

Background: Deregulation of EGF receptor (EGFR) signaling leads to cellular transformation.
Results: Reduction of net charge in the EGFR juxtamembrane region generates constitutively active EGFR, and activity is maintained when this receptor is ER-retained.
Conclusion: Wholly ER-retained, constitutively active EGFRs can drive transformation through PI3K activation.
Significance: The polybasic juxtamembrane sequence plays an important role in regulating EGFR signaling leading to oncogenic transformation.
Keywords: Endoplasmic Reticulum (ER), Epidermal Growth Factor Receptor (EGFR), Oncogene, PI 3-Kinase (PI3K), Transformation
Abstract
Deregulation of ErbB receptor-tyrosine kinases is a hallmark of many human cancers. Conserved in the ErbB family is a cluster of basic amino acid residues in the cytoplasmic juxtamembrane region. We found that charge-silencing mutagenesis within this juxtamembrane region of the epidermal growth factor receptor (EGFR) results in the generation of a mutant receptor (EGFR Mut R1-6) that spontaneously transforms NIH 3T3 cells in a ligand-independent manner. A similar mutant with one additional basic residue, EGFR Mut R1-5, fails to exhibit ligand-independent transformation. The capacity of EGFR Mut R1-6 to mediate this transformation is maintained when this mutant is retained in the endoplasmic reticulum via a single point mutation, L393H, which we describe. We show that EGFR Mut R1-6 with or without L393H exhibits enhanced basal tyrosine phosphorylation when ectopically expressed, and the ligand-independent transforming activity of EGFR Mut R1-6 is sensitive to inhibition of EGFR kinase activity and is particularly dependent on PI3K and mTOR activity. Similar to EGFR Mut R1-6/L393H in NIH 3T3 cells, EGFR variant type III, a highly oncogenic mutant form of EGFR linked to human brain cancers, confers transforming activity while it is wholly endoplasmic reticulum-retained in U87 cells. Our findings highlight the importance of the polybasic juxtamembrane sequence in regulating the oncogenic potential of EGFR signaling.
Introduction
Receptor-tyrosine kinases (RTKs)2 initiate some of the most complex signaling networks in cells (1). To maintain normal cellular homeostasis, RTK signaling must be tightly regulated. Loss or perturbation of this regulation leads to defects in cellular processes that give rise to oncogenic transformation (2). Oncogenic mutations in RTKs can lead to their overexpression, constitutive activation, and/or mislocalization in cells.
The ErbB family and in particular, ErbB1, or the epidermal growth factor receptor (EGFR) serves as a prototypic model for RTK signaling. EGFR is a type I transmembrane glycoprotein consisting of an extracellular ligand binding segment, a single transmembrane segment, and an intracellular segment that contains a juxtamembrane (JX) region, a tyrosine kinase domain (TKD), and a C-terminal tail. Signaling by EGFR is initiated after its ligand-induced dimerization, which leads to TKD-dependent phosphorylation of the C-terminal segments of two interacting receptors (3). The ∼37-residue cytoplasmic JX region of EGFR has emerged as a key regulator of receptor function (4–6). Several intrinsic sorting signals have been mapped to this JX region (7–9), which also contains a basic calmodulin binding sequence (10) that additionally interacts with acidic phospholipids (11–13). In an active receptor dimer, the C-terminal halves of the JX region have been shown to clasp two interacting EGFRs together in a conformation that promotes their activation (4, 5), whereas the N-terminal halves of these domains stabilize this interaction via the formation of an antiparallel helical dimer (5). Recent evidence and molecular dynamics simulations suggest that basic residues in the N-terminal JX region facilitate insertion of adjacent leucine amino acid side chains into the plasma membrane (PM) bilayer in inactive EGFR, and these hydrophobic side chains are expelled to allow formation of the helical dimer in the active state (6, 14).
EGFR plays important roles in cell differentiation, proliferation, and epithelial organogenesis (15). Changes in its expression and regulation, due to gene duplication events (16–19), in-frame deletions (20, 21), and point mutations (4, 21), have been linked to tumor progression. EGFR variant type III (EGFRvIII) is an example of an in-frame deletion of EGFR found frequently in glioblastoma multiformes as well as in non-small cell lung carcinomas and breast and ovarian cancers. Expression of this tumor-specific mutant receptor occurs in one-third of glioblastoma multiformes and is an independent negative prognostic indicator of survival (22). It results from deletion of exons 2–7 of EGFR (801 bp), which encompasses much of the ligand binding domain. This mutant exhibits constitutive tyrosine phosphorylation (23) and downstream signaling through the PI3K pathway (24) to JNK (25). Although it has been reported that EGFRvIII is completely (26) or partially (27) mislocalized intracellularly, this remains an area of controversy (28–30). An additional question we address in the present study is whether a constitutively active variant EGFR that is fully retained in the ER can cause cellular transformation.
Motivated by the hypothesis that electrostatic binding of the polybasic JX region of EGFR to acidic phospholipids restricts access of the kinase domain to substrate tyrosines, we demonstrate that sufficient reduction of the net charge in the JX region of wild-type (WT) EGFR results in constitutive activation of the receptor. Furthermore, we show that this activity is maintained when the receptor is retained in the ER via a novel point mutation that we describe. Importantly, we find that this ER-retained mutant EGFR exhibits constitutive autophosphorylation in the absence of ligand binding and induces cellular transformation by preferentially signaling through phosphatidylinositol 3-kinase (PI3K) and mTOR. In addition, we provide evidence that the highly oncogenic mutant EGFRvIII is wholly retained in the ER of U87 glioblastoma cells and is robustly phosphorylated at this intracellular membrane, demonstrating that this EGFR deletion mutant, implicated in human brain cancers, can be oncogenic in an intracellular location. Our findings provide strong evidence that constitutively active EGFRs are capable of sending transforming signals from the ER and, furthermore, that the cytoplasmic JX polybasic sequence regulates spontaneous EGFR activation and cell transformation.
EXPERIMENTAL PROCEDURES
Materials
All cell culture reagents, EGF, and precast gels for blotting were from Invitrogen. FuGENE HD was from Roche Applied Sciences. The anti-N-terminal (clone LA1) and anti-C-terminal (clone E235) human EGFR antibodies (used for immunochemistry) as well as the PVDF filters were from Millipore Corp. The anti-protein disulfide isomerase (PDI) mAb was from Affinity Bioreagents. All Alexa dye-conjugated secondary antibodies were from Invitrogen, and the Cy5-conjugated anti-rabbit IgG was from Jackson ImmunoResearch Laboratories. The antibodies that recognize the phosphorylated forms of Akt, c-Jun, and Erk as well as phospho-specific EGFR antibody (1068), anti-EGFR (used for blotting), anti-Akt, and anti-c-Jun antibodies were from Cell Signaling. The anti-actin antibody was from LabVision/Thermo. HRP-conjugated secondary antibodies used for blotting were from GE Healthcare. LY294002, PD98059, and rapamycin were from EMDBiosciences. Gefitinib (Iressa) was from Selleck Chemicals. The endoglycosidase H (Endo H) and peptide N-glycosidase F (PNGase F) as well as restriction enzymes (DpnI, XbaI, EcoRI, MfeI), and the Phusion High-Fidelity DNA Polymerase were from New England Biolabs. The anti-GFP mAb and monoclonal anti-EGFR (clone 225, Cetuximab) as well as any chemical not noted otherwise were purchased from Sigma.
Expression Plasmids
The human EGFR-GFP construct has been described previously (31). To generate the juxtamembrane mutants, site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene) or Phusion High-Fidelity DNA Polymerase (New England Biolabs). Before generation of stable cell lines, the WT and mutant EGFR constructs were cloned into the pKH3 expression vector (Addgene) using the Xba1 and EcoR1 restriction sites. Primers containing the XbaI and MfeI restriction sites were annealed to the N and C termini, respectively, of the EGFR constructs due to the presence of an EcoRI restriction site within EGFR. The particular restriction sites were chosen to eliminate the triple-HA tag, present in the pkH3 vector, from the final protein product. EGFR-vIII cDNA was obtained in an MSCV-XZ066 vector (Addgene). The EGFR-vIII-pkH3 construct was made as described above for the other EGFR constructs.
Cell Culture
Rat RBL-2H3 mast cells were grown in minimum Eagle's medium containing 20% (v/v) fetal bovine serum (FBS) (Atlanta Biologicals) and 10 μg/ml gentamicin sulfate as described previously (32). In preparation for imaging and flow cytometry, cells were plated at 25–50% confluency into 35-mm MatTek wells (MatTek Corp.) or 60-mm dishes, respectively. After ∼20 h, RBL-2H3 cells were transfected with either mutant or wild-type versions of EGFR. These constructs were transfected using FuGENE HD (Roche Applied Sciences) per the manufacturer's instructions, with modification to enhance transfection efficiency in the RBL cells previously described (32). Cells were processed for imaging or flow cytometry 24 h after transfection.
Mouse NIH 3T3 cells were grown in DMEM containing 10% (v/v) calf serum. In preparation for imaging, cells were plated at 50% confluency into 35-mm MatTek wells. After ∼20 h, cells were transfected with either mutant or wild-type versions of EGFR. These constructs were transfected using FuGENE HD (Roche Applied Sciences) per the manufacturer's instructions. U87 cells were grown in DMEM supplemented with 10% (v/v) FBS. Where indicated, NIH 3T3 or U87 cells were treated with 0.1 or 0.02 μg/ml EGF. To generate stable cell lines, NIH 3T3 or U87 cells were transfected with the various pkH3 vectors either alone or incorporating EGFR constructs using FuGENE HD (Roche Applied Sciences). Transfected cells were maintained in DMEM supplemented with 10% calf serum (NIH 3T3) or 10% FBS (U87) and 2 μg/ml puromycin (Invitrogen). After 10–14 days, puromycin-resistant colonies were selected (NIH 3T3) or pooled (U87) and subcultured in DMEM supplemented with 10% calf serum (NIH 3T3) or 10% FBS (U87) and 0.5 μg/ml puromycin. The clones/lines were then assessed for expression of each respective EGFR construct.
Confocal Fluorescence Microscopy
NIH 3T3 and RBL-2H3 cells were washed once with buffered salt solution (20 mm HEPES, 135 mm NaCl, 1.8 mm CaCl2, 2 mm MgCl2, 5.6 mm glucose, 1 mg/ml BSA, pH 7.4), fixed using 4% paraformaldehyde with 0.1% glutaraldehyde, permeabilized (or not) with either 1% v/v Triton X-100 or 0.05% w/v saponin, and labeled for 1 h with the specified antibodies in phosphate-buffered saline (PBS) with 10 mg/ml BSA and 0.01% w/v sodium azide (PBS/BSA). Images were collected using an upright Leica TCS SP2 laser scanning confocal microscope (Leica Microsystems, Exton, PA) with a 63 × 0.9 NA, HCX APO L U-V-I water-immersion objective.
Flow Cytometry
RBL-2H3 cells were harvested, washed once with buffered salt solution, fixed using 4% paraformaldehyde with 0.1% glutaraldehyde, and washed once with PBS/BSA. Cells expressing GFP-tagged wild-type or mutant EGFR constructs were labeled with the appropriate antibodies for 1 h in PBS/BSA. Samples were evaluated using a BD Biosciences LSR II flow cytometer, and data were analyzed using BD FACSDiva software. Analysis was gated to include single cells on the basis of forward and side light-scatter, and data from single-color samples were used to determine the gates for positive fluorescence from each fluorophore.
Immunoblot Analysis
Cells were washed in PBS, incubated in lysis buffer (25 mm Tris, pH 7.4, 100 mm NaCl, 1 mm EDTA, 1% (v/v) Triton 100, 1 mm DTT, 1 mm sodium orthovanadate, 1 mm β-glycerol phosphate, 1 μg/ml leupeptin, and 1 μg/ml aprotinin), and supernatants were retained following microcentrifuge centrifugation. Protein concentrations of the whole cell lysates were determined using the Bio-Rad DC protein assay. The whole-cell lysates (35 μg/lane) were resolved by SDS/PAGE, and the proteins were transferred to PVDF membranes. The filters were blocked in 10% BSA diluted in 20 mm Tris, 135 mm NaCl, and 0.02% Tween 20 and then incubated with the indicated primary antibodies diluted in the same buffer. The primary antibodies were detected with HRP-conjugated secondary antibodies followed by exposure to ECL reagent (Invitrogen).
Endo H Sensitivity Assay
NIH 3T3 or U87 cells were lysed as above, and whole-cell lysates were divided into three equal portions that then were treated or not with Endo H (33) or PNGase F according to the manufacturer's instructions (New England Biolabs). Samples were incubated for 24 h at 37°C, and the digestion reactions were stopped by heating at 95 °C for 5 min after the addition of SDS-PAGE sample buffer. Samples were analyzed by immunoblotting.
Anchorage-independent Growth Assays
NIH 3T3 cells stably overexpressing vector only (control), WT EGFR, EGFR Mut R1-5, EGFR Mut R1-6, EGFR L393H, or EGFR R1-6/L393H were plated at densities of 8.5 × 103 cells/ml in medium containing 0.3% agarose with or without EGF or the specified inhibitors onto underlays composed of growth medium containing 0.6% agarose in 6-well dishes. The soft agar cultures were re-fed with fresh medium (including EGF or specified inhibitors) every 5th day for 15–20 days, at which time the colonies that had formed were counted. Each of the assays was performed at least three times, and the results were averaged and expressed ± S.D. Experiments using parental U87 cells and stably expressing EGFRvIII cells were performed similarly, except 3 × 103 cell/ml were plated in medium containing 0.3% agarose, and colonies were enumerated after 6 days, including re-feeding with fresh medium ± inhibitor on day 3.
Data Analysis
Statistical analyses were determined using GraphPad Prism (GraphPad Software, La Jolla, CA) using Student's t test, with p ≤ 0.05 considered statistically significant.
RESULTS
Mutations in EGFR Influence Its Trafficking from the ER to the Plasma Membrane
In an attempt to understand the functional roles of the polybasic JX cytoplasmic sequence in EGFR-mediated cell signaling (5, 34–36), we sequentially mutated these basic residues to alanines (Fig. 1A). Based on the work of others (11, 12), we hypothesized that charge silencing within the JX sequence of EGFR would release regulatory electrostatic interactions with membrane lipids and thereby promote the activity of the TKD (see “Discussion”). Whereas WT EGFR-GFP is found almost exclusively at the PM when expressed in RBL mast cells, reduction in the net charge of the JX region from +8 to +3 (EGFR Mut R1-6) initially appeared to result in complete intracellular retention. However, sequencing revealed that unintentional mutagenesis occurred during the construction of Mut R1-6 and that a single point mutation, L393H, in the EGFR extracellular domain is responsible for the complete intracellular retention (EGFR Mut R1-6/L393H). When this single point mutation was made in the context of WT EGFR (wt EGFR L393H), trafficking was similarly prevented, as evidenced by both confocal imaging (Fig. 1B) and quantitative flow cytometry (Fig. 1, C and E). Notably, none of these mutations (L393H or JX) significantly altered the protein expression levels (Fig. 1D).
FIGURE 1.

A single point mutation L393H within the N-terminal segment of EGFR inhibits its capacity to traffic to the plasma membrane. A, wild-type and mutant EGFR-GFP constructs generated. The JX sequence in EGFR is shown, with basic amino acids in bold and mutated residues underlined. (TM = transmembrane). B, expression of WT and mutant EGFR-GFP constructs in suspended RBL mast cells. Transiently expressing cells were fixed and labeled with an anti-N-terminal EGFR antibody followed by Alexa 647-anti-IgG. Scale bar, 10 μm. C, representative flow cytometric scatter plots. RBL-2H3 cells transiently expressing GFP-tagged EGFR constructs were harvested, fixed, and labeled with anti-N-terminal EGFR antibody followed by Alexa 647-anti-IgG. Cells were gated on positive GFP fluorescence (blue and purple populations), and Alexa 647 fluorescence was analyzed to determine PM localization (purple population). D, EGFR-GFP constructs express at similar levels. The average GFP fluorescence (total EGFR fluorescence) is from the data plotted in E. UN, untransfected; LH, L393H point mutation; n.s, not significant; a.u., arbitrary units. E, quantification of the effect of mutation of the polybasic JX segment of EGFR and the L393H point mutation (LH) on biosynthetic trafficking. The ratio of PM-localized EGFR fluorescence to total EGFR fluorescence for mutants is normalized to WT, and error bars indicate ± S.D. of three independent experiments in which ≥8000 cells expressing each construct were analyzed.
Strong evidence that EGFR L393H and EGFR Mut R1-6/L393H localize to the ER is provided by immunofluorescent colocalization with the resident lumenal ER protein, PDI (Fig. 2A), and by complete sensitivity of EGFR Mut R1-6/L393H to digestion by Endo H (Fig. 2B). Endo H acts by cleaving N-linked oligosaccharide chains near their site of attachment to the protein, and once a protein has trafficked to the Golgi, further oligosaccharide processing results in resistance to Endo H digestion. PNGase F removes all glycosylations and serves as a control for these assays. When lysates from cells expressing EGFR Mut R1-6/L393H are subjected to Endo H digestion, the Endo H-sensitive band is the same molecular weight as the PNGase F-sensitive band, thus supporting the contention that EGFR Mut R1-6/L393H contains only N-linked oligosaccharide chains and has not trafficked beyond the ER. In contrast, WT EGFR and EGFR Mut R1-6 (without L393H) show only a small amount of Endo H sensitivity (Fig. 2B), consistent with confocal imaging and flow cytometry results showing predominant surface localization for both of these proteins (Fig. 1). Immunoblot analysis shows that EGFR Mut R1-6/L393H retained in the ER is intact in RBL cells.3 ER-retained EGFR Mut R1-6/L393H appears to be oriented correctly across the ER membrane, with its C-terminal domain extending into the cytosol; confocal imaging of cells in which the PM (but not the ER) was selectively permeabilized with a low concentration of saponin showed that these receptors are labeled with anti-GFP antibody (Fig. 3). Collectively, these results show that EGFR Mut R1-6/L393H is confined to the ER, whereas EGFR Mut R1-6 is largely expressed at the PM.
FIGURE 2.
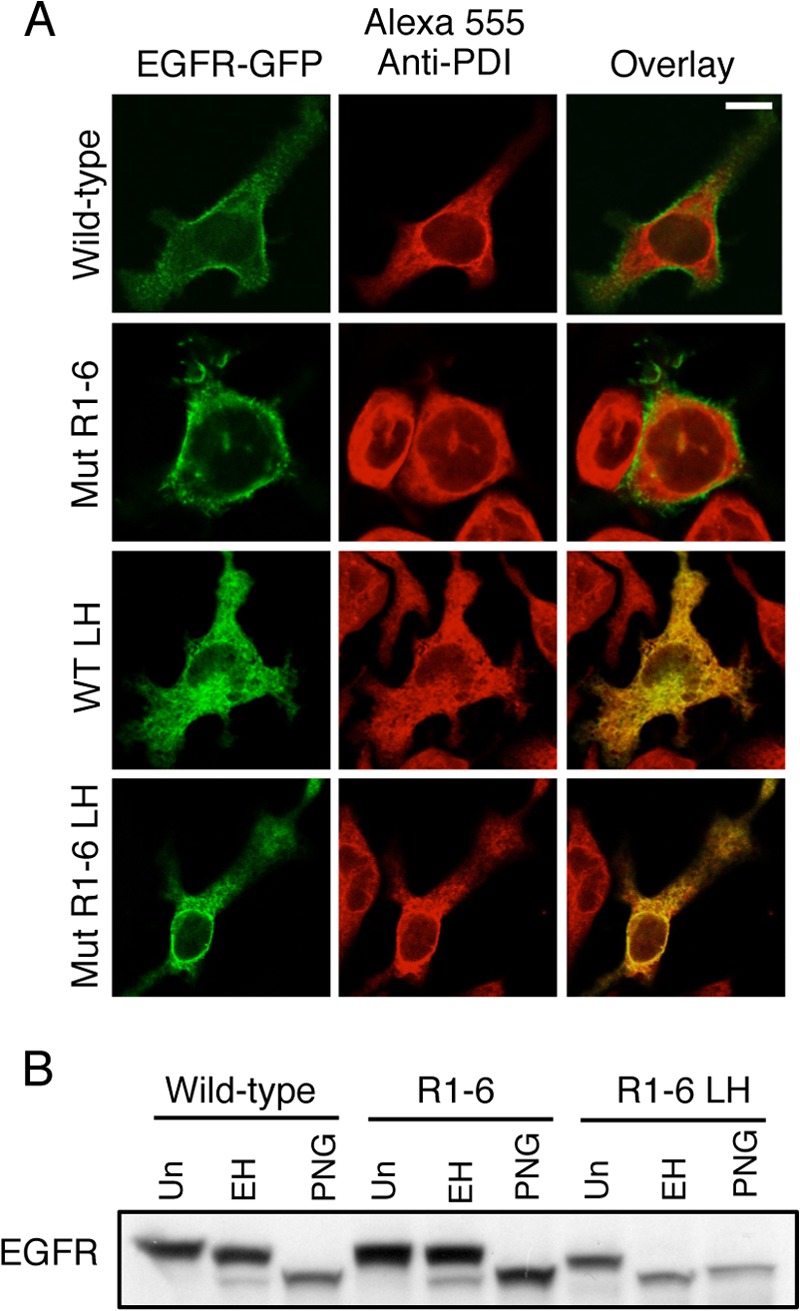
EGFR-GFP Mut R1-6/L393H is retained and intact within the ER. A, attached RBL mast cells transiently expressing WT EGFR-GFP, EGFR-GFP Mut R1-6, EGFR-GFP L393H, or EGFR-GFP Mut R1-6/L393H were fixed, permeabilized, and labeled with an anti-PDI antibody followed by Alexa555-anti-mouse IgG to label the ER. Scale bar, 10 μm. B, whole cell lysates from RBL mast cells transiently expressing either WT EGFR, EGFR Mut R1-6, or EGFR Mut R1-6/L393H (LH) were untreated (Un), treated with Endo H (EH), or treated with PNGase F (PNG) for 24 h before Western blot analysis with an anti-EGFR antibody.
FIGURE 3.
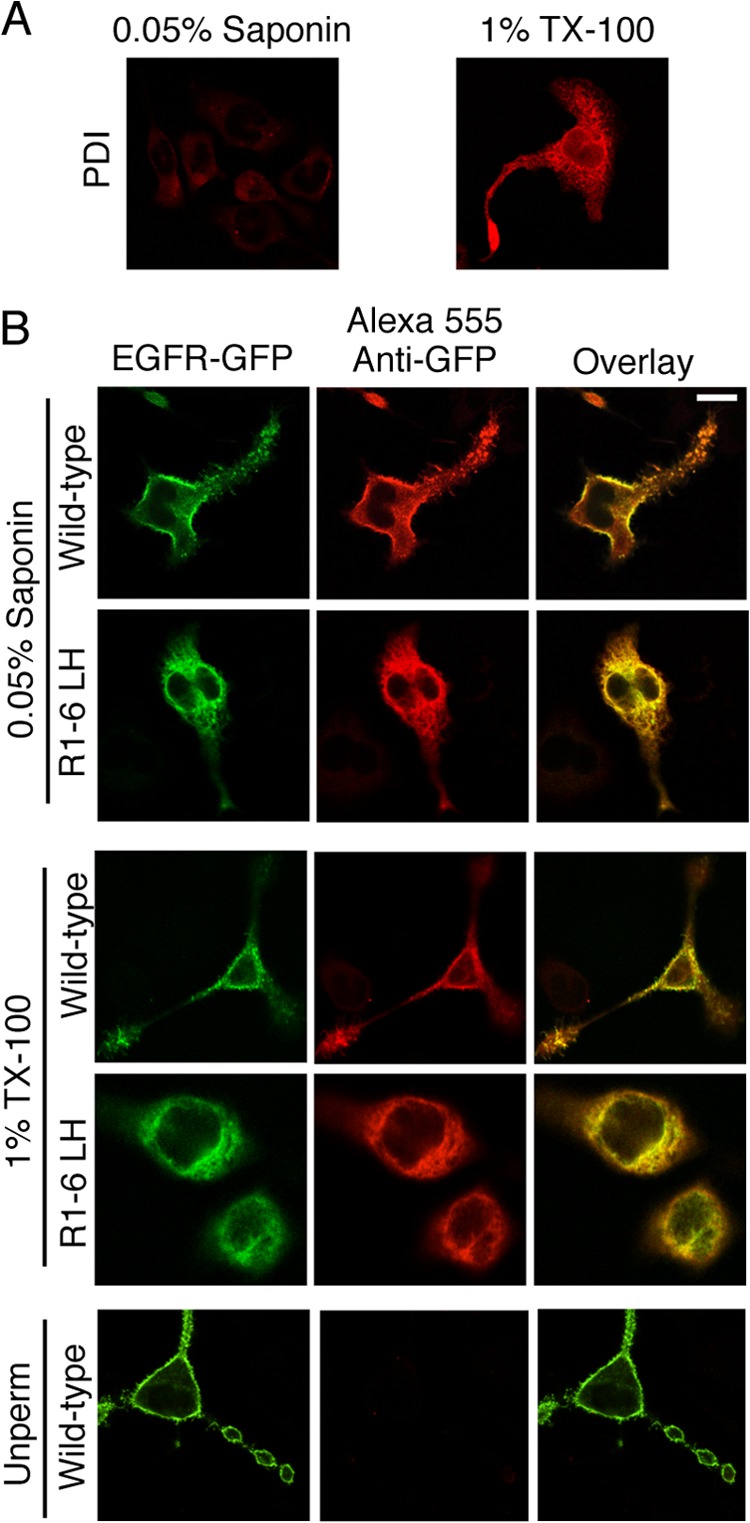
EGFR-GFP Mut R1-6/L393H is correctly oriented across the ER membrane. A, the lumen of the ER, marked by PDI, is accessed when 1% Triton X-100 is used as a permeabilization agent but not with 0.05% saponin, which permeabilizes the PM without permeabilizing internal membranes. RBL-2H3 cells were fixed, permeabilized as indicated, and labeled with an anti-PDI mAb followed by Alexa 555-anti-mouse IgG. B, RBL-2H3 cells transiently expressing WT EGFR-GFP or EGFR-GFP Mut R1-6/L393H (R1-6 LH) were fixed, permeabilized (or not) as indicated, and labeled with anti-GFP mAb followed by Alexa555-anti-mouse IgG to label the C terminus of the expressed EGFR construct. The GFP epitope on the EGFR-GFP Mut R1-6/L393H C terminus is labeled after permeabilization (Unperm) with either 0.05% saponin or Triton X-100, consistent with cytoplasmic exposure. Scale bar, 10 μm.
To investigate the basis for retention of EGFR L393H, we mutated Leu-393 to amino acids other than histidine. We found that the causative factor for retention is loss of leucine 393, as replacement of this residue with alanine also results in complete retention in the ER, and replacement with a phenylalanine results in substantially reduced PM association (Fig. 4). Whereas replacement of Leu-393 with histidine or alanine results in ER retention, we think this is not due to gross misfolding of the protein; EGFR acquires the capacity to bind ligand after exiting from the ER (37), and neither the ER-localized WT receptor nor EGFR Mut R1-6/L393H bind the conformation-specific antibody, Cetuximab (Fig. 5). However, EGFR L393F, which does traffic from the ER, binds Cetuximab once it is Golgi- or PM-localized (Fig. 5), indicating that mutation of Leu-393 per se does not globally disrupt the folding of the extracellular EFGR region.
FIGURE 4.
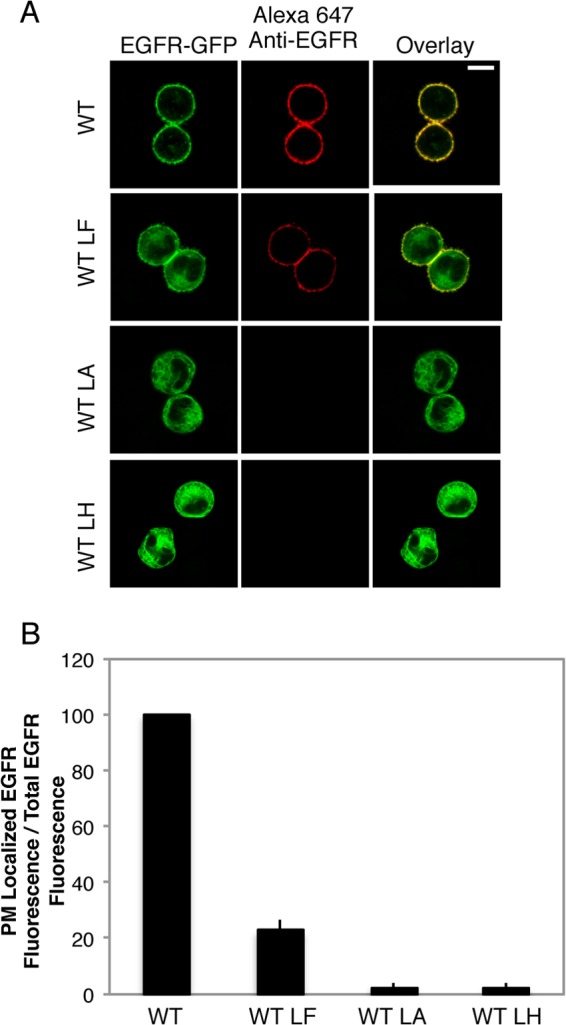
EGFR-GFP L393A was ER-retained, whereas EGFR-GFP L393F showed reduced PM association. A, representative confocal images of WT and mutant EGFR-GFP constructs in suspended RBL mast cells. Transiently expressing cells were fixed and labeled with an anti-N-terminal EGFR antibody followed by Alexa 647-anti-IgG to reveal surface expression. Scale bar, 10 μm. LH, L393H; LF, L393F; LA, L393A. B, quantification of the effect of the L393H, L393F, and L393A point mutations on biosynthetic trafficking. RBL-2H3 cells transiently expressing GFP-tagged EGFR constructs were harvested, fixed, and labeled with anti-N-terminal EGFR antibody followed by Alexa 647-anti-IgG, then analyzed by flow cytometry to determine the ratio of PM-localized EGFR fluorescence to total-EGFR fluorescence. Data from mutants is normalized to WT, and error bars indicate ± S.D. of three independent experiments in which ≥8000 cells expressing each construct were analyzed.
FIGURE 5.
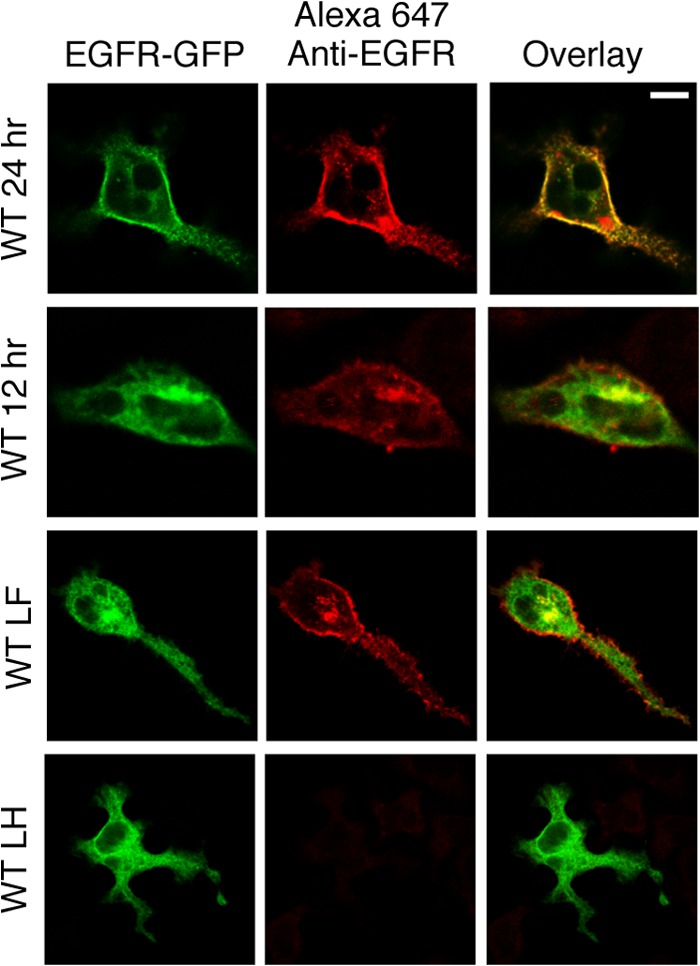
Wt EGFR and EGFR L393F bind Cetuximab only after they exit the ER. RBL-2H3 cells transiently expressing WT EGFR-GFP, EGFR-GFP L393F (LF), or EGFR-GFP L393H (LH) for either 12 h or 24 h were fixed, permeabilized, and labeled with the anti-EGFR mAb Cetuximab followed by Alexa 647-anti-mouse IgG to label the EGF binding segment of the expressed EGFR construct. Specific labeling was observed once the receptors exit the ER, and EGFR L393F bound Cetuximab as well as WT EGFR in both the Golgi region and PM. Scale bar, 10 μm.
Both EGFR Mut R1-6 and ER-retained EGFR Mut R1-6/L393H Transform Cells Independently of EGF
A previous study showed that individual mutations of five of the six basic residues to alanine significantly reduced the constitutive tyrosine kinase activity of a cytoplasmic intracellular domain construct (4). In contrast, we hypothesized that charge silencing within the JX sequence of full-length EGFR would release regulatory electrostatic interactions with acidic phospholipids at the membrane and thereby promote the activity of the TKD. Thus, we tested the possibility that EGFR Mut R1-6 would promote cell transformation due to constitutive activation by generating NIH 3T3 cell lines stably overexpressing WT EGFR, EGFR Mut R1-5, EGFR Mut R1-6, EGFR L393H, and EGFR Mut R1-6/L393H and comparing their anchorage-independent growth. We selected a NIH 3T3 subline that has an unusually low expression of endogenous EGFR (38), and the EGFR constructs we used for stable expression contained no GFP tag.
Selected clones stably expressing WT EGFR, EGFR Mut R1-5, and EGFR Mut R1-6 were found to express their respective EGFR proteins to similar extents, whereas the level of ER-retained EGFR Mut R1-6/L393H is somewhat lower and that of EGFR L393H is further reduced (Fig. 6A). However, in all five cases robust overexpression of EGFR was observed to be far above the endogenous level of EGFR expressed by the parent NIH 3T3 cell line (vector control; Fig. 6A). Furthermore, cellular distributions of these constructs, monitored with immunostaining, are similar to their GFP-tagged counterparts transiently expressed in RBL-2H3 cells (compare Figs. 1B and 6B). Similarly, Endo H sensitivity supports the conclusion that these stably expressed EGFR proteins localize in the NIH 3T3 cells similarly to their transiently expressed counterparts in RBL-2H3 cells; WT EGFR and EGFR Mut R1-6 are largely trafficked out of the ER, whereas EGFR mutants that contain L393H are localized exclusively in the ER (Figs. 6C and 2B).
FIGURE 6.
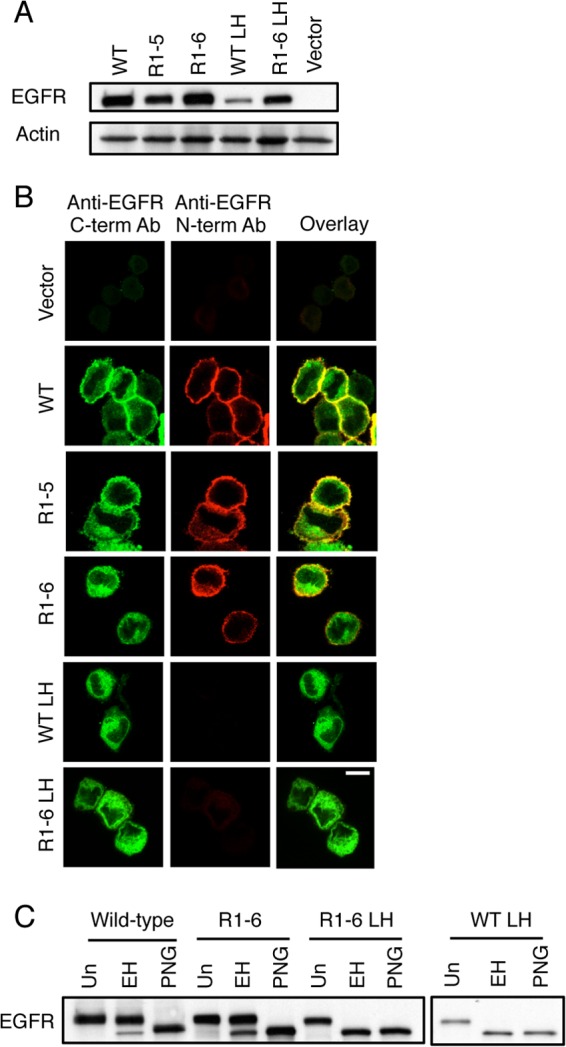
EGFR Mut R1-6/L393H is retained in the ER when stably expressed in NIH 3T3 cells. A, whole cell lysates of NIH 3T3 cells stably expressing WT EGFR, EGFR Mut R1-5, EGFR Mut R1-6, EGFR L393H (LH), EGFR Mut R1-6/L393H, or vector only were subjected to Western blot analysis using an anti-EGFR antibody and an anti-actin antibody (loading control). B, confocal images of NIH 3T3 cells stably expressing vector only, WT EGFR, EGFR R1-5, EGFR Mut R1-6, EGFR L393H, or EGFR Mut R1-6/L393H. Before imaging cells were fixed and labeled with an anti-N-terminal EGFR antibody followed by Alexa 647 anti-mouse IgG, then washed, permeabilized, and labeled with anti-C-terminal EGFR antibody followed by Alexa 488-anti-rabbit IgG. Scale bar, 10 μm. C, whole cell lysates from NIH 3T3 cells stably expressing either the WT EGFR, EGFR Mut R1-6, EGFR Mut R1-6/L393H, or EGFR L393H were untreated (Un) or were treated with Endo H (EH) or with PNGase F (PNG) for 24 h before Western blot analysis with an anti-EGFR antibody.
We tested the stably expressed receptors for their capacity to induce anchorage-independent growth, a reliable indicator of transformation (39). Although the NIH 3T3 cells stably expressing vector alone express small amounts of endogenous EGFR, these levels are not sufficient to mediate anchorage-independent growth even when stimulated with EGF. Overexpression of WT EGFR results in robust transformation but only in the presence of EGF (Fig. 7, A and B). Similarly, overexpressed EGFR Mut R1-5 transforms NIH 3T3 cells in an EGF-dependent manner. However, stably expressed EGFR Mut R1-6 stimulates anchorage-independent growth of NIH 3T3 cells even in the absence of EGF. Furthermore, EGFR Mut R1-6/L393H retains the capacity to transform cells in a ligand-independent manner, whereas EGFR L393H is incapable of transforming NIH 3T3 fibroblasts in the absence or presence of ligand. The transforming capacity of EGFR Mut R1-6 and EGFR Mut R1-6/L393H is observed in multiple clones, and the degree of colony formation by these clones correlates with EGFR Mut R1-6/L393H expression levels (Fig. 7, C and D). In addition, EGFR Mut R1-6, which expresses to a higher level than EGFR Mut R1-6/L393H (Fig. 6A), forms more colonies than ER-retained EGFR Mut R1-6/L393H in soft agar in the presence and absence of EGF (Fig. 7, A and B). Collectively, these results show that the charge-silencing mutation R1-6 in the JX region leads to structural alterations in EGFR that cause ligand-independent transformation by receptors at the PM and the ER.
FIGURE 7.

ER-retained EGFR MutR1-6 transforms cells in a growth factor-independent manner. A, NIH 3T3 fibroblasts stably expressing vector, WT EGFR, EGFR Mut R1-5, EGFR Mut R1-6, EGFR L393H (LH), or EGFR Mut R1-6/L393H incubated with or without EGF (100 ng/ml) were cultured in soft agar. The data shown represent the mean ± S.D. for colonies per 2.25 cm2 from at least three independent experiments. B, representative images of the colonies quantified in A. Scale bar, 100 μm. C, two different clones of NIH 3T3 fibroblasts stable expressing EGFR Mur R1-6 cultured in soft agar as in A (Mean ± S.D. from three independent experiments). Western blot analysis using an anti-EGFR antibody was performed on whole cell lysates from the same two clones without or with EGF stimulation to determine relative expression levels. D, three different clones of NIH 3T3 fibroblasts stably expressing EGFR Mut R1-6/L393H cultured in soft agar as in A (mean ± S.D. from three independent experiments). Western blot analysis using an anti-EGFR antibody was performed on whole cell lysates from the same three clones and vector control cells without or with EGF stimulation to determine relative expression levels. The blot is representative of trends observed in multiple experiments.
We observe that EGF consistently enhances the number of colonies formed in NIH 3T3 cells stably expressing EGFR Mut R1-6/L393H by a small, but statistically significant, amount (Fig. 7A). Thus, we tested the possibility that a small population of EGFR Mut R1-6/L393H, below the level of detection by imaging (Fig. 6B) and Endo H sensitivity assays (Fig. 6C), is trafficked to the PM. Anchorage-independent growth assays were carried out on the cells expressing EGFR Mut R1-6/L393H in the presence of the anti-EGFR antibody Cetuximab (mAb 225), which prevents the transition of EGFR to its active conformation (40). Ligand-independent transformation mediated by EGFR Mut R1-6/L393H that is due to a PM-associated pool would be expected to be reduced in the presence of this antibody. However, the addition of Cetuximab did not decrease transformation mediated by EGFR Mut R1-6/L393H under conditions in which it significantly inhibited transformation by WT EGFR (Fig. 8). These findings suggest that the enhanced transformation seen in NIH 3T3 cells stably expressing the EGFR Mut R1-6/L393H in the presence of EGF is due to signaling by endogenous EGFR at the PM that synergizes with constitutively active EGFR Mut R1-6/L393H at the ER (see “Discussion”).
FIGURE 8.
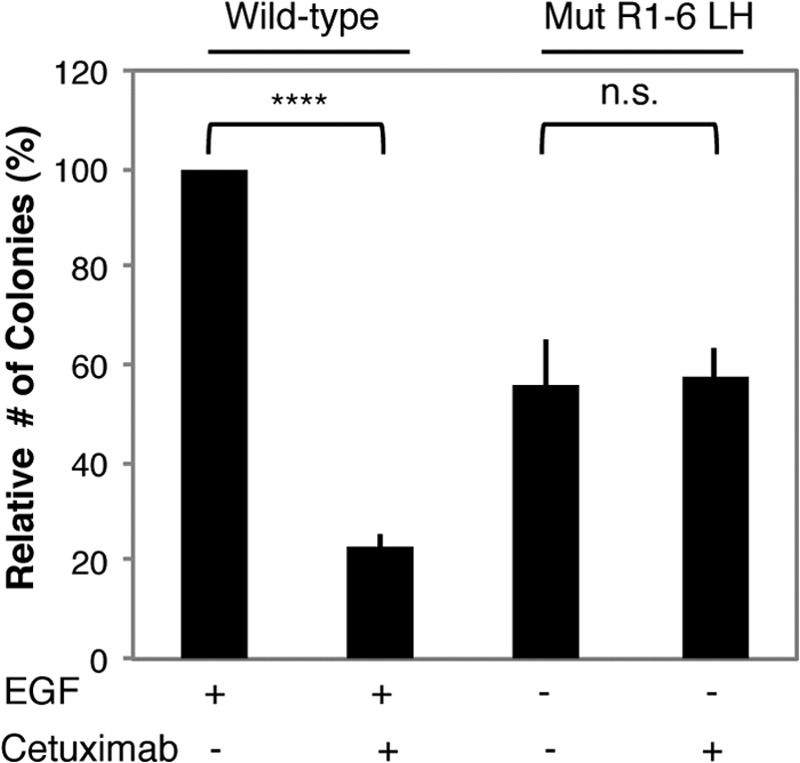
Anti-EGFR Cetuximab does not inhibit transformation by EGFR Mut R1-6/L393H. NIH 3T3 cells stably expressing WT EGFR or EGFR Mut R1-6/L393H (LH) were cultured in soft agar in the presence or absence of Cetuximab (5 μg/ml) and/or EGF (20 ng/ml). Mean ± S.D. is from three independent experiments, normalized to WT EGFR + EGF. ****, p < 0.01; n.s., not significant.
Transformation Mediated by EGFR Mut R1-6 Requires Its Kinase Activity
We investigated whether EGFR Mut R1-6 and EGFR Mut R1-6/L393H are constitutively active. We found that treatment with the EGFR-tyrosine kinase inhibitor gefitinib (Iressa) at 2.5 μm eliminates transformation of NIH 3T3 cells expressing either EGFR Mut R1-6 or EGFR Mut R1-6/L393H and by EGF-stimulated NIH 3T3 cells overexpressing WT EGFR (Fig. 9A). The effects of gefitinib on these stable cell lines is dose-dependent, with IC50 values of <2 μm gefitinib.3 We observed significant basal phosphorylation of both EGFR Mut R1-6 and EGFR Mut R1-6/L393H by Western blotting of whole cell lysates from cells expressing these mutant receptors using anti-phospho-EGFR (Tyr-1068) antibody. The phosphorylation of EGFR Mut R1-6/L393H is not detectably enhanced upon EGF addition, and it decreases substantially upon treatment with gefitinib (Fig. 9B). These results suggest that the ligand-independent transformation mediated by EGFR Mut R1-6/L393H is consistent with its constitutive activity. Wt EGFR (and EGFR L393H)3 exhibit lower levels of constitutive phosphorylation at residue 1068, and Endo H digestion confirms that the WT EGFR and EGFR Mut R1-6, which become robustly phosphorylated at Tyr-1068 upon the addition of ligand, are largely at the PM (Fig. 9C). In contrast, Tyr-1068-phosphorylated EGFR Mut R1-6/L393H is retained in the ER (Fig. 9C), providing additional evidence that receptor signaling from this location leads to transformation.
FIGURE 9.
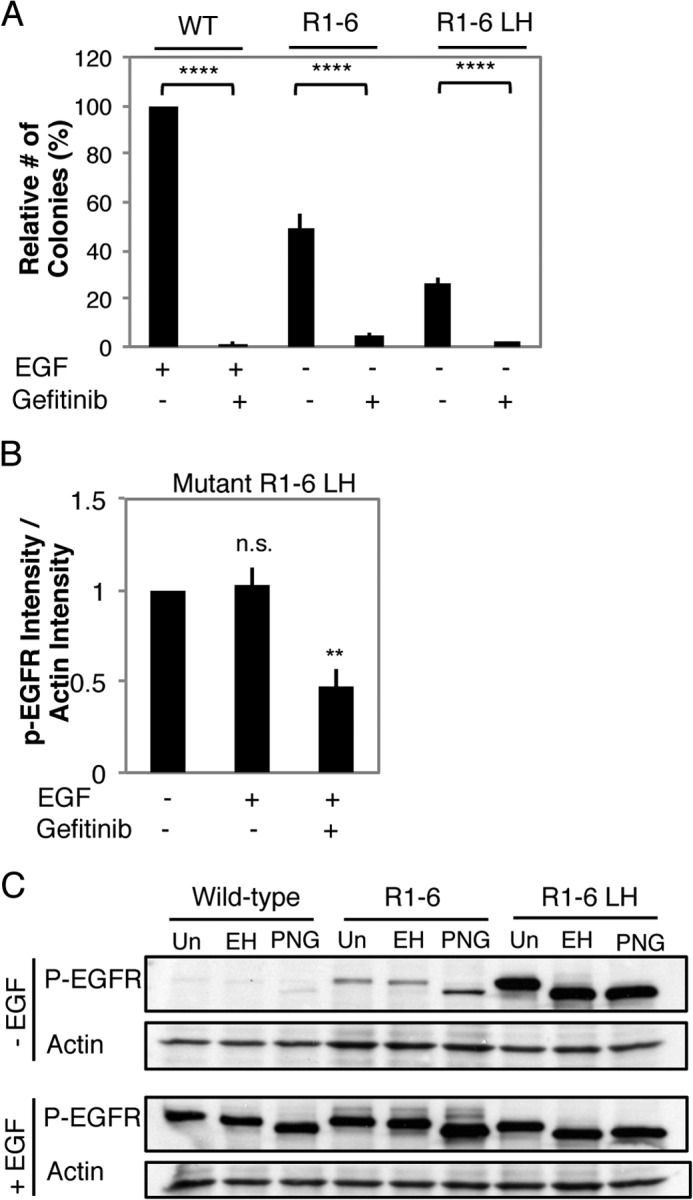
Transformation by EGFR Mut R1-6 and ER-retained EGFR Mut R1-6/L393H and their ligand-independent phosphorylation are EGFR kinase-dependent. A, inhibition of colony formation of NIH 3T3 cells stably expressing WT EGFR, EGFR Mut R1-6, or EGFR Mut R1-6/L393H (LH) by the EGFR kinase inhibitor gefitinib (2.5 μm). Data shown are the mean ± S.D. from three independent experiments normalized to WT EGFR + EGF. ****, p < 0.0001. B, quantification of phospho-Tyr-1068 EGFR detected in the whole cell lysates from serum-starved NIH 3T3 cells expressing EGFR Mut R1-6/L393H treated as shown (gefitinib = 5 μm). Data were averaged from four independent experiments. Error bars show S.E., **, p < 0.01; n.s., not significant. C, serum-starved NIH 3T3 cells stably expressing either WT EGFR, EGFR Mut R1-6, or EGFR Mut R1-6/L393H were treated without or with EGF as in Fig. 3. Lysates (equal protein content) were untreated (Un) or were treated with Endo H (EH) or with PNGase F (PNG) for 24 h before Western blot analysis using anti-phospho-Tyr-1068-EGFR and anti-actin antibodies.
The ER-retained EGFR Mut R1-6/L393H Signals through the PI3K Pathway
To determine the signaling pathway(s) responsible for mediating the ligand-independent transforming activity of EGFR Mut R1-6/L393H, we focused our investigation on proteins that interact with EGFR via phospho-Tyr-1068, including PI3K. Fig. 10 summarizes results comparing the activity of this pathway in the NIH 3T3 cells stably expressing WT EGFR to those expressing EGFR Mut R1-6/L393H. Cells expressing EGFR Mut R1-6/L393H exhibit higher basal levels (without EGF stimulation) of activated Akt than cells expressing WT EGFR (Fig. 10, A and B), and this activation is prevented by treatment with the PI3K inhibitor LY294002 at 10 μm (Fig. 10A). Treatment with a concentration of LY294002 (5 μm) that only partially decreases EGF-dependent WT EGFR-mediated anchorage-independent cell growth abolishes colony formation mediated by EGFR Mut R1-6/L393H, suggesting greater sensitivity of transformation by this mutant to PI3K inhibition (Fig. 10C). Similarly, transformation mediated by EGFR Mut R1-6/L393H is more susceptible to rapamycin, an inhibitor that interferes with the capacity of PI3K to signal through mTOR, than transformation mediated by WT EGFR (Fig. 10D). Cells expressing EGFR Mut R1-6 without L393H also exhibit increased susceptibility to PI3K (Fig. 10C) and mTOR inhibition (Fig. 10D) as compared with WT EGFR-expressing cells. However, they are not as susceptible as cells expressing EGFR Mut R1-6/L393H. In contrast, cells expressing EGFR Mut R1-6/L393H do not show elevated levels of phosphorylated Erk compared with WT EGFR-expressing and vector control cells with or without EGF stimulation, and treatment with the MEK inhibitor PD98059 inhibits transformation mediated by WT EGFR and EGFR Mut R1-6/L393H to a similar extent (Fig. 11). These results provide a consistent picture that the transformation caused EGFR Mut R1-6 and EGFR Mut R1-6/L393H is mediated primarily by the PI3K-mTOR signaling axis.
FIGURE 10.
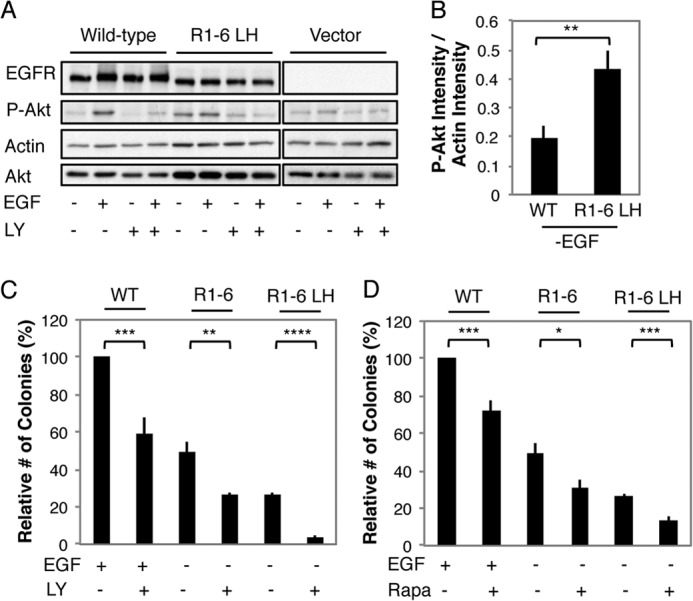
Transformation mediated by ER-retained EGFR Mut R1-6/L393H depends on PI3K signaling. A, whole cell lysates from serum-starved NIH 3T3 cells stably expressing WT EGFR or EGFR Mut R1-6/L393H (LH) and treated as indicated were immunoblotted with antibodies specific for phospho-Akt S473, total Akt, total EGFR, or actin (loading control). Before lysing the cells, EGF was added for 5 min at 100 ng/ml in the presence or absence of 10 μm LY294002 (LY). B, quantification of Akt phosphorylation in cells expressing WT EGFR or EGFR Mut R1-6/393H in the absence of EGF. Data were averaged from five independent experiments including those shown in A (error bars show S.E.; **, p < 0.01). C, the effect of the PI3K inhibitor LY294002 (5 μm) on the capacity of WT EGFR-, EGFR Mut R1-6-, or EGFR Mut R1-6/393H-expressing cells to grow in soft agar. The data shown represent the mean ± S.D. from three independent experiments, normalized to WT EGFR + EGF. **, p < 0.01; ****, p < 0.0001. D, the effect of mTOR inhibition on the capacity of WT EGFR-, EGFR Mut R1-6-, or EGFR Mut R1-6/L393H-expressing cells to grow in soft agar was assessed by adding rapamycin (Rapa, 25 nm) to soft agar cultures. The mean ± S.D. is from three independent experiments, normalized to WT EGFR + EGF. * p < 0.05; ***, p < 0.001.
FIGURE 11.

Contribution of Erk pathway to EGFR Mut R1-6/L393H-mediated transformation. A, whole cell lysates of serum-starved NIH 3T3 cells stably expressing vector control, WT EGFR, or EGFR Mut R1-6/L393H (LH) were treated or not with EGF (100 ng/ml) ± gefitinib (5 μm) and immunoblotted with antibodies against phospho-Erk, total EGFR, and actin (loading control). B, inhibition of transformation by the MEK inhibitor PD98059 (50 μm). The mean ± S.D. is from three independent experiments, normalized to WT EGFR + EGF. ***, p < 0.001.
EGFRvIII Localizes to the ER Where It Is Constitutively Active in U87 Cells
It is becoming increasingly clear that the subcellular localization of a signaling protein can influence signaling outputs (41). Although EGFR R1-6/L393H has not been observed clinically, it has been suggested that other cancer-causing EGFR mutants, such as EGFRvIII, are ER-localized (26). Because the subcellular distribution of EGFRvIII in cancer cells has been controversial, we investigated this question by generating U87 human glioblastoma cells that stably express EGFRvIII. Similar to EGFR Mut R1-6/L393H in NIH 3T3 cells, we found that EGFRvIII in U87 cells is robustly phosphorylated on tyrosine 1068 independent of EGF, and Akt and c-Jun, components of the PI3K signaling pathway, are also basally phosphorylated (Fig. 12A). U87 cells expressing EGFRvIII show enhanced transformation compared with vector control, as revealed by anchorage-independent growth assays in soft agar, and this colony formation is inhibited by treatment with gefitinib (Fig. 12B). Immunostaining of EGFRvIII with an antibody specific for the cytoplasmic C terminus of EGFR showed a clear reticulate pattern that colocalizes with PDI (Fig. 12C), and ER localization of EGFRvIII in U87 cells was further confirmed by complete sensitivity of this mutant receptor to digestion by Endo H (Fig. 12D). Most importantly, Tyr-1068-phosphorylated EGFRvIII is Endo H-sensitive, corresponding to ER localization, whereas Tyr-1068-phosphorylated WT EGFR is not, consistent with PM localization of this receptor (Fig. 12D). These results provide evidence that the human oncogene, EGFRvIII, like EGFR R1-6/L393H, can signal from the ER in a ligand-independent manner.
FIGURE 12.

EGFRvIII stably expressed in U87 glioblastoma cells is robustly phosphorylated, enhances cell transformation, and is localized to the ER. A, parental U87 cells and those stably expressing EGFRvIII were treated without or with 100 ng/ml EGF for 5 min. Whole cell lysates were electrophoresed, and Western blots were probed with antibodies that recognize EGFR, the activated form of EGFR (phospho-Tyr-1068-EGFR), P-Akt (T308), P-c-Jun, total Akt, or total c-Jun. B, parental U87 cells and those stably expressing EGFRvIII were cultured in soft agar without EGF with or without gefitinib (20 μm). The data shown represent the mean ± S.D. for colonies per 2.25 cm2 from at least three independent experiments. ***, p < 0.001; ****, p < 0.0001. C, parental U87 cells and those stably expressing EGFRvIII were fixed, permeabilized, and labeled with an anti-C-terminal EGFR antibody followed by Alexa 488-anti-rabbit IgG. Cells were then labeled with an anti-PDI antibody followed by Alexa555-anti-mouse IgG to label the ER. Scale bar, 10 μm. D, whole cell lysates from parental U87 cells and those stably expressing EGFRvIII treated without or with EGF (100 ng/ml) were untreated (Un) or treated with Endo H (EH), or with PNGase F (PNG) for 24 h before Western blot analysis using an anti-EGFR antibody or an anti-phospho-Tyr-1068-EGFR antibody.
DISCUSSION
Signal generation from phosphorylated RTKs that are activated by their respective ligands at the PM has been a tenet in the field of receptor biology since the early 1980s (42). The findings that many epithelial tumors overexpress RTKs, particularly members of the ErbB family, and that they are key contributors in cancer progression (1, 43) led to an explosion of a field that continues to grow at a rapid pace today. By the end of that decade it was understood that, like overexpression, mutations in RTKs can lead to dysregulated signaling. A prominent example in human brain cancers is EGFRvIII, a deletion mutant lacking exons 2–7 (20). Although a number of other cancer-causing mutations within surface-localized RTKs have been described previously (3), impaired receptor trafficking both to and from the plasma membrane has been recognized more recently as yet another possible mechanism of dysregulation (44, 45).
Prompted by the hypothesis that the basic residues in the JX region of EGFR negatively regulate kinase domain interactions, we sequentially mutated these residues to alanines. We found that by reducing the net charge of the polybasic sequence within the JX region of EGFR from +8 to +3 we generated a receptor (EGFR Mut R1-6) that is constitutively active and can transform cells even when retained in the ER by a point mutation (L393H; Figs. 7 and 9). We showed that EGFR Mut R1-6/L393H receptors are intracellularly retained using both confocal microscopy and quantitative flow cytometry (Figs. 1 and 6), and we identified the site of retention as the ER both by colocalization with the ER-marker PDI (Fig. 2A) and by Endo H digestion (Figs. 2B and 6C). Similarly, we found that EGFRvIII stably expressed in U87 cells is completely Endo H-sensitive (Fig. 12D) and that its immunolabeling colocalizes with PDI (Fig. 12C), consistent with its ER localization. Furthermore, we show that both EGFRvIII (Fig. 12D) and EGFR Mut R1-6/L393H (Fig. 9C) are constitutively phosphorylated in the ER and signal primarily through the PI3K pathway to mediate cell transformation, which is sensitive to gefitinib (Figs. 9A and 12B).
Although ER-retained RTK mutants have been described previously (26, 46), these mutants all possess a large deletion of the original protein sequence. Novel to this study is the discovery that a single amino acid substitution of leucine 393 for histidine results in the retention of EGFR in the ER. The causative factor for retention seems to be the loss of leucine 393, as we show that replacement of this residue with alanine also results in complete ER retention, and replacement with a phenylalanine results in dramatically reduced PM association (Fig. 4). Leucine 393 is located in domain III of the extracellular region of EGFR, which resembles other leucine-rich repeat proteins (47, 48). We hypothesize that replacement of leucine 393 with a histidine or alanine residue disrupts the closely packed hydrophobic core of the leucine-rich repeat, and this disruption results in a structural change that in turn disallows EGFRs containing this mutation from exiting the ER. Interestingly, human ErbB3 and ErbB4 both have a hydrophobic phenylalanine at position 393 (48), consistent with the observed tolerance for this substitution in EGFR.
Although the L393H point mutation is primarily responsible for the ER retention described herein, we consistently observe a modest reduction in the trafficking of EGFR Mut R1-6 to the PM. Based on quantitative flow cytometry, we observed a ∼35% decrease in the ratio of PM-localized to total expressed EGFR Mut R1-6 as compared with WT EGFR (Fig. 1E), and it appears that this is due to partial retention in the ER, based on confocal imaging (Figs. 1B and 6B) and Endo H digestion assays (Fig. 6C). EGFR contains a canonical di-acidic ER exit motif downstream from an YXXφ targeting sequence in its C terminus (49). It has been demonstrated in other proteins, such as the vesicular stomatitis viral glycoprotein (vsv-g), that both these sequences are important for efficient ER exit (50). We hypothesize that, by mediating protein-lipid interactions, the polybasic JX region of EGFR positions this potential ER exit motif for efficient recognition by the COPII trafficking machinery. In preliminary experiments to test this hypothesis, we generated a mutant in which the residues defined to be important for ER exit within these two motifs were replaced with alanines. Initial results suggest that the trafficking rate of this mutant is very similar to that of Mut R1-6,3 supporting the possibility that the JX region is functionally linked to this downstream ER exit motif.
It has been suggested that electrostatic binding of the JX region and a TKD face to acidic lipids in the PM restrict access of the kinase domain to substrate tyrosines in an inactive conformation (11). Recently reported molecular dynamics simulations provide further support that basic residues in the JX region of EGFR can associate electrostatically with acidic lipids and contribute to burying hydrophobic residues of this JX region of non-activated EGFRs in the membrane (6). Based on these observations, we hypothesized that an EGFR mutant lacking these basic amino acids has a more easily accessible kinase domain that signals without regulation. Consistent with this view, we show that stable expression of EGFR Mut R1-6 confers the capacity of NIH 3T3 cells to form colonies under anchorage-independent and ligand-independent conditions irrespective of its localization at the PM or ER (Fig. 7). Notably, stable expression of EGFR Mut R1-5 does not result in ligand-independent transformation, indicating that reduction of the net charge of the polybasic JX region from +8 to +3 is the minimal change necessary for constitutive kinase activity and oncogenic potential of EGFR Mut R1-6. Interestingly, the number of colonies formed by EGFR Mut R1-6/L393H-expressing cells increases a small but significant extent in the presence of EGF (Fig. 7A). This may be explained by the small amount of EGFR that is endogenous to the parental NIH 3T3 cells, which is not sufficient to promote colony formation in soft agar in response to EGF but may synergize with the large number of constitutively active Mut R1-6/L393H EGFRs. This synergy may involve additional activation of Erk by EGF (Fig. 11).
Consistent with a constitutively activated state, EGFR Mut R1-6/L393H is phosphorylated at residue Tyr-1068 independent of the addition of EGF (Fig. 9, B and C). We are currently investigating the nature of this constitutive phosphorylated state, including the possibility that EGFR Mut R1-6/L393H spontaneously forms active dimers in the ER. Activation of EGFRvIII has been suggested to be independent of dimer formation (51); thus, understanding the basis for activation of EGFR Mut R1-6/L393H could reveal a potentially novel mechanism for EGFR dimerization that could be more generally relevant for cell transformation by EGFR variants. The N-terminal portion of the JX sequence containing the basic residues we have mutated has been shown to form an anti-parallel α-helical dimer important in EGF-activated EGFR (5, 14). Consistent with this, we find that EGFR Mut R1-6 exhibits reduced Ca2+ signaling in response to EGF compared with its WT counterpart.3
Constitutively phosphorylated at Tyr-1068 and oriented correctly across the ER membrane (Fig. 3), EGFR Mut R1-6/L393H appears poised to requisition the canonical cytosolic signaling proteins that normally interact with EGF-stimulated EGFR. Adaptors that interact with Tyr(P)-1068, Grb2 and Gab2 (52, 53), lead to Ras-Erk/MAPK and PI3K pathways, respectively. We found that EGFR Mut R1-6/L393H signals primarily through the PI3K pathway; colony formation mediated by Mut R1-6/L393H EGFR is particularly sensitive to treatment with the PI3K inhibitor LY294002 (Fig. 10B) and to mTOR inhibition by rapamycin (Fig. 10D). Furthermore, we found that Akt is basally phosphorylated in EGFR Mut R1-6/L393H-expressing cells (Fig. 10, A and B). In contrast, Erk phosphorylation is not increased in these cells compared with those expressing WT EGFR or vector only (Fig. 11A). Although inhibition of Erk by PD98059 decreases colony formation mediated by EGFR Mut R1-6/L393H, this decrease is similar to that for WT EGFR in the presence of EGF (Fig. 11B), suggesting a contribution from endogenous EGFR to the transformation phenotype. Interestingly, EGFRvIII has been shown to signal similarly through the PI3K pathway while only slightly activating the Ras-Erk/MAP kinase pathway (24, 54). Determining how ER-retained EGFR mutants selectively commandeer the PI3K pathway merits further study. We note that all Ras isoforms are tethered to membranes through an isoprenyl moiety together with either palmitoylation or a polybasic sequence (55). In contrast, PI3K and its downstream mediator Akt are cytosolic proteins that are recruited through protein-protein or protein-lipid interactions. This disposition likely increases their intracellular accessibility for activation by ER-membrane-localized EGFR Mut R1-6/L393H or EGFRvIII receptors.
Our results confirm that wholly ER-retained, constitutively active EGFRs can drive cellular transformation. In a clinical context, cancers mediated by similarly retained mutant EGFRs will likely be more optimally treated by inhibitors that access their intracellular location. Our results further reveal that the polybasic JX sequence confers a regulatory role in EGFR signaling important for oncogenic cell transformation.
Acknowledgments
We thank Dr. John Koland (University of Iowa) for the EGFR-EGFP construct, Dr. Cynthia Leifer (Cornell University) for assistance with Endo H assays, and Carol Bayles for maintaining the Cornell Imaging Facility. The Cytometry core is supported in part by the Empire State Stem Cell Fund, New York State Department of Health Contract 123456.
This work was supported, in whole or in part, by National Institutes of Health Chemistry/Biology Interface Training Grant 5T32GM008500 (to K. B.) and Grant R01AI022449 (NIAID). This work was also supported by a PhRMA Foundation Predoctoral Fellowship in Pharmacology/Toxicology (to K. B.).
K. L. Bryant, unpublished observations.
- RTK
- receptor-tyrosine kinase
- JX
- juxtamembrane
- TKD
- tyrosine kinase domain
- mTOR
- mammalian target of rapamycin
- Akt
- protein kinase B
- PDI
- protein disulfide isomerase
- Endo H
- endoglycosidase H
- PNGase F
- peptide N-glycosidase F
- PM
- plasma membrane
- ER
- endoplasmic reticulum.
REFERENCES
- 1. Lemmon M. A., Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blume-Jensen P., Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 3. Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 [DOI] [PubMed] [Google Scholar]
- 4. Red-Brewer M., Choi S. H., Alvarado D., Moravcevic K., Pozzi A., Lemmon M. A., Carpenter G. (2009) The juxtamembrane region of the EGF receptor functions as an activation domain. Mol. Cell 34, 641–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jura N., Endres N. F., Engel K., Deindl S., Das R., Lamers M. H., Wemmer D. E., Zhang X., Kuriyan J. (2009) Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 137, 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arkhipov A., Shan Y., Das R., Endres N. F., Eastwood M. P., Wemmer D. E., Kuriyan J., Shaw D. E. (2013) Architecture and membrane interactions of the EGF receptor. Cell 152, 557–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morrison P., Chung K. C., Rosner M. R. (1996) Mutation of di-leucine residues in the juxtamembrane region alters EGF receptor expression. Biochemistry 35, 14618–14624 [DOI] [PubMed] [Google Scholar]
- 8. He C., Hobert M., Friend L., Carlin C. (2002) The epidermal growth factor receptor juxtamembrane domain has multiple basolateral plasma membrane localization determinants, including a dominant signal with a poly proline core. J. Biol. Chem. 277, 38284–38293 [DOI] [PubMed] [Google Scholar]
- 9. Hsu S. C., Hung M. C. (2007) Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J. Biol. Chem. 282, 10432–10440 [DOI] [PubMed] [Google Scholar]
- 10. Martín-Nieto J., Villalobo A. (1998) The human epidermal growth factor receptor contains a juxtamembrane calmodulin-binding site. Biochemistry 37, 227–236 [DOI] [PubMed] [Google Scholar]
- 11. McLaughlin S., Smith S. O., Hayman M. J., Murray D. (2005) An electrostatic engine model for autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB) family. J. Gen. Physiol. 126, 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sengupta P., Bosis E., Nachliel E., Gutman M., Smith S. O., Mihályné G., Zaitseva I., McLaughlin S. (2009) EGFR juxtamembrane domain, membranes, and calmodulin. Kinetics of their interaction. Biophys. J. 96, 4887–4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Michailidis I. E., Rusinova R., Georgakopoulos A., Chen Y., Iyengar R., Robakis N. K., Logothetis D. E., Baki L. (2011) Phosphatidylinositol-4,5-bisphosphate regulates epidermal growth factor receptor activation. Pflugers Arch. 461, 387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Endres N. F., Das R., Smith A. W., Arkhipov A., Kovacs E., Huang Y., Pelton J. G., Shan Y., Shaw D. E., Wemmer D. E., Groves J. T., Kuriyan J. (2013) Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 152, 543–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carpenter G. (2000) The EGF receptor. A nexus for trafficking and signaling. Bioessays 22, 697–707 [DOI] [PubMed] [Google Scholar]
- 16. Xu Y. H., Richert N., Ito S., Merlino G. T., Pastan I. (1984) Characterization of epidermal growth factor receptor gene expression in malignant and normal human cell lines. Proc. Natl. Acad. Sci. U.S.A. 81, 7308–7312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Libermann T. A., Nusbaum H. R., Razon N., Kris R., Lax I., Soreq H., Whittle N., Waterfield M. D., Ullrich A., Schlessinger J. (1985) Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313, 144–147 [DOI] [PubMed] [Google Scholar]
- 18. King C. R., Kraus M. H., Williams L. T., Merlino G. T., Pastan I. H., Aaronson S. A. (1985) Human tumor cell lines with EGF receptor gene amplification in the absence of aberrant sized mRNAs. Nucleic Acids Res. 13, 8477–8486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kraus M. H., Popescu N. C., Amsbaugh S. C., King C. R. (1987) Over-expression of the EGF receptor-related proto-oncogene erbB-2 in human mammary tumor cell lines by different molecular mechanisms. EMBO J. 6, 605–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Humphrey P. A., Wong A. J., Vogelstein B., Friedman H. S., Werner M. H., Bigner D. D., Bigner S. H. (1988) Amplification and expression of the epidermal growth factor receptor gene in human glioma xenografts. Cancer Res. 48, 2231–2238 [PubMed] [Google Scholar]
- 21. Lynch T. J., Bell D. W., Sordella R., Gurubhagavatula S., Okimoto R. A., Brannigan B. W., Harris P. L., Haserlat S. M., Supko J. G., Haluska F. G., Louis D. N., Christiani D. C., Settleman J., Haber D. A. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 [DOI] [PubMed] [Google Scholar]
- 22. Heimberger A. B., Hlatky R., Suki D., Yang D., Weinberg J., Gilbert M., Sawaya R., Aldape K. (2005) Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 11, 1462–1466 [DOI] [PubMed] [Google Scholar]
- 23. Huang H. S., Nagane M., Klingbeil C. K., Lin H., Nishikawa R., Ji X. D., Huang C. M., Gill G. N., Wiley H. S., Cavenee W. K. (1997) The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 272, 2927–2935 [DOI] [PubMed] [Google Scholar]
- 24. Moscatello D. K., Holgado-Madruga M., Emlet D. R., Montgomery R. B., Wong A. J. (1998) Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J. Biol. Chem. 273, 200–206 [DOI] [PubMed] [Google Scholar]
- 25. Antonyak M. A., Moscatello D. K., Wong A. J. (1998) Constitutive activation of c-Jun-N-terminal kinase by a mutant epidermal growth factor receptor. J. Biol. Chem. 273, 2817–2822 [DOI] [PubMed] [Google Scholar]
- 26. Ekstrand A. J., Liu L., He J., Hamid M. L., Longo N., Collins V. P., James C. D. (1995) Altered subcellular location of an activated and tumour-associated epidermal growth factor receptor. Oncogene 10, 1455–1460 [PubMed] [Google Scholar]
- 27. Gupta P., Han S. Y., Holgado-Madruga M., Mitra S. S., Li G., Nitta R. T., Wong A. J. (2010) Development of and EGFRvIII specific recombination antibody. BMC Biotechnol. 10, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wikstrand C. J., McLendon R. E., Friedman A. H., Bigner D. D. (1997) Cell surface localization and density of the tumor associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 57, 4130–4140 [PubMed] [Google Scholar]
- 29. Johns T. G., Mellman I., Cartwright G. A., Ritter G., Old L. J., Burgess A. W., Scott A. M. (2005) The antitumor monoclonal antibody 806 recognizes a high-mannose form of the EGF receptor that reaches the cell surface when cells overexpress the receptor. FASEB J. 19, 780–782 [DOI] [PubMed] [Google Scholar]
- 30. Cvrljevic A. N., Akhavan D., Wu M., Martinello P., Furnari F. B., Johnston A. J., Guo D., Pike L., Cavenee W. K., Scott A. M., Mischel P. S., Hoogenraad N. J., Johns T. G. (2011) Activation of Src induces mitochondrial localization of de2–7EGFR (EGFRvIII) in glioma cells. Implications for glucose metabolism. J. Cell Sci. 124, 2938–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Monick M. M., Cameron K., Staber J., Powers L. S., Yarovinsky T. O., Koland J. G., Hunninghake G. W. (2005) Activation of the epidermal growth factor receptor by respiratory syncytial virus results in increased inflammation and delayed apoptosis. J. Biol. Chem. 280, 2147–2158 [DOI] [PubMed] [Google Scholar]
- 32. Gosse J.A., Wagenknecht-Wiesner A., Holowka D., Baird B. (2005) Transmembrane sequences are determinants of immunoreceptor signaling. J. Immunol. 175, 2123–2131 [DOI] [PubMed] [Google Scholar]
- 33. Maley F., Trimble R. B., Tarentino A. L., Plummer T. H., Jr. (1989) Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 180, 195–204 [DOI] [PubMed] [Google Scholar]
- 34. Aifa S., Miled N., Frikha F., Aniba M. R., Svensson S. P., Rebai A. (2006) Electrostatic interactions of peptides flanking the tyrosine kinase domain in the epidermal growth factor receptor provides a model for intracellular dimerization and autophosphorylation. Proteins 62, 1036–1043 [DOI] [PubMed] [Google Scholar]
- 35. Sato T., Pallavi P., Golebiewska U., McLaughlin S., Smith S. O. (2006) Structure of the membrane reconstituted transmembrane-juxtamembrane peptide EGFR (622–660) and its interaction with Ca2+/calmodulin. Biochemistry 45, 12704–12714 [DOI] [PubMed] [Google Scholar]
- 36. Thiel K. W., Carpenter G. (2007) Epidermal growth factor receptor juxtamembrane region regulated allosteric tyrosine kinase activation. Proc. Natl. Acad. Sci. U.S.A. 104, 19238–19243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gamou S., Shimagaki M., Minoshima S., Kobayashi S., Shimizu N. (1989) Subcellular localization of the EGF receptor maturation process. Exp. Cell Res. 183, 197–206 [DOI] [PubMed] [Google Scholar]
- 38. Wikstrand C. J., Hale L. P., Batra S. K., Hill M. L., Humphrey P. A., Kurpad S. N., McLendon R. E., Moscatello D., Pegram C. N., Reist C. J. (1995) Monoclonal antibodies against EGFRvllI are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 55, 3140–3148 [PubMed] [Google Scholar]
- 39. Macpherson I., Montagnier L. (1964) Agar suspension culture for the selective assay of cells transformed by polyoma virus. Virology 23, 291–294 [DOI] [PubMed] [Google Scholar]
- 40. Li S., Schmitz K. R., Jeffrey P. D., Wiltzius J. J., Kussie P., Ferguson K. M. (2005) Structural basis for inhibition of the epidermal growth factor receptor by Cetuximab. Cancer Cell 7, 301–311 [DOI] [PubMed] [Google Scholar]
- 41. Kholodenko B. N., Hancock J. F., Kolch W. (2010) Signalling ballet in space and time. Nat. Rev. Mol. Cell Biol. 11, 414–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ushiro H., Cohen S. (1980) Identification of phosphotyrosine as a product of epidermal growth factor-activated protein kinase in A-431 cell membranes. J. Biol. Chem. 255, 8363–8365 [PubMed] [Google Scholar]
- 43. Merlino G. T., Xu Y. H., Ishii S., Clark A. J., Semba K., Toyoshima K., Yamamoto T., Pastan I. (1984) Amplification and enhanced expression of the epidermal growth factor receptor gene in A431 human carcinoma cells. Science 224, 417–419 [DOI] [PubMed] [Google Scholar]
- 44. Choudhary C., Olsen J. V., Brandts C., Cox J., Reddy P. N., Böhmer F. D., Gerke V., Schmidt-Arras D. E., Berdel W. E., Müller-Tidow C., Mann M., Serve H. (2009) Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol. Cell 36, 326–339 [DOI] [PubMed] [Google Scholar]
- 45. Carpenter G., Liao H.J. (2009) Trafficking of receptor tyrosine kinases to the nucleus. Exp. Cell Res. 315, 1556–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Privalsky M.L., Bishop J.M. (1984) Subcellular localization of the v-erb-B protein, the product of a transforming gene of avian erythroblastosis virus. Virology 135, 356–368 [DOI] [PubMed] [Google Scholar]
- 47. Bajaj M., Waterfield M. D., Schlessinger J., Taylor W. R., Blundell T. (1987) On the tertiary structure of the extracellular domains of the epidermal growth factor and insulin receptors. Biochim. Biophys. Acta 916, 220–226 [DOI] [PubMed] [Google Scholar]
- 48. Ward C. W., Garrett T. P. (2001) The relationship between the L1 and L2 domains of the insulin and epidermal growth factor receptors and leucine-rich repeat modules. BMC Bioinformatics 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nishimura N., Balch W.E. (1997) A di-acidic signal required for selective export from the endoplasmic reticulum. Science 277, 556–558 [DOI] [PubMed] [Google Scholar]
- 50. Sevier C. S., Weisz O. A., Davis M., Machamer C. E. (2000) Efficient export of the vesicular stomatitis virus g protein from the endoplasmic reticulum requires a signal in the cytoplasmic tail that induces both tyrosine-based and di-acidic motifs. Mol. Biol. Cell 11, 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chu C. T., Everiss K. D., Wikstrand C. J., Batra S. K., Kung H. J., Bigner D. D. (1997) Receptor dimerization is not a factor in the signaling activity of a transforming variant epidermal growth factor receptor (EGFRvIII). Biochem. J 324, 855–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rojas M., Yao S., Lin Y. Z. (1996) Controlling epidermal growth factor (EGF)-stimulated Ras activation in intact cells by a cell permeable peptide mimicking phosphorylated EGF receptor. J. Biol. Chem. 271, 27456–27461 [DOI] [PubMed] [Google Scholar]
- 53. Kong M., Mounier C., Wu J., Posner B. I. (2000) Epidermal growth factor-induced phosphatidylinositol 3-kinase activation and DNA synthesis. J. Biol. Chem. 275, 36035–36042 [DOI] [PubMed] [Google Scholar]
- 54. Moscatello D. K., Montgomery R. B., Sundareshan P., McDanel H., Wong M. Y., Wong A. J. (1996) Transformational and altered signal transduction by a naturally occurring mutant EGF receptor. Oncogene 13, 85–96 [PubMed] [Google Scholar]
- 55. Hancock J. F., Paterson H., Marshall C. J. (1990) A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63, 133–139 [DOI] [PubMed] [Google Scholar]