

Abstract
The structure of human Methylenetetrahydrofolate Reductase (MTHFR) is not known either by NMR or by X-ray methods. Phosphorylation seems to play an important role in the functioning of this flavoprotein. MTHFR catalyzes an irreversible reaction in homocysteine metabolism. Phosphorylation decreases the activity of MTHFR by enhancing the sensitivity of the enzyme to SAdenosylmethione. Two common polymorphisms in MTHFR, Ala222Val and Glu429Ala, can result in a number of vascular diseases. Effects of the Glu429Ala polymorphism on the structure of human MTHFR remain undetermined due to limited structural information. Hence, structural models of the MTHFR mutants were constructed using I-TASSER and assessed by PROCHECK, DFIRE and Verify3D tools. A mechanism is further suggested for the decreased activity of the Ala222Val and Glu429Ala mutants due to a decrease in number of serine phosphorylation sites using information gleaned from the molecular models. This provides insights for the understanding of structure-function relationship for MTHFR.
Keywords: Methylenetetrahydrofolate Reductase; in silico modeling; hyperhomocysteinemia; 5,10-Methylenetetrahydrofolate; Methylenetetrahydrofolate Reductase phosphorylation
Background
Methylenetetrahydrofolate reductase (MTHFR) plays an important role in processing of amino acids. It converts 5, 10 Methylenetetrahydrofolate to 5-Methyltetrahydrofolate which provides the methyl group for methylation of homocysteine to methionine by the enzyme Methionine Synthase [1]. High plasma homocysteine levels (hyperhomocysteinemia) are associated with increased risk for cardiovascular disease and neural tube defects in humans. Treatment with Folic Acid decreases homocysteine levels and dramatically reduces the incidence of neural tube defects. The flavoprotein, MTHFR is a likely target for the action of Folic Acid. MTHFR is a homodimer. Each domain can be separated by partial digestion of the native enzyme with trypsin. It contains a bound FAD prosthetic group on each domain and uses NADPH as reducing agent. An S-adenosylmethione-binding site is present in the regulatory domain. The binding of S-adenosylmethione allosterically inhibits the activity of the enzyme.
MTHFR gene codes for a protein of 656 amino acids with a predicted molecular mass of 74.5 kDa [2]. MTHFR uses NADPH as a reducing agent and FAD as a cofactor in mammalian cells [3]. Eukaryotic MTHFR comprises a catalytic domain of about 40 kDa at the N-terminus and a C-terminus regulatory domain of about 37 kDa. The C-terminus of human MTHFR contains the S-Adenosylmethionine-binding site which regulates the Methionine level in cells [4]. This C-terminus domain is not present in MTHFR from E. coli. Hence, NADH-dependent MTHFR from E. coli is insensitive to S-Adenosylmethionine [5].
Both human and other eukaryotic MTHFR exist as homodimers, whereas MTHFR of E. coli is a homotetramer [6–7]. The homodimers can be separated by partial digestion of the native enzyme with trypsin [6]. Each homodimer contains two noncovalently bound FAD molecules per dimer. SAdenosylmethionine plays an important role in keeping the enzyme in dimeric form [8]. MTHFR can also be phosphorylated which decreases its activity by ~20% and allows it to be more easily inhibited by the S-Adenosylmethionine [9]. As mentioned before, MTHFR contains a bound flavin cofactor and uses NADPH. A→V mutation in human (A222V) and in E. coli (A177V) increases the rate of dissociation of FAD leading to the structural changes in the enzyme [10]. Structural changes in the MTHFR enzyme lead to many diseases such as Homocystinuria [11], Coronary artery disease [12], Neural tube defects [13], thyroid dysfunction [14] and colorectal cancer [15], etc. Protein structure prediction is one of the most important areas in the fields of structural bioinformatics, computational biology and drug design. The current study was carried out to predict the structure of human MTHFR for a better understanding of the mechanism of the enzyme. The structure of human MTHFR was predicted using different online servers and then evaluated for further analysis.
Methodology
Sequence of MTHFR and Template Selection:
The sequence of MTHFR (P42898) was retrieved from Expasy server [16] in FASTA format. PSI BLAST [17] was used to retrieve homologous sequences of existing protein structures in the Protein Data Bank. Fifteen sequences of different species showing some homology were retrieved using PSI-BLAST. Phylogenetic analysis was carried out to obtain information about evolutionary relationship between the different species and as well as to find out if structural homologs were available in PDB for template-based modeling using the ClustalW server [18].
Homology modeling:
For tertiary structure prediction of human MTHFR, different methodologies were adopted. Homology modeling was performed using different online servers for 3D structure prediction such as CPH Models [19], EsyPred3D [20], HMMstr/Rosetta [21–22], Swiss Model [23] and Protein Structure Prediction Server (PS) [24]. Then I-TASSER [25–27] server was used for threading purposes. In this method the target sequences are first threaded using a representative PDB structure library to search for the possible folds by Profile- Profile Alignment (PPA), Hidden Markov Model, PSI-BLAST profiles, Needleman-Wunch and Smith-Waterman alignment algorithms. I-TASSER server predicted the human MTHFR protein models from which five best models were selected after refinement.
Model Validation:
For evaluation purposes, we used DFIRE [28], Verify3D [29], and Procheck [30–31] programs. DFIRE is a statistical, potentialbased program that uses a distance-scaled finite ideal-gas reference state. DFIRE is used to assess non-bonding interactions in the protein model. A lower energy indicates that a model is closer to the native conformation. The Verify3D method analyzes the compatibility of an atomic model of the protein with its amino acid sequence. The scores range from -1 to +1 (-1 being unacceptable and +1 being an acceptable score). The PROCHECK suite of program was used to assess the stereochemical quality of a given protein structure. The results were also confirmed using the link: http://nihserver.mbi.ucla.edu /SAVES/.
Model Analysis:
For the predicting the number of domains, DOMAC server [32–35] was used. The human MTHFR was predicted to have two domains. To predict phosphorylation sites, Netphos 2.0 [36] sever was used. Also identified in the structure are Tryptic cleavage site, FAD binding site at the N-terminus, NADPH and S-Adenosylmethionine-binding sites. Amino acid sequences of mutant proteins were obtained by introducing mutations in the sequence of the wild type manually. The three mutant sequences were constructed: (a) by replacing Alanine with Valine at position 222 in the protein sequence; (b) by replacing Glutamate with Alanine at the position 429 in the protein sequence; and (c) by replacing both Alanine at position 222 and Glutamate at position 429 with Valine and Alanine, respectively. After introducing the mutations in the sequences, the tertiary structures were predicted by I-TASSER. The protein models were visualized by the use of VMD software [37].
Results
MTHFR protein sequence was used for multiple sequence alignment and retrieval of homologous sequences of experimentally determined 3-D structures present in the Protein Data Bank. Table 1 (see supplementary material) shows the percentage identity of the similar sequences and their PDB (Protein Data Bank) ID codes of known structures. It was noticed that experimentally determined structures were available for proteins sequences from E. coli and Thermus thermophilus only in the Protein Data Bank. Figure 1 shows the Phylogenetic relationship between the selected species. It was found out that the acid acid sequences of both E. coli and Thermus thermophilus MTHFR are very different from the human MTHFR sequence. The complete models were not predicted by homology modeling-based protein structure prediction servers because of the unavailability of the required templates in the Protein Data Bank (PDB). These servers were able to predict the structure of MTHFR from residue 1 to 340, but the rest of the structure was not predicted. Five models of the protein structure were generated by the I-TASSER server after refinement using the threading technique and were visualized by VMD software (Figure 2).
Figure 1.
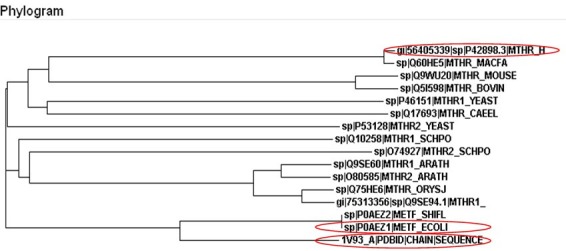
Phylogenetic relationship of Human MTHFR protein sequence (P42898) with MTHFR protein sequence of 15 selected species. The phylogram shows that Human MTHFR posseses greater similarity with Macaca fascicularis (SwissProt ID: Q60HE5). Only two structures of MTHFR from the selected species are experimentally determined having their structures available in the Protein Data Bank. These two species are E. coli (PDB code: 1b5t) and Thermus thermophilus (PDB code: 1v93).
Figure 2.
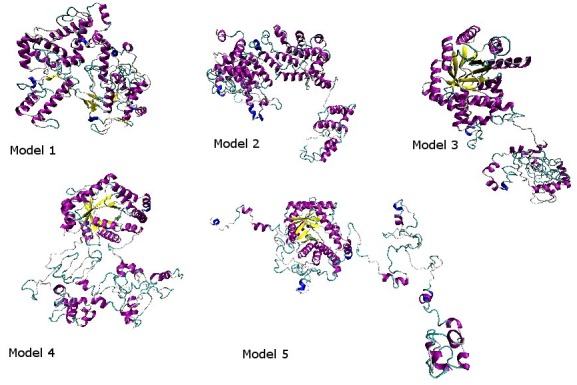
The models 1-5 are generated from the I-TASSER server and visualized by the VMD.
The five complete models obtained from I-TASSER server were evaluated for the one best model. Table 2 (see supplementary material) shows the structural evaluation results using DFIRE, Verify3D and Procheck. The DFIRE program uses energy value to evaluate the structures. According to these results the lowest energy is of model number 1. The Verify3D program assigned the highest positive score to model number 4. To analyze the stereochemical quality of the predicted structure Procheck software was used. According to Procheck results, model number 1 is the most stable because it has less area in the disallowed region (2.3% as compared to the other models). Model number 1 was finally used for further analysis based on energy, stereochemical results and I-TASSER structure prediction server ranking.
Two domains were predicted by the DOMAC server. The domain prediction was made by the template-based methods using the protein templates available in Protein Data Bank (figure 3A. B) shows the protein as a homodimer. The first domain stretches from amino acid 1 to 342 while the second domain is from amino acid 343 to 656. The phosphorylation sites predicted by the Motif Scan servers are shown in the figure 4 (A-B). Figure 5 (A, B) shows the FAD-binding site of the catalytic domain, S-Adenosylmethione-binding site, Trypsin cleavage site (KRREED) and a conserved amino acid motif (VRPIFW) in the regulatory domain. Two possible NADPHbinding sites are shown in (Figure 6).
Figure 3.
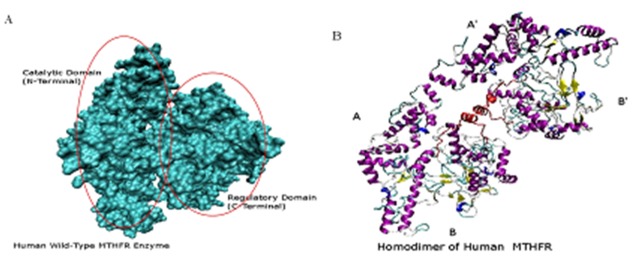
(A) Two domains (Surf view) of wild-type Human MTHFR structure. (B) Homodimer view of Human MTHFR.
Figure 4.
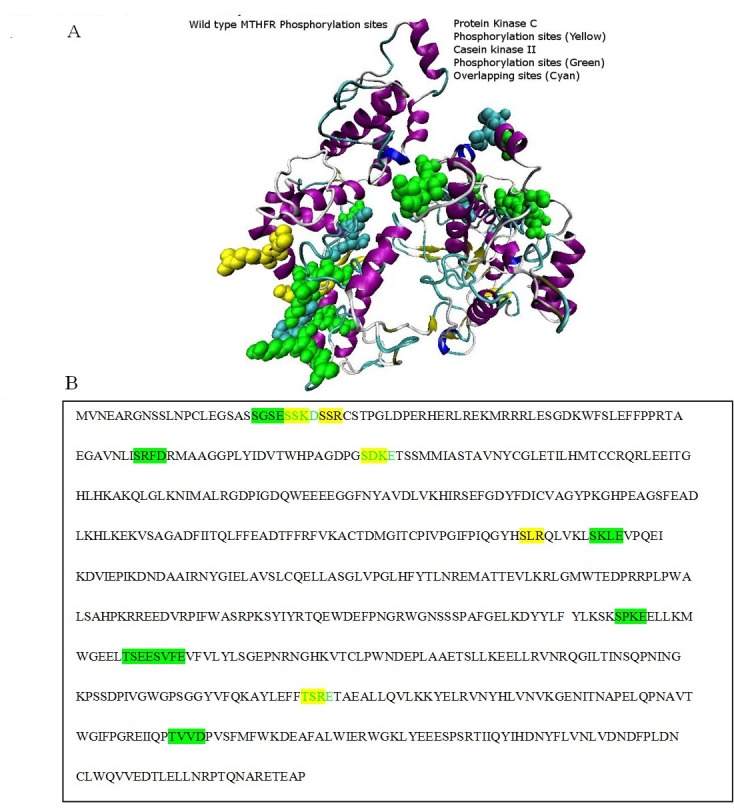
Phosphorylation binding sites: A) Protein Kinase C phosphorylation sites are colored in yellow, Casein Kinase II phosphorylation sites are colored in green, while the overlapping phosphorylation sites of the both enzymes are colored in cyan as show in the figure A; B) Protein Kinase C phosphorylation sequence is colored in yellow, Casein Kinase II phosphorylation sequence is colored in green, while the green colored sequence highlighted with yellow background is the overlapping sequence of both the enzymes.
Figure 5.
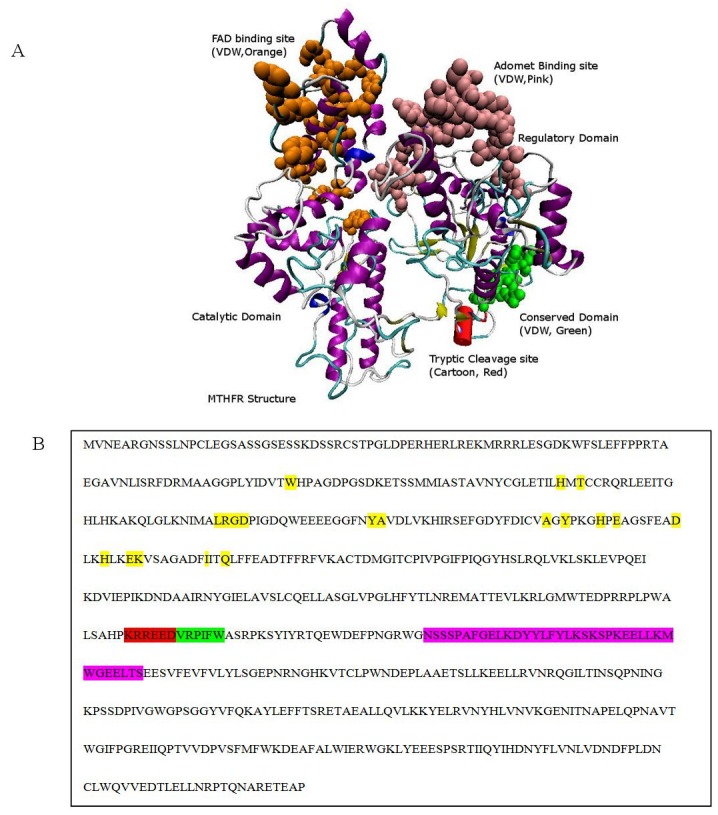
Validation by Experimental Evidences: A) FAD binding site at the N-terminus, S-Adenosylmethione-binding site in the regulatory domain, tryptic cleavage, and a conserved domain is shown in the wild type MTHFR; B) MTHFR sequence showing the FAD binding site in yellow color, Adomet binding site in the pink color, tryptic cleavage site in the red color and the conserved domain in the green color.
Figure 6.
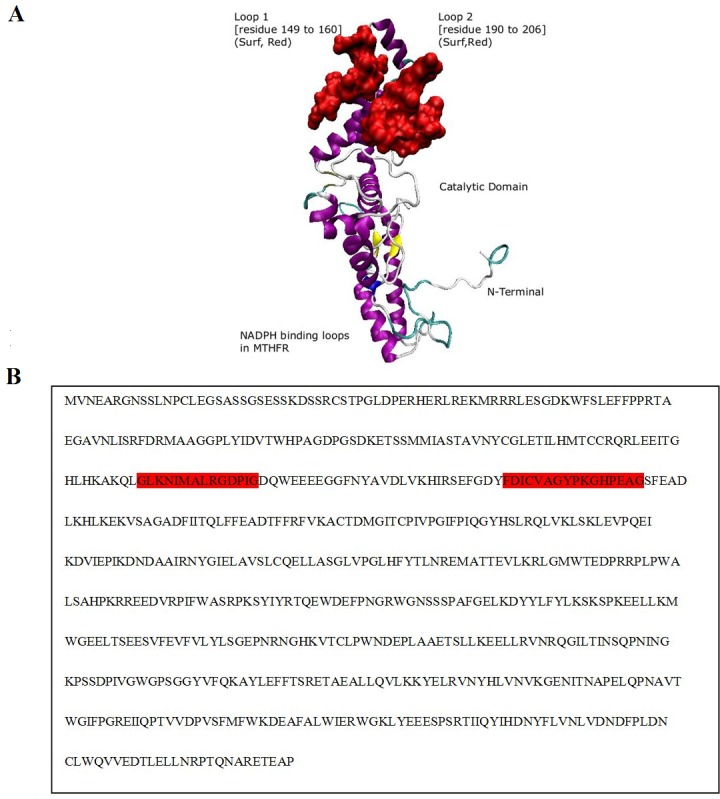
NADPH-binding sites: A) Structural view of supposed NADPH binding sites Loop 1 and Loop 2 are colored in red to indicate the NADPH-binding sites; B) The MTHFR sequence from 149 to 160 residues indicates the loop 1 region while sequence from 190 to 206 residues (colored in gray) indicates the loop 2 region.
After selecting the best 3-D model of the wild type MTHFR, its amino acid sequence was mutated and then the structures of the three mutated structures were predicted using the I-TASSER server. After this step, different core sites were highlighted which can be affected due to mutations (Figure 7). A total of 22 Serine, 6 Threonine and 3 Tyrosine phosphorylation sites were predicted by NetPhos 2.0 server for the wild-type Human MTHFR. There were differences found in Serine phosphorylation sites, which were 21 in both mutated sequences having a single amino acid mutation and 20 sites were predicted in MTHFR sequence having both the mutations. Figure 8 (A-D) shows the phosphorylation sites in normal and mutated structures predicted by NetPhos 2.0 server. The FADbinding sites in both wild type and mutant are shown in the (Figure 9). S-Adenosylmethione-binding site mutations are also visible in the structure (Figure 10). The complete view of the model is shown in (Figure 11).
Figure 7.

A) Structural alignment of Normal (Blue) and Mutated (Purple) amino acids of MTHFR. Residue number 222 of both types is shown (CPK view). Normal amino acid Alanine is shown in the lime color while mutated amino acid Valine is shown in the red color; B) Structural alignment of the normal and mutated amino acids of MTHFR. Glutamic Acid is shown in the lime color which is replaced by Alanine in the mutated structure; C) Close look of A222V mutated structure. Blue colored helix shows the normal structure while purple colored helix represents the mutated structure; D) Close look of E429A mutated structure. Blue colored helix shows the normal structure while purple colored helix represents the mutated structure.
Figure 8.

Comparison of Phosphorylation sites of normal and mutated sites: A) Wild-type Human MTHFR model with active Serine Phosphorylation residue at its position 219; B) Wild type MTHFR with active Serine residue at its position 427 and 430; C)
Figure 9.
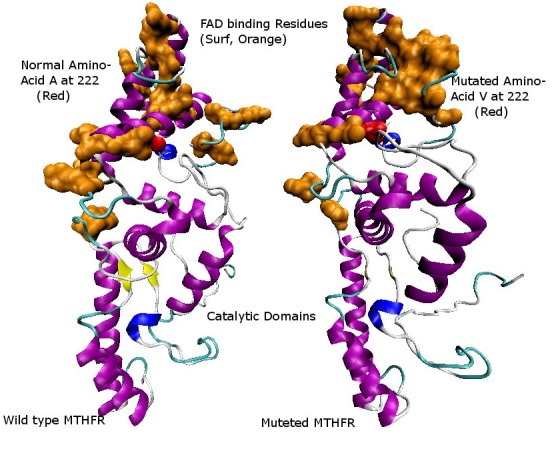
Comparison of FAD-binding sites in normal and mutated structures. FAD-binding region is highlighted in orange color present in the catalytic region. The structural perturbance can be seen very clearly in the figure due to the effect of mutations.
Figure 10.
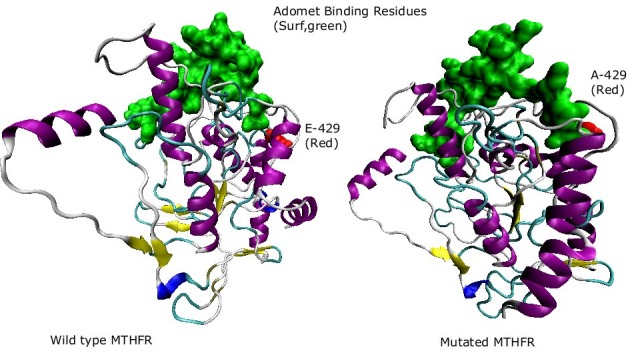
S-Adenosylmethione-binding sites of both mutatnats are shown in the figure. The difference between the two sites can be visualized clearly. S-Adenosylmethione-binding site in the mutated structure is distorted as compared to the normal structure.
Figure 11.
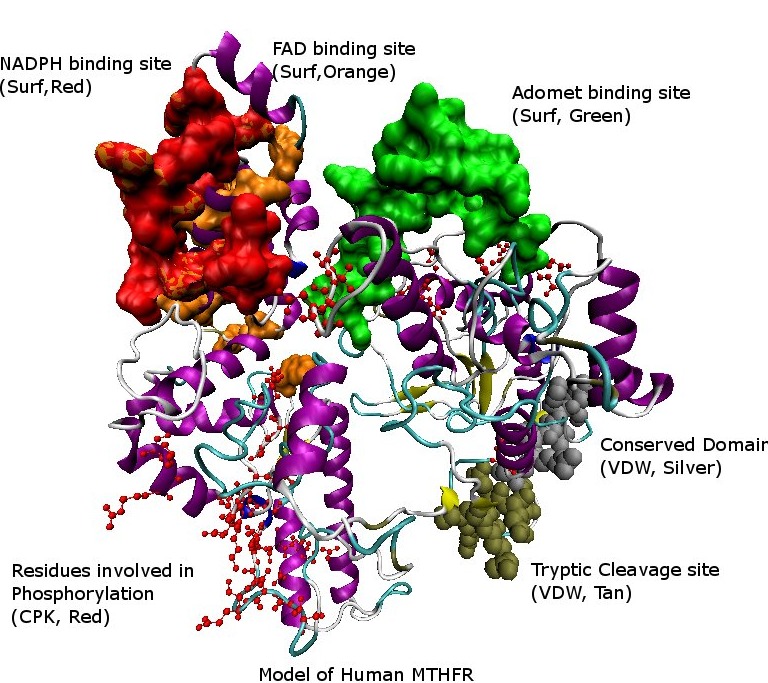
A model of the Human MTHFR structure showing different sites by color-coding in the structure.
Discussion
in silico methods were used to determine the structural difference between the wild type and mutant human MTHFR enzyme. The predicted structures show β8α8 barrel catalytic domains that bind FAD. In order to understand the underlining factors responsible for the malfunction of mutants of MTHFR, structural studies were carried out by using different bioinformatics tools and software. The sequence alignment and phylogenetic analysis was carried out using PSI-BLAST and ClustalW software. Only two experimentally determined structures were present in the Protein Data Bank (PDB), namely from E. coli (PDB code: 1b5t) [38] and from Thermus thermophilus (PDB code:1v93) [39, 40]. Using these two known structures, homology modeling for tertiary structure prediction was carried out. Only the structure of first domain of Human MTHFR was predicted from homology modeling results based on the available templates of E. coli and Thermus thermophilus in the Protein Data Bank. To predict the complete structure of Human MTHFR, the threading technique was applied. I-TASSER server predicted the complete structural results by threading. The models predicted by this server were used for further analysis.
The X-ray analysis of E. coli MTHFR provides a model for the catalytic domain that is shared by all MTHFRs. This domain is a β8α8 barrel that binds FAD in a novel fashion [38]. Ala177, which corresponds to Ala222 in human MTHFR, is located near the bottom of the barrel and is distant from FAD. The mutation A177V does not affect Km or kcat but instead increases the propensity for bacterial MTHFR to lose its essential FAD cofactor. Folate derivatives protect the wild-type and mutant E. coli enzymes against the loss of FAD and also against thermal inactivation.
According to the known structure of bacterial MTHFR enzyme, FAD-binding residues are located near the N-terminus of MTHFR [38]. It was observed that these FAD-binding residues have a compact structure for binding of FAD cofactor in the wild-type human MTHFR. When these residues were colorcoded in the predicted structure of Ala222Val mutant, it was observed that the FAD-binding groove appeared broadened. This result indicates that FAD binding in the catalytic domain was disturbed which resulted in the distortion of the original structure of wild-type human MTHFR. Trypsin cleavage of native enzyme was also shown in the wild-type human MTHFR structure, which also confirmed the two domains in the structure.
According to Yamada et al [9], phosphorylation is crucial for the regulation of human MTHFR. It acts as an on and off switch for the expression of MTHFR. NetPhos 2.0 server was used for the prediction of phosphorylation sites and it predicted a total of 22 Serine, 6 Threonine and 3 Tyrosine sites. It was analyzed that there was no change in the phosphorylation sites of Threonine and Tyrosine residues and only the Serine phosphorylation sites were perturbed. In mutated Ala222Val and Glu429Ala mutants, a total of 21 Serine active sites were predicted which were one less than the total sites predicted in the wild type MTHFR enzyme. It was found that double mutants, containing both Ala222Val and Glu429Ala mutations, a lower number of serine phosphorylation sites are present as compared to the singly mutated structures mentioned before. Cysteine phosphorylation sites were also affected by mutations. These sites were absent in the mutated region.
Conclusion
It has been shown that the mutated amino acids of human MTHFR affect the phosphorylation sites in the protein structure by affecting the FAD-binding sites. Phosphorylation seems to play a significant role in the functioning of the wild-type enzyme. However, the structural basis for this observation is not clear. Hence, molecular models of the MTHFR mutants were developed to elucidate the thermodynamically unfavorable observations in comparison to the wild type. This provides insights to the understanding of structure-function relationship of MTHFR mutants and its wild type for developing treatments in hyperhomocysteinemia, homocystinuria, thyroid dysfunction and colorectal cancer.
Supplementary material
Acknowledgments
We would like to acknowledge Dr. S.M. Junaid Zaidi (Rector CIIT), Dr. Raheel Qamar (Dean, Faculty of Research and Development), Dr. Fauzia Yusuf Hafeez (Chairperson, Department of Biosciences), Dr. Mahmood A. Kayani (Head, Department of Biosciences), Dr. Farah Mustafa, and Dr. Shahid Nadeem Chohan for providing best research facilities at COMSATS Institute of Information Technology (CIIT), Islamabad, Pakistan and their efforts for pursuing research in the field of Bioinformatics.
Footnotes
Citation:Shahzad et al, Bioinformation 9(18): 929-936 (2013)
References
- 1.Zhang Z, et al. Eur J Med Chem. 2012;58:228. doi: 10.1016/j.ejmech.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 2.Goyette P, et al. Nat Genet. 1994;7:195. doi: 10.1038/ng0694-195. [DOI] [PubMed] [Google Scholar]
- 3.Kutzbach C, Stokstad EL. Biochim Biophys Acta. 1971;250:459. doi: 10.1016/0005-2744(71)90247-6. [DOI] [PubMed] [Google Scholar]
- 4.Sumner J, et al. J Biol Chem. 1986;261:7697. [PubMed] [Google Scholar]
- 5.Roje S, et al. J Biol Chem. 2002;277:4056. doi: 10.1074/jbc.M110651200. [DOI] [PubMed] [Google Scholar]
- 6.Matthews RG, et al. J Biol Chem. 1984;259:11647. [PubMed] [Google Scholar]
- 7.Sheppard CA, et al. J Bacteriol. 1999;181:718. doi: 10.1128/jb.181.3.718-725.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jencks DA, Matthews RG. J Biol Chem. 1987;262:2485. [PubMed] [Google Scholar]
- 9.Yamada K, et al. Proc Natl Acad Sci USA. 2005;102:10454. doi: 10.1073/pnas.0504786102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamada K, et al. Proc Natl Acad Sci USA. 2001;98:14853. doi: 10.1073/pnas.261469998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacques PF, et al. Circulation. 1996;93:7. [Google Scholar]
- 12.Friso S, et al. Clin Exp Med. 2002;2:7. doi: 10.1007/s102380200001. [DOI] [PubMed] [Google Scholar]
- 13.Peng F, et al. Int J Mol Med. 2001;8:509. doi: 10.3892/ijmm.8.5.509. [DOI] [PubMed] [Google Scholar]
- 14.Hustad S, et al. Am J Clin Nutr. 2004;80:1050. doi: 10.1093/ajcn/80.4.1050. [DOI] [PubMed] [Google Scholar]
- 15.Le Marchand L, et al. Cancer Epidemiol Biomarkers Prev. 2005;14:1198. doi: 10.1158/1055-9965.EPI-04-0840. [DOI] [PubMed] [Google Scholar]
- 16.Gasteiger E, et al. Nucleic Acids Res. 2003;31:3784. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altschul SF, et al. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson JD, et al. Nucleic Acids Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nielsen M, et al. In Press - NAR web issue. 2010 [Google Scholar]
- 20.Lambert C, et al. Bioinformatics. 2002;18:1250. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 21.Bystroff C, Shao Y. Bioinformatics. 2002;18:S54. doi: 10.1093/bioinformatics/18.suppl_1.s54. [DOI] [PubMed] [Google Scholar]
- 22.Bystroff C, et al. J Mol Biol. 2002;301:173. doi: 10.1006/jmbi.2000.3837. [DOI] [PubMed] [Google Scholar]
- 23.Arnold K, et al. Bioinformatics. 2006;22:195. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 24.Chen CC, et al. BMC Bioinformatic. 2009;10:366. doi: 10.1186/1471-2105-10-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y. Proteins. 2007;69:108. [Google Scholar]
- 26.Wu S, Zhang Y. Nucleic Acids Res. 2007;35:3375. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou H, Zhou Y. Protein Sci. 2002;11:2714. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luthy R, et al. Nature. 1992;356:83. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 29.Vuister GW, et al. J Biomol NMR. 2013 [Google Scholar]
- 30.Morris AL, et al. Proteins. 1992;12:345. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 31.Cheng J. Nucleic Acids Res. 2007;35:W354. doi: 10.1093/nar/gkm390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tress M, et al. Proteins. 2007;69:137. doi: 10.1002/prot.21675. [DOI] [PubMed] [Google Scholar]
- 33.Larson J, et al. J Virol. 2007;81:6957. doi: 10.1128/JVI.02207-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadowski MI. Proteins. 2013;81:253. doi: 10.1002/prot.24181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blom N, et al. J Mol Biol. 1999;294:1351. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 36.Pagni M, et al. Nucleic Acids Res. 2007;35:W433. doi: 10.1093/nar/gkm352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humphrey W, et al. J Mol Graph. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 38.Guenther BD, et al. Nat Struct Biol. 1999;6:359. doi: 10.1038/7594. [DOI] [PubMed] [Google Scholar]
- 39. http://www.pdb.org/pdb/explore/explore.do?structureId=1V93.
- 40.Pejchal R, et al. Biochemistry. 2006;45:4808. doi: 10.1021/bi052294c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.