y. Effects of mexiletine on delayed afterdepolarization and triggered activity. Heart Vessels. 1990;5:140–145. doi: 10.1007/BF02059908. doi:10.1007/BF02059908. [DOI] [PubMed] [Google Scholar]
- 164.Vollmer B, Meuter C, Janssen PAJ. R56865 prevents electrical and mechanical signs of ouabain intoxication in guinea-pig papillary muscle. Eur J Pharmacol. 1987;142:137–140. doi: 10.1016/0014-2999(87)90663-7. doi:10.1016/0014-2999(87)90663-7. [DOI] [PubMed] [Google Scholar]
- 165.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz AL, Kirchhof P, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. doi:10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, et al. A beta(iv)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120:3508–3519. doi: 10.1172/JCI43621. doi:10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Aiba T, Hesketh GG, Liu T, Carlisle R, Villa-Abrille MC, O'Rourke B, et al. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc Res. 2010;85:454–463. doi: 10.1093/cvr/cvp324. doi:10.1093/cvr/cvp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, et al. Ca2+/Calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem. 2012;287:19856–19869. doi: 10.1074/jbc.M111.322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, et al. CaMKII-based regulation of voltage-gated Na+ channel function in cardiac disease. Circulation. 2012;126:2084–2094. doi: 10.1161/CIRCULATIONAHA.112.105320. doi:10.1161/CIRCULATIONAHA.112.105320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Guo T, Zhang T, Ginsburg KS, Mishra S, Brown JH, Bers DM. CaMKIIδC slows [Ca]i decline in cardiac myocytes by promoting Ca sparks. Biophys J. 2012;102:2461–2470. doi: 10.1016/j.bpj.2012.04.015. doi:10.1016/j.bpj.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Curran J, Brown KH, Santiago DJ, Pogwizd S, Bers DM, Shannon TR. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase II. J Mol Cell Cardiol. 2010;49:25–32. doi: 10.1016/j.yjmcc.2010.03.013. doi:10.1016/j.yjmcc.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mines GR. On circulating excitations in heart muscles and their possible relation to tachycardia and fibrillation. Trans R Soc Can. 1914;8:43–52. [Google Scholar]
- 173.Di Diego JM, Antzelevitch C. Pinacidil-induced electrical heterogeneity and extrasystolic activity in canine ventricular tissues. Does activation of ATP-regulated potassium current promote phase 2 reentry? Circulation. 1993;88:1177–1189. doi: 10.1161/01.cir.88.3.1177. doi:10.1161/01.CIR.88.3.1177. [DOI] [PubMed] [Google Scholar]
- 174.Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm. 2007;4:964–972. doi: 10.1016/j.hrthm.2007.03.036. doi:10.1016/j.hrthm.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Galinier M, Vialette J-C, Fourcade J, Cabrol P, Dongay B, Massabuau P, et al. QT interval dispersion as a predictor of arrhythmic events in congestive heart failure. Importance of aetiology. Eur Heart J. 1998;19:1054–1062. doi: 10.1053/euhj.1997.0865. doi:10.1053/euhj.1997.0865. [DOI] [PubMed] [Google Scholar]
- 176.Yan G-X, Wu Y, Liu T, Wang J, Marinchak RA, Kowey PR. Phase 2 early afterdepolarization as a trigger of polymorphic ventricular tachycardia in acquired long-QT syndrome. Direct evidence from intracellular recordings in the intact left ventricular wall. Circulation. 2001;103:2851–2856. doi: 10.1161/01.cir.103.23.2851. doi:10.1161/01.CIR.103.23.2851. [DOI] [PubMed] [Google Scholar]
- 177.Sicouri S, Glass A, Ferreiro M, Antzelevitch C. Transseptal dispersion of repolarization and its role in the development of Torsades de Pointes arrhythmias. J Cardiovasc Electrophysiol. 2010;21:441–447. doi: 10.1111/j.1540-8167.2009.01641.x. doi:10.1111/j.1540-8167.2009.01641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sidorov VY, Uzelac I, Wikswo JP. Regional increase of extracellular potassium leads to electrical instability and reentry occurrence through the spatial heterogeneity of APD restitution. Am J Physiol Heart Circ Physiol. 2011;301:H209–H220. doi: 10.1152/ajpheart.01141.2010. doi:10.1152/ajpheart.01141.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Clayton RH, Holden AV. Dispersion of cardiac action potential duration and the initiation of re-entry: a computational study. Biomed Eng Online. 2005;4:11. doi: 10.1186/1475-925X-4-11. doi:10.1186/1475-925X-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Aslanidi OV, Boyett MR, Dobrzynski H, Li J, Zhang H. Mechanisms of transition from normal to reentrant electrical activity in a model of rabbit atrial tissue: interaction of tissue heterogeneity and anisotropy. Biophys J. 2009;96:798–817. doi: 10.1016/j.bpj.2008.09.057. doi:10.1016/j.bpj.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yamauchi M, Watanabe E, Yasui K, Takeuchi H, Terasawa T, Sawada K, et al. Prevention of ventricular extrasystole by mexiletine in patients with normal QT intervals is associated with a reduction of transmural dispersion of repolarization. Int J Cardiol. 2005;103:92–97. doi: 10.1016/j.ijcard.2004.09.016. doi:10.1016/j.ijcard.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 182.Shimizu W, Antzelevitch C. Cellular and ionic basis for t-wave alternans under long-QT conditions. Circulation. 1999;99:1499–1507. doi: 10.1161/01.cir.99.11.1499. doi:10.1161/01.CIR.99.11.1499. [DOI] [PubMed] [Google Scholar]
- 183.Restivo M, Caref EB, Kozhevnikov DO, El-Sherif N. Spatial dispersion of repolarization is a key factor in the arrhythmogenicity of long QT syndrome. J Cardiovasc Electrophysiol. 2004;15:323–331. doi: 10.1046/j.1540-8167.2004.03493.x. doi:10.1046/j.1540-8167.2004.03493.x. [DOI] [PubMed] [Google Scholar]
- 184.Milberg P, Reinsch N, Wasmer K, Monnig G, Strypmann J, Osada N, et al. Transmural dispersion of repolarization as a key factor of arrhythmogenicity in a novel intact heart model of LQT3. Cardiovasc Res. 2005;65:397–404. doi: 10.1016/j.cardiores.2004.10.016. doi:10.1016/j.cardiores.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 185.Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long-QT syndrome: effects of beta-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation. 1998;98:2314–2322. doi: 10.1161/01.cir.98.21.2314. doi:10.1161/01.CIR.98.21.2314. [DOI] [PubMed] [Google Scholar]
- 186.Nieminen T, Nanbu DY, Datti IP, Vaz GR, Tavares CAM, Pegler JRM, et al. Antifibrillatory effect of ranolazine during severe coronary stenosis in the intact porcine model. Heart Rhythm. 2011;8:608–614. doi: 10.1016/j.hrthm.2010.11.029. doi:10.1016/j.hrthm.2010.11.029. [DOI] [PubMed] [Google Scholar]
- 187.Garfinkel A, Kim Y-H, Voroshilovshy O, Qu Z, Kil JR, Lee M-H, et al. Preventing ventricular fibrillation by flattening cardiac restitution. Proc Natl Acad Sci USA. 2000;97:6061–6066. doi: 10.1073/pnas.090492697. doi:10.1073/pnas.090492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, et al. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med. 2001;7:1021–1027. doi: 10.1038/nm0901-1021. doi:10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- 189.Fabritz L, Kirchhof P, Franz MR, Nuyens D, Rossenbacker T, Ottenhof A, et al. Effect of pacing and mexiletine on dispersion of repolarisation and arrhythmias in deltaKPQ SCN5A (long QT3) mice. Cardiovasc Res. 2003;57:1085–1093. doi: 10.1016/s0008-6363(02)00839-8. doi:10.1016/S0008-6363(02)00839-8. [DOI] [PubMed] [Google Scholar]
- 190.Fredj S, Lindegger N, Sampson KJ, Carmeliet P, Kass RS. Altered Na+ channels promote pause-induced spontaneous diastolic activity in long QT syndrome type 3 myocytes. Circ Res. 2006;99:1225–1232. doi: 10.1161/01.RES.0000251305.25604.b0. doi:10.1161/01.RES.0000251305.25604.b0. [DOI] [PMC free article] [PubMed] [Google Scholar]