

Background: The role of argininosuccinate lyase (ASL) transcripts in disease variability is unclear.
Results: The most common ASL transcript variants decrease the functional enzymatic activity after co-expression with wild type or mutant ASL.
Conclusion: ASL transcripts expressed at high levels can contribute to the variable phenotype in ASL-deficient patients.
Significance: A new explanation of the molecular basis adds to our understanding of the clinical variability in patients.
Keywords: Computer Modeling, Metabolic Diseases, Recombinant Protein Expression, Transfection, Urea Cycle, Argininosuccinate Lyase (ASL), Argininosuccinic Aciduria (ASA), Homomeric/Heterotetrameric Protein, Transcript Variants
Abstract
Argininosuccinic aciduria (ASA) is an autosomal recessive urea cycle disorder caused by deficiency of argininosuccinate lyase (ASL) with a wide clinical spectrum from asymptomatic to severe hyperammonemic neonatal onset life-threatening courses. We investigated the role of ASL transcript variants in the clinical and biochemical variability of ASA. Recombinant proteins for ASL wild type, mutant p.E189G, and the frequently occurring transcript variants with exon 2 or 7 deletions were (co-)expressed in human embryonic kidney 293T cells. We found that exon 2-deleted ASL forms a stable truncated protein with no relevant activity but a dose-dependent dominant negative effect on enzymatic activity after co-expression with wild type or mutant ASL, whereas exon 7-deleted ASL is unstable but seems to have, nevertheless, a dominant negative effect on mutant ASL. These findings were supported by structural modeling predictions for ASL heterotetramer/homotetramer formation. Illustrating the physiological relevance, the predominant occurrence of exon 7-deleted ASL was found in two patients who were both heterozygous for the ASL mutant p.E189G. Our results suggest that ASL transcripts can contribute to the highly variable phenotype in ASA patients if expressed at high levels. Especially, the exon 2-deleted ASL variant may form a heterotetramer with wild type or mutant ASL, causing markedly reduced ASL activity.
Introduction
Argininosuccinate lyase (ASL2; EC 4.3.2.1; OMIM *608310) catalyzes the reversible hydrolytic cleavage of argininosuccinate into arginine and fumarate and contributes to the removal of waste nitrogen and biosynthesis of arginine within the urea cycle in ureotelic species (1). ASL is also involved in the arginine-citrulline cycle as part of a multiprotein complex required for production of nitric oxide (2) as well as in other pathways (Fig. 1).
FIGURE 1.
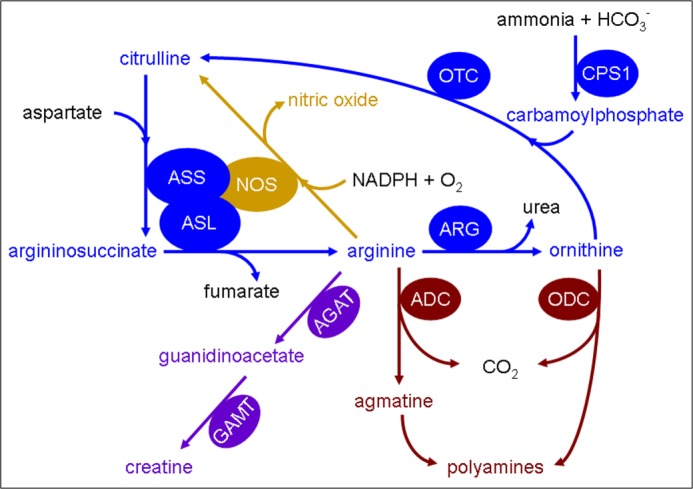
Involvement of argininosuccinate lyase in various metabolic and biochemical pathways. Shown is a schematic illustration of the involvement of ASL in various metabolic and biochemical reactions. Colors are used to show the affiliation of metabolites and enzymes to different pathways: urea cycle (blue), nitric oxide synthesis (gold), polyamine synthesis (maroon), and creatine synthesis (purple). Enzymes are depicted in ovals: arginine decarboxylase (ADC), arginine:glycine amidinotransferase (AGAT), arginase (ARG); argininosuccinate lyase (ASL), argininosuccinate synthetase (ASS), carbamoylphosphate synthetase 1 (CPS1), guanidinoacetate methyltransferase (GAMT), nitric-oxide synthase (NOS), ornithine decarboxylase (ODC), and ornithine transcarbamylase (OTC).
The human ASL gene is located on chromosome 7q11.21 (3, 4) and comprises 16 exons encoding 464 amino acids (5, 6). The resulting monomers have a predicted molecular mass of ∼52 kDa and form a homotetrameric functional enzyme with four active sites (7). ASL has significant homology to δ-crystallin with an amino acid sequence identity of 64–71% between human ASL and various δ-crystallins (8, 9). The δ-crystallins are major structural components of avian and reptilian eye lenses and show significant ASL enzyme activity in duck and chicken (9, 10). Human ASL is expressed predominantly in liver (11) but is also detected in many other tissues, including kidney (12), small intestine (13, 14), pancreas and muscle (15), heart (16), brain (17, 18), skin fibroblasts (19), and erythrocytes (20).
Mutations in the ASL gene result in an autosomal recessive disorder known as argininosuccinic aciduria (ASA; synonymous ASL deficiency, ASLD; OMIM number 207900) (21), which is the second most common disorder in the urea cycle, with an estimated incidence of ∼1 per 70,000 live births (22). The clinical and biochemical phenotype of ASA is highly variable ranging from asymptomatic cases with only a biochemical phenotype (23–25), some of them diagnosed through newborn screening, to severe neonatal-onset hyperammonemic encephalopathy (26, 27). The molecular basis for the diversity of ASA is not fully understood, and several explanations have been suggested, including tissue-specific ASL expression (27, 28), genetic heterogeneity at the ASL locus (29), intragenic complementation (7, 30–32), different levels of residual ASL activity (33, 34), the developmental control of the ASL gene by DNA methylation (35), and alternative splicing events at the ASL locus leading to frequent exon deletions (5, 36, 37).
In this study, we explored the role of naturally occurring ASL transcript variants in the formation and function of the ASL homotetramer to better understand the phenotypic variability of ASA. By combining computational structural analysis using molecular dynamic (MD) simulations and eukaryotic (co-)expression of wild type (WT) with the most common transcript variants formed by deletions of exon 2 or 7, we could show that exon 2-deleted (ex2del) or exon 7-deleted (ex7del) ASL has a dominant negative effect on the ASL activity after co-expression with wild type or mutant ASL, respectively. Suggesting a physiological role of transcript variants, RNA analysis revealed a predominant expression of ex7del ASL in two ASA patients identified with heterozygosity for the ASL mutant p.E189G. Taken together, these findings suggest that the frequent occurrence of ASL transcript variants, when they are expressed at high levels, can be a factor contributing to the highly variable clinical and biochemical phenotype of ASA. In particular, the effect may be even more striking in a stable mutant, such as the ex2del ASL variant, because it may form a heterotetramer with ASL wild type or naturally occurring missense mutations (i.e. sequence alterations with a disease-causing role found in ASL-deficient patients), contributing to reduced ASL activity.
EXPERIMENTAL PROCEDURES
ASL Transcript Expression in Different Tissues
A panel of cDNAs from 17 different human tissues comprising ASL patient fibroblasts and 16 other tissues (Multiple Tissue cDNA Panel Human I and II, Clontech, Mountain View CA) was used for the amplification of full-length ASL as well as shorter RNA fragments. In addition, short ASL cDNA fragments derived from skin fibroblasts of 24 ASA patients with 10 different genotypes were amplified. Specific oligonucleotides ASL-FL-F (5′-acgaggaaccgcccaacat-3′) (forward) and ASL-FL-R (5′-tgcctctccagtccctgactgt-3′) (reverse) were used for amplification of full-length ASL; primers ASL-SF-F (5′-acactatccgtgcggccagg-3′) (forward, derived from ASL 5′-UTR sequence) and ASL-SF-R (5′-tgagatgggtcatgcacagcg-3′) (reverse, derived from ASL exon 10 sequence) were used for amplification of a short fragment containing exons 1–9 with a fragment size of 864 bp. Hot start PCR was performed at an annealing temperature of 60 °C (62 °C for primer ASL-SF-F with ASL-SF-R) for 38 cycles (42 cycles for primer ASL-SF-F with ASL-SF-R) using HOT FIREPol® DNA polymerase (Solis Biodyne, Tartu, Estonia) or HotStarTaq DNA Polymerase (Qiagen GmbH, Hilden, Germany) prior to gel electrophoresis. PCR products representing probable splice variants were confirmed by sequencing using the BigDye Terminator cycle sequencing kit version 1.1 (ABI sequence, Applied Biosystems). Patients or their legal guardians had consented to the use of the cultured skin fibroblasts for research purposes when the skin biopsy was taken, and cDNA samples were anonymized prior to their use in this study.
Patient Characteristics and RNA Studies
The 19-year-old male patient, offspring of non-consanguineous parents, was first identified at the age of 13 years with mild hyperammonemia (250 μmol/liter, normal <50), confusion, and irritability during an episode of gastroenteritis. The diagnosis of ASA was made based on characteristic urine metabolites and underlined by typical signs and symptoms such as mild hepatomegaly but normal liver function, trichorrhexis nodosa, cognitive impairment (IQ 67), and a natural avoidance of protein-rich food since early childhood. In addition, there was persistent mild elevation of plasma citrulline (between 80 and 150 μmol/liter, normal <60). Under treatment with l-arginine (150 mg/kg/day) and mild protein restriction, he had been stable since age 13 apart from a single mild metabolic decompensation at age 17 years, again during gastroenteritis with maximum ammonia (230 μmol/liter).
To confirm the diagnosis, DNA sequencing of the ASL gene by standard methods (6) as well as array comparative genomic hybridization (38) for exclusion of a deletion on the second allele revealed only a single heterozygous mutation in exon 7 (c.566A→G, p.E189G), which was not present on the maternal allele (also, no other mutation was found in the mother in DNA or RNA), whereas the mutation was found in a heterozygous state in paternal DNA. To identify the second mutant allele, RT-PCR was performed using RNA (PrimeScript II 1st Strand cDNA Synthesis kit, TaKaRa Bio) derived from a 3-day full blood culture treated with phytohemagglutinin and cycloheximide (39). For control RT-PCR, cDNAs derived from lymphocytes, liver, and fibroblasts were used. The patient's cDNA was amplified in full-length ASL as well as in a short fragment comprising exons 1–9, as described above. In addition, primers ASL-SF-F and ASL-SF-R7 (5′-ctccccaggggcaggacattg-3′) (reverse, derived from ASL exon 7 sequence) were used for amplification of a short fragment containing exons 1–7 (fragment size of 670 bp) to confirm mutation c.566A→G in this transcript. PCR products were sequenced as described above.
In another patient (aged 12 years) with late onset ASA and a similar clinical and biochemical situation, the mutation p.E189G was found in a heterozygous state, and the same DNA and RNA investigations were performed as above. To estimate expression levels of transcripts, densitometry analysis of RT-PCR products on gel electrophoresis and of bands detected by Western blotting was performed by using Carestream Molecular Imaging software (Carestream Health). The genetic studies were done after written informed consent of the patient and his legal guardians was obtained.
Generation of Wild Type, Mutant ASL p.E189G, and Exon 2- or 7-deleted Splice Variant Constructs
Full-length ASL cDNA (1395 bp) and ex7del ASL transcript variant (1317 bp) were cut from pCR2.1 carrying the WT ASL (33) and pCMV6-XL4 carrying the ex7del ASL cDNA (OriGene, Rockville, MD) using restriction enzymes BamHI and NotI (both from New England Biolabs (Beverly, MA)), respectively. Restriction products were cloned into the expression vector pcDNA3 (Invitrogen), yielding pcDNA3-ASL-WT (P-WT) and pcDNA3-ASL-ex7del (P-ex7del), respectively. Oligonucleotides ASL-ex2del-F (5′-atccggatccatggcctcggaggtggctgaggagtgggcc-3′) (forward, consisting of a BamHI site, the 12 nucleotides of ASL exon 1, and 18 nucleotides from 5′ exon 3) and ASL-ex2del-R (5′-cctctagatgcatgctcgagcggccgctatatctaggc-3′) (reverse, with a NotI site added to an ASL 3′-UTR sequence) were used to amplify the exon 2-deleted ASL cDNA from P-WT. The obtained PCR product was gel-purified and cloned into the above expression vector, yielding pcDNA3-ASL-ex2del (P-ex2del). The mutant p.E189G (c.566A→G) was constructed as pcDNA3-ASL-E189G (P-E189G) based on P-WT by site-directed mutagenesis (Phusion Site-directed Mutagenesis Kit, Finnzymes (Espoo, Finland)) according to the manufacturer's protocol. All established constructs were confirmed by sequencing as above.
Expression and Co-expression of ASL Constructs in Human Embryonic Kidney 293T Cells
Three different mammalian cell lines (COS-1, HeLa, and 293T) were tested for their background ASL activity before establishing the eukaryotic ASL expression system. ASL expression levels after transfection with P-WT were determined by Western blot. Cells were grown in Dulbecco's modified Eagle's medium + GlutaMAX (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic/antimycotic solution (both from PAA (Pasching, Austria)) and maintained in an incubator containing 5% CO2 at 37 °C in a humidified atmosphere.
Based on the lowest ASL background activity, 293T cells were considered as ideal for the ASL expression system in this study. The cells were transiently (48 h) transfected with a total of 7 μg of the construct P-WT, P-ex2del, or P-ex7del in 60-mm dishes using LipofectamineTM LTX and PLUSTM reagents according to the manufacturer's instructions (Invitrogen). A total of 7 or 10.5 μg of plasmids was used for co-transfection with two or three plasmids; if not indicated otherwise, we used the same amount of the respective constructs (3.5 μg each) in co-transfectants. For the co-transfection of P-WT and P-ex2del at different ratios, 1.75 μg of P-WT and 1.75 μg of P-ex2del was used for the ratio 1:1; 1.75 μg of P-WT and 3.5 of μg P-ex2del for 1:2; 1.75 μg of P-WT and 8.75 μg of P-ex2del for 1:5; 3.5 μg of P-WT and 1.75 μg of P-ex2del for 2:1; and 8.75 μg of P-WT and 1.75 μg of P-ex2del for 5:1. The empty vector (EV) pcDNA3 was used either as negative control or to set up the same amounts of total plasmids for co-transfection.
RNA was isolated from the cells transiently transfected with P-WT, P-ex2del, or P-ex7del, respectively, using the QIAamp RNA blood minikit according to the manufacturer's protocol (Qiagen GmbH, Hilden, Germany). The concentration of nucleic acids was determined by a NanoDrop spectrophotometer. RT-PCR was performed as a standard protocol using primers ASL-SF-F3 (5′-atccacacagccaatgagcgc-3′) (forward, derived from ASL exon 3 sequence) and ASL-SF-R for amplification of a short fragment containing exons 4–9 with a fragment size of 535 bp.
Protein Extraction, Western Blot Analysis, and Immunoprecipitation
Cells were harvested and lysed in Lubrol WX lysis buffer containing 0.15% (w/v) of Lubrol WX (Sigma) and 10 mm Tris-HCl (pH 8.6). Human liver tissue (n = 3, shock-frozen needle biopsy samples for diagnostic purposes in non-ASLD patients after informed consent for scientific use was obtained) was homogenized in complete Nonidet P-40 (Roche Applied Science) lysis buffer containing 1% Nonidet P-40, 50 mm Tris-HCl (pH 8), 125 mm NaCl, 1 mm EDTA, and protease inhibitors (1× Complete EDTA-free + 1 mm of PMSF) (Roche Applied Science) in a prechilled glass grinder by quickly grinding on ice. Cell lysates and liver homogenates were then centrifuged at maximum speed at 4 °C for 15 min. Protein concentrations in the supernatants (equivalent to cell extracts) were determined by the method of Lowry (40) using bovine serum albumin (BSA) as a standard.
Western blotting was performed as described previously (41). 30 μg of total protein of cell extracts was separated by 10% denaturing SDS-PAGE or native PAGE (without SDS in the Laemmli loading buffer and electrophoresis buffer), and subsequently transferred to nitrocellulose transfer membranes (Whatman GmbH, Dassel, Germany). The primary polyclonal antibody anti-ASL (GeneTex, Irvine, CA), recognizing ASL residues 13–261 according to the manufacturer, was used at a dilution of 1:1000, and the horseradish peroxidase (HRP)-conjugated secondary antibody anti-rabbit (Santa Cruz Biotechnology, Inc.) was used at a dilution of 1:5000. Antibodies against β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (both from Santa Cruz Biotechnology) served as loading controls. Protein detection was done using ECL reagents (GE Healthcare) for chemiluminescent labeling.
Fibroblasts derived from three ASA patients as well as one control were cultured under the same conditions as for 293T cells. Immunoprecipitation analysis was done as a standard protocol using 5 mg of total protein of fibroblasts, followed by Western blot analysis as described above. HRP-conjugated mouse anti-rabbit IgG (L27A9, Cell Signaling Technology, Inc. (Danvers, MA)), which does not recognize the denatured and reduced rabbit IgG heavy or light chains on Western blot, was used as secondary antibody at a working dilution of 1:2000.
Measurement of ASL Enzymatic Activity
The ASL enzyme activity was determined spectrophotometrically in cell extracts after transient transfection or co-transfection of P-WT, P-ex2del, and/or P-ex7del, using a coupled assay with arginase and measuring urea production as described before (33). Briefly, 100 μl of 34 mm argininosuccinate (argininosuccinic acid disodium salt hydrate) in water and 100 μl of arginase (50 units) (both from Sigma-Aldrich) in 66.7 mm phosphate buffer (11.1 mm potassium dihydrogenphosphate and 55.6 mm disodium hydrogenphosphate, pH 7.5) were incubated at 37 °C for 5 min. Then 40 μl of cell extract (3–14 μg of total protein) and 10 μl of phosphate buffer were incubated with the above reagents at 37 °C for 30 min. The reaction was stopped by adding perchloric acid at a final concentration of 2%. The ASL enzyme activities are given as mIU/mg total protein. The residual ASL activities of splicing variants were determined as a percentage of ASL-WT or ASL-WT with EV activity (percentage) in each (co-)transfection under the same conditions, respectively. All assays were carried out in triplicate for at least three independent co-transfection experiments.
Structure Modeling
Three-dimensional Protein Model and in Silico Mutagenesis of ASL
The tetrameric three-dimensional structural model of ASL (NCBI NP_000039.2, Uniprot P04424) sequence (amino acids 1–464) was built using the ASL structure (PDB entry 1K62) as template. Model building was performed with the programs YASARA (42) and WHATIF (43). Side chains in newly built parts were optimized by MD simulations. The geometry information for the tetramer was extracted from the original crystallographic data. The final model was refined by a 1000-ps (MD) simulation using an AMBER 2003 force field and checked with the programs WHAT_CHECK (44), WHATIF (43), and Verify3D (45, 46) and Ramachandran plot analysis (47, 48). To create the exon 2 and exon 7 deletions, we first made the FASTA sequence files of modified proteins and performed model building using both the monomer and tetramer structures and performed refinements as described for WT protein. Structures were depicted with PyMOL (Schrödinger, LLC, New York). Structural properties of the proteins were calculated by YASARA and WHATIF, and general protein parameters were calculated with ExPASy protein tools (available from the ExPASy Web site).
Molecular Dynamics Simulation for Model Refinement
The MD simulations were performed using an AMBER03 force field (42). The simulation cell was filled with water, pH was fixed to 7.4, and the AMBER03 (49) electrostatic potentials were evaluated for water molecules in the simulation cell and adjusted by the addition of sodium and chloride ions. The final MD simulations were then run with the AMBER03 force field at 298 K, 0.9% NaCl, and pH 7.4 for 1000 ps to refine the models. The best models were selected for analysis and evaluation of the effect of exon deletions on monomer and tetramer structures.
Statistics
Statistical analyses were done using percentages of ASL-WT activities by one-way analysis of variance with the program GraphPad Prism 4 (GraphPad Software, San Diego, CA) to describe the differences of ASL activities between cells co-expressing ASL-WT with EV and cells co-expressing ASL-WT with the transcript variants. Differences were considered as significant if the p value was <0.05.
RESULTS
Expression of Wild Type ASL and of Transcript Variants in Different Tissues
To investigate whether the expression of ASL transcript variants is tissue-dependent and to confirm the reported occurrence of transcripts with deletions of exon 2 or 7 (5, 36, 37), we amplified ASL cDNA either in full-length cDNA (data not shown) or in short cDNA fragments comprising exons 1–9 from 17 different human tissues (only 16 tissues shown in Fig. 2A) and a series of 24 skin fibroblast cell lines from ASA patients representing 10 different genotypes (Fig. 2B). In all tissues, WT ASL cDNA (short fragment with 864 bp) could be detected, and in addition, in most tissues shorter transcript variants (786 and 669 bp) could be detected (Fig. 2, A and B). ASL-WT cDNA was predominantly found in liver (Fig. 2A, lane 5) and kidney (Fig. 2A, lane 7), but expression was detected in all tissues. Furthermore, a similar expression pattern of ASL transcript variants was detected at a low level in all cDNAs investigated for short fragments (Fig. 2, A and B). Sequencing analysis of the shorter fragments identified them as exon 2-deleted (669 bp) or exon 7-deleted (786 bp) transcripts.
FIGURE 2.

Occurrence of transcript variants of the ASL gene in different tissues. PCR products representing short fragments of ASL cDNA from 16 different tissues (A), from cultured skin fibroblasts derived from 12 ASA patients (B), from lymphocytes derived from two ASA patients and one patient's mother (C), and from controls' lymphocytes, livers, and fibroblasts as well as lymphocytes derived from one ASA patient and his mother (D). A, lane 1, skin fibroblast; lane 2, brain; lane 3, placenta; lane 4, lung; lane 5, liver; lane 6, skeletal muscle; lane 7, kidney; lane 8, pancreas; lane 9, spleen; lane 10, thymus; lane 11, prostate; lane 12, testis; lane 13, ovary; lane 14, small intestine; lane 15, colon; lane 16, peripheral leukocytes. B, lanes 1–12 (obtained from three different investigations), skin fibroblasts from 12 ASA patients with 10 different genotypes. C, lane 1, mother of patient in lane 2; lanes 2 and 3, two ASA patients; lane 4, control. D, lanes 1–15, five controls' lymphocytes (lanes 1–5), livers (lane 6–10), and fibroblasts (lanes 11–15); lane 16, lymphocytes from one patient (same patient as in Fig. 2C, lane 2); lane 17, lymphocytes from mother of patient in lane 16 (same as in Fig. 2C, lane 1). E, sequencing chromatogram of PCR product from lane 2 in Fig. 2C. F, sequencing chromatogram of PCR product from lane 3 in C. M, peqGOLD 100-bp DNA ladder plus (Peqlab, Erlangen, Germany); M′, 100-bp DNA ladder (Solis BioDyne, Tartu, Estonia). Gel electrophoresis was done in 1% agarose.
RNA Studies Reveal Predominant Expression of Exon 7-deleted ASL Transcript Variant in ASA Patients
In RNA from both patients, ASL-ex7del variant (786 bp) was predominantly expressed (Fig. 2, C (lanes 2 and 3) and D (lane 16)). Estimation of expression levels by densitometry yielded similar levels of the mutant variant (114% for Fig. 2C (lane 2) and 83% for Fig. 2C (lane 3)) when compared with WT ASL (864 bp) in lymphocyte control (Fig. 2C (lane 4), set to 100%) but much higher levels (330% for Fig. 2C (lane 2) and 182% for Fig. 2C (lane 3)) if compared with the full-length band in the same patients (Fig. 2C (lanes 2 and 3), set to 100%). Notably, the full-length band was expressed much less (34% for Fig. 2C (lane 2) and 46% for Fig. 2C (lane 3)) compared with WT in lymphocyte control (Fig. 2C (lane 4), set to 100%). Expression levels of transcript variants in controls (Fig. 2, C (lane 4) and D (lanes 1–15)) and also in the patient's mother (Fig. 2, C (lane 1) and D (lane 17)) were much lower in all tissues compared with WT (lymphocytes 9%, liver 6%, and fibroblasts 10%, as shown in Fig. 2D, lanes 1–5, 6–10, and 11–15, respectively). Sequencing confirmed that the variants represented ex2del or ex7del transcripts. To facilitate further sequencing, only a small fragment comprising exons 1–7 was amplified using a reverse primer derived from exon 7. Hereby, the mutation c.566A→G was confirmed in a hemizygous state in one patient (Fig. 2, C (lane 2) and E) and in a heterozygous state in the other patient (Fig. 2, C (lane 3) and F).
Endogenous ASL Expression in Mammalian Cell Lines and in Human Liver
To establish an ASL expression system in mammalian cells lacking endogenous but allowing for high ectopic ASL expression, we transiently transfected P-WT as well as EV pcDNA3 into COS-1, 293T, and HeLa cells, respectively. Western blot analysis indicated that 293T cells fulfilled the above criteria in an optimal way, whereas COS-1 cells showed endogenous ASL expression and HeLa cells failed to express ASL after transfection (Fig. 3A). These results were further confirmed by analysis of ASL activity (ASL endogenous activity in 293T cells shown in Table 1).
FIGURE 3.

Expression or co-expression of ASL wild type and transcript variants in different mammalian cell lines and human livers. Western blot analysis of ASL expression in cell extracts of COS-1, 293T, and HeLa cells (A); in human livers (B); or in transfected 293T cells (C–G). 30 μg of total protein was separated by 10% SDS-PAGE (A, B, D, F, G, and top panel in C) or by native PAGE (E and middle panel in C) for analyzing ASL expression. A, NT, non-transfected; wt, pcDNA3-ASL-WT (P-WT). B, lanes 1–3, human liver samples from three controls. C, ASL expression in 293T cells non-transfected (NT) or transfected with EV, P-WT, pcDNA3-ASL-ex2del (P-ex2del), or pcDNA3-ASL-ex7del (P-ex7del), respectively. D, (co-)expression of ASL monomers in 10% SDS-PAGE (ASL-WT, 52 kDa; ASL-ex2del, 45 kDa). Lane 1, P-WT with EV; lane 2, P-WT with P-ex2del and EV; lane 3, P-WT with P-ex7del and EV; lane 4, P-WT with P-ex2del and P-ex7del; lane 5, P-ex2del; lane 6, P-ex7del; lane 7, EV. E, (co-)expression of ASL homotetramers in 10% native PAGE (ASL-WT, ∼200 kDa; ASL-ex2del, ∼180 kDa). Lanes 1–7, same as in D. Asterisks, nonspecific bands. F, ASL expression in 293T cells co-transfected with P-WT and P-ex2del at different ratios. Lane 1, P-WT (1.75 μg); lane 2, 1:1; lane 3, 1:2; lane 4, 1:5; lane 5, P-WT (3.5 μg); lane 6, 2:1; lane 7, P-WT (8.75 μg); lane 8, 5:1; lane 9, ASl-ex2del (3.5 μg). G, (co-)expression of ASL mutant p.E189G (P-E189G) with transcript variants. Lane 1, P-WT with EV; lane 2, P-E189G with EV; lane 3, P-WT with P-E189G; lane 4, P-WT with P-ex2del; lane 5, P-WT with P-ex7del; lane 6, P-E189G with P-ex2del; lane 7, P-E189G with P-ex7del; lane 8, P-ex2del; lane 9, P-ex7del. Asterisks, nonspecific bands. β-Actin (42 kDa) and GAPDH (37 kDa) served as loading control.
TABLE 1.
Residual ASL activities in 293T cells (co-)transfected with recombinant ASL-WT and transcript variants
Data were obtained under standard conditions (13.6 mm argininosuccinate) after at least three independent experiments. ASL activity in cells (co-)expressing ASL-WT or ASL-WT with EV is set to 100% in each transfection (7 μg of plasmid) or co-transfection (total 10.5 μg of plasmids, each 3.5 μg) under the same condition, respectively. NT, non-transfected cells; EV: empty vector.
| 293T whole cell extracts | NT | EV | ex2del | ex7del | WT | WT + EV | WT + EV + ex2del | WT + EV + ex7del | WT + ex2del + ex7del | ex2del + ex7del + EV |
|---|---|---|---|---|---|---|---|---|---|---|
| ASL activity in mIU/mg total proteina | 1.2 ± 1.9 | 1.6 ± 2.2 | 3.2 ± 3.0 | 1.9 ± 1.4 | 830.2 ± 78.8 | 520.1 ± 297.5 | 307.5 ± 151.4 | 466.1 ± 232.7 | 305.3 ± 201.7 | 3.2 ± 1.5 |
| ASL activity in % of ASL-WTa | 0.4 ± 0.4 | 0.3 ± 0.2 | 0.5 ± 0.4 | 0.4 ± 0.2 | 100.0 ± 7.8 | 100.0 ± 2.6 | 62.5b ± 7.2 | 94.2 ± 9.4 | 55.5b ± 7.2 | 0.7 ± 0.3 |
a Mean S.D. (in triplicate).
b Significant difference compared with ASL-WT activity (p < 0.05).
Although ASL splice variants (ASL-ex7del and ASL-ex2del) were present in all cDNAs from various tissues (Fig. 2, A–D), there was no corresponding signal detected when human liver homogenates from controls were investigated by Western blot analysis (Fig. 3B). Likewise, immunoprecipitation did not yield a detectable WT signal when patient and control fibroblasts were used (data not shown). This points toward and is probably explained by the low level of ASL expression in this cell type, which is obviously not suited for further investigation of ASL wild type and mutants on the protein level.
Expression and Co-expression of ASL Wild Type, Mutant, and Transcript Variants in 293T Cells
To study the role of naturally occurring ASL splice variants in wild type as well as in mutant p.E189G, we first introduced diverse ASL recombinant constructs P-WT, P-E189G, P-ex2del, and P-ex7del into 293T cells to (co-)express ASL WT or mutant p.E189G, ex2del, or ex7del ASL, respectively. At the protein level, expression of ASL-WT or ASL-ex2del was detected either as monomers (Fig. 3, C (top, lanes 3 and 4) and D (top, lanes 1 and 5), respectively) or as homotetramer (Fig. 3, C (middle, lanes 3 and 4) and E (top, lanes 1 and 5), respectively). Moreover, ASL-ex2del could also be co-expressed with ASL-WT as well as with mutant p.E189G (Fig. 3, D–G). Densitometry analysis of the detectable bands by Western blotting showed that ASL-WT was less expressed in cells co-expressed with transcript variants (71.7% in co-transfectant with ASL-ex2del (Fig. 3D, lane 2), 88.7% in co-transfectant with ASL-ex7del (Fig. 3D, lane 3), and 71.2% in co-transfectant with ASL-ex2del and ASL-ex7del in Fig. 3D (lane 4); all done after normalization according to expressed loading control GAPDH, respectively) compared with co-expression with EV (Fig. 3D (lane 1), set to 100%). Additionally, ASL-ex2del was expressed at higher levels (55.4, 53.6, 60, or 61%) than ASL-WT (44.6, 46.4, or 40%) or mutant p.E189G (39%) (cells co-transfected with ASL-ex2del and ASL-WT (Fig. 3, A (lanes 2 and 4) and G (lane 4)) and cells co-transfected with ASL-ex2del and p.E189G in Fig. 3G (lane 6)). However, ASL-ex7del was not found (Fig. 3, C (lane 5), D, E (top, lane 6), and G (lane 9), respectively), although ASL-ex7del-RNA derived from cells transfected with P-ex7del could be detected by RT-PCR (data not shown). These results suggest that deletion of ASL exon 2 results in a truncated but stable ASL protein, whereas deletion of ASL exon 7 probably leads to an unstable protein.
Structure of ASL Monomers and Homotetramer
To study the structural implications of exon 2 and 7 deletions, we made computational structural models of exon-deleted ASL variants based on known structures of ASL available from the RCSB database. We first performed a PhiBlast search of the PDB database with the modified amino acid sequences to create a custom position-specific scoring matrix that was then used in further runs of PhiBlast searches to identify structurally similar sequences. Then a secondary structure prediction of the original WT sequence was used to perform the structure-based alignment of the sequences (Fig. 4). Aligned sequences were loaded in the programs YASARA and WHATIF for generating structural models. Some of the loops in exon-deleted sequences were modeled separately by scanning a library of loop databases. For our model building, we used the information available in the PDB_REDO database, which re-refines the old structures in the PDB database using the latest methods based on original structural data deposited in the PDB and corrects the errors in structures found in the PDB database. This is achieved by employing current state of the art refinement methods and software to the older crystallographic data. For the ASL structure (PDB code 1K62), the overall Ramachandran plot appearance improved from −2.514 to −0.803 for the optimized entry, and the total number of bumps/structural clashes was reduced from 150 in the original entry to 54 in the optimized structure, χ-1/χ-2 rotamer normality improved from −2.186 to −0.576, first generation packing quality improved from 0.148 to 0.466, backbone conformation improved from −0.369 to −0.168, and the R-free value changed from 0.2290 to 0.1909 in the fully optimized version used by us.
FIGURE 4.

Positions of exons 2 and 7 in secondary structure of the ASL protein. The helices are in yellow, sheets in green, and turns in cyan; exons 2 and 7 are depicted in magenta. Amino acids are colored based on their chemical properties with aspartic and glutamic acid in red, arginines and lysines in blue, and aromatic amino acids in green.
The ASL structure is composed of residues 5–464 of the ASL protein. The enzymatically active ASL tetramer is formed by four identical subunits of ASL, comprising mainly α helices, and contains three structurally distinct domains. Domains 1 and 3 have similar topology and contain two helix-turn-helix motifs, whereas domain 2 has nine helices, of which five helices form the core monomer structure in an up-down-up-down-up sequence (Fig. 5A). Two sets of dimers come together in an antiparallel manner to form the tetramer (Figs. 5A and 6A). The core of the homotetramer is composed of a four-helix bundle, with each monomeric subunit contributing one helix to the central core, and the tetramer is held together by hydrophobic interactions between the four central helices as well as ionic interactions between arginines and glutamic acid residues on two distinct dimeric structures.
FIGURE 5.

Monomeric structure of wild type and models of ASL exon 2- or exon 7-deleted transcript variant. A, monomeric structure of WT ASL. ASL monomer has three distinct subdomains, domains 1 and 3 have similar structure and topology with two helix-turn-helix motifs in a perpendicular arrangement. Domain 2 has nine helices, and five of them form the central five-helix bundle with up-down-up-down-up topology. B, monomeric structural model of ex2del ASL. Two critical helices that are part of domain 1 (indicated by a black arrow) and contribute to the active site are missing in the ex2del variant of ASL. C, monomeric structural model of ex7del ASL. Exon 7 residues are part of domain 2 in the ASL monomer comprising the central five-helix bundle. Deletion of exon 7 results in a disordered central core with one of the five central helices partially replaced by an unstructured loop (indicated by a black arrow). N, N terminus; C, C terminus.
FIGURE 6.

Tetrameric structure of wild type and models of ASL exon 2- or exon 7-deleted transcript variant. A, tetrameric structure of WT ASL showing the positions of active site histidine 160 and exons 2 and 7. The structure is shown as a ribbon model in light blue; amino acids contributed by exon 2 are colored in red, whereas amino acids contributed by exon 7 are colored in magenta. The active site residues are colored in green with His-160 at the catalytic center shown a sphere model. B, location of exon 2 in relation to the active site. In the ASL homotetramer, active site residues are contributed by three different subunits. The ASL structure is shown as a ribbon model with different subunits colored in green, cyan, magenta, and yellow. The green and cyan subunits form a dimer and join with another dimer formed by magenta and yellow subunits to form the tetramer. Active site residues from one of the sites are shown as solid surfaces, whereas the catalytic center (His-160) is shown as spheres. Residues contributed by exon 2 are shown in red. C, role of the N terminus tail in stabilizing the active site. The ASL homotetramer is shown as a solid surface model with different subunits colored in green, cyan, magenta, and yellow. Residues contributed by exon 2 are shown in red. The N terminus tail surrounds the active site and provides the stability required for binding of substrate. D, conjectural prediction of exon 2-deleted ASL based on homology modeling. The core structure of the ASL homotetramer can still be formed in the absence of N terminus residues contributed by exon 2. The model of the ex2del tetrameric complex is similar to the WT ASL, and the central four-helix bundle comprising the tetramer remains intact, resulting in a stable structure. However, the loss of the N terminus required for substrate binding will result in an enzymatically inactive protein. E, the WT ASL tetramer structure showing the locations of residues contributed by exon 7. The homotetramer structure is shown as a ribbon model with different subunits colored in green, cyan, magenta, and yellow. The amino acids contributed by exon 7 are shown as surface models in two of the adjacent dimeric structures that form the tetramer. Chains shown in green and yellow are from two different dimers that join together to form the tetramer. The His-160 residue at the catalytic center is shown as spheres. F, a space-filled surface model of homotetrameric ASL structure showing the role of exon 7 in tetramer formation. Interactions between charged residues on adjacent dimeric ASL subunits stabilize the tetrameric structure. G, a structural model of exon 7 deleted variant of ASL transcript. One subunit of the exon 7-deleted variant is superimposed on the WT ASL homotetramer to show the structural differences. The protein is shown as a ribbon model with the exon 7-deleted variant colored in gray, whereas WT subunits in the tetramer are colored in green, cyan, magenta, and yellow. The residues involved in charge-based interaction located on two adjacent subunits in the WT protein are shown as sticks in the larger oval red dotted box. The secondary structure elements altered in the exon 7-deleted variant are marked in a smaller oval box. H, a close-up of the structural changes resulting from the exon 7 deletion. The exon 7-deleted variant (gray) and WT (green) are superimposed, and residues contributed by exon 7 in the WT protein are shown in red. Two of the central helices in the WT protein are altered to flexible loops in the exon 7-deleted variants, resulting in an unstable protein due to the requirement of core helices in domain 2 of the ASL monomers for overall stability. The resultant protein was found to be highly unstable and unlikely to participate in tetramer formation.
Role of Exon 2 Residues
The amino acid residues contributed by exon 2 (residues 5–69) form the N terminus of the ASL protein (Figs. 4 and 5A) and are needed for the enzymatic activity because the previously described R12Q mutation in exon 2 has been shown to result in loss of activity. Two of the helices in domain 2 of ASL monomeric structure are formed by residues from exon 2 and are missing in the ex2del variant of ASL (Fig. 5B). Most of the N-terminal chain in the ASL monomer is flexible, and during molecular dynamic simulations, it was found to fluctuate during the whole run, indicating a dynamic arrangement in both the monomeric and tetrameric form. There were few contacts between the N-terminal loops of any individual subunit and other subunits in the tetrameric structure, indicating that it probably does not influence tetramer formation. The role of N terminus residues on the catalytic activity is not clear, but one side of the active site is covered by these residues (Fig. 6, B and C). The flexible nature of amino acids 5–18 suggests that although exact conformation of the N terminus does not influence the overall tetrameric structure or active site of the enzyme (Fig. 6, B and C), the structural requirements for substrate binding and retention in the active site may depend on an intact N terminus. Residues 23–32 of the ASL have been proposed to be important for substrate binding (50, 51).
From our computational analysis, we found that exon 2-deleted protein can form a stable monomer (Fig. 5B) and homotetramer (Fig. 6D), which was confirmed by Western blots showing a smaller sized complex from the cell-based assays (Fig. 3, C–G). However, such a homotetramer will be devoid of activity due to loss of N-terminal residues needed for substrate binding. Therefore, although an exon 2-deleted transcript of ASL is still capable of expressing a stable monomer that can form a tetrameric complex, the loss of residues involved in catalysis means that such a complex cannot be enzymatically active. It is possible that ASL-WT and ex2del monomers can form heterotetramers with different combinations of the two transcript variants. Because the active site of the ASL protein is formed by contributions from three different monomeric subunits, each unit of exon 2-deleted transcript variants in the heterotetramer will result in loss of one active site, without compromising the overall structure. Because the N-terminal residues affected by deletion of exon 2 do not participate in the formation of ASL dimers, we propose that a combination of WT, ex2del, and WT-ex2del versions of the dimers is generated, which will then further oligomerize to form the tetrameric structures. WT and ex2del variants of the monomers may have a preference for similar sized proteins and only form homodimers, which will result in at least two functional active sites in a 2:2 heterotetramer. Functional data from cell-based assays that show that even a 1:5 combination of WT-ex2del ASL retained about 30% activity (Fig. 7C) seem to support this model. A combination of WT-ex2del dimers may also give similar activities.
FIGURE 7.

Analysis of recombinant ASL activities. Shown is ASL enzymatic activity analysis in non-transfected or (co-)transfected 293T cell extracts. Each 4.8–14 μg of total protein of cell extracts was used for the enzyme assay. The residual ASL activities are represented as a percentage of ASL-WT activity (percentage of ASL-WT or of ASL-WT with EV). A, 293T cells were transiently transfected with 7 μg of P-WT (wt), P-ex2del (ex2del), or P-ex7del (ex7del), respectively. NT, non-transfected. Non-transfected cells and those transfected with EV served as negative control. B, 293T cells were transiently co-transfected with 10.5 μg of total plasmids at a ratio of 1:1 (each 3.5 μg) of P-WT to P-ex2del or P-ex7del plasmid, respectively. C, 293T cells were transiently co-transfected with 7 μg of total plasmids at a ratio of 1:1 (each 3.5 μg) of P-WT to P-ex2del or P-ex7del and of P-E189G to P-ex2del or P-ex7del plasmid, respectively. D, ASL enzymatic activity in 293T cell extracts co-transfected with P-WT and P-ex2del at different ratios. 3–12 μg of cell extracts was used for ASL enzyme analysis depending on the amount of P-WT used for the co-transfections. For co-transfections, different ratios of ASL-WT to ex2del splice variant (1:1, 1:2, 1:5, 2:1, or 5:1) were used, respectively. The residual activity of each co-transfectant compared with ASL-WT under the same condition is indicated as a percentage of ASL-WT activity (percentage of ASL-WT co-transfected with EV to use identical amounts of total plasmids and of P-WT for all experiments). EV was used to set up the same amount of total plasmids for the co-transfections. Levels of significance are given as p values obtained by one-way analysis of variance using GraphPad software from triplicate measurements of at least three independent experiments, respectively. Differences were considered as significant if the p value was <0.05. Error bars, S.D.
Role of Exon 7 Residues
Residues 175–200 are contributed by exon 7 in the WT ASL transcript. Exon 7-deleted variant was found to be capable of forming a monomeric structure still composed of three distinct domains, similar to that of WT ASL (Fig. 5C). Although the core of the homotetramer of ASL is formed by α helices of monomers formed by residues 291–314, we found ionic interactions between adjacent dimeric subunits in the tetramer to stabilize the structure (Fig. 6, E and F). From the molecular dynamic simulations and structural analysis, we found that charge pair interactions between Arg-193 on one subunit and Glu-189 and Glu-185 of another adjacent subunit in the tetramer stabilize the structure (Fig. 6G). The loss of ionic interactions contributed by exon 7 residues will result in an unstable tetramer. Computational potential energy of WT and ex7del tetramers supported this theory with an increase in potential energy for the ex7del tetramer (−76,378 kJ/mol for ex7del compared with −95,229 kJ/mol for WT) (Table 2). Moreover, a change in core structure of the monomers was also observed with the loss of two central helices and formation of a structurally unstable loop in the middle of the helical core (Figs. 5C and 6, G and H). Such a change is likely to impact the stability of the monomers and may result in an unstable protein that will probably be degraded, especially in the absence of a stable tetrameric complex. Significantly, due to this unstructured loop formation, the accessible surface area of the ex7del variant increased significantly (65,259 Å2 compared with 53,534 Å2 for the WT), whereas the solvent-accessible surface remained similar (326,476 Å3 compared with 322,934 Å3 for the WT), which further points toward structural instability of ex7del variant (Table 2). This was also further supported by Western blots showing no detectable ASL-ex7del expression (Fig. 3, C–E and G).
TABLE 2.
Computational determination of structural properties of ASL-WT, ASL-ex2del, and ASL-ex7del proteins
Computational parameters were determined with WHATIF and YASARA.
| ASL-WT | ASL-ex2del | ASL-ex7del | |
|---|---|---|---|
| Molecular mass | 51,658 Da | 44,471 Da | 48,733 Da |
| Theoretical pI | 6.04 | 5.90 | 5.76 |
| Solvent-accessible surface | 322,934 Å3 | 296,898 Å3 | 326,476 Å3 |
| Accessible surface area | 53,534 Å2 | 55,904 Å2 | 65,259 Å2 |
| Potential energy | −95,229 kJ/mol | −77,219 kJ/mol | −76,378 kJ/mol |
| Radius of gyration | 36.3 Å | 36.1 Å | 36.9 Å |
| Electrostatic solvation energy | −29,769 kJ/mol | −24,454 kJ/mol | −27,781 kJ/mol |
| Electrostatic potential | −14.89 kJ/mol | −20.4 kJ/mol | −43.07 kJ/mol |
Residual ASL Enzymatic Activities in (Co-)transfected 293T Cell Extracts
To determine whether the expressed ASL splice variants have any residual enzyme activity, we performed ASL enzyme activity assays with the cell extracts used for Western blot analysis (summary of data in Table 1, Table 3, and Fig. 7). There was no relevant background activity in cells transfected with the empty vector or non-transfected cells (Fig. 7A, columns 1 and 2, respectively). No significant residual activity was detected in exon 2- and exon 7-deleted ASL splice variants (Fig. 7A, columns 3 and 4, respectively), whereas ASL-WT transfection yielded high enzymatic activity (Fig. 7A, column 5).
TABLE 3.
Residual ASL activities in 293T cells co-transfected with recombinant ASL-WT, mutant p.E189G, and transcript variants
Data were obtained under standard conditions (13.6 mm argininosuccinate) after three independent experiments. ASL activity in cells co-expressing ASL-WT with EV is set to 100% in each co-transfection (total 7 μg of plasmids, each 3.5 μg) under the same conditions. All measurements were in triplicate.
| 293T whole cell extracts | WT + EV | WT + ex2del | WT + ex7del | E189G + EV | E189G + ex2del | E189G + ex7del | WT + E189G |
|---|---|---|---|---|---|---|---|
| Mean ASL activity in mIU/mg total protein (mean ± S.D.) | 450.2 ± 38.3 | 286.1 ± 5.5 | 363.5 ± 15.0 | 389.1 ± 30.8 | 78.4 ± 26.4 | 257.2 ± 10.5 | 883.8 ± 80.0 |
| Mean ASL activity in % of ASL-WT + EV (mean ± S.D.) | 100.0 ± 7.1 | 61.9a ± 5.8 | 88.5 ± 11.5 | 88.4 ± 11.9 | 16.6a ± 5.3 | 57.5a ± 3.2 | 197.6 ± 8.9 |
a Significant differences compared with ASL-WT activity (p < 0.05).
To study the effect of ASL splice variants on the function of the ASL homotetramer and the possibility of a heterotetramer formation with reduced ASL activity that may contribute to the biochemical and clinical variability in ASA patients, we measured the ASL activity after co-transfections. The residual ASL activities in cells co-expressing P-WT and P-ex2del (Fig. 7, B and C, column 2) or co-expressing P-WT, P-ex2del, and P-ex7del (Fig. 7B, column 4) showed a significant decrease to 62.5 ± 7.2 or 55.5 ± 7.2% (mean ± S.D.), respectively, of cells co-transfected with P-WT and EV (Fig. 7, B and C (column 1), and Table 1). Cells co-transfected with P-WT and P-ex7del (Fig. 7, B and C, column 3) displayed no decrease of ASL activity (94.2 ± 9.4% of ASL WT). Cells co-transfected with P-ex2del and P-ex7del (Fig. 7B, column 5) showed no relevant residual ASL activity. Furthermore, the reduced level of residual ASL activities exhibited no significant difference between cells co-expressing P-WT and P-ex2del and cells co-expressing P-WT, P-ex2del, and P-ex7del (Fig. 7B, column 4). Interestingly, cells expressing only the ASL mutant P-E189G had a similar level of ASL-WT activity (Fig. 7C, column 4), but cells co-expressing P-WT and P-E189G showed 2-fold levels of WT activity (Fig. 7C, column 7). However, the residual activity in cells co-transfected with P-E189G and P-ex2del or co-transfected with P-E189G and P-ex7del (Fig. 7D, column 5 or 6) exhibited a significant decrease to 16.6 ± 5.3 or 57.5 ± 3.2% (mean ± S.D.), respectively, of cells co-transfected with P-WT and EV (Table 3). These findings indicate that the truncated protein caused by deletion of ASL exon 2, although showing no relevant residual activity, has a dominant negative effect on the ASL activity after co-expression with P-WT or P-E189G. In contrast, deletion of exon 7 seems to have no significant effect on ASL-WT activity after co-expression with P-WT, because it probably forms an unstable protein according to Western blot analysis (Fig. 3, C–E and G) and computational predictions (Figs. 5C and 6 (G and H)), but it nevertheless has a potential dominant negative effect on ASL mutant activity after co-expression with P-E189G (Fig. 7C, column 6).
In order to further assess whether the negative effect of the simultaneous expression of ex2del mutant on ASL-WT is dose-dependent in vitro, we performed transient co-transfections with P-WT and P-ex2del at different ratios (1:1, 1:2, 1:5, 2:1, or 5:1). The co-expression of ASL-WT and ASL-ex2del at the different ratios above could be detected by Western blot analysis (Fig. 3F). In cells co-expressing ASL-WT and ASL-ex2del with a higher proportion of mutant DNA, we observed a significant gradual decrease of ASL activity when compared with cells co-expressing WT and EV (Fig. 7D). Moreover, cells co-expressing ASL-WT and ASL-ex2del with a higher proportion of WT DNA showed a significant gradual increase of ASL activity (Fig. 7D). These findings indicate that the negative effect of ASL-ex2del on the WT activity is dose-dependent.
DISCUSSION
To date, the mechanism of the broad biochemical and clinical heterogeneity in ASA patients still remains to be fully explained because it is not just the result of different genotypes. Possible hypotheses include ASL tissue-specific expression (27, 28), genetic variability (29), intragenic complementation (7, 30–32), the DNA methylation status (35), and the frequent occurrence of alternative splicing variants at the ASL locus (5, 36, 37). In addition, several hormones, such as glucocorticosteroids and insulin, influence the regulation of ASL mRNA (52). Recently, a complex formation with the enzymes argininosuccinate synthetase and nitric-oxide synthase required for nitric oxide synthesis was described (2), adding further to the complexity of this, obviously not only, urea cycle enzyme. In the present study, we were interested in the tissue dependence of ASL transcript variants and their role for the clinical and biochemical phenotype in ASA.
RT-PCR yielded comparable expression levels for the detected transcript variants in 17 different human tissues (Fig. 2), suggesting that the expression of transcript variants is not tissue-dependent. These findings are consistent with earlier reports, in which full-length ASL expression was studied in 11 different human tissues (53) and with previous reports on the frequent skipping of exons 2 or 7 (5, 36).
However, it remains pivotal to understand whether the frequent transcript variants in this gene indeed play a role in ASA patients at physiological levels or if they are present at higher concentrations. To do so, we further investigated the expression levels of transcript variants as well as their enzymatic characteristics in vitro. Establishment of an ASL expression system in 293T cells (Fig. 3A) allowed us to investigate the ASL activities after ectopically expressing or co-expressing ASL-WT and one ASL mutant (p.E189G) and/or splicing variants. Immunoblotting showed non-detectable ASL expression in cells transfected with the ex7del construct (Fig. 3, C–E and G), yielding unchanged ASL activity levels after co-expression of ASL-WT and ASL-ex7del (Fig. 7, B and C, and Table 1) but having a potential dominant negative effect on the ASL mutant activity after co-expression of ASL mutant p.E189G and ASL-ex7del (Fig. 7C and Table 3). Moreover, the observation of a 2-fold increase in the ASL activity after co-expressing P-WT and P-E189G (Fig. 7C, column 7) further supports the view that the negative effect on ASL mutant activity in cells co-transfected with P-E189G and P-ex7del is not caused by the mutant itself. Computational structure modeling revealed that the ex7del variant was capable of forming a monomeric structure with three distinct domains, similar to that of ASL-WT (Fig. 5C). As predicted by structural analysis, ex7del may interfere with tetramer formation and seriously impact the stability of the core monomeric structure (Fig. 5), leading to premature degradation of the monomers. This may explain the undetectable ASL-exdel7 monomers after co-expression with ASL-WT or with p.E189G (Fig. 3, D and G). Nevertheless, the ex7del ASL monomer may still form a heterotetramer with other monomers before degradation occurs, especially with easily accessible mutants, such as p.E189G, which then would result in reduced ASL activity. It should be noted that exon 7 contains 78 bases, allowing formation of an in-frame mutant ASL protein (5). This finding is somewhat different from that of an earlier study (54), in which a small amount of a 49 kDa band in addition to the expected ASL-WT band on Western blots was speculated to be an exon 7 skipping product that may be due to proteolysis (5). However, the details of the possible degradation of ex7del ASL were not investigated in this study.
Although exon 2 skipping was observed in previous studies (29, 36), these authors did not determine the molecular basis and impact of this deletion. As one of the main findings of our investigations, we could show that exon 2-spliced ASL can form a stable truncated protein (Fig. 3, C–G), but it lacks any relevant activity (Fig. 7A and Table 1). This finding is consistent with our prediction explored by computational structure modeling that exon 2 is in close vicinity to the active site (Fig. 6, B and C), and its deletion is probably deleterious. Surprisingly, this stable truncated mutant protein has a dominant negative effect on the ASL activity after co-expressing ASL-WT as well as ASL-p.E189G with ASL-ex2del (Fig. 7, B and C); in the latter case, this was even more relevant. Furthermore, this negative effect is ASL-ex2del DNA dose-dependent (Fig. 7D). The reduced ASL activity may result from the formation of a heterotetramic structure between ASL-WT and ASL-ex2del mutant protein as well as homotetrameric ex2del variants that are enzymatically inactive, competing for the substrate, depending on their proportions. Because three different subunits of ASL contribute to the active site in the tetrameric structures, different combinations of WT and ex2del variant subunits may combine to form tetramers with three, two, one, or zero active sites. It is possible that proteins of different size resulting from WT or exon 2-deleted transcripts may have a preference for each other when forming the dimeric units before joining to form tetramers. In such a scenario, only 2:2 versions of the heterotetramers may be the preferred assembly in addition to the fully functional and non-functional molecules with four or zero active sites. Due to a lack of liver samples derived from ASA patients, we could not show whether a different amount of ASL-ex2del mutant protein is expressed in the ASA patients, which may interact with ASL-WT to form a heterotetramer, leading to a different level of residual ASL activity.
Although this study clearly shows that transcript variants under physiological conditions are only present at low concentrations (Fig. 2, A–D), the situation was found to be different in two patients affected by late onset ASA (Figs. 1D (lane 16) and 2C (lanes 2 and 3)). Both patients were shown to carry the same known ASL missense mutation (p.E189G) with low residual ASL activity in red blood cells found in one of the patients (25) and absent activity after bacterial overexpression (33) but significant residual activity in yeast experiments (55). Although the mutation was found in a heterozygous state by genomic DNA sequencing, the level of the ASL-ex7del transcript variant observed by RNA studies was much higher (Fig. 2, C (lanes 2 and 3) and D (lane 16)). Under standard PCR conditions (38 cycles), no full-length ASL transcript was amplified, but a faint full-length band was detected after 42 PCR cycles, indicating the low expression of this transcript. Sequencing confirmed the mutation c.566A→G in a hemizygous state in one patient (Fig. 2E), suggesting that the ASL WT allele in this particular patient is completely subject to alternative splicing, and hence, no WT transcript is expressed, whereas in the other patient heterozygosity was found (Fig. 2F). We suggest here that the high expression level of the ASL-ex7del variant contributed to the ASA phenotype in the patients in addition to the missense mutation. From the finding in these patients, we deduce that the same phenomenon may also play a role in other patients possibly affected by any of the transcript variants (such as patient 16 in Ref. 25 with no mutation found but skipping of exon 5).
In conclusion, this study suggests that transcript variants of ASL exist at high frequency in different tissues and play a role in the clinical and biochemical variability in ASA patients in whom they are expressed at high levels. If this occurs with an increased expression level of a stable expressed truncated variant, such as the ASL-ex2del variant, the effect may be even more prominent because the likelihood of stable mutant homo- or heterotetramer formation increases, impairing overall ASL activity. Although the unstable splice variant ASL-ex7del has no effect on ASL-WT activity, it probably shows a dominant negative effect on ASL mutant activity. The findings from this study expand our knowledge of the molecular background in ASA patients, rendering further studies into transcript variants necessary, especially in patients with no or only single mutations.
Acknowledgments
We acknowledge the technical assistance provided by Dana Leiteritz (Zurich, Switzerland). We thank Drs. C. Giunta and D. Coelho (Zurich, Switzerland) for helpful suggestions during planning of experiments. We also acknowledge Dr. Lee-Jun C. Wong (Baylor College of Medicine, Houston, TX) for performing array comparative genomic hybridization.
This work was supported by Swiss National Science Foundation Grants 310030_127184/1 (to J. H.) and 31003A-134926 (to A. V. P.) and a grant from Schweizerische Mobiliar Genossenschaft Jubiläumsstiftung (to A. V. P.).
- ASL
- argininosuccinate lyase
- ASA
- argininosuccinic aciduria
- EV
- empty vector pcDNA3
- P-E189G
- pcDNA3-ASL-E189G
- P-WT
- pcDNA3-ASL-WT
- ex2del
- exon 2-deleted
- P-ex2del
- pcDNA3-ASL-ex2del
- ex7del
- exon 7-deleted
- P-ex7del
- pcDNA3-ASL-ex7del
- MD
- molecular dynamics
- PDB
- Protein Data Bank.
REFERENCES
- 1. Brusilow S., Horwich A. (2001) Urea cycle enzymes. in The metabolic and molecular bases of inherited disease (Scriver C., Beaudet A., Sly W., Valle D., eds) pp. 1909–1963, 8th Ed., McGraw-Hill, New York [Google Scholar]
- 2. Erez A., Nagamani S. C., Shchelochkov O. A., Premkumar M. H., Campeau P. M., Chen Y., Garg H. K., Li L., Mian A., Bertin T. K., Black J. O., Zeng H., Tang Y., Reddy A. K., Summar M., O'Brien W. E., Harrison D. G., Mitch W. E., Marini J. C., Aschner J. L., Bryan N. S., Lee B. (2011) Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 17, 1619–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Todd S., McGill J. R., McCombs J. L., Moore C. M., Weider I., Naylor S. L. (1989) cDNA sequence, interspecies comparison, and gene mapping analysis of argininosuccinate lyase. Genomics 4, 53–59 [DOI] [PubMed] [Google Scholar]
- 4. Naylor S. L., Klebe R. J., Shows T. B. (1978) Argininosuccinic aciduria. Assignment of the argininosuccinate lyase gene to the pter to q22 region of human chromosome 7 by bioautography. Proc. Natl. Acad. Sci. U.S.A. 75, 6159–6162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abramson R. D., Barbosa P., Kalumuck K., O'Brien W. E. (1991) Characterization of the human argininosuccinate lyase gene and analysis of exon skipping. Genomics 10, 126–132 [DOI] [PubMed] [Google Scholar]
- 6. Linnebank M., Tschiedel E., Häberle J., Linnebank A., Willenbring H., Kleijer W. J., Koch H. G. (2002) Argininosuccinate lyase (ASL) deficiency. Mutation analysis in 27 patients and a completed structure of the human ASL gene. Hum. Genet. 111, 350–359 [DOI] [PubMed] [Google Scholar]
- 7. Turner M. A., Simpson A., McInnes R. R., Howell P. L. (1997) Human argininosuccinate lyase. A structural basis for intragenic complementation. Proc. Natl. Acad. Sci. U.S.A. 94, 9063–9068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matsubasa T., Takiguchi M., Amaya Y., Matsuda I., Mori M. (1989) Structure of the rat argininosuccinate lyase gene. Close similarity to chicken δ-crystallin genes. Proc. Natl. Acad. Sci. U.S.A. 86, 592–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Piatigorsky J., O'Brien W. E., Norman B. L., Kalumuck K., Wistow G. J., Borras T., Nickerson J. M., Wawrousek E. F. (1988) Gene sharing by δ-crystallin and argininosuccinate lyase. Proc. Natl. Acad. Sci. U.S.A. 85, 3479–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Piatigorsky J., Horwitz J. (1996) Characterization and enzyme activity of argininosuccinate lyase/δ-crystallin of the embryonic duck lens. Biochim. Biophys. Acta 1295, 158–164 [DOI] [PubMed] [Google Scholar]
- 11. Ratner S. (1973) Enzymes of arginine and urea synthesis. Adv. Enzymol. Relat. Areas Mol. Biol. 39, 1–90 [DOI] [PubMed] [Google Scholar]
- 12. Ratner S., Petrack B. (1953) The mechanism of arginine synthesis from citrulline in kidney. J. Biol. Chem. 200, 175–185 [PubMed] [Google Scholar]
- 13. De Jonge W. J., Dingemanse M. A., de Boer P. A., Lamers W. H., Moorman A. F. (1998) Arginine-metabolizing enzymes in the developing rat small intestine. Pediatr. Res. 43, 442–451 [DOI] [PubMed] [Google Scholar]
- 14. Flynn N. E., Meininger C. J., Kelly K., Ing N. H., Morris S. M., Jr., Wu G. (1999) Glucocorticoids mediate the enhanced expression of intestinal type II arginase and argininosuccinate lyase in postweaning pigs. J. Nutr. 129, 799–803 [DOI] [PubMed] [Google Scholar]
- 15. Walker J. B. (1958) Role for pancreas in biosynthesis of creatine. Proc. Soc. Exp. Biol. Med. 98, 7–9 [DOI] [PubMed] [Google Scholar]
- 16. Pisarenko S. I., Minkovskiı̆ E. B., Studneva I. M. (1980) [Urea synthesis in the myocardium]. Biull. Eksp. Biol. Med. 89, 165–168 [PubMed] [Google Scholar]
- 17. Ratner S., Morell H., Carvalho E. (1960) Enzymes of arginine metabolism in brain. Arch. Biochem. Biophys. 91, 280–289 [DOI] [PubMed] [Google Scholar]
- 18. Jones M. E., Anderson A. D., Anderson C., Hodes S. (1961) Citrulline synthesis in rat tissues. Arch. Biochem. Biophys. 95, 499–507 [DOI] [PubMed] [Google Scholar]
- 19. O'Brien W. E., Barr R. H. (1981) Argininosuccinate lyase. Purification and characterization from human liver. Biochemistry 20, 2056–2060 [DOI] [PubMed] [Google Scholar]
- 20. Tomlinson S., Westall R. G. (1964) Argininosuccinic aciduria. Argininosuccinase and arginase in human blood cells. Clin. Sci. 26, 261–269 [PubMed] [Google Scholar]
- 21. Allan J. D., Cusworth D. C., Dent C. E., Wilson V. K. (1958) A disease, probably hereditary characterised by severe mental deficiency and a constant gross abnormality of aminoacid metabolism. Lancet 1, 182–187 [DOI] [PubMed] [Google Scholar]
- 22. Brusilow S. W., Maestri N. E. (1996) Urea cycle disorders. Diagnosis, pathophysiology, and therapy. Adv. Pediatr. 43, 127–170 [PubMed] [Google Scholar]
- 23. Wong L. T., Hardwick D. F., Applegarth D. A., Davidson A. G. (1979) Review of metabolic screening program of Children's Hospital, Vancouver, British Columbia. 1971–1977. Clin. Biochem. 12, 167–172 [DOI] [PubMed] [Google Scholar]
- 24. Ficicioglu C., Mandell R., Shih V. E. (2009) Argininosuccinate lyase deficiency. Longterm outcome of 13 patients detected by newborn screening. Mol. Genet. Metab. 98, 273–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mercimek-Mahmutoglu S., Moeslinger D., Häberle J., Engel K., Herle M., Strobl M. W., Scheibenreiter S., Muehl A., Stöckler-Ipsiroglu S. (2010) Long-term outcome of patients with argininosuccinate lyase deficiency diagnosed by newborn screening in Austria. Mol. Genet. Metab. 100, 24–28 [DOI] [PubMed] [Google Scholar]
- 26. Erez A., Nagamani S. C., Lee B. (2011) Argininosuccinate lyase deficiency-argininosuccinic aciduria and beyond. Am. J. Med. Genet. C Semin. Med. Genet. 157C, 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Glick N. R., Snodgrass P. J., Schafer I. A. (1976) Neonatal argininosuccinic aciduria with normal brain and kidney but absent liver argininosuccinate lyase activity. Am. J. Hum. Genet. 28, 22–30 [PMC free article] [PubMed] [Google Scholar]
- 28. Perry T. L., Wirtz M. L., Kennaway N. G., Hsia Y. E., Atienza F. C., Uemura H. S. (1980) Amino acid and enzyme studies of brain and other tissues in an infant with argininosuccinic aciduria. Clin. Chim. Acta 105, 257–267 [DOI] [PubMed] [Google Scholar]
- 29. Barbosa P., Cialkowski M., O'Brien W. E. (1991) Analysis of naturally occurring and site-directed mutations in the argininosuccinate lyase gene. J. Biol. Chem. 266, 5286–5290 [PubMed] [Google Scholar]
- 30. Howell P. L., Turner M. A., Christodoulou J., Walker D. C., Craig H. J., Simard L. R., Ploder L., McInnes R. R. (1998) Intragenic complementation at the argininosuccinate lyase locus. Reconstruction of the active site. J. Inherit. Metab. Dis. 21, Suppl. 1, 72–85 [DOI] [PubMed] [Google Scholar]
- 31. McInnes R. R., Shih V., Chilton S. (1984) Interallelic complementation in an inborn error of metabolism. Genetic heterogeneity in argininosuccinate lyase deficiency. Proc. Natl. Acad. Sci. U.S.A. 81, 4480–4484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Walker D. C., Christodoulou J., Craig H. J., Simard L. R., Ploder L., Howell P. L., McInnes R. R. (1997) Intragenic complementation at the human argininosuccinate lyase locus. Identification of the major complementing alleles. J. Biol. Chem. 272, 6777–6783 [DOI] [PubMed] [Google Scholar]
- 33. Engel K., Vuissoz J. M., Eggimann S., Groux M., Berning C., Hu L., Klaus V., Moeslinger D., Mercimek-Mahmutoglu S., Stöckler S., Wermuth B., Häberle J., Nuoffer J. M. (2012) Bacterial expression of mutant argininosuccinate lyase reveals imperfect correlation of in-vitro enzyme activity with clinical phenotype in argininosuccinic aciduria. J. Inherit. Metab. Dis. 35, 133–140 [DOI] [PubMed] [Google Scholar]
- 34. Trevisson E., Burlina A., Doimo M., Pertegato V., Casarin A., Cesaro L., Navas P., Basso G., Sartori G., Salviati L. (2009) Functional complementation in yeast allows molecular characterization of missense argininosuccinate lyase mutations. J. Biol. Chem. 284, 28926–28934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Renouf S., Fairand A., Husson A. (1998) Developmental control of argininosuccinate lyase gene by methylation. Biol. Neonate 73, 190–197 [DOI] [PubMed] [Google Scholar]
- 36. Linnebank M., Homberger A., Rapp B., Winter C., Marquardt T., Harms E., Koch H. G. (2000) Two novel mutations (E86A, R113W) in argininosuccinate lyase deficiency and evidence for highly variable splicing of the human argininosuccinate lyase gene. J. Inherit. Metab. Dis. 23, 308–312 [DOI] [PubMed] [Google Scholar]
- 37. Walker D. C., McCloskey D. A., Simard L. R., McInnes R. R. (1990) Molecular analysis of human argininosuccinate lyase. Mutant characterization and alternative splicing of the coding region. Proc. Natl. Acad. Sci. U.S.A. 87, 9625–9629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Landsverk M. L., Wang J., Schmitt E. S., Pursley A. N., Wong L. J. (2011) Utilization of targeted array comparative genomic hybridization, MitoMet, in prenatal diagnosis of metabolic disorders. Mol. Genet. Metab. 103, 148–152 [DOI] [PubMed] [Google Scholar]
- 39. Kretz R., Hu L., Wettstein V., Leiteritz D., Häberle J. (2012) Phytohemagglutinin stimulation of lymphocytes improves mutation analysis of carbamoylphosphate synthetase 1. Mol. Genet. Metab. 106, 375–378 [DOI] [PubMed] [Google Scholar]
- 40. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 41. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 42. Krieger E., Darden T., Nabuurs S. B., Finkelstein A., Vriend G. (2004) Making optimal use of empirical energy functions. Force-field parameterization in crystal space. Proteins 57, 678–683 [DOI] [PubMed] [Google Scholar]
- 43. Vriend G. (1990) WHAT IF. A molecular modeling and drug design program. J. Mol. Graph. 8, 52–56, 29 [DOI] [PubMed] [Google Scholar]
- 44. Hooft R. W., Vriend G., Sander C., Abola E. E. (1996) Errors in protein structures. Nature 381, 272. [DOI] [PubMed] [Google Scholar]
- 45. Bowie J. U., Lüthy R., Eisenberg D. (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253, 164–170 [DOI] [PubMed] [Google Scholar]
- 46. Lüthy R., Bowie J. U., Eisenberg D. (1992) Assessment of protein models with three-dimensional profiles. Nature 356, 83–85 [DOI] [PubMed] [Google Scholar]
- 47. Ramachandran G. N., Ramakrishnan C., Sasisekharan V. (1963) Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7, 95–99 [DOI] [PubMed] [Google Scholar]
- 48. Hooft R. W., Sander C., Vriend G. (1997) Objectively judging the quality of a protein structure from a Ramachandran plot. Comput. Appl. Biosci. 13, 425–430 [DOI] [PubMed] [Google Scholar]
- 49. Liu H., Elstner M., Kaxiras E., Frauenheim T., Hermans J., Yang W. (2001) Quantum mechanics simulation of protein dynamics on long timescale. Proteins 44, 484–489 [DOI] [PubMed] [Google Scholar]
- 50. Vallée F., Turner M. A., Lindley P. L., Howell P. L. (1999) Crystal structure of an inactive duck δ II crystallin mutant with bound argininosuccinate. Biochemistry 38, 2425–2434 [DOI] [PubMed] [Google Scholar]
- 51. Sampaleanu L. M., Yu B., Howell P. L. (2002) Mutational analysis of duck δ2 crystallin and the structure of an inactive mutant with bound substrate provide insight into the enzymatic mechanism of argininosuccinate lyase. J. Biol. Chem. 277, 4166–4175 [DOI] [PubMed] [Google Scholar]
- 52. Husson A., Renouf S., Fairand A., Buquet C., Benamar M., Vaillant R. (1990) Expression of argininosuccinate lyase mRNA in foetal hepatocytes. Regulation by glucocorticoids and insulin. Eur. J. Biochem. 192, 677–681 [DOI] [PubMed] [Google Scholar]
- 53. Neill M. A., Aschner J., Barr F., Summar M. L. (2009) Quantitative RT-PCR comparison of the urea and nitric oxide cycle gene transcripts in adult human tissues. Mol. Genet. Metab. 97, 121–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Simard L., O'Brien W. E., McInnes R. R. (1986) Argininosuccinate lyase deficiency. Evidence for heterogeneous structural gene mutations by immunoblotting. Am. J. Hum. Genet. 39, 38–51 [PMC free article] [PubMed] [Google Scholar]
- 55. Doimo M., Trevisson E., Sartori G., Burlina A., Salviati L. (2012) Yeast complementation is sufficiently sensitive to detect the residual activity of ASL alleles associated with mild forms of argininosuccinic aciduria. J. Inherit. Metab. Dis. 35, 557–558 [DOI] [PubMed] [Google Scholar]