

Abstract
We have reported that compounds containing a bi-aryl linked unit (Ar-X-Ar′) modulated Na+ currents by promoting slow inactivation and fast inactivation processes and by inducing frequency (use)-dependent inhibition of Na+ currents. These electrophysiological properties have been associated with the mode of action of several antiepileptic drugs. In this study, we demonstrate that the readily accessible (biphenyl-4-yl)methylammonium chlorides (compound class B) exhibited a broad range of anticonvulsant activities in animal models and in the maximal electroshock seizure test the activity of (3′-trifluoromethoxybiphenyl-4-yl)methylammonium chloride (8) exceeded that of phenobarbital and phenytoin upon oral administration to rats. Electrophysiological studies of 8 using mouse catecholamine A– differentiated cells and rat embryonic cortical neurons confirmed that 8 promoted slow and fast inactivation in both cell types but did not affect the frequency (use)-dependent block of Na+ currents.
Lacosamide1(1) is a first-in-class antiepileptic drug (AED) that has been introduced in 34 countries, including the US, as an adjunctive therapy for the treatment of partial-onset seizures.2 Whole-cell, patch-clamp electrophysiology showed that 1 reduced Na+ channel availability by a mechanism consistent with its increasing the transition of Na+ channels to the slow-inactivated state without affecting the fast inactivation process.3-5 (For an alternative mechanism where the agent blocks Na+ channel fast-inactivated channels with very slow kinetics, see reference 6). We demonstrated that lacosamide analogs in which the N-benzyl amide group was extended by an additional aryl unit to give compound class A exhibited pronounced anticonvulsant activity in proven rodent seizure models.7 Electrophysiologic examination of A in neuronal-like catecholamine A–differentiated (CAD) cells showed that these compounds promoted Na+ channel slow inactivation and that several compounds were 40-80–fold more potent than 1. 8 Interestingly, we found that members of compound class A, unlike 1, affected Na+ channel fast inactivation and exhibited frequency (use)-dependent inhibition of Na+ channel firing. We investigated the origin of A's increased potency, compared with 1, for Na+ channel slow inactivation and showed that both the core “lacosamide unit” and the “bi-aryl linked unit” (Fig. 1) promoted slow inactivation.9
Figure 1.
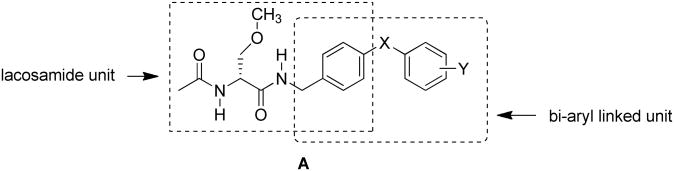
Key structural units in compound class A that affect Na+ channel slow inactivation.
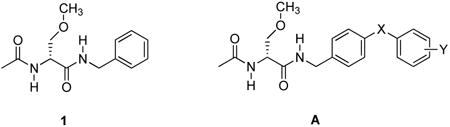
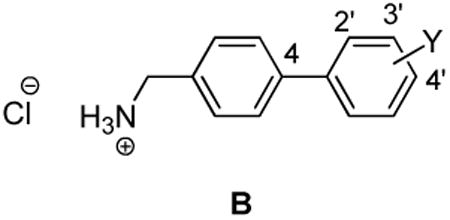
In this study, we asked if compounds conforming to the bi-aryl linked unit exhibited anticonvulsant activity in rodents. Here, we focus on substituted (biphenyl-4-yl)methylammonium chlorides (B) wherein the aryl linker (X) is a single bond. We report that compound class B showed a broad profile of anticonvulsant activity, and when a selected compound of class B was given orally to rats potent activity in the maximal electroshock seizure10 (MES) model that was comparable to the clinical agents phenobarbital and phenytoin was seen.11
Results and Discussion
Choice of Compounds
We selected 10 (biphenyl-4-yl)methylammonium chlorides (B) in which the substituent (Y) and the site of substitution on the terminal aryl unit were varied (Table 1, compounds 2–11). Both electron-withdrawing and electron-donating substituents were chosen. In most instances, the groups were placed at either the 3′ (5, 7, 8, 12, 13) or the 4′ (6, 8, 11) position since earlier structure-activity relationship studies on 112 and compound class A7 derivatives showed substitution at these sites provided compounds with excellent anticonvulsant activities. In the case of the trifluoromethoxy (CF3O) substituent, we prepared the 2′, 3′, and 4′ regioisomers (9–11). The compounds were purified and tested as their hydrochloride salts.
Table 1. Anticonvulsant Activities of (Biphenyl-4-yl)methylammonium Chlorides (B)a.
| Cmpd No. |
|
mice (ip)b [hour] | ratc [hour] | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| MESd | 6 Hze | scMetf | Toxg | ip | oral | ||||
|
| |||||||||
| MESd | Toxh | MESd | Toxh | ||||||
| 2 | Y = H | 30∼100 [0.25-0.5] | ∼100 [0.5-1.0] | >300 [0.5-4.0] | >100 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] |
| 3 | Y = 3′-F | >50 [0.25-4.0] | ∼50 [1.0] | ∼50 [2.0] | >100 [0.25-4.0] | ∼65 [0.25] | >65 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] |
| 4 | Y = 4′-F | ∼50 [4.0] | <100 [1.0-4.0] | >50 [0.25-4.0] | >50 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] |
| 5 | Y = 3′-Cl | <100 [0.25, 1.0] | <100 [0.5-4.0] | <100 [0.5-2.0] | >100 [0.25-4.0] | ∼65 [0.25-0.5] | ∼65 [0.25-0.5] | >30 [0.25-4.0] | >30 [0.25-4.0] |
| 6 | Y = 4′-Cl | >50 [0.25-4.0] | <100 [1.0-4.0] | >50 [0.25-4.0] | >50 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] |
| 7 | Y = 2′-OCF3 | 40 [0.25] (28-59) | <50 [0.25-1.0] | >120 [0.25] | 72 [0.25] (59-84) | <30 [0.25-2.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] |
| 8 | Y = 3′-OCF3 | 25 [0.5] (20-34) | 43 [1.0] (35-53) | 81 [4.0] (60-118) | 81 [6.0] (58-99) | 16 [2.0] (15-18) | 72 [2.0] (46-79) | 8.7 [2.0] (5.1-13.3) | >500 [0.25-24.0] |
| 9 | Y = 4′-OCF3 | ∼50 [0.5-2.0] | <100 [2.0] | >50 [0.25-4.0] | >50 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] |
| 10 | Y = 3′-OCH3 | <100 [0.25] | ∼100 [1.0] | >100 [0.5-4.0] | >100 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] |
| 11 | Y = 3′-C(O)OCH3 | ∼100 [0.5] | >300 [0.5-2.0] | NDi | 100-300 [0.5-2.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] | >30 [0.25-4.0] |
| lacosamide j(1) | 4.5 [0.5] (3.7-5.5) | 27 [0.25] (26-28) | 3.9 [2.0] (2.9-6.2) | >500 | |||||
| phenytoin k | 9.5 [2.0] (8.1-10) | 66 [2.0] (53-72) | 30 [40] (22-39) | ||||||
| phenobarbital k | 22 [1.0] (15-23) | 69 [0.5] (63-73) | 9.1 [5.0] (7.6-12) | 61 [0.5] (44-96) | |||||
| valproate k | 270 [0.25] (250-340) | 430 [0.25] (370-450) | 490 [0.5] (350-730) | 280 [0.5] (190-350) | |||||
The compounds were tested through the auspices of the NINDS ASP.
The compounds were administered intraperitoneally (ip) to mice. ED50 and TD50 values are in milligrams per kilogram. Numbers in parentheses are 95% confidence intervals. A dose-response curve was generated for all compounds that displayed sufficient activity. The dose-effect data for these compounds was obtained at the “time of peak effect” (indicated in hours in the brackets).
The compounds were administered either ip or orally (po) to rats.
MES = Maximal electroshock seizure test.
6 Hz = 6 Hz psychomotor seizure test (32 mA).
scMet = subcutaneous pentylenetetrazol test.
Tox = rotorod test.
Tox = behavioral toxicity.
ND = not determined.
Chemistry
The (biphenyl-4-yl)methylammonium chlorides 2–11 were prepared using Suzuki coupling13 of 4-bromobenzyl amine (12) with the appropriate, commercially available substituted phenylboronic acids (13–21) to give the amines, which were then were immediately converted to their hydrochloride salts 3–11 (Scheme 1). For hydrochloride salt 2, we treated a commercial sample of 4-(phenyl)benzylamine with HCl in dioxane.
Scheme 1. Synthesis of (Biphenyl-4-yl)methylammonium Chlorides (B).

Pharmacological Activity
Compounds 2–11 were tested for anticonvulsant activity at the Anticonvulsant Screening Program (ASP), of the National Institute of Neurological Disorders and Stroke (NINDS) at the U.S. National Institutes of Health. Screening was performed using the procedures described by Stables and Kupferberg.14 The anticonvulsant activity data from the MES,10 psychomotor 6 Hz,15 and scMetrazol16 (scMet) tests are summarized in Table 1, along with similar results obtained for 1 and the clinical AEDs phenytoin,11 valproate,11 and phenobarbital.11 All compounds were administered intraperitoneally (ip) to mice and ip or orally (po) to rats. For compounds that showed significant activity, we report the 50% effective dose (ED50) values obtained in quantitative screening evaluations. We also provide the median doses for 50% neurological impairment (TD50) in mice (rotorod test17) and in rats18 (behavioral toxicity effects).
Compounds 5 and 8 displayed activities (50–100 mg/kg) in mice (ip) in the three seizure models (MES, 6 Hz, scMet), while 2, 3, 4, 7, 9, and 10 displayed activities (30–100 mg/kg) in two of the three assays. The observed seizure protection of 3, 5, and 8 in the scMet model was interesting since 112 and compounds belonging to class A7 did not display anticonvulsant activity in this model. The broad whole-animal pharmacological profile for several Bs suggested that these compounds exert their anticonvulsant activities through multiple pathways. For the trifluoromethoxy-substituted compounds 7–9, we found that the 3′-trifluoromethoxy derivative 8 was the most potent in the MES test (mice, ip). Finally, for 2–11, we did not observe a clear trend on the effect of the electronic properties of the terminal aryl substituent (Y) in 2–11 on anticonvulsant activity.
In mice (ip), 8 was among the most active (biphenyl-4-yl)methylammonium chlorides. We observed ED50 values of 25 mg/kg, 43 mg/kg, and 81 mg/kg in the MES, 6 Hz, and scMet seizure models, respectively. When 8 was tested in the MES model in rats (po), the ED50 value was 8.7 mg/kg. Compound 8 showed no neurotoxicity in rats (po) at doses as high as 500 mg/kg, providing a protective index (PI = TD50/ED50) of >57. The oral activity of 8 exceeded that of phenytoin,11 phenobarbital,11 and valproate11 and was approximately twofold less active than 1.1 Similar activity for 8 was observed in the rat after ip administration (MES ED50 = 16 mg/kg; TD50 = 72 mg/kg). When tested in the iv Metrazol test18 (mice, ip) at 25 mg/kg and 81 mg/kg there was no statistical difference from the control group in seizure threshold.
The excellent activity observed for 8 led us to examine its cellular activity by patch-clamp electrophysiology using CAD cells. We have previously shown that CAD cells express endogenous tetrodotoxin-sensitive Na+ currents with rapid activation and inactivation kinetics upon membrane depolarization and are likely mediated by NaV1.7, NaV1.1, and NaV1.3 channels.5,9 Moreover, we found that the Na+ channel properties of 1 in CAD cells5 were similar to those reported in cultured neurons and mouse N1E-115 neuroblastoma cells.3 Accordingly, we used readily accessible CAD cells to evaluate the effect of 8 on neuronal function, recognizing in advance that CAD cells do not express the same complement of Na+ channels expressed in central nervous system (CNS) neurons. We found that 8 preferentially promoted Na+ channel slow inactivation (Fig. 2A-D). The Na+ slow inactivation IC50 value at −50 mV was 2.7 μM, which was approximately 30-times more potent than 1 (IC50 = 85 μM).5 We chose the potential of −50 mV for three reasons: (1) a large fraction of the channels undergo steady-state inactivation, which involves contributions from slow and fast inactivation pathways,19,20 where −50 mV is within the steep voltage-dependence range for each; (2) it is near the resting membrane potential and approaches the action potential firing threshold for CNS neurons,21 where slow inactivation appears to be physiologically relevant during sustained subthreshold depolarizations;22 and (3) changes in the Na+ channel availability near −50 mV can impact the overlap of Na+ current activating and inactivating under steady-state conditions.19,22
Figure 2. Effect on steady-state slow inactivation state of Na+currents in mouse CAD cells by 8.
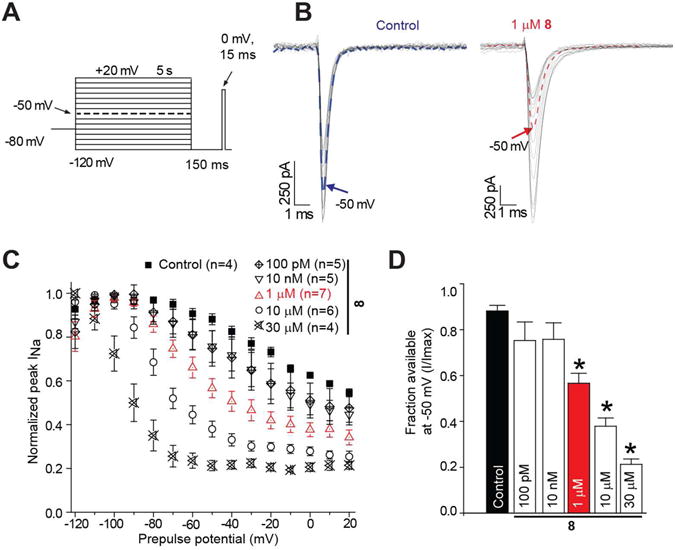
A. Currents were evoked by 5 s prepulses between −120 mV and −20 mV and then fast-inactivated channels were allowed to recover for 150 ms at a hyperpolarized pulse to −120 mV. The fraction of channels available at 0 mV was analyzed. B. Representative current traces from CAD cells in the absence (control, 0.1% DMSO) or presence of 100 μM of compounds as indicated. The blue (control) and red (8) traces represent the current at −50 mV. C. Summary of steady-state slow activation curves for CAD cells treated with DMSO (control) or 10 μM of 8. D. Summary of the fraction of current available at −50 mV for CAD cells treated with DMSO (control) or 10 μM of 8. Asterisks (*) indicate statistically significant differences in fraction of current available between control and the indicated concentrations of 8 (p < 0. 05, Student's t-test). Data are from 4-7 cells per condition.
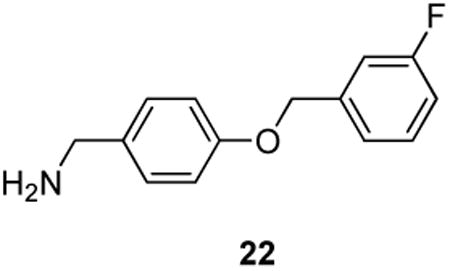
We found that 8 did not affect Na+ channel steady-state activation, defined as the relationship between voltage and a shift of channels from closed to open confirmations, but did modify steady-state fast inactivation, defined as the relationship between voltage and a shift of channel gating from open to inactivated confirmations over several hundred milliseconds (Fig. 3). Steady-state fast inactivation was assessed using a previously described protocol designed to induce a fast-inactivated state.5,8,9 Cells were held at −80 mV, stepped to inactivating prepulse potentials ranging from −120 to −10 mV (in 10-mV increments) for 500 ms, then the cells were stepped to 0 mV for 20 ms to measure the available current (Fig. 3, top left protocol). A 500-ms conditioning pulse was used because it allowed all of the endogenous channels to transition to a fast-inactivated state at all potentials examined. Steady-state, fast inactivation curves of Na+ currents from DMSO-treated and various concentrations of 8-treated CAD cells were well fitted with a single Boltzmann function (R2 > 0.935 for all conditions) and are illustrated in Figure 3 (leftmost curves). The V1/2 value for inactivation of 0.1% DMSO-treated cells was −71.6 ± 0.6 mV (n=6), which was significantly different from the V1/2 values of all concentrations of 8-treated CAD cells (p > 0.05; ANOVA with a post-hoc Dunnett's test). The 1 μM concentration of 8 caused a significant hyperpolarizing shift of ∼11.2 mV while the 30 μM concentration of 8 caused a significant hyperpolarizing shift of ∼26.5 mV with no commensurate significant changes in slope values compared with control cells. The slopes of fast inactivation were not affected by 8. Steady-state activation, as measured by 15 ms depolarizing pulses from −70 mV to +80 mV (in 10-mV increments) produced equivalent V1/2 and slope values in all conditions. Finally, we found that 8 did not exhibit frequency (use)-dependent inhibition of Na+ currents as currents recorded from cells treated with 10 μM 8 displayed no statistically significant differences in trend or amplitude compared with control (Fig. 4). The whole-cell, patch-clamp electrophysiology for 8 mirrored aspects observed for 22 in CAD cells.9 For 22, the IC50 value for Na+ channel slow inactivation was 2.1 μM, and like 8, it affected fast inactivation. However, 22 displayed frequency (use)-dependent blockage of Na+ currents, while 8 did not.
Figure 3. Effects of 8 on activation and inactivation properties of Na+currents in mouse CAD cells.
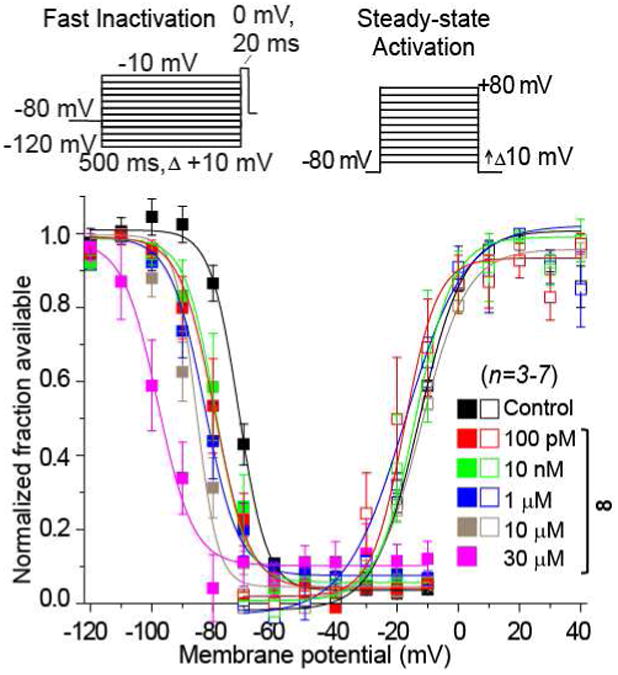
Values for V1/2, the voltage of half-maximal activation and steady-state fast inactivation and the slope factors (k) were derived from Boltzmann distribution fits to the individual recordings and averaged to determine the mean (± SEM) voltage dependence of activation and fast inactivation. The voltage protocol used to evoke current responses for each protocol is shown above the fits. Representative Boltzmann fits for activation and steady-state inactivation for CAD cells treated with 0.1% DMSO (control) and indicated concentrations of 8 are shown.
Figure 4. Lack of effect on frequency-dependent block by 8 of Na+currents in mouse CAD cells.

The frequency dependence of block was examined by holding cells at the hyperpolarized potential of −80 mV and evoking currents at 10 Hz by 20-ms test pulses to −10 mV (Inset middle). Representative overlaid traces are illustrated by pulses 1, 10, 20, and 30 for control (predrug) and in the presence of 8 (1 μM). Summary of average frequency-dependent decrease in current amplitude (± SEM) produced by control (Pre-drug) or by the presence of 8 (1 μM) (p > 0.05, one-way ANOVA with Dunnett's post-hoc test). Data are from 5–7 cells per condition.
We also tested the activity of 8 in rat embryonic cortical neurons. These neurons typically express Na+ channel isoforms NaV1.1, NaV1.2, NaV1.3, and NaV1.6.24 The slow inactivation, steady-state inactivation, fast inactivation, and use-dependence of Na+ currents in cortical neurons grown for 7-10 days in vitro (Fig. 5A, D, G, and J) were examined using protocols mostly similar to those described for CAD cells with a few exceptions noted in the legend to Figure 5. A single concentration (20 μM) of 8 was chosen as it represented almost 8– fold the IC50 value for slow inactivation based on CAD cell data. At this concentration, the extent of slow inactivation induced by 8 was significantly greater than neurons treated with the vehicle DMSO: 0.17 ± 0.03 (n=5) versus 0.41 ± 0.0.05 (n=7), respectively (p>0.05, one-way ANOVA Fig. 5B, C). Steady-state activation was also unchanged between the two conditions with the V1/2 and k values being statistically similar (Fig. 5E, F). Steady-state, fast inactivation curves of Na+ currents from DMSO-treated and 20 μM 8-cortical neurons were well fitted with a single Boltzmann function (R2 > 0.995 for both conditions) and are illustrated in Figure 5I. The V1/2 value for inactivation of 0.1% DMSO-treated cells was −53.9 ± 1.2 mV (n=4), which was significantly different from the V1/2 value of −69.6 ± 1.2 mV (n=5) for 8-treated neurons (p < 0.05; ANOVA with a post-hoc Dunnett's test). The 20 μM concentration of 8 caused a significant hyperpolarizing shift of ∼15.7 mV with no commensurate significant changes in slope values compared with control cells. Finally, we found that 8 did not exhibit frequency (use)-dependent inhibition of Na+ currents in cortical neurons (Fig. 5K, L).
Figure 5. Evaluation of 8 on biophysical properties of Na+currents in rat embryonic cortical neurons.
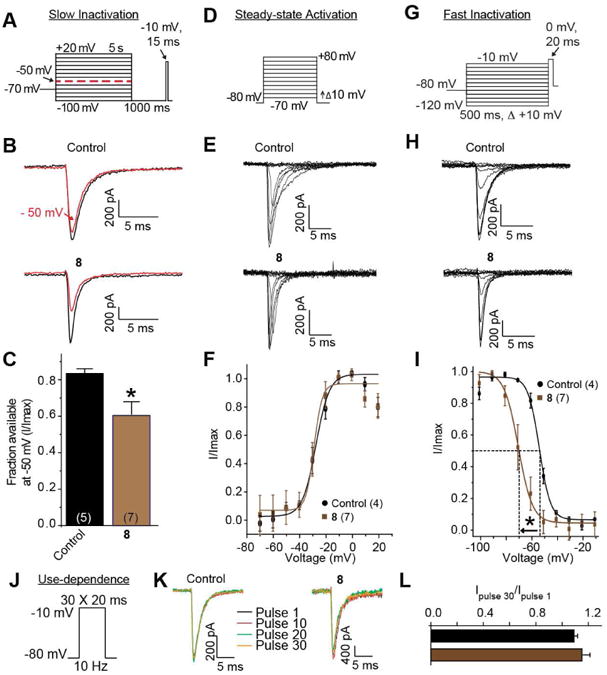
A. Currents were evoked by 5 s prepulses between −100 mV and +20 mV and then fast-inactivated channels were allowed to recover for 1000 ms at a hyperpolarized pulse to -100 mV. The fraction of channels available at 0 mV was analyzed. B. Representative current traces from cortical neurons in the absence (control, 0.1% DMSO) or presence of 20 μM8. The black and red traces represent the current at −50 mV. C. Summary of the fraction of current available at −50 mV for cortical neurons treated with DMSO (control) or 20 μM of 8. Values for V1/2, the voltage of half-maximal activation (D-F) and steady-state fast inactivation (G-I) and the slope factors (k) were derived from Boltzmann distribution fits to the individual recordings and averaged to determine the mean (± SEM) voltage dependence of activation and fast inactivation. The voltage protocol used to evoke current responses for each protocol is shown above the fits. Representative Boltzmann fits for activation and steady-state inactivation for cortical neurons treated with 0.1% DMSO (control) and 20 μM of 8 are shown. Fast-inactivation was significantly affected by 20 μM of 8. J-L. Use-dependence was not affected by 20 μM of 8. Details are similar to those in legend to Figure 4. Five to seven cells were tested in these experiments.
Compound 8, and the other agents in this study, contained a biphenyl unit. This motif is considered a privileged substructure and has been shown to bind to many proteins, including G-protein-coupled receptors.25-27 Accordingly, we determined the binding of 2–11 at UNC's Psychoactive Drug Screening Program against 43 receptors. We observed appreciable binding of most compounds at 10 μM to several serotonin (e.g., 5-HT2A, 5-HT2B, 5-HT5A, 5-HT6, 5-HT7) and adrenergic (e.g., alpha 2A, 2B, 2C) receptors, the DAT, NET, and SERT transporters, and the sigma-1 and -2 receptors (Supplementary Table 1). The importance of these interactions on the observed anticonvulsant activities has not been determined.
Conclusions
Our previous finding that the bi-aryl linked unit (Fig. 1) promoted Na+ channel slow inactivation in CAD cells9 led to the discovery that compounds conforming to class B exhibited anticonvulsant activities in proven whole animal seizure models. Structurally, B is exceedingly simple, readily synthesized, and soluble in water (≥300 μM). The anticonvulsant activity for 8 in the MES seizure model (rat, po) rivaled that of established AEDs. Compound 8 appears to exert its activity, in part, by inhibiting Na+ channel currents.
Experimental Section
General Methods
The general methods used in this study are identical to those previously reported.7 The compounds were checked by TLC, 1H NMR, and 13C NMR, MS, and elemental analyses. The analytical results, except for 6, are within ± 0.40% of the theoretical value. The NMR and analytical data confirmed the purity of the products was ≥95%.
(Biphenyl-4-yl)methylammonium Chloride28 (2)
To a solution of 4-(phenyl)benzylamine (1.00 g, 5.5 mmol) in CH2Cl2 (50 mL) was added an HCl solution in dioxane dropwise (1.5 mL, 4 N) with stirring at room temperature (1 h). The resulting precipitate was filtered, washed with hexanes, dried in vacuo to give 2 (1.03 g, 86%) as a white solid: Rf = 0.00 (hexanes/EtOAc 1/1); mp 301-303 °C (lit.28 mp 308-310 °C); 1H NMR (CDCl3, CD3OD) δ4.14 (s, CH2N), 4.62-4.78 (br s, NH3), 7.32-7.41 (m, ArH), 7.42-7.48 (m, 2 ArH), 7.49 (d, J = 8.4 Hz, 2 ArH), 7.59 (d, J = 8.8 Hz, 2 ArH), 7.68 (d, J = 8.4 Hz, 2 ArH); 13C NMR (CDCl3, CD3OD) δ42.7 (CH2N), 126.5, 127.3, 127.4, 128.5, 128.9, 131.3, 139.7, 141.9 (8 ArC); LRMS (ES+) 184.00 [M - Cl]+ (calcd for C13H14N+ 184.11). Anal. Calcd. for C13H14ClN: C, 71.07; H, 6.42; Cl, 16.14; N, 6.38. Found: C, 70.95; H, 6.44; Cl, 16.24; N, 6.34.
(3′-Fluorobiphenyl-4-yl)methylammonium Chloride (3)
To a solution of 12 (1.21 g, 6.50 mmol) in acetonitrile (65 mL) was added 3-fluorophenylboronic acid (13) (1.00 g, 7.15 mmol), tetrakis(triphenylphosphine)palladium(0) (0.38 g, 0.32 mmol), and aqueous 2 N K2CO3 (16.2 mL). The resulting mixture was sparged with Ar with stirring (30 min) and then stirred again at 90 °C under Ar (16 h). The reaction mixture was filtered and evaporated in vacuo. The resulting residue was diluted with EtOAc (50 mL) and washed with H2O (2 × 50 mL) and with saturated aqueous brine solution (2 × 50 mL). The organic layer was dried (Na2SO4) and concentrated in vacuo to give the amine as a yellow oil. To a solution of the amine in ethyl acetate was added aqueous concentrated HCl (0.8 mL) with stirring at room temperature (1 h) to give a precipitate. To the resulting mixture, H2O was added and then the aqueous layer separated. The aqueous layer was basified with aqueous 4 N NaOH and extracted with CH2Cl2 (2×), and the combined CH2Cl2 layers dried (Na2SO4) and evaporated in vacuo. The resulting oil was diluted in CH2Cl2 and then 4 N HCl in dioxane added. The resulting precipitate was filtered and washed with hexanes to give 3 (1.20 g, overall yield 77%) as a white solid: Rf = 0.00 (EtOAc/hexanes 1/1); mp 301-304 °C; 1H NMR (CD3OD) δ4.18 (s, CH2N), 7.06-7.20 (m, ArH), 7.30-7.39 (m, ArH), 7.43-7.48 (m, 2 ArH), 7.58 (d, J = 8.2 Hz, 2 ArH), 7.71 (d, J = 8.2 Hz, 2 ArH); 13C NMR (CD3OD) δ44.5 (CH2N), 115.1 (d, J = 20.2 Hz), 115.9 (d, J = 20.2 Hz), 124.4, 129.2, 131.2, 132.3 (d, J = 8.8 Hz), 134.6, 142.4 (d, J = 7.6 Hz), 144.4 (d, J = 7.6 Hz), 165.2 (d, J = 243.0 Hz) (10 ArC); LRMS (ES+) 201.95 [M -Cl]+ (calcd for C13H13FN+ 202.10). Anal. Calcd. for C13H13ClFN: C, 65.69; H, 5.51; Cl, 14.91; F, 7.99; N, 5.89. Found: C, 65.68; H, 5.45; Cl, 14.77; F, 7.81; N, 5.74.
(4′-Fluorobiphenyl-4-yl)methylammonium Chloride (4)
Employing the procedure for 3 and using 12 (5.00 g, 26.9 mmol), acetonitrile (100 mL), 4-fluorophenylboronic acid (14) (3.77 g, 26.9 mmol), tetrakis(triphenylphosphine)palladium(0) (1.55 g, 1.35 mmol), and aqueous 2 N K2CO3 (53.8 mL) gave the free amine29 as a yellow oil. The amine was treated with aqueous concentrated HCl (2.5 mL) and then purified to give 4 (4.99 g, overall yield 78%) as a white solid: Rf = 0.00 (EtOAc/hexanes 1/1); mp 302-303 °C; 1H NMR (DMSO-d6) δ4.06 (s, CH2N), 7.28-7.34 (m, 2 ArH), 7.61-7.75 (m, 6 ArH), 8.70 (s, NH3); 13C NMR (DMSO-d6) δ41.7 (CH2N), 115.8 (d, J = 21.3 Hz, C3′, C5′), 126.7 (ArC), 128.7 (d, J = 8.0 Hz, C2′, C6′), 129.6, 133.3 (2 ArC), 136.0 (d, J = 3.7 Hz, C1′), 139.1 (ArC), 162.0 (d, J = 243.2 Hz, C4′); HRMS (ESI+) 202.1022 [M - Cl]+ (calcd for C13H13FN+ 202.1022). Anal. Calcd. for C13H13ClFN: C, 65.69; H, 5.51; Cl, 14.91; F, 7.99; N, 5.89. Found: C, 65.55; H, 5.54; Cl, 14.79; F, 7.82; N, 5.88.
(3′-Chlorobiphenyl-4-yl)methylammonium Chloride (5)
Employing the procedure for 3 and using 12 (2.16 g, 11.63 mmol), acetonitrile (116 mL), 3-chlorophenylboronic acid (15) (4.21 g, 26.9 mmol), tetrakis(triphenylphosphine)palladium(0) (1.55 g, 1.35 mmol), and aqueous 2 N K2CO3 (53.8 mL) gave the free amine30 as a yellow oil. The amine was treated with aqueous concentrated HCl (2.5 mL) and then purified to give 5 (4.37 g, overall yield 64%) as a white solid: Rf = 0.00 (EtOAc/hexanes 1/1); mp 264-265 °C; 1H NMR (DMSO-d6) δ 4.07 (s, CH2N), 7.43-7.76 (m, 8 ArH), 8.75 (s, NH3Cl); 13C NMR (DMSO-d6) δ 40.5 (CH2N), 125.4, 126.3, 126.9, 127.4, 129.7, 130.8, 133.8, 134.0, 138.5, 141.6 (10 ArC); LRMS (ES+) 218.0 [M - Cl]+ (calcd for C13H13ClN+ 218.1). Anal. Calcd. for C13H13Cl2N·0.18 H2O: C, 60.66; H, 5.23; Cl, 27.55; N, 5.44. Found: C, 60.30; H, 5.12; Cl, 27.16; N, 5.49.
(4′-Chlorobiphenyl-4-yl)methylammonium Chloride (6)
Employing the procedure for 3 and using 12 (2.16 g, 11.63 mmol), acetonitrile (116 mL), 4-chlorophenylboronic acid (16) (2.00 g, 12.79 mmol), tetrakis(triphenylphosphine)palladium(0) (0.68 g, 0.58 mmol), and aqueous 2 N K2CO3 (29.1 mL) gave the free amine as a yellow oil. The amine was treated with aqueous concentrated HCl (0.5 mL) and then purified to give 6 (1.00 g, overall yield 100%) as a white solid: Rf = 0.00 (EtOAc/hexanes 1/1); mp 298-302 °C; 1H NMR (CD3OD) δ 4.19 (s, CH2N), 7.42-7.46 (m, 2 ArH), 7.56-7.63 (m, 4 ArH), 7.67-7.71 (m, 2 ArH); 13C NMR (CD3OD) δ 40.5 (CH2N), 125.1, 126.1, 126.6, 127.3, 130.4, 131.4, 136.6, 138.4 (8 ArC); HRMS (ESI+) 218.0764 [M - Cl]+ (calcd for C13H13ClN+ 218.0736).
(2′-Trifluoromethoxybiphenyl-4-yl)methylammonium Chloride31 (7)
Employing the procedure for 3 and using 12 (1.65 g, 8.9 mmol), acetonitrile (100 mL), 2-trifluoromethoxyphenylboronic acid (17) (2.00 g, 9.8 mmol), tetrakis(triphenylphosphine)palladium(0) (0.51 g, 0.44 mmol), and aqueous 2 N K2CO3 (17.7 mL) gave the free amine as a yellow oil. The amine was treated with aqueous concentrated HCl (2.3 mL) and then purified to give 7 (1.70 g, overall yield 57%) as a white solid: Rf = 0.00 (hexanes/EtOAc 1/1); mp 175-179 °C; 1H NMR (CDCl3, CD3OD) δ 4.15 (s, CH2N), 4.23-4.40 (br s, NH3), 7.35-7.46 (m, 5 ArH), 7.53-7.57 (br s, 3 ArH); 13C NMR (CDCl3, CD3OD) δ 43.0 (CH2N), 121.4, 127.2, 128.7, 129.1, 129.9, 131.2, 132.0, 134.3, 137.9, 146.0 (10 ArC), the OCF3 resonance was not detected and was believed to overlap with nearby peaks; LRMS (ES+) 268.01 [M - Cl]+ (calcd for C14H13F3NO+ 268.09). Anal. Calcd. for C14H13ClF3NO: C, 55.37; H, 4.31; Cl, 11.67; F, 18.77; N, 4.61. Found: C, 55.48; H, 4.39; Cl, 11.48; F, 18.51; N, 4.56.
(3′-Trifluoromethoxybiphenyl-4-yl)methylammonium Chloride (8)
Employing the procedure for 3 and using 12 (1.56 g, 8.4 mmol), acetonitrile (85 mL), 3-trifluoromethoxyphenylboronic acid (18) (2.00 g, 8.4 mmol), tetrakis(triphenylphosphine)palladium(0) (0.38 g, 0.4 mmol), and aqueous 1 N K2CO3 (30 mL) gave the free amine as a yellow oil. The amine was treated with aqueous concentrated HCl (0.5 mL) and then purified to give 8 (1.00 g, overall yield 70%) as a white solid: Rf = 0.00 (EtOAc/hexanes 1/1); mp 218-219 °C; 1H NMR (CD3OD) δ 4.10 (s, CH2N), 7.38-7.40 (m, 1 ArH), 7.63-7.79 (m, 7 ArH), 8.81-8.85 (br s, NH3Cl); 13C NMR (CD3OD) δ 51.4 (CH2), 128.8, 129.5 (2 ArC), 129.8 (q, J = 254.9 Hz, OCF3), 135.5, 136.6, 139.4, 140.6, 143.9, 148.0, 151.6, 158.6 (8 ArC). Anal. Calcd. for C14H13ClF3NO: C, 55.37; H, 4.31; Cl, 11.67; F, 18.77; N, 4.61. Found: C, 55.36; H, 4.31; Cl, 11.55; F, 18.58; N, 4.55.
(4′-Trifluoromethoxybiphenyl-4-yl)methylammonium Chloride32 (9)
Employing the procedure for 3 and using 12 (1.65 g, 8.9 mmol), acetonitrile (100 mL), 4-trifluoromethoxyphenylboronic acid (19) (2.00 g, 9.8 mmol), tetrakis(triphenylphosphine)palladium(0) (0.51 g, 0.44 mmol), and aqueous 2 N K2CO3 (17.7 mL) gave the free amine as a yellow oil. The amine was treated with aqueous concentrated HCl (2.3 mL) and then purified to give 9 (2.18 g, overall yield 74%) as a white solid: Rf = 0.00 (hexanes/EtOAc 1/1); mp 281-288 °C; 1H NMR (CDCl3, CD3OD) δ 4.14 (s, CH2N), 4.24 (s, NH3), 7.31 (d, J = 8.8 Hz, 2 ArH), 7.55 (d, J = 8.4 Hz, 2 ArH), 7.60-7.68 (m, 4 ArH); 13C NMR (CDCl3, CD3OD) δ 42.9 (CH2N), 121.2, 127.6, 128.4, 129.4, 131.9, 138.8, 146.0, 148.9 (8 ArC), the OCF3 resonance was not detected and was believed to overlap with nearby peaks; HRMS (ESI+) 290.0763 [M-HCl+Na]+ (calcd for C14H13F3NO+ 290.0769). Anal. Calcd. for C14H13ClF3NO·0.08C6H14: C, 55.99; H, 4.58; Cl, 11.41; F, 18.35; N, 4.51. Found: C, 56.03; H, 4.22; Cl, 11.03; F, 18.52; N, 4.50.
(3′-Methoxybiphenyl-4-yl)methylammonium Chloride (10)
Employing the procedure for 3 and using 12 (1.12 g, 6.0 mmol), acetonitrile (60 mL), 3-methoxyphenylboronic acid (20) (1.00 g, 6.6 mmol), tetrakis(triphenylphosphine)palladium(0) (0.35 g, 0.3 mmol), and aqueous 2 N K2CO3 (15.0 mL) gave the free amine as a yellow oil. The amine30 was treated with aqueous concentrated HCl (1.6 mL) and then purified to give 10 (0.80 g, overall yield 54%) as a white solid: Rf = 0.00 (hexanes/EtOAc 1/1); mp 231-234 °C; 1H NMR (DMSO-d6) δ 3.83 (OCH3), 4.05 (CH2N), 6.95 (dd, J = 2.2, 8.3 Hz, ArH), 7.20 (s, ArH), 7.25 (d, J = 8.4 Hz, ArH), 7.39 (t, J = 7.8 Hz, ArH), 7.50 (d, J = 8.0 Hz, 2 ArH), 7.72 (d, J = 8.0 Hz, 2 ArH), 8.40-8.58 (br s, NH3); 13C NMR (DMSO-d6) δ 42.3 (CH2N), 55.6 (OCH3), 112.7, 113.7, 119.4, 127.3, 129.9, 130.4, 133.9, 140.5, 141.5, 160.2 (10 ArC); LRMS (ES+) 214.09 [M - Cl]+ (calcd for C14H16NO+ 214.12). Anal. Calcd. for C13H13Cl2N: C, 67.33; H, 6.46; Cl, 14.20; N, 5.61. Found: C, 67.05; H, 6.44; Cl, 13.94; N, 5.58.
(3′-Methoxycarbonylbiphenyl-4-yl)methylammonium Chloride32 (11)
Employing the procedure for 3 and using 12 (0.86 g, 4.6 mmol), acetonitrile (40 mL), 3-methoxycarbonylphenylboronic acid (21) (1.00 g, 5.6 mmol), tetrakis(triphenylphosphine)palladium(0) (0.27 g, 0.2 mmol), and aqueous 2 N K2CO3 (5 mL) gave the free amine as a yellow oil. The amine was treated with aqueous concentrated HCl (1.2 mL) and then purified to give 11 (0.71 g, overall yield 55%) as a white solid: Rf = 0.00 (hexanes/EtOAc 1/1); mp 215-218 °C; 1H NMR (CD3OD) δ 3.90 (OCH3), 4.08 (CH2N), 7.58-7.70 (m, 3 ArH), 7.77 (d, J = 8.0 Hz, 2 ArH), 7.98 (dd, J = 1.0, 8.0 Hz, 2 ArH), 8.21 (s, ArH), 8.40-8.60 (br s, NH3); 13C NMR (CD3OD) δ 41.8 (CH2N), 52.3 (OCH3), 127.0, 127.1, 128.3, 129.7, 129.8, 130.4, 131.6, 133.8, 139.1, 140.0 (10 ArC), 166.2 (C(O)); LRMS (ES+) 242.03 [M - Cl]+ (calcd for C15H16NO2+ 242.12). Anal. Calcd. for C15H16ClNO2·0.2H2O: C, 64.03; H, 5.88; Cl, 12.06; N, 4.98. Found: C, 63.65; H, 5.82; Cl, 11.84; N, 4.79.
Pharmacology
Compounds were screened under the auspices of the National Institutes of Health's ASP. Experiments were performed in male rodents (albino Carworth Farms No. 1 mice (ip), albino Sprague-Dawley rats (ip, po)). Housing, handling, and feeding were in accordance with recommendations contained in the Guide for the Care and Use of Laboratory Animals. Anticonvulsant activity was established using the MES test,10 6 Hz,15 and the scMet test,16 according to previously reported methods.1
Catecholamine A–Differentiated (CAD) Cells
CAD cells were grown at 37 °C and in 5% CO2 (Sarstedt, Newton, NC) in Ham's F12/EMEM (GIBCO, Grand Island, NY), supplemented with 8% fetal bovine serum (Sigma, St. Louis, MO) and 1% penicillin/streptomycin (100% stocks, 10,000U/mL penicillin G sodium and 10,000 μg/mL streptomycin sulfate).5,8 Cells were passaged every 6–7 days at a 1:25 dilution.
Cortical Neurons
Rat cortical neuron cultures were prepared from cortices dissected from embryonic day 19 brains exactly as described.33,34
Electrophysiology
Whole-cell voltage clamp recordings were performed at room temperature on CAD cells and cortical neurons using an EPC 10 Amplifier (HEKA Electronics, Lambrecht/Pfalz Germany). Electrodes were pulled from thin-walled borosilicate glass capillaries (Warner Instruments, Hamden, CT) with a P-97 electrode puller (Sutter Instrument, Novato, CA) such that final electrode resistances were 1–2 MΩ when filled with internal solutions. The internal solution for recording Na+ currents contained (in mM): 110 CsCl, 5 MgSO4, 10 EGTA, 4 ATP Na2-ATP, 25 HEPES (pH 7.2, 290–310 mOsm/L). The external solution contained (in mM): 100 NaCl, 10 tetraethylammonium chloride (TEA-Cl), 1 CaCl2, 1 CdCl2, 1 MgCl2, 10 D-glucose, 4 4-AP, 0.1 NiCl2, 10 HEPES (pH 7.3, 310-315 mOsm/L). Whole-cell capacitance and series resistance were compensated with the amplifier. Series resistance error was always compensated to be less than ± 3 mV. Cells were considered only when the seal resistance was less than 3 MΩ;. Linear leak currents were digitally subtracted by P/4.
Data Acquisition and Analysis
Signals were filtered at 10 kHz and digitized at 10–20 kHz. Analysis was performed using Fitmaster and origin8.1 (OriginLab Corporation, MA, USA). For activation curves, conductance (G) through Na+ channels was calculated using the equation G= I/(Vm -Vrev), where Vrev is the reversal potential, Vm is the membrane potential at which the current was recorded and I is the peak current. Activation and inactivation curves were fitted to a single-phase Boltzmann function G/Gmax = 1/{1+exp[(V–V50)/k]}, where G is the peak conductance, Gmax is the fitted maximal G, V50 is the half-activation voltage, and k is the slope factor. Additional details of specific pulse protocols are described in the results text or figure legends.
Statistical Analyses
Differences between means were compared by either paired or unpaired, two-tailed Student's t-tests or an analysis of variance (ANOVA), when comparing multiple groups (repeated measures whenever possible). If a significant difference was determined by ANOVA, then a Dunnett's or Tukey's post-hoc test was performed. Data are expressed as mean ± SEM, with p<0.05 considered as the level of significance.
Supplementary Material
Supplementary Table 1. Receptor Binding Profiles for Compounds 2-11.a
aData represent mean % inhibition (N = 4 determinations) for compound tested at receptor subtypes. Significant inhibition is considered > 50%. In cases where negative inhibition (-) is seen, this represents a stimulation of binding. Occasionally, compounds at high concentrations will non-specifically increase binding. The default concentration for primary binding experiments is 10 μM. b Compound number. ND; not determined.
Acknowledgments
This work is supported by a grant from the North Carolina Biotechnology Center (Technology Enhancement Grant) administered by the University of North Carolina-Chapel Hill and awards from the Indiana Clinical and Translational Sciences Institute funded, in part by a Project Development Team Grant Number (RR025761) from the National Institutes of Health, National Center for Research Resources, Clinical and Translational Sciences Award, the Indiana State Department of Health – Spinal Cord and Brain Injury Fund (A70-9-079138 to R.K.), a National Scientist Development Award from the American Heart Association (SDG5280023 to R.K.), and a Neurofibromatosis New Investigator Award from the Department of Defense Congressionally Directed Military Medical Research and Development Program (NF1000099 to R.K.). We thank the NINDS and the ASP at the National Institutes of Health with Drs. Tracy Chen and Jeffrey Jiang for kindly performing the pharmacological studies via the ASP's contract site at the University of Utah with Drs. H. Wolfe, H.S. White, and K. Wilcox. We express our appreciation to Dr. Bryan L. Roth and Mr. Jon Evans at the National Institute of Mental Health (NIMH) Psychoactive Drug Screening Project for performing in vitro receptor binding studies. This work was supported by NIMH Psychoactive Drug Screening Program, Contract No. HHSN-271-2008-00025-C (NIMH-PDSP). The NIMH PDSP is directed by Bryan Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. The content is solely the responsibility of the authors and does not represent the official views of the National Center for Research Resources, National Institutes of Health. Harold Kohn has a royalty-stake position in 1.
Abbreviations
- AED
antiepileptic drug
- ASP
Anticonvulsant Screening Program
- CAD
catecholamine A-differentiated
- CF3O
trifluoromethoxy
- ED50
effective dose (50%)
- IC50
concentration at which half of the channels have transitioned to a slow-inactivated state
- ip
intraperitoneally
- MES
maximal electroshock seizure
- NINDS
National Institutes of Neurological Disorders and Stroke
- PI
protective index
- po
orally
- scMet
scMetrazol
- TD50
neurological impairment (toxicity, 50%)
- TEA-Cl
tetraethylammonium chloride
- V1/2
voltage of half-maximal activation
Footnotes
Supporting Information Available: Receptor binding assay profile for compounds 2 – 11 against 43 receptors. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Choi D, Stables JP, Kohn H. Synthesis and anticonvulsant activities of N-benzyl-2-acetamidopropionamide derivatives. J Med Chem. 1996;39:1907–1916. doi: 10.1021/jm9508705. [DOI] [PubMed] [Google Scholar]
- 2.Perucca E, Yasothan U, Clincke G, Kirkpatrick P. Lacosamide. Nature Rev Drug Disc. 2008;7:973–974. doi: 10.1038/nrd2764. [DOI] [PubMed] [Google Scholar]
- 3.Errington AC, Stohr T, Heers C, Lees G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol Pharmacol. 2008;73:157–169. doi: 10.1124/mol.107.039867. [DOI] [PubMed] [Google Scholar]
- 4.Sheets PL, Heers C, Stoehr T, Cummins TR. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide, lidocaine and carbamazepine. J Pharmacol Exp Ther. 2008;326:89–99. doi: 10.1124/jpet.107.133413. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Park KD, Salome C, Wilson SM, Stables JP, Liu R, Khanna R, Kohn H. Development and characterization of novel derivatives of the antiepileptic drug lacosamide that exhibit far greater enhancement in slow inactivation of voltage-gated sodium channels. ACS Chem Neurosci. 2011;2:90–106. doi: 10.1021/cn100089b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuo CC, Bean BP. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol Pharmacol. 1994;46:716–725. [PubMed] [Google Scholar]
- 7.Salome C, Salome-Grosjean E, Stables JP, Kohn H. Merging the structural motifs of functionalized amino acids and α-aminoamides: compounds with significant anticonvulsant activities. J Med Chem. 2010;53:3756–3771. doi: 10.1021/jm100185c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Wilson SM, Brittain JM, Ripsch MS, Salome C, Park KD, White FA, Khanna R, Kohn H. Merging structural motifs of functionalized amino acids and α-aminoamides results in novel anticonvulsant compounds with significant effects on slow and fast inactivation of voltage-gated sodium channels and in the treatment of neuropathic pain. ACS Chem Neurosci. 2011;2:317–332. doi: 10.1021/cn200024z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King AM, Yang XF, Wang Y, Dustrude ET, Barbosa C, Due MR, Piekarz AD, Wilson SM, White FA, Salome C, Cummins TR, Khanna R, Kohn H. Identification of the benzyloxyphenyl pharmacophore: A structural unit that promotes sodium channel slow inactivation. ACS Chem Neurosci. 2012;3:1037–1049. doi: 10.1021/cn300129d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy RH, Mattson R, Meldrum B. Antiepileptic Drugs. 4th. Raven Press; New York: 1995. [Google Scholar]
- 11.Porter RJ, Cereghino JJ, Gladding GD, Hessie BJ, Kupferberg HJ, Scoville B, White BG. Antiepileptic drug development program. Cleveland Clin Q. 1984;51:293–305. doi: 10.3949/ccjm.51.2.293. [DOI] [PubMed] [Google Scholar]
- 12.Salome C, Salome-Grosjean E, Park KD, Morieux P, Swendiman R, DeMarco E, Stables JP, Kohn H. Synthesis and anticonvulsant activities of (R)-N-(4′-substituted)benzyl 2-acetamido-3-methoxypropionamides. J Med Chem. 2010;53:1288–1305. doi: 10.1021/jm901563p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 14.Stables JP, Kupferberg HG. In: Molecular and Cellular Targets for Antiepileptic Drugs. Avanzini G, Tanganelli P, Avoli M, editors. John Libbey; London: 1977. pp. 191–198. [Google Scholar]
- 15.Barton ME, Klein BD, Wolff HH, White HS. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epil Res. 2001;47:217–227. doi: 10.1016/s0920-1211(01)00302-3. [DOI] [PubMed] [Google Scholar]
- 16.Swinyard EA. Laboratory evaluation of antiepileptic drugs: review of laboratory methods. Epilepsia. 1969;10:107–119. doi: 10.1111/j.1528-1157.1969.tb03838.x. [DOI] [PubMed] [Google Scholar]
- 17.Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc. 1957;46:208–209. doi: 10.1002/jps.3030460322. [DOI] [PubMed] [Google Scholar]
- 18.White HS, Woodhead JH, Wilcox KS, Stables JP, Kupferberg HJ, Wolf HH. General Principles: Discovery and Preclinical Development of Antiepileptic Drugs. In: Levy RH, Mattson RH, Meldrum BS, Perruca E, editors. Antiepileptic Drugs. 5th. Lippincott, Williams and Wilkins; Philadelphia, PA: 2002. pp. 36–48. [Google Scholar]
- 19.Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol. 1952;116:497–506. doi: 10.1113/jphysiol.1952.sp004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rudy B. Slow inactivation of the sodium conductance in squid giant axons. Pronase resistance. J Physiol. 1978;283:1–21. doi: 10.1113/jphysiol.1978.sp012485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- 22.Do MTH, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: Modulation by slow inactivation. Neuron. 2003;39:109–120. doi: 10.1016/s0896-6273(03)00360-x. [DOI] [PubMed] [Google Scholar]
- 23.Vilin YY, Ruben PC. Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem Biophys. 2001;35:171–190. doi: 10.1385/CBB:35:2:171. [DOI] [PubMed] [Google Scholar]
- 24.Goldin AL. Resurgence of sodium channel research. Ann Rev Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 25.Horton DA, Bourne GT, Smythe ML. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 26.Bleicher KH, Green LG, Martin RE, Rogers-Evans M. Ligand identification for G-protein-coupled receptors: a lead generation perspective. Curr Opin Chem Biol. 2004;8:287–296. doi: 10.1016/j.cbpa.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 27.Corbett DF, Heightman TD, Moss SF, Bromidge SM, Coggon SA, Longley MJ, Roa AM, Williams JA, Thomas DR. Discovery of a potent and selective 5-ht5A receptor antagonist by high-throughput chemistry. Bioorg Med Chem Letters. 2005;15:4014–4018. doi: 10.1016/j.bmcl.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 28.Goldschmidt St, Modderman P. Biphenyl derivatives II. Basic 4-Biphenyl compounds. Rec Trav Chim Pays-Bas Belg. 1950;69:1109–1117. [Google Scholar]
- 29.Galiano S, Ceras J, Cirauqui N, Perez S, Juanenea L, Rivera G, Aldana I, Monge A. Novel series of substituted biphenylmethyl urea derivatives as MCH-R1 antagonists for the treatment of obesity. Bioorg Med Chem. 2007;15:3896–3911. doi: 10.1016/j.bmc.2007.02.049. [DOI] [PubMed] [Google Scholar]
- 30.Bhuniya D, Umrani D, Dave B, Salunke D, Kukreja G, Gundu J, Naykodi M, Shaikh NS, Shitole P, Kurhade S. Discovery of a potent and seelctive small molecule hGPR91 antagonist. Bioorg Med Chem Letters. 2011;21:3596–3602. doi: 10.1016/j.bmcl.2011.04.091. [DOI] [PubMed] [Google Scholar]
- 31.Guo Z, Orth P, Zhu Z, Mazzola RD, Chan TY, Vaccaro HA, McKittrick B, Kozlowski JA, Lavey BJ, Zhou G, Paliwal S, Wong S, Shih N, Ting PC, Rosner KE, Shipps GW, Jr, Siddiqui MA, Belanger DB, Dai C, Li D, Girijavallabhan VM, Popovici-Muller J, Yu W, Zhao L. Preparation of tartaric acid functional compounds for the treatment of inflammatory disorders. PCT Int Appl. 2005 WO 2005121130 A2 20051222. [Google Scholar]
- 32.Okada I, Takizawa E, Kikutake K, Fukuchi T. Preparation of pyrazolecarboxamide derivatives as pest control agents, fungicides, and acaricidesagea. PCT Int Appl. 2002 WO 2002083647 A1 20021024. [Google Scholar]
- 33.Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsing response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated Ca2+ channels. J Biol Chem. 2009;284:31375–31390. doi: 10.1074/jbc.M109.009951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brittain JM, Wang Y, Eruvwetere O, Khanna R. Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Lett. 2012;586:3813–3818. doi: 10.1016/j.febslet.2012.09.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Receptor Binding Profiles for Compounds 2-11.a
aData represent mean % inhibition (N = 4 determinations) for compound tested at receptor subtypes. Significant inhibition is considered > 50%. In cases where negative inhibition (-) is seen, this represents a stimulation of binding. Occasionally, compounds at high concentrations will non-specifically increase binding. The default concentration for primary binding experiments is 10 μM. b Compound number. ND; not determined.