

Background: ROP18 is a Toxoplasma secreted Ser/Thr protein kinase important for acute virulence.
Results: The crystal structure of the unphosphorylated ROP18 kinase domain was determined in complex with an ATP analog.
Conclusion: The structure is inconsistent with a previously proposed model of autoinhibition and identifies an additional ligand binding site important for virulence.
Significance: Structure-function studies of ROP18 will aid development of novel drugs against toxoplasmosis.
Keywords: Crystal Structure, Protein Kinases, Protein Phosphorylation, Protozoan, Signal Transduction, Toxoplasma Gondii, Rhoptry Protein
Abstract
At least a third of the human population is infected with the intracellular parasite Toxoplasma gondii, which contributes significantly to the disease burden in immunocompromised and neutropenic hosts and causes serious congenital complications when vertically transmitted to the fetus. Genetic analyses have identified the Toxoplasma ROP18 Ser/Thr protein kinase as a major factor mediating acute virulence in mice. ROP18 is secreted into the host cell during the invasion process, and its catalytic activity is required for the acute virulence phenotype. However, its precise molecular function and regulation are not fully understood. We have determined the crystal structure of the ROP18 kinase domain, which is inconsistent with a previously proposed autoinhibitory mechanism of regulation. Furthermore, a sucrose molecule bound to our structure identifies an additional ligand-binding pocket outside of the active site cleft. Mutational analysis confirms an important role for this pocket in virulence.
Introduction
Toxoplasma gondii is a highly prevalent protozoan intracellular parasite that is estimated to have infected a third of the human population. The organism can cause severe disease in immunocompromised individuals, as well as those suffering from AIDS and hematopoietic malignancies (1–5). Primary infection of women during pregnancy with vertical transmission to the fetus can result in a wide range of birth defects. Studies in mice have demonstrated a critical role for neutrophils during the early immune response to T. gondii infection in limiting the parasite burden and lethality (6), and this has serious implications for patients with defective neutrophil-mediated killing of pathogens. In North America and Europe, the majority of T. gondii isolates from patients can be grouped into three major clonal lineages, which show dramatic differences in virulence in mice (7). Type I strains are acutely virulent with inoculations of single parasites being lethal in mice (LD100 ∼1), whereas type II strains show intermediate virulence (LD50 ∼103 parasites) and type III strains are relatively avirulent (LD50 ≥104 parasites). To identify genetic loci underlying the differences in virulence between these strains, pairwise crosses between these strains have been conducted, in which the F1 progeny were genotyped and scored for their virulence in mice. In one study, a single locus, corresponding to the ROP18 gene, was identified and accounted for >90% of the difference in virulence between type I and type III strains (8). The ROP18 gene was also one of five virulence-associated loci identified in a type II × type III cross (9). Type III strains show a 10,000-fold lower level of the ROP18 mRNA transcript relative to the other two strains and are effectively ROP18 null at the level of protein expression. This is likely due to a 2.1-kb region within the promoter region of the type III ROP18 allele that is not present in the type I or II alleles (9–11). Expression of either type I or II ROP18 alleles in a type III strain resulted in a ∼104-fold decrease in LD50 (8, 9). ROP18 is a member of the rhoptry kinase (ROPK)7 family of protein kinases (12). Many members of the ROPK family are catalytically inactive pseudokinases lacking key active site residues as exemplified by the prototypic member, ROP2 (13). In marked contrast, ROP18 is an active Ser/Thr kinase (14), and its catalytic activity is required to confer virulence (8). How the catalytic activity of ROP18 is regulated, and the basis for ROP18 substrate selection, remain unknown.
Attachment of T. gondii to a host cell triggers the secretion of multiple proteins from specialized organelles (micronemes, rhoptries, and dense granules) at the apical end of the parasite (15, 16). This is followed by active entry into the host cell and enclosure of the parasite within a parasitophorous vacuole (PV). The PV provides a protective environment for parasite growth and replication inside the host cell. Upon secretion, a number of the rhoptry proteins, including ROP18, localize to the host cytosolic face of the parasitophorous vacuole membrane (PVM), where they are well positioned to play important yet poorly understood roles at the host-pathogen interface that affect virulence determinants such as parasite growth and survival (14, 15). The recent identification of the interferon γ (IFNγ) inducible immunity-related p47 GTPases (IRGs) as substrates of ROP18 has provided some mechanistic insights into the molecular functions of one such rhoptry-secreted virulence factor (17, 18). IFNγ induction of the IRG family of proteins is an important component of the early innate immune response in mice against intracellular pathogens such as T. gondii. A major host resistance mechanism contributing to the clearance of the avirulent strains of T. gondii within IFNγ-stimulated host cells involves the sequential loading of multiple IRG proteins at the PVM shortly after infection. The accumulation of IRG proteins at the PVM results in the rupture of the PV and release of the parasites into the host cytosol, where they are destroyed (19, 20). Recent studies have also revealed that mice deficient in p65 guanylate-binding proteins are defective in IFNγ-mediated suppression of T. gondii intracellular growth and recruitment of IRGs to the PVM (21–23). Virulent type I strains avoid destruction by inhibiting IRG accumulation on the PVM and are thus able to aggressively expand within the host. ROP18 was recently shown to directly phosphorylate IRG proteins to impair their accumulation on the PVM (17, 18). Given that the IRG resistance system is absent in host species such as humans and cats (19), other types of host defense mechanisms likely exist that may be compromised by ROP18 and other virulence factors. One such mechanism involves the host transcription factor ATF6β, which up-regulates multiple genes in response to endoplasmic reticulum stress. ATF6β-deficient mice showed increased sensitivity to infection by avirulent ROP18-deficient parasites, and ROP18 was recently reported to phosphorylate ATF6β leading to its proteasome-dependent degradation (24).
To further our understanding of ROP18 regulation and function, we have solved the crystal structure of its kinase domain and identified features important for virulence in mice. Our structure is the first determined for a catalytically active rhoptry kinase and builds on data from previously published crystal structures of the pseudokinase domains of ROP2 (PDB accession codes 2W1Z and 3DZO), ROP8 (PDB code 3BYV), and ROP5 (PDB code 3Q60) (25–27). Unlike ROP18, all three of these ROPK proteins do not function by phosphorylating downstream targets. Our structure of the ROP18 kinase domain disproves a previously proposed autoinhibitory mechanism (26). Importantly, the structure also identifies a novel second ligand binding pocket adjacent to the active site, which is critical for ROP18-mediated virulence and may provide an additional substrate specificity determinant or allosteric regulatory site.
EXPERIMENTAL PROCEDURES
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The MIT Committee on Animal Care (assurance number A-3125-01) approved all protocols. All mice were maintained in specific pathogen-free conditions, and all efforts were made to minimize suffering.
Production of Recombinant Proteins
The ROP18 kinase domain (residues 187–554) was cloned into pETDL4.0, a modified pET28a vector (Novagen), which incorporated a series of N-terminal fusion tags consisting of: (His6)-(λ-phosphatase)-(glutatathione S-transferase)-(maltose-binding protein)-(Streptococcus protein B1 domain)-(tobacco etch virus cleavage site) (His6- λ-phosphatase-GST-MBP-Gb1). Inclusion of λ-phosphatase in the fusion tags allowed production of a catalytically active wild type ROP18 kinase domain free of any autophosphorylation. The protein was expressed in Escherichia coli Rosetta 2 (Novagen). For large scale preparations, the protein was first purified on a nickel SepahroseTM Fast Flow column (GE Healthcare Life Sciences), followed by an amylose column (New England Biolabs). Tobacco etch virus protease was then added to cleave the N-terminal fusion tags, which were subsequently removed by passage of the protein through a GSH-agarose column and a final gel filtration step. The protein was then concentrated and frozen in aliquots in liquid nitrogen for storage at −80 °C. Small scale preparations of WT and mutant ROP18 kinase domain constructs were expressed as His6-GST-MBP-Gb1 fusions and batch purified on amylose beads and used directly for in vitro kinase assays.
Crystallization and Data Collection
The ROP18 kinase domain was crystallized by hanging drop vapor diffusion, in which 1 μl of 10 mg/ml of protein in 15 mm Tris, pH 8, 0.15 m NaCl, 1 mm AMP-PNP, 2 mm MgSO4 was mixed with 1 μl of precipitant solution and equilibrated over a reservoir of precipitant solution. Native crystals were grown using a precipitant solution containing 0.56 m ammonium sulfate, 15% PEG 3350, 0.1 m BisTris, pH 5.5, and 20 mm isopropyl 1-thio-β-d-galactopyranoside. For holmium co-crystals of the ROP18 kinase domain, HoCl3 was substituted for MgSO4 (28) in the protein solution, and the precipitant solution contained 2.66 mm HoCl3, 0.28 m ammonium sulfate, 13% PEG 3350, 64 mm BisTris, pH 5.5, and 36 mm MES, pH 6. Crystals typically appeared overnight and grew to full size after a few days.
Native crystals were cryo-protected by stepwise transfer to 0.56 m ammonium sulfate, 15% PEG 3350, 0.1 m BisTris, pH 5.5, 30% (w/v) sucrose, 1 mm AMP-PNP, and 2 mm MgSO4. Holmium co-crystals were cryo-protected by stepwise transfer to 10 mm HoCl3, 0.28 m ammonium sulfate, 13% PEG 3350, 70 mm BisTris, pH 5.5, 30 mm MES, pH 6, 33.3% sucrose, and 1 mm AMP-PNP. Cryo-protected crystals were flash cooled in a nitrogen gas stream at 100 K, and stored in liquid nitrogen. Data were collected on the 24-ID-E and 24-ID-C beam lines at the Advanced Photon Source, Argonne National Laboratory using an ADSC Quantum 315 detector. Data were processed using HKL 2000 (29) and the CCP4 software suite (30).
Structure Determination and Refinement
The two holmium sites were located using the automated Patterson search implemented in CNS 1.21 (31, 32) and used for phasing in SHARP (33). The multi-wavelength anomalous diffraction phases from data collected from two separate holmium co-crystals were combined using SigmaA (30, 34) and subsequently improved by solvent flattening using RESOLVE (35, 36). The model was built using XtalView (37, 38) and refined using PHENIX (39). The final model contains a single residue (Lys-479) in the disallowed region of the Ramachandran plot. This strained conformation is likely stabilized by the adjacent disulfide bond between Cys-478 and Cys-497. Figures were created using Molscript (40), PyMOL (41), and XtalView (37, 38). Electrostatic potentials were calculated with DelPhi (42, 43).
In Vitro Kinase Reactions
The ROP18 kinase motif was determined using a positional scanning peptide library array as previously published (44, 45) and a reaction buffer consisting of 10 mm HEPES, pH 7.5, 0.5 m NaCl, 10 mm DTT, 10 mm MgSO4, and 50 μm cold ATP. Each individual reaction contained 300 ng of untagged recombinant ROP18 kinase domain (residues 187–554) and ∼1 μCi of [γ-32P]ATP. The kinase reactions were incubated at 30 °C for 8 h and then spotted on a SAM2® biotin capture membrane (Promega). The phosphorylation content of each peptide library in the array was quantified by phosphorimage analysis with the program YIMP8 and sequence logos were generated from normalized background-corrected intensities of the peptide library spots using POSTSCRIPT files generated by Visual Basic code adapted from the original PSSM (position-specific scoring matrix) logo code from Shaw and co-workers (46), which contains POSTCRIPT code adapted from MAKELOGO from Schneider and Stephens (47).
Kinase assays using fixed sequence peptide substrates were conducted at room temperature using either 69 ng of untagged WT ROP18 kinase domain or 150 ng of His6-GST-MBP-GB1-tagged WT or mutant ROP18 kinase domain, and a reaction buffer consisting of 50 mm HEPES, pH 7.5, 0.5 m NaCl, 10 mm DTT, 10 mm MgSO4, and 60 μm cold ATP. Each reaction contained 0.5 mm peptide substrate and 2–10 μCi of [γ-32P]ATP. Reactions were stopped by spotting on Whatman P81 phosphocellulose paper, which were then dried and washed with 0.425% phosphoric acid until no significant radioactivity remained in the washes. Radioactivity captured on P81 filters was then quantified by phosphorimage analysis with ImageQuant 5.2 software (Molecular Dynamics). Immunoprecipitation kinase assays of HA-tagged full-length ROP18 constructs were performed as previously described (48). Kinase activities were normalized for the amounts of ROP18 protein immunoprecipitated (Fig. 5B).
FIGURE 5.
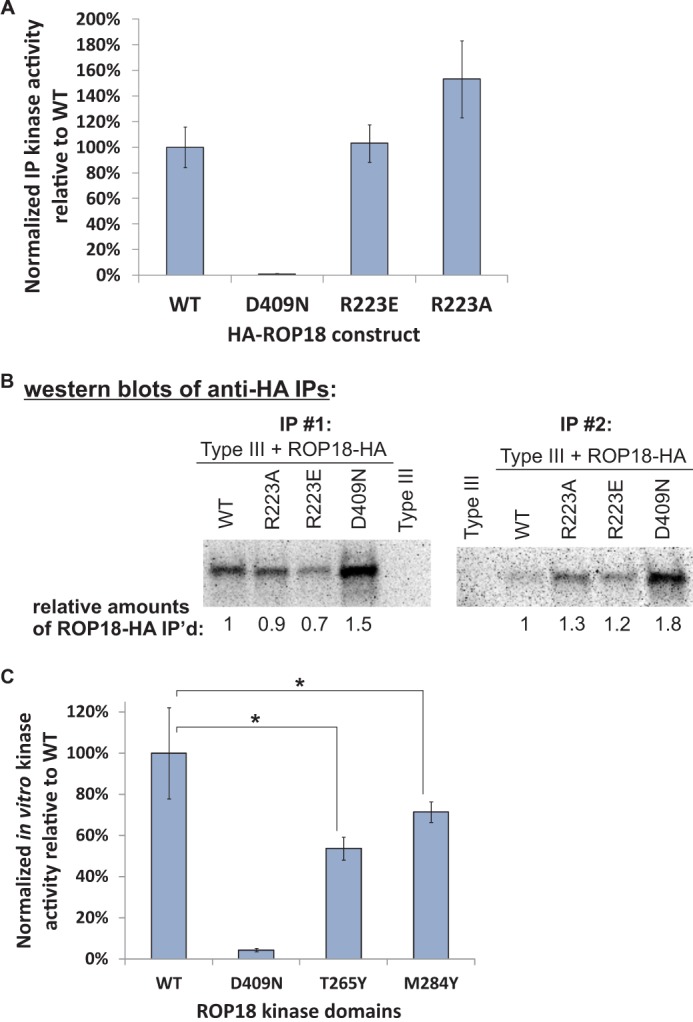
Kinase activities of ROP18 basic pocket mutants relative to WT. A, immunoprecipitation (IP) kinase assays of WT, R223A, R223E, and D409N HA-tagged full-length ROP18. The data are averaged from the results of two biological replicate assays. The relative kinase activities were normalized for differences in the amounts of ROP18 immunoprecipitated (based on Western blotting). B, Western blots showing relative levels of immunoprecipitated HA-tagged ROP18 constructs as determined by probing with an anti-HA antibody and subsequently used for immunoprecipitation kinase assays in A. C, in vitro kinase assays show decreased activity for the T265Y and M284Y mutants relative to the WT ROP18 kinase domain. An equal amount of recombinant ROP18 protein was used in each reaction, and data were averaged from two biological replicates, each assayed in triplicate. For both A and C an optimal peptide substrate (NH3-KKKKKWISEHTRYFF-CONH2) was used, a kinase-dead D409N mutant served as a negative control, and error bars show S.D.
Generation of Transgenic Parasites
The coding region of ROP18, along with putative promoter (742 bp upstream of the ATG start codon), from type I T. gondii genomic DNA was amplified by PCR (forward 5′-CACCAGATTCGAAACGCGGAAGTA-3′; reverse 5′-TTACGCGTAGTCCGGGACGTCGTACGGGTATTCTGTGTGGAGATGTTCCTGCTGTTC-3′). The HA tag sequence was included in the reverse primer. Specific ROP18 mutants were generated using the megaprimer method with splice overlap extension (49, 50). The ROP18 cassette was cloned into pENTR/D-TOPO (Invitrogen) and subsequently cloned in pTKO-att (51) via LR recombination. The pTKO-att-ROP18 vector was linearized by digestion with NdeI (New England Biolabs) and transfected into CEPHXGPRT− C22 parasites that express luciferase and GFP by electroporation. This was performed in 2-mm cuvettes (Bio-Rad Laboratories) with 2 mm ATP (MP Biomedicals) and 5 mm GSH (EMD) in a Gene Pulser XCell (Bio-Rad) with the following settings: 25 microfarads, 1.25 kV, and ∞ Ω. Stable integrants were selected in media with 25 mg/ml of mycophenolic acid (Axxora) and 25 mg/ml of xanthine (Alfa Aesar) and afterward cloned by serial dilution. Expression of the ROP18 constructs was confirmed by immunofluorescence using an anti-HA tag antibody (Roche Applied Science). Rhoptry localization and PVM localization upon secretion were confirmed for all constructs, with at least two independent clones for each mutant, and expression levels of the ROP18 constructs were quantitated by Western blotting (Fig. 4A). Parasites were cultured by passage onto monolayers of human foreskin fibroblasts at 37 °C with 5% CO2 as previously described (51).
FIGURE 4.

Mutations within the ROP18 basic pocket reveal biochemical functions of ROP18 in addition to its kinase activity that are required for virulence. A, representative blot showing relative levels of HA-tagged ROP18 constructs in parasite lysates as determined by probing with a rat anti-HA antibody. Toxoplasma Sag1 was used as a loading control. B, quantitation of the Western blot data averaged from two biological replicates, with HA-tagged ROP18 levels normalized to the corresponding Sag1 levels. The B5.2 clone was not used in our analyses due to its exceptionally high HA-tagged ROP18 expression level relative to the other WT-expressing clones. C, survival of C57BL/6 mice infected intraperitoneal with type III parasites, type III parasites complemented with wild-type ROP18, or type III parasites complemented with the indicated mutant ROP18 through day 30 after injection. D, mouse survival analysis as determined in C showing no significant differences in the outcome after including the B5.2 high level WT ROP18 expressing clone. E and F, in vivo growth of type III parasites complemented with wild-type ROP18 or with the indicated ROP18 mutants, 5 days post-injection, as measured by luciferase imaging. E, one representative mouse infected with each parasite strain. F, average total flux of all infected mice for each parasite strain. Error bars show S.E.
Mouse Infections
Female C57BL/6 mice that were 6–10 weeks old (Jackson Laboratories, Bar Harbor, ME), were used for all experiments. For intraperitoneal infection, tachyzoites were grown in vitro and extracted from host cells by passage through a 27-gauge needle, washed twice in PBS, and quantified with a hemocytometer. Parasites were diluted in PBS, and mice were inoculated intraperitoneally with 5000 tachyzoites of each strain (in 100 μl) with a 28-gauge needle. Plaque assays demonstrated injection of similar viable parasite numbers. 15–26 mice were infected per parasite strain and 2 different clones were used for each recombinant parasite. Figures represent the combined survival curves of these 2 different clones. To image mice infected with a parasite strain expressing the enzyme luciferase, mice were injected intraperitoneally with 3 mg of firefly d-luciferin dissolved in PBS, anesthetized with isoflurane, and imaged with an IVIS Spectrum-bioluminescent and fluorescent imaging system (Xenogen Corporation). Images were processed and analyzed with Living Image software. The MIT Committee on Animal Care approved all protocols. All mice were maintained in specific pathogen-free conditions, in accordance with institutional and federal regulations.
Irgb6 Parasitophorous Vacuole Coating Assays
Parasites were allowed to invade monolayers of MEF cells grown on coverslips that were previously incubated for 24 h with or without 1000 units of IFNγ. After 20 min, non-invading parasites were washed away with PBS 3 times, and the infection proceeded for 1 h. The cells were then fixed with 3% (v/v) formaldehyde in PBS for 20 min at room temperature, permeabilized with 0.2% (v/v) saponin, and blocked in PBS with 3% (w/v) BSA and 5% (v/v) FBS. Coverslips were incubated with a goat polyclonal antibody against mouse Irgb6 (TGTP A-20, Santa Cruz Biotechnology, 1:100 dilution), for 1 h, and fluorescent secondary antibody coupled with Alexa Fluor 594 and Hoechst dye were used for antigen and DNA visualization, respectively. Coverslips were mounted on a glass slide with Vectashield (Vector Laboratories), and photographs were taken using NIS-Elements software (Nikon) and a digital camera (Coolsnap EZ; Roper Scientific) connected to an inverted fluorescence microscope (model eclipse Ti-S; Nikon).
Statistical Methods
Statistical analysis was done using GraphPad Prism software version 6.0b (GraphPad Software Inc., La Jolla, CA). Unless otherwise stated, one-way analysis of variance was used for comparison of results from different ROP18 constructs. p values <0.05 were considered significant (*).
Accession Code
Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession code 4JRN.
RESULTS
ROP18 Kinase Is Not Autoinhibited by the N-terminal Subdomain
To facilitate biochemical and structural analysis of the ROP18 kinase domain, we cloned an N terminally truncated construct consisting of residues 187–554 (numbering based on the first ATG codon of CAJ27113) into a modified pET28a-based vector (see “Experimental Procedures”). This construct was expressed as a soluble and catalytically active protein in E. coli, suitable for in vitro kinase assays and structural analysis.
The crystal structure of the unphosphorylated ROP18 kinase domain was determined by multi-wavelength anomalous diffraction using a holmium derivative. The structure was refined against a native data set to 2.7-Å resolution. Data and refinement statistics are summarized in Table 1. The structure adopts the canonical protein kinase domain-fold (52) with a predominantly α-helical C-terminal lobe and a smaller N-terminal lobe containing a central β-sheet (Fig. 1A). The kinase domain of ROP18 shows significant overall structural deviations from those of ROP22W1Z (1.9 Å root mean square deviation over 315 Cα pairs), ROP23DZO (2.0 Å root mean square deviation over 299 Cα pairs), ROP83BYV (2.0 Å root mean square deviation over 306 Cα pairs for 3BYV), and ROP53Q60 (2.8 Å root mean square deviation over 280 Cα pairs). In particular, the N-terminal subdomain (a conserved structural feature of ROPKs consisting of αN1, αN2, and βN) of ROP18 contains a disordered linker region between αN1 and αN2 (residues 211–217), which contradicts a previously proposed regulatory function for this N-terminal subdomain in ROP18 (26). In both the ROP23DZO and ROP83BYV structures, the αN1-αN2 linker can be seen encroaching into the ATP binding pocket and is stabilized by an Arg-Glu salt bridge (26), which sterically occludes ATP binding (Fig. 1B). The previously published homology model of ROP18 (based on the ROP83BYV structure), assumed a similar conformation adopted by the αN1-αN2 linker, with side chains protruding into the active site to preclude ATP binding. It was further proposed that autophosphorylation of serine and threonine residues within helix αN2 and on strand β1 (adjacent to strand βN) would induce conformational changes within the N-terminal subdomain to relieve this autoinhibition (26). The physiological relevance of these autophosphorylation sites (Ser-221, Thr-229, Thr-249, Thr-251, and Thr-293, renumbered based on the first ATG codon of CAJ27113) as reported for bacterially expressed recombinant kinase domain is unclear and do not match any of the ROP18 phosphopeptides detected in recent phosphoproteomic studies of T. gondii proteins purified from parasites at the infection-competent tachyzoite stage (53, 54). Additionally, the Arg-Glu salt bridge within the ATP-binding pockets of ROP23DZO and ROP83BYV is not conserved in any of the other available ROPK structures, and consequently this autoinhibitory conformation of the αN1-αN2 linker is not observed in the structure of a different ROP2 isoform (ROP22W1Z), the ROP53Q60 structure or our ROP18 structure (Fig. 1B). In particular, our crystal structure of unphosphorylated ROP18 with AMP-PNP bound at the ATP binding pocket rules out any requirement of autophosphorylation for ATP binding. The crystals were grown from unphosphorylated recombinant protein that had been expressed in E. coli as a fusion construct with λ-phosphatase (see “Experimental Procedures”). Both the electron density map and mass spectrometry data (41464.3 Da predicted, 41464 Da measured) on the purified recombinant protein show no evidence of any covalent modification.
TABLE 1.
Data collection, phasing and refinement statistics
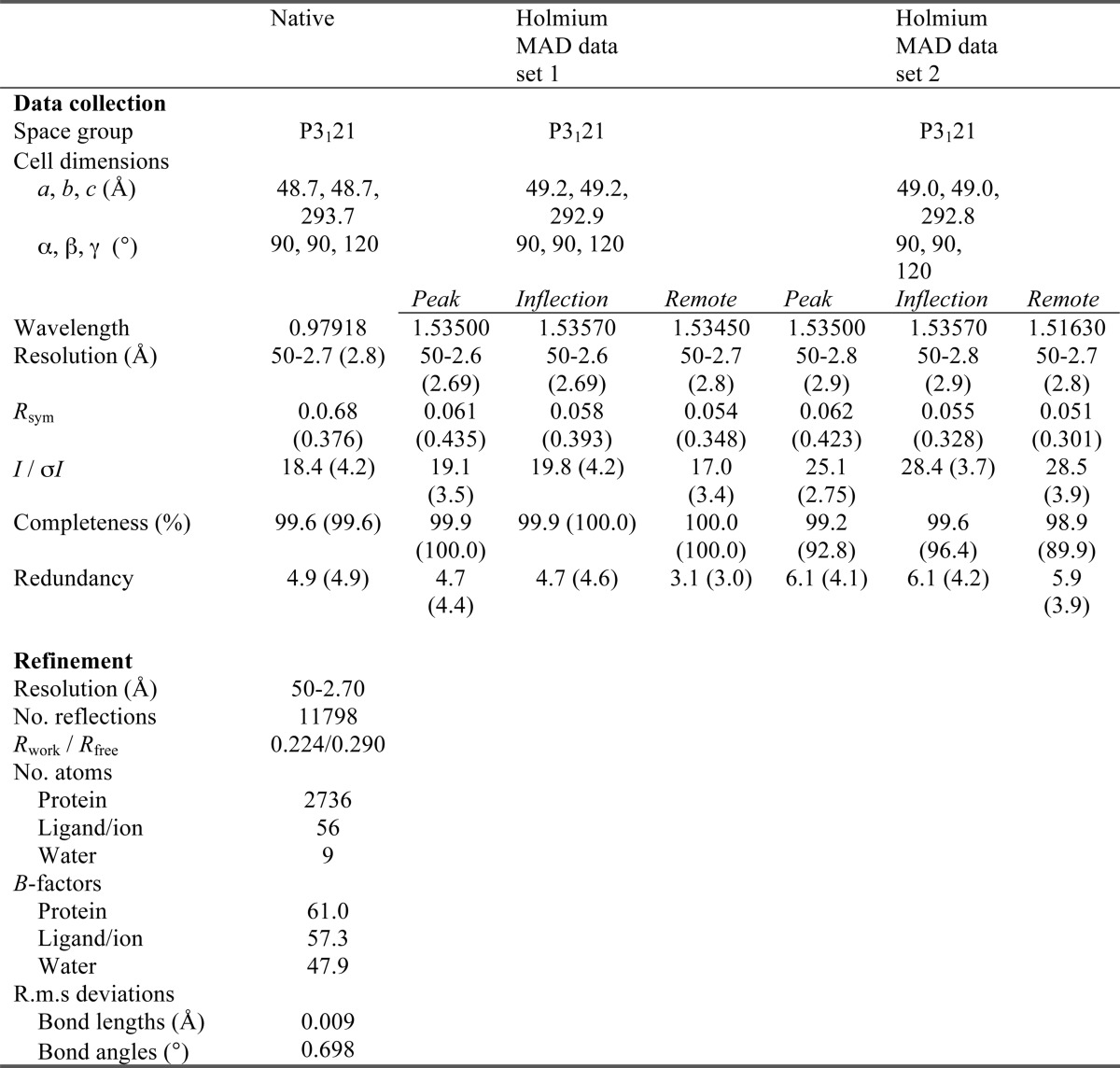
* Values in parentheses are for highest resolution shell.
FIGURE 1.
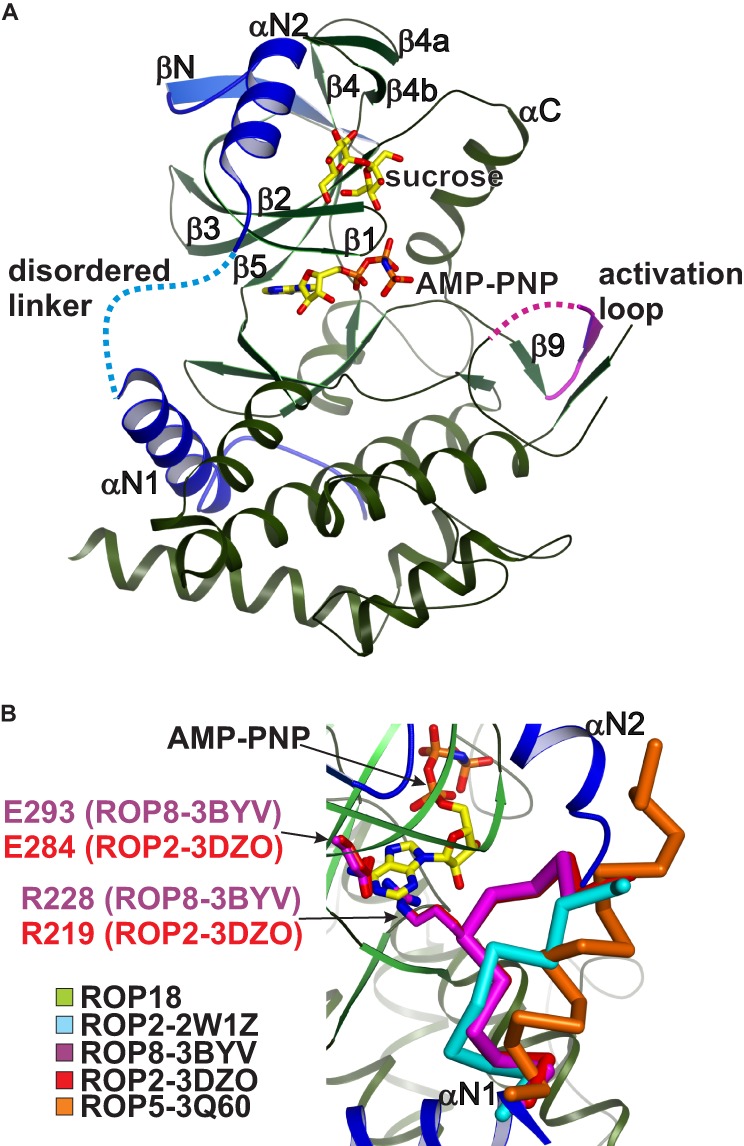
Crystal structure of the ROP18 kinase domain. A, schematic rendering of the ROP18 kinase domain with the N-terminal subdomain highlighted in blue and disordered regions indicated by dotted lines. The bound sucrose and AMP-PNP molecules are shown in stick rendering with yellow carbons. B, close-up view of the αN1-αN2 linker within the N-terminal subdomain. Although this linker is disordered in ROP18, in ROP2–3DZO and ROP8–3BYV the linker adopts an autoinhibitory conformation (red and purple Cα traces, respectively) stabilized by an Arg-Glu salt bridge (stick rendering) that precludes ATP binding. The corresponding regions from ROP2–2W1Z and ROP5–3Q60 are shown as cyan and orange Cα traces, respectively.
The ROP18 Kinase Motif Has Broad Sequence Selectivity
To probe the substrate selectivity of ROP18, we determined its optimal substrate phosphorylation motif (Fig. 2A) using a positional scanning peptide library array (44, 45). The resulting motif is complex, but can be generalized as: (X)-(X, not E)-(X)-(E)-(H)-(T)-(R/mixed, not P and not negatively charged)-(Ar)-(Ar)-(Ar), where the phosphoacceptor is underlined, X denotes any amino acid residue, and Ar denotes an aromatic residue (Fig. 2B). As previously reported (17), peptide libraries containing a fixed Thr at positions −5 to +3 relative to the phosphoacceptor are strongly phosphorylated by the ROP18 kinase domain. However, we interpret this as due to phosphorylation of, rather than selection for, the fixed Thr residues at those positions (Fig. 2C).
FIGURE 2.
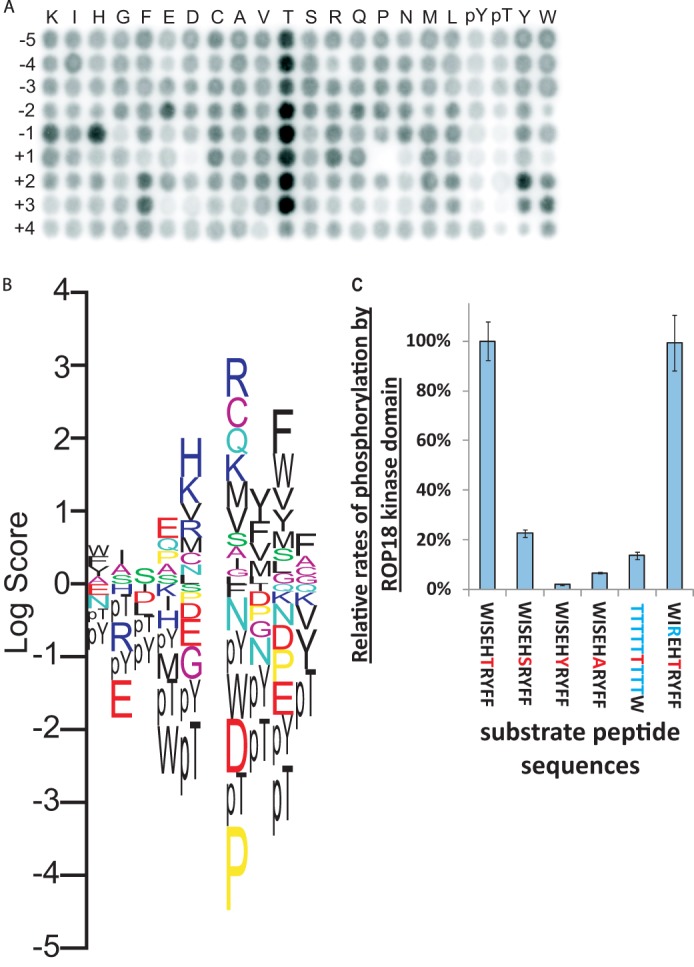
ROP18 peptide substrate specificity. A, unbiased determination of ROP18 peptide substrate specificity using an oriented peptide library array to probe amino acid preferences at positions from −5 to +4 relative to the phosphoacceptor site (position 0). B, motif logo generated from integrated spot intensities from A. C, comparison of specific peptides as in vitro substrates for WT ROP18 kinase domain. All peptides contained an N-terminal pentalysine tag (to facilitate capture on P81 phosphocellulose filter paper) and were C-terminally amidated. The phosphoacceptor residue is colored red, whereas changes from the optimal sequence (leftmost) are colored blue. Assays were performed in triplicate, and error bars represent S.D. The peptide substrate containing Thr fixed at the −5 to +3 positions is a poor substrate for the ROP18 kinase domain relative to an optimal peptide (NH3-KKKKKWISEHTRYFF-CONH2) substrate, indicating that Thr is in fact not generally favored around the phosphoacceptor site, but instead likely functions as a second phosphoacceptor residue in these positions. Moreover, Thr is greatly favored at the phosphoacceptor position versus Ser, Tyr, or Ala. Therefore, the apparent preference for Thr at multiple positions around the phosphoacceptor site is likely accounted for by the generally broad target sequence selectivity of ROP18 and its strong preference for phosphorylating Thr. The substitution of the -3 Ser with Arg did not significantly affect the activity assayed, indicating that phosphorylation of the -3 Ser did not contribute to the detected activity.
ROP18 was recently reported to phosphorylate multiple IRG proteins, including Irgb6 and Irga6. For Irgb6, the single ROP18 phosphorylation site (GAAP-Thr(P)-89-GAIE) previously identified (17) deviates substantially from the optimal ROP18 substrate phosphorylation motif. In contrast, the two phosphorylation sites identified within Irga6 (GAAK-Thr(P)-102-GVVE and VVEV-Thr(P)-108-MER) more closely matches the ROP18 substrate phosphorylation motif (Table 2), and consistent with this, Irga6 was more strongly phosphorylated by ROP18 relative to Irgb6 (18). The ROP18 phosphorylation sites within these IRG proteins are located within the Switch I region. An analysis of the Switch I peptide sequences for IRG proteins from hamster (Cricetulus griseus), mouse (Mus musculus), and rat (Rattus norvegicus) using Scansite (55, 56) did not reveal that the sequences from more T.gondii-susceptible species (hamster and mouse) showed a better overall match with the ROP18 kinase motif determined in this study relative to those from the T. gondii-resistant rat (Table 2). This observation suggests that factors other than the local sequence context may influence the ability of ROP18 to phosphorylate these sites, and/or that other proteins in addition to the IRGs may contribute to resistance against T. gondii infection in rats. The requirement of ROP5 for ROP18-mediated virulence and the ability of ROP5 to interact with and disrupt the recruitment of IRG proteins suggests that ROP5 may be an additional factor in determining the relationship between IRG polymorphism and susceptibility to T. gondii (48).
TABLE 2.
Scansite matching of phosphorylation sites within Switch I region of IRG proteins with ROP18 kinase motif
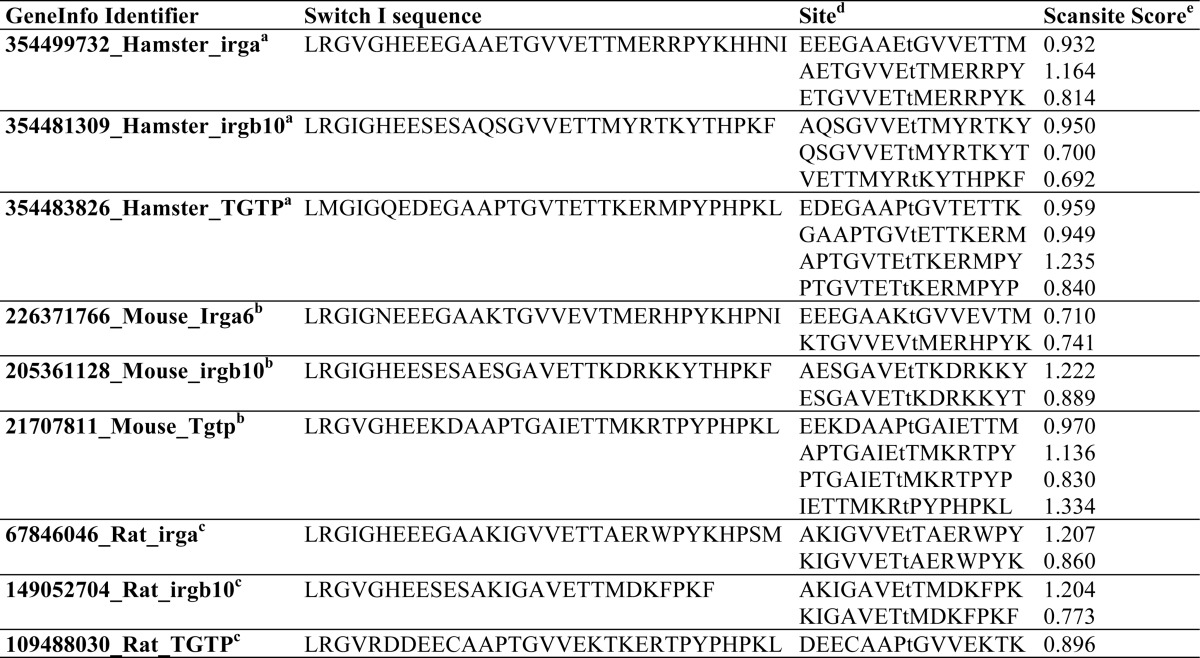
a C. griseus.
b M. musculus.
c R. norvegicus.
d Phospho-accepting Thr residue is in lower case.
e Lower scores indicate a better match, with 0 corresponding to a perfect match with the ROP18 kinase motif determined in Fig. 3.
The Crystal Structure of the ROP18 Kinase Domain Reveals a Potential Ligand-binding Pocket Adjacent to the Active Site
The multiwavelength anomalous diffraction-phased electron density map revealed a sucrose molecule bound within a shallow pocket on the N-terminal lobe of the ROP18 kinase domain (Fig. 3). Sucrose, which was included as a cryoprotectant during flash cooling of the crystals, was the only buffer component that was consistent with the size and shape of the strong non-protein electron density (Fig. 3B). This putative ligand-binding pocket is lined by elements of helix αN2 strands β1 and β2, the G-loop, and strands β3 and β4b. Binding of sucrose within this pocket buries 567 Å2 of surface area. The sucrose molecule is hydrogen bonded to Ser-260, Gly-261 (backbone carbonyl), Thr-265 and Arg-347, and makes van der Waals contacts with Leu-224, Ile-227, the carbonyl carbon of Ala-264, Met-284, and the Cα of Ser-285 (Fig. 3C). A distinctive feature of this pocket is a positively charged region at one end, corresponding to the side chains of Arg-223 and Arg-347, and we hereafter refer to this pocket as the ROP18 “basic pocket.” The basic pocket is not conserved in the other ROPK family crystal structures, due to the presence of side chains that protrude into the region corresponding to the ROP18 basic pocket in ROP2, ROP8, and ROP5 (25–27).
FIGURE 3.
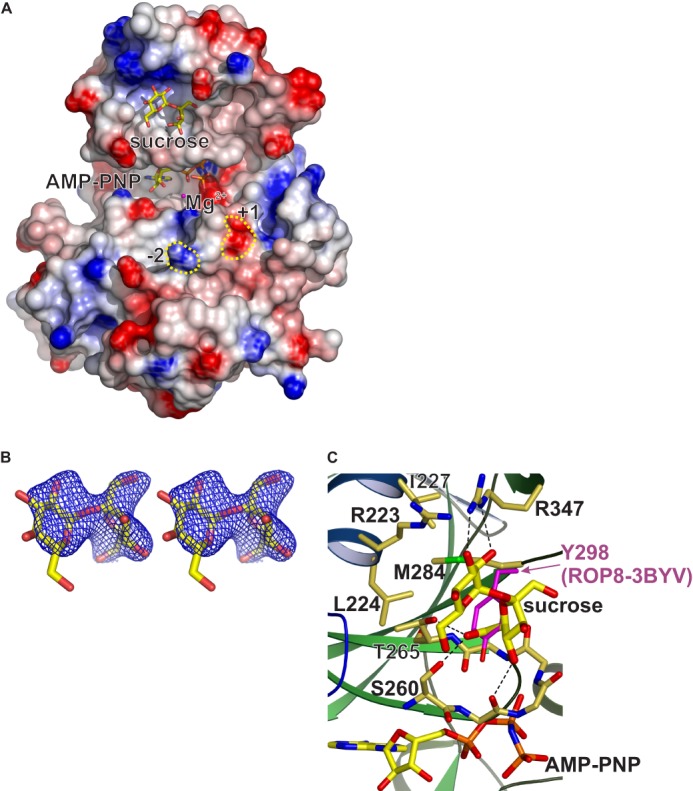
Bound sucrose molecule in the ROP18 kinase domain crystal structure identifies a potential ligand-binding pocket on the N-terminal lobe. A, molecular surface with positive and negative electrostatic potentials colored blue and red, respectively. Sucrose and AMP-PNP are shown in stick rendering with yellow carbons. Regions corresponding to the −2 and +1 positions relative to the phosphoacceptor position of the substrate are highlighted with yellow dotted lines. B, electron density (figure of merit weighted Fo map with solvent flattened experimental phases from two independent multiwavelength anomalous diffraction experiments) for the sucrose molecule contoured at 1σ to 2.6-Å resolution. C, close-up view of the basic pocket with sucrose-interacting side chains shown in stick rendering and hydrogen bonds indicated by dashed lines.
Given that sucrose is not known to be present in mammalian cells, it is unlikely to be a physiological ligand of ROP18 in vivo. However, the proximity of the bound sucrose molecule to the active site raised the possibility that the basic pocket may have physiologically relevant ligand(s), and that binding of such ligand(s) may contribute to substrate recognition or affect kinase activity by inducing conformational changes in the active site. Neither sucrose nor glucose at concentrations up to 100 mm affected ROP18 kinase activity in in vitro assays (data not shown). We suspected that the positively charged region within the ROP18 basic pocket may favor binding of ligands with negatively charged substituents. We therefore tested a number of candidate small molecule compounds with phosphate groups, including sugars, nucleotides, and phospholipids, for an effect on the in vitro activity of the ROP18 kinase domain. However, none of the compounds tested had a significant effect on kinase activity at physiologically relevant concentrations (data not shown). It remains possible that kinase activity may be affected by binding of a different (and more relevant) ligand, however, this cannot be tested directly without knowledge of what this ligand may be. We therefore tested the functional significance of this pocket by mutating residues within this region. To test the importance of this positive charge, Arg-223 was mutated to Ala or Glu, with the expectation that the R223E mutation would neutralize the positive charge in the basic pocket by forming a salt bridge with Arg-347. Because Arg-347 is hydrogen bonded to Thr-345 and with the backbone carbonyl of Val-340 on strand β4a, Arg-347 mutations were predicted to destabilize the folding in this region and were therefore not tested. In addition, Thr-265 and Met-284 were mutated to Tyr to introduce a bulky aromatic side chain that would partially fill the interior of the pocket. It is worth noting that in the ROP2 and ROP8 crystal structures, the position corresponding to Met-284 in ROP18 is occupied by Tyr, which would sterically clash with a bound sucrose molecule as seen in our ROP18 structure (Fig. 3C).
Our mutational analysis of the ROP18 basic pocket identifies the region around Arg-223 (positioned ∼20 Å from the γ-phosphate group of AMP-PNP) as being crucial for the ability of ROP18 to mediate virulence in mice. HA-tagged ROP18 constructs were expressed in type III parasites, and expression levels in multiple clones were determined by Western blotting (Fig. 4A). Wild type clones were selected for analysis such that the average WT expression level was similar or below those of the ROP18 mutants (Fig. 4B), and exclusion of the outlier WT clone B5.2 did not significantly alter the mouse survival analysis (Fig. 4, C and D). Expression of the R223A and R223E mutants in type III parasites resulted in greatly reduced virulence with 50 and 80% of the mice still surviving 30 days post-injection, respectively (Fig. 4C). Expression of the M284Y ROP18 mutant in type III parasites also resulted in a significant reduction of virulence in mice with ∼50% survival 30 days post-injection (Fig. 4C). The R223A, R223E, and M284Y mutations in ROP18 also reduced the rate of parasite growth within the host relative to WT (Fig. 4E and F), although the differences did not reach statistical significance (p values of 0.10, 0.10, and 0.09, respectively). In contrast, the T265Y mutation did not affect virulence or parasite growth in mice relative to WT (Fig. 4, A–C). Given its more peripheral position within the basic pocket, it is possible that the Tyr-265 substitution does not effectively occlude the key ligand binding region. The 4-fold increase in survival of mice infected with R223E ROP18-expressing parasites (80%) versus mice infected with WT ROP18-expressing parasites (20%) at 30 days post-injection (Fig. 4C) is unlikely to be accounted for by the statistically insignificant 35% reduction (p = 0.25) in expression level (Fig. 4B). We therefore tested the effect of mutating Arg-223 on kinase activity. In vitro kinase assays were performed with HA-tagged full-length WT, R223E, R223A, and D409N ROP18 proteins immunoprecipitated from parasites using an optimal peptide substrate (Fig. 5A), and kinase activities were normalized for the amounts of ROP18 protein immunoprecipitated (Fig. 5B). Interestingly both the R223E and R223A mutants were slightly more active relative to WT, although the differences were not statistically significant (p values of 0.90 and 0.089, respectively). We also tested the effects of the T265Y, M284Y, and D409N mutations on ROP18 kinase activity using purified recombinant ROP18 protein and found that both mutations reduced the normalized in vitro activity of the kinase domain relative to WT (p values of 0.0005 and 0.0258, respectively, Fig. 5C). In all cases, a kinase dead (D409N) control indicated no contaminating kinase activity toward the substrate peptide. The lack of an effect of the T265Y mutation on virulence despite the reduced kinase activity may have been due in part to the 2-fold greater expression level relative to WT (Fig. 4B). That the effects of the basic pocket mutants on virulence could not be explained by their effects on kinase activity or differences in their expression levels in parasites relative to WT suggests that the basic pocket is likely to have a biochemical function important for ROP18-mediated pathogenicity.
Acute Virulence Conferred by ROP18 Involves Multiple Downstream Targets Requiring Distinct Modes of Recognition
ROP18 promotes the survival and rapid expansion of T. gondii in host cells (including early responding immune cells such as inflammatory monocytes and macrophages), and this has been shown to be required for acute virulence in mice. A key mechanism of parasite clearance within host cells involves the sequential recruitment of multiple members of the IRGs to the PVM, which leads to the rupture of the vacuole and destruction of the parasites (19). Irgb6 is among the first IRGs recruited to the PVM, and has recently been shown to be phosphorylated by ROP18, which blocks its accumulation on the PVM to prevent IRG-mediated rupture of the PV (17). We therefore tested the ability of our ROP18 mutants to inhibit Irgb6 accumulation on the PVM in MEFs infected with T. gondii (Fig. 6, A and B). To elicit a robust IRG response, the MEFs were pretreated with IFNγ to up-regulate IRG expression. Expression of WT ROP18 in the type III strain dramatically reduced the proportion of PVs coated with Irgb6 from 50 to 14% (p = 0.0008). Parasites expressing the M284Y and T265Y mutants were the least effective in blocking Irgb6 recruitment, with 34 and 32% of PVs coated with Irgb6, respectively (p values of 0.025 and 0.052, respectively). Interestingly, the R223A and R223E mutations, which resulted in greater reductions in virulence relative to the T265Y and M284Y mutations, resulted in a smaller defect in the ability of ROP18 to block Irgb6 recruitment, with 23 and 20% of PVs coated with Irgb6 for the R223A and R223E ROP18 expressing parasites, respectively, and were not statistically different from WT. The strong reduction in virulence caused by the Arg-223 mutations is therefore unlikely to be accounted for by reduced phosphorylation of Irgb6 alone. These results indicate that in addition to Irgb6, ROP18 likely phosphorylates or interacts with other targets (such as Irga6) important for acute virulence (18), and that Arg-223 (which is positioned in a potential ligand-binding pocket outside of the active site) is required for interaction with some of these targets.
FIGURE 6.
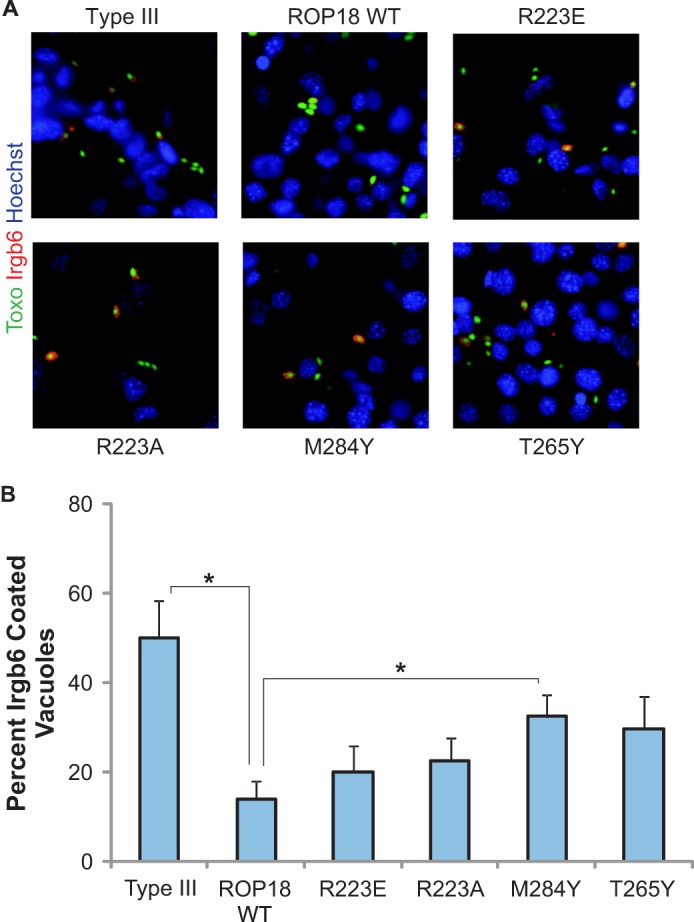
Parasites expressing ROP18 constructs mutated within the basic pocket are less effective in inhibiting Irgb6 accumulation at the PVM. A, immunofluorescence localization of Irgb6 (red) in WT MEFs stimulated for 24 h with 1000 units of IFNγ and infected for 1 h with type III, type III + ROP18WT, and type III strains expressing the indicated ROP18 construct. Parasites expressed GFP (green). B, quantification of Irgb6 localization on the parasite-containing vacuole for type III parasites and type III parasites expressing the indicated ROP18. Mean ± S.E., n = 3 experiments (* indicates p < 0.05).
DISCUSSION
We have determined the structure of the unphosphorylated ROP18 kinase domain in complex with AMP-PNP. Our structure is inconsistent with the published ROP18 homology model, which predicted an autoinhibitory αN1-αN2 linker with the side chains of Gln-214 and Gln-216 protruding into the ATP binding pocket (26). Recombinant ROP18 kinase domain was previously found to be autophosphorylated at sites within helix αN2 (Ser-221 and Thr-229) and strand β1 (Thr-249 and Thr-251), and it was proposed that phosphorylation of these residues would induce conformational changes that would displace the αN1-αN2 linker to relieve autoinhibition. Mutation of these serine and threonine residues to alanine was reported to drastically reduce kinase activity, presumably by abrogating autophosphorylation of these sites (26). It is unclear why mutation of these residues to alanine affected kinase activity, given that our structure is inconsistent with ATP binding requiring phosphorylation of any of these residues. Furthermore, the side chain hydroxyls of these serine and threonine residues are not within hydrogen bonding distance to any of the surrounding protein atoms, although it remains possible that these residues are involved in hydrogen bonding networks mediated by water molecules not visible due to the limited resolution of our x-ray data. Such hydrogen bonding interactions would be disrupted by mutation of these residues to Ala, which could reduce kinase activity by destabilizing or perturbing the structure.
Our crystal structure also does not readily explain the increased activity reported for the Q214A and Q216A mutants (26). It is unlikely that these residues impede ATP-binding as proposed, given that they are located within the disordered αN1-αN2 linker region. It is possible that the Q214A and Q216A mutations may facilitate improved binding of protein substrates in the in vitro autophosphorylation and myelin basic protein phosphorylation assays reported.
The ROP18 kinase substrate phosphorylation motif presented here shows a broad substrate specificity. The relatively low sequence selectivity of the ROP18 kinase motif suggests that other specificity determinants may exist to ensure that ROP18 is properly targeted to its physiological substrates.
The basic pocket on the N-terminal lobe of the ROP18 kinase domain that we identified reveals a potential ligand interaction site that may provide this additional substrate selectivity determinant and/or a site of allosteric regulation. Despite considerable experimental effort, however, it is currently unclear what the physiological ligand for this pocket may be. The proximity of this pocket to the active site, however, suggests several potential mechanisms of how ligand binding at this pocket may regulate substrate specificity and/or kinase activity. Although sucrose does not significantly affect kinase activity in vitro, the reduced activity resulting from the R223E mutation in the context of full-length ROP18 demonstrates the potential for an allosteric function for the basic pocket upon binding of a suitable ligand to affect kinase activity. The basic pocket may also provide a substrate interaction site outside of the active site required for recognition of a subset of ROP18 substrates, and suggests that post-translational modifications such as glycosylation, may be important in substrate selection. Such a role for the basic pocket could also be a contributing factor to the severely reduced virulence resulting from the Arg-223 and Met-284 mutations. This mode of substrate recognition is analogous to the binding mode of eIF2α to protein kinase R (PKR), in which substrate specificity is not determined by the immediate sequence context surrounding the phosphoacceptor site, but by substrate docking onto helix αG, which is peripheral to the active site. The crystal structure of the PKR-eIF2α complex shows eIF2α binding to helix αG in a configuration that positions the segment containing the Ser-51 phosphorylation site on eIF2α within the PKR active site (57). However, this segment of eIF2α is disordered in the crystal structure, which is consistent with the complete lack of sequence specificity of PKR for this region (58). The basic pocket on ROP18 may provide a peripheral substrate binding/recognition site similar to helix αG of PKR, which could confer higher substrate specificity than is indicated by the kinase motif. In light of the strong reduction of virulence by the Arg-223 mutations, which only mildly affect Irgb6 accumulation to PVs, it is likely that ROP18 phosphorylates a number of proteins critical for virulence in addition to the IRG proteins, and that the potential substrate recognition function of the basic pocket may be required for recognition of some of these additional ROP18 substrates. This may be of particular importance in host species that lack IRG family proteins.
It remains possible that N-terminal regions of ROP18 not included in our structural studies of the kinase domain may affect the kinase activity and/or substrate targeting of ROP18. In support of this, the host endoplasmic reticulum-bound transcription factor ATF6β was recently reported as an additional ROP18 target critical for host resistance against T. gondii infection, and a direct interaction was reported between the C terminus of ATF6β, and residues 147 to 164 of ROP18, which are N-terminal to the kinase domain (24). ATF6β phosphorylation by ROP18 at Thr residues mediated its proteasome-dependent degradation, resulting in decreased expression of its target genes, although the specific residues phosphorylated were not identified. It was unclear if the N-terminal region of ROP18 directly binds to the C-terminal region of ATF6β in vivo as proposed, given that ATF6 proteins are anchored in the endoplasmic reticulum membrane with their C termini within the endoplasmic reticulum lumen, and that ROP18 is localized on the cytoplasmic face of the PVM. Additionally, the experiments using their N terminally truncated ROP18 constructs are difficult to interpret. The ΔN2-ROP18 construct lacked the N-terminal region required for PVM localization (59). Loss of PVM localization would be expected to impair targeting of ROP18 to PVM-localized substrates, such as IRGs (60, 61). The severely truncated Δ240 ROP18 construct (missing the first 240 residues) lacked a significant portion of the kinase domain (as can be seen from our crystal structure), such that the Δ240 ROP18 construct would not be expected to fold properly.
The interaction of sucrose with the basic pocket in the N-terminal lobe of the kinase domain involves multiple hydrogen bonds and van der Waals contacts, and hints at the potential for binding small molecule metabolites (e.g. carbohydrates) or post-translational modifications on proteins (e.g. glycosylation). A recent study identified 132 proteins that were enriched by serial lectin affinity chromatography from a T. gondii membrane extract (62). ROP18 was among the 15 rhoptry proteins identified, but the serial lectin affinity chromatography purification was performed under native conditions, and it remains unclear how many of the proteins identified in this study were indeed glycosylated and directly bound by the lectins or were enriched by serial lectin affinity chromatography due to their association with glycoproteins. However, the results of this study indicate that glycosylation may be a more prominent post-translational modification in T. gondii than previously appreciated, and increases the likelihood that glycosylation may be involved in ROP18 substrate recognition.
Our results showing the dramatically reduced virulence of type III parasites expressing R223A and R223E ROP18 mutants indicate a critical role for the positive charge in this region of the basic pocket, which may optimally bind ligands containing negatively charged substituents, such as carboxylate or phosphate groups. Interestingly, crystal structures of CK2α have revealed a hydrophobic pocket on the N-terminal lobe, capable of binding glycerol and the small molecule ATP-competitive inhibitor 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (63, 64). This pocket coincides with the site of interaction with the non-catalytic CK2β subunit, and a biochemical screen for inhibitors of CK2 subunit association have identified a podophyllotoxine indolo analog (W16) that binds at the CK2 subunit interface and allosterically inhibits the catalytic activity of CK2α (65). Binding of CK2β alters the substrate preference and the subcellular localization of CK2α (66, 67), and it has been proposed that CK2 function may be regulated by small molecule metabolites in vivo or small molecule inhibitors that inhibit CK2 subunit association (63–65). Drawing on these parallels with CK2α, an additional potential role for the basic pocket in ROP18 may be as a site of interaction with protein interaction partners or substrates, which can be regulated by small molecule metabolite binding. Our functional studies have identified Arg-223 as a critical residue for the ability of ROP18 to confer acute virulence and indicate the potential for small molecule ligands targeting this region to abrogate virulence by disrupting ROP18 function. Sucrose (in the context of our ROP18 structure) may provide a suitable starting template for the rational development of such compounds that could provide a novel and potentially more specific mode of kinase inhibition than what is typically achieved with conventional ATP-competitive inhibitors.
Acknowledgments
We thank the staff at the NE-CAT 24-ID-E and 24-ID-C beam lines at the Advanced Photon Source synchrotron facility at Argonne National Laboratory for assistance with x-ray data collection, and Tobias Ehrenberger for help with the Scansite analyses. Sequencing and mass spectrometry data, and substrate peptides were obtained from the Koch Institute Swanson Biotechnology Center.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-AI080621 (to J. P. J. S.) and R01-GM52981, P50-GM68762, and R01-GM104047 (to M. B. Y.) and a Massachusetts Life Sciences Center New Investigator Award (to J. P. J. S.).
The atomic coordinates and structure factors (code 4JRN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
D. Liu and M. B. Yaffe, unpublished data.
- ROPK
- rhoptry kinase
- AMP-PNP
- adenylyl-β,γ-imidodiphosphate
- PV
- parasitophorous vacuole
- PVM
- parasitophorous vaculolar membrane
- IFNγ
- interferon γ
- IRG
- immunity-related p47 GTPases
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Dubey J. P., Jones J. L. (2008) Toxoplasma gondii infection in humans and animals in the United States. Int. J. Parasitol. 38, 1257–1278 [DOI] [PubMed] [Google Scholar]
- 2. Hill D., Dubey J. P. (2002) Toxoplasma gondii. Transmission, diagnosis and prevention. Clin. Microbiol. Infect. 8, 634–640 [DOI] [PubMed] [Google Scholar]
- 3. Joiner K. A., Dubremetz J. F. (1993) Toxoplasma gondii, a protozoan for the nineties. Infect. Immun. 61, 1169–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones J. L., Kruszon-Moran D., Sanders-Lewis K., Wilson M. (2007) Toxoplasma gondii infection in the United States, 1999 2004, decline from the prior decade. Am. J. Trop. Med. Hyg. 77, 405–410 [PubMed] [Google Scholar]
- 5. Montoya J. G., Liesenfeld O. (2004) Toxoplasmosis. Lancet 363, 1965–1976 [DOI] [PubMed] [Google Scholar]
- 6. Bliss S. K., Gavrilescu L. C., Alcaraz A., Denkers E. Y. (2001) Neutrophil depletion during Toxoplasma gondii infection leads to impaired immunity and lethal systemic pathology. Infect. Immun. 69, 4898–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sibley L. D., Ajioka J. W. (2008) Population structure of Toxoplasma gondii. Clonal expansion driven by infrequent recombination and selective sweeps. Annu. Rev. Microbiol. 62, 329–351 [DOI] [PubMed] [Google Scholar]
- 8. Taylor S., Barragan A., Su C., Fux B., Fentress S. J., Tang K., Beatty W. L., Hajj H. E., Jerome M., Behnke M. S., White M., Wootton J. C., Sibley L. D. (2006) A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314, 1776–1780 [DOI] [PubMed] [Google Scholar]
- 9. Saeij J. P., Boyle J. P., Coller S., Taylor S., Sibley L. D., Brooke-Powell E. T., Ajioka J. W., Boothroyd J. C. (2006) Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314, 1780–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gajria B., Bahl A., Brestelli J., Dommer J., Fischer S., Gao X., Heiges M., Iodice J., Kissinger J. C., Mackey A. J., Pinney D. F., Roos D. S., Stoeckert C. J., Jr., Wang H., Brunk B. P. (2008) ToxoDB. An integrated Toxoplasma gondii database resource. Nucleic Acids Res. 36, D553–D556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kissinger J. C., Gajria B., Li L., Paulsen I. T., Roos D. S. (2003) ToxoDB. Accessing the Toxoplasma gondii genome. Nucleic Acids Res. 31, 234–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lim D. C., Cooke B. M., Doerig C., Saeij J. P. (2012) Toxoplasma and Plasmodium protein kinases. Roles in invasion and host cell remodelling. Int. J. Parasitol. 42, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peixoto L., Chen F., Harb O. S., Davis P. H., Beiting D. P., Brownback C. S., Ouloguem D., Roos D. S. (2010) Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe 8, 208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. El Hajj H., Lebrun M., Arold S. T., Vial H., Labesse G., Dubremetz J. F. (2007) ROP18 is a rhoptry kinase controlling the intracellular proliferation of Toxoplasma gondii. PLoS Pathog 3, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boothroyd J. C., Dubremetz J. F. (2008) Kiss and spit. The dual roles of Toxoplasma rhoptries. Nat. Rev. Microbiol. 6, 79–88 [DOI] [PubMed] [Google Scholar]
- 16. Dubey J. P., Lindsay D. S., Speer C. A. (1998) Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin. Microbiol. Rev. 11, 267–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fentress S. J., Behnke M. S., Dunay I. R., Mashayekhi M., Rommereim L. M., Fox B. A., Bzik D. J., Taylor G. A., Turk B. E., Lichti C. F., Townsend R. R., Qiu W., Hui R., Beatty W. L., Sibley L. D. (2010) Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 8, 484–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Steinfeldt T., Könen-Waisman S., Tong L., Pawlowski N., Lamkemeyer T., Sibley L. D., Hunn J. P., Howard J. C. (2010) Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol. 8, e1000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Howard J. C., Hunn J. P., Steinfeldt T. (2011) The IRG protein-based resistance mechanism in mice and its relation to virulence in Toxoplasma gondii. Curr. Opin. Microbiol. 14, 414–421 [DOI] [PubMed] [Google Scholar]
- 20. Martens S., Parvanova I., Zerrahn J., Griffiths G., Schell G., Reichmann G., Howard J. C. (2005) Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 1, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamamoto M., Okuyama M., Ma J. S., Kimura T., Kamiyama N., Saiga H., Ohshima J., Sasai M., Kayama H., Okamoto T., Huang D. C., Soldati-Favre D., Horie K., Takeda J., Takeda K. (2012) A cluster of interferon-γ-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity 37, 302–313 [DOI] [PubMed] [Google Scholar]
- 22. Selleck E. M., Fentress S. J., Beatty W. L., Degrandi D., Pfeffer K., Virgin H. W., 4th, Macmicking J. D., Sibley L. D. (2013) Guanylate-binding protein 1 (Gbp1) contributes to cell-autonomous immunity against Toxoplasma gondii. PLoS Pathog 9, e1003320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Degrandi D., Kravets E., Konermann C., Beuter-Gunia C., Klümpers V., Lahme S., Wischmann E., Mausberg A. K., Beer-Hammer S., Pfeffer K. (2013) Murine guanylate binding protein 2 (mGBP2) controls Toxoplasma gondii replication. Proc. Natl. Acad. Sci. U.S.A. 110, 294–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamamoto M., Ma J. S., Mueller C., Kamiyama N., Saiga H., Kubo E., Kimura T., Okamoto T., Okuyama M., Kayama H., Nagamune K., Takashima S., Matsuura Y., Soldati-Favre D., Takeda K. (2011) ATF6beta is a host cellular target of the Toxoplasma gondii virulence factor ROP18. J. Exp. Med. 208, 1533–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Labesse G., Gelin M., Bessin Y., Lebrun M., Papoin J., Cerdan R., Arold S. T., Dubremetz J. F. (2009) ROP2 from Toxoplasma gondii. A virulence factor with a protein-kinase fold and no enzymatic activity. Structure 17, 139–146 [DOI] [PubMed] [Google Scholar]
- 26. Qiu W., Wernimont A., Tang K., Taylor S., Lunin V., Schapira M., Fentress S., Hui R., Sibley L. D. (2009) Novel structural and regulatory features of rhoptry secretory kinases in Toxoplasma gondii. EMBO J. 28, 969–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reese M. L., Boothroyd J. C. (2011) A conserved non-canonical motif in the pseudoactive site of the ROP5 pseudokinase domain mediates its effect on Toxoplasma virulence. J. Biol. Chem. 286, 29366–29375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ku S. Y., Smith G. D., Howell P. L. (2007) ADP-2Ho as a phasing tool for nucleotide-containing proteins. Acta Crystallogr. D Biol. Crystallogr. 63, 493–499 [DOI] [PubMed] [Google Scholar]
- 29. Otwinowski Z., Minor W. (1997) in Methods in Enzymology, Macromolecular Crystallography, part A (Carter C. W., Jr., Sweet R. M., ed) Vol. 276, pp. 307–326, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 30. Collaborative Computation Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D. Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 31. Brunger A. T. (2007) Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 32. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system. A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 33. Bricogne G., Vonrhein C., Flensburg C., Schiltz M., Paciorek W. (2003) Generation, representation and flow of phase information in structure determination. Recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 59, 2023–2030 [DOI] [PubMed] [Google Scholar]
- 34. Read R. J. (1986) Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A Found. Crystallogr. 42, 140–149 [Google Scholar]
- 35. Terwilliger T. C. (2003) SOLVE and RESOLVE. Automated structure solution and density modification. Methods Enzymol. 374, 22–37 [DOI] [PubMed] [Google Scholar]
- 36. Terwilliger T. (2004) SOLVE and RESOLVE. Automated structure solution, density modification and model building. J. Synchrotron Radiat. 11, 49–52 [DOI] [PubMed] [Google Scholar]
- 37. McRee D. E., Israel M. (2008) XtalView, protein structure solution and protein graphics, a short history. J. Struct. Biol. 163, 208–213 [DOI] [PubMed] [Google Scholar]
- 38. McRee D. E. (1999) XtalView/Xfit. A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125, 156–165 [DOI] [PubMed] [Google Scholar]
- 39. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kraulis P. J. (1991) MOLSCRIPT. A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24, 946–950 [Google Scholar]
- 41. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA [Google Scholar]
- 42. Rocchia W., Alexov E., Honig B. (2001) Extending the applicability of the nonlinear Poisson-Boltzmann equation. Multiple dielectric constants and multivalent ions. J. Phys. Chem. B 105, 6507–6514 [Google Scholar]
- 43. Rocchia W., Sridharan S., Nicholls A., Alexov E., Chiabrera A., Honig B. (2002) Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies. Applications to the molecular systems and geometric objects. J. Comput. Chem. 23, 128–137 [DOI] [PubMed] [Google Scholar]
- 44. Alexander J., Lim D., Joughin B. A., Hegemann B., Hutchins J. R., Ehrenberger T., Ivins F., Sessa F., Hudecz O., Nigg E. A., Fry A. M., Musacchio A., Stukenberg P. T., Mechtler K., Peters J. M., Smerdon S. J., Yaffe M. B. (2011) Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci. Signal. 4, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hutti J. E., Jarrell E. T., Chang J. D., Abbott D. W., Storz P., Toker A., Cantley L. C., Turk B. E. (2004) A rapid method for determining protein kinase phosphorylation specificity. Nat. Methods 1, 27–29 [DOI] [PubMed] [Google Scholar]
- 46. Fujii K., Zhu G., Liu Y., Hallam J., Chen L., Herrero J., Shaw S. (2004) Kinase peptide specificity. Improved determination and relevance to protein phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 101, 13744–13749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schneider T. D., Stephens R. M. (1990) Sequence logos. A new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Niedelman W., Gold D. A., Rosowski E. E., Sprokholt J. K., Lim D., Farid Arenas A., Melo M. B., Spooner E., Yaffe M. B., Saeij J. P. (2012) The rhoptry proteins ROP18 and ROP5 mediate Toxoplasma gondii evasion of the murine, but not the human, interferon-γ response. PLoS Pathog. 8, e1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horton R. M., Hunt H. D., Ho S. N., Pullen J. K., Pease L. R. (1989) Engineering hybrid genes without the use of restriction enzymes. Gene splicing by overlap extension. Gene 77, 61–68 [DOI] [PubMed] [Google Scholar]
- 50. Kammann M., Laufs J., Schell J., Gronenborn B. (1989) Rapid insertional mutagenesis of DNA by polymerase chain reaction (PCR). Nucleic Acids Res. 17, 5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rosowski E. E., Lu D., Julien L., Rodda L., Gaiser R. A., Jensen K. D., Saeij J. P. (2011) Strain-specific activation of the NF-κB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J. Exp. Med. 208, 195–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dar A. C., Wybenga-Groot L. E., Sicheri F. (2005) The Eukaryotic Protein Kinase Domain in Modular Protein Domains (Cesarini G., Gimona M., Sudol M., Yaffe M., eds), Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, 10.1002/3527603611.ch9 [DOI] [Google Scholar]
- 53. Nebl T., Prieto J. H., Kapp E., Smith B. J., Williams M. J., Yates J. R., 3rd, Cowman A. F., Tonkin C. J. (2011) Quantitative in vivo analyses reveal calcium-dependent phosphorylation sites and identifies a novel component of the Toxoplasma invasion motor complex. PLoS Pathog. 7, e1002222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Treeck M., Sanders J. L., Elias J. E., Boothroyd J. C. (2011) The phosphoproteomes of Plasmodium falciparum and Toxoplasma gondii reveal unusual adaptations within and beyond the parasites' boundaries. Cell Host Microbe 10, 410–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Obenauer J. C., Cantley L. C., Yaffe M. B. (2003) Scansite 2.0. Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yaffe M. B., Leparc G. G., Lai J., Obata T., Volinia S., Cantley L. C. (2001) A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat. Biotechnol. 19, 348–353 [DOI] [PubMed] [Google Scholar]
- 57. Dar A. C., Dever T. E., Sicheri F. (2005) Higher-order substrate recognition of eIF2α by the RNA-dependent protein kinase PKR. Cell 122, 887–900 [DOI] [PubMed] [Google Scholar]
- 58. Lu J., O'Hara E. B., Trieselmann B. A., Romano P. R., Dever T. E. (1999) The interferon-induced double-stranded RNA-activated protein kinase PKR will phosphorylate serine, threonine, or tyrosine at residue 51 in eukaryotic initiation factor 2α. J. Biol. Chem. 274, 32198–32203 [DOI] [PubMed] [Google Scholar]
- 59. Reese M. L., Boothroyd J. C. (2009) A helical membrane-binding domain targets the Toxoplasma ROP2 family to the parasitophorous vacuole. Traffic 10, 1458–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Melo M. B., Jensen K. D., Saeij J. P. (2011) Toxoplasma gondii effectors are master regulators of the inflammatory response. Trends Parasitol. 27, 487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fentress S. J., Steinfeldt T., Howard J. C., Sibley L. D. (2012) The arginine-rich N-terminal domain of ROP18 is necessary for vacuole targeting and virulence of Toxoplasma gondii. Cell Microbiol. 14, 1921–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Luo Q., Upadhya R., Zhang H., Madrid-Aliste C., Nieves E., Kim K., Angeletti R. H., Weiss L. M. (2011) Analysis of the glycoproteome of Toxoplasma gondii using lectin affinity chromatography and tandem mass spectrometry. Microbes Infect. 13, 1199–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Raaf J., Brunstein E., Issinger O. G., Niefind K. (2008) The CK2α/CK2β interface of human protein kinase CK2 harbors a binding pocket for small molecules. Chem. Biol. 15, 111–117 [DOI] [PubMed] [Google Scholar]
- 64. Raaf J., Issinger O. G., Niefind K. (2009) First inactive conformation of CK2α, the catalytic subunit of protein kinase CK2. J. Mol. Biol. 386, 1212–1221 [DOI] [PubMed] [Google Scholar]
- 65. Laudet B., Moucadel V., Prudent R., Filhol O., Wong Y. S., Royer D., Cochet C. (2008) Identification of chemical inhibitors of protein-kinase CK2 subunit interaction. Mol. Cell Biochem. 316, 63–69 [DOI] [PubMed] [Google Scholar]
- 66. Meggio F., Boldyreff B., Marin O., Marchiori F., Perich J. W., Issinger O. G., Pinna L. A. (1992) The effect of polylysine on casein-kinase-2 activity is influenced by both the structure of the protein/peptide substrates and the subunit composition of the enzyme. Eur. J. Biochem. 205, 939–945 [DOI] [PubMed] [Google Scholar]
- 67. Filhol O., Nueda A., Martel V., Gerber-Scokaert D., Benitez M. J., Souchier C., Saoudi Y., Cochet C. (2003) Live-cell fluorescence imaging reveals the dynamics of protein kinase CK2 individual subunits. Mol. Cell. Biol. 23, 975–987 [DOI] [PMC free article] [PubMed] [Google Scholar]