

Abstract
Mitogen-activated protein kinases (MAPKs; ERK1/2, p38, JNK, and ERK5) have evolved to transduce environmental and developmental signals (growth factors, stress) into adaptive and programmed responses (differentiation, inflammation, apoptosis). Almost 20 years ago, it was discovered that MAPKs contain a docking site in the C-terminal lobe that binds a conserved 13-16 amino acid sequence known as the D- or KIM-motif (kinase interaction motif). Recent crystal structures of MAPK:KIM-peptide complexes are leading to a precise understanding of how KIM sequences contribute to MAPK selectivity. In addition, new crystal and especially NMR studies are revealing how residues outside the canonical KIM motif interact with specific MAPKs and contribute further to MAPK selectivity and signaling pathway fidelity. In this review, we focus on these recent studies, with an emphasis on the use of NMR spectroscopy, isothermal titration calorimetry and small angle X-ray scattering to investigate these processes.
Keywords: MAP kinase, ERK, p38, JNK, structure, kinase interaction motif, KIM, D-motif, NMR, SAXS, PTP, DUSP
Introduction
Protein phosphorylation1,2 is one of the key mechanisms used to communicate external signals from the membrane to the nucleus. In particular, mitogen-activated protein kinases (MAPKs; ERK1/2, p38, JNK and ERK5) have evolved to transduce environmental and developmental signals (growth factors, stress) into adaptive and programmed responses (differentiation, inflammation, apoptosis).3,4 The importance of protein phosphorylation by MAPKs is well-illustrated by the many inherited or acquired human diseases that stem from abnormalities in MAPK signaling pathways, including Parkinson's, inflammatory disorders and cancer.4,5 As might be expected, MAPKs are ubiquitously expressed, making them conceivable but difficult targets for drug treatments.6 However, MAPK activation is finely tuned in a cell-type specific and temporal manner, making MAPK regulators much more attractive targets for drug development.7 These regulators include: (1) upstream kinases, (2) downstream phosphatases, and (3) scaffolding proteins.8,9
The MAPK pathways consist of three components: (1) a MAP kinase kinase kinase (MAP3K), (2) a MAP kinase (MAP2K), and (3) a MAPK. Stimulation of the pathway results in the eventual activation of the MAPK by dual phosphorylation of a threonine and a tyrosine residue (T-X-Y) located in their phosphorylation loop.10 This activation is controlled by MAP2Ks (e.g. MEK1/2 for ERK; MKK3/6 for p38 and MKK4/7 for JNK). While the kinase cascades that direct MAPK activation appear stereotypic, the guidance and fine-tuning that direct cell- and situation-specific responses are governed by MAPK-specific phosphatases and scaffolding proteins. For example, MAPKs are dephosphorylated by dual specificity phosphatases (DUSPs),11 kinase interaction motif protein tyrosine phosphatases (KIM-PTPs)12 and serine/threonine protein phosphatases (e.g. PP2A). Recently, MAPK scaffolds (e.g., KSR) have also been shown to play a key role in modulating the strength and duration of MAPK activation.13,14 A comprehensive understanding of how the activity of MAPKs is finely controlled by MAPK regulatory proteins can only be obtained by understanding how these proteins interact at a molecular level. In this review, we summarize recent crystallographic and biomolecular NMR spectroscopy studies and highlight how these studies provide fundamental new structural insights into these interactions.
Mitogen-Activated Protein Kinases
Mitogen-activated protein kinases (MAPKs) are serine/threonine-specific kinases. In mammals, the MAPKs fall into distinct groups, including the extracellular signal-regulated kinases 1 and 2 (ERK1/2), c-Jun amino terminal kinases 1-3 (JNK1/2/3), p38 (isoforms α, β, γ, and δ) and ERK5.15 MAPKs are also present in lower organisms, such as yeast (i.e., Fus3).16 MAPKs have a bi-lobed architecture, with a five-stranded β-sheet in the N-terminal lobe and six α-helices in the C-terminal lobe.17,18 Almost 20 years ago, it was discovered that MAPKs contain a docking site in the C-terminal lobe that binds a conserved 13–16 amino acid sequence known as the D- or KIM-motif (kinase interaction motif).19 The KIM-motif has a well-established consensus sequence (R/K)2–3-X2–6-ΦA-X-ΦB (Φ is any hydrophobic residue; Fig. 1).
Figure 1.
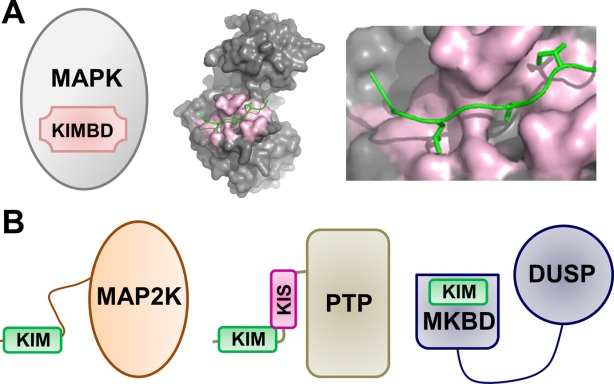
MAPKs and MAPK regulatory proteins. (A) Cartoon of the MAPK KIM binding domain (KIMBD); p38 bound to the MEF2A KIM peptide (PDBID 1LEW). (B) Cartoons of MAPK regulatory proteins, including MAP2Ks (left), KIM-PTPs (middle) and DUSPs (right).
The majority of MAPK regulatory proteins investigated thus far contain KIMs.20–22 For example, the MKKs MEK1/2, MKK3/6, MKK4/7, and STE7, representing the MAP2Ks of ERK, p38, JNK, and FUS3, respectively, possess a flexible N-terminal extension, which includes the ∼15 amino acid KIM, followed by their dual-specificity kinase domain [Fig. 1(B), left].23,24 Furthermore, the KIM-PTPs [Fig. 1(B), center; hematopoietic tyrosine phosphatase, HePTP, immune system specific;25 striatum-enriched phosphatase, STEP, brain specific;26 STEP-like PTP, PTPSL, brain specific27) also possess an N-terminal unstructured extension, which includes the KIM, followed by their C-terminal phosphatase domain (PTP). The KIM-PTPs regulate the activity of MAPKs by both protein localization (by retaining the MAPKs in the cytosol, the “resting” state) and dephosphorylation of their active sites (the “active” state).28 Notably, MAPK scaffolding proteins (JNK-interacting protein, JIP1;29 phosphoprotein enriched in astrocytes 15, PEA-1530) and substrates (Ribosomal protein S6 kinase alpha-1, RSK1;31 MAP kinase-activated protein kinase 2, MK2;32 and myocyte enhancer factor 2A, MEF2a33) also contain linear KIM domain, which mediates MAPK binding in the cell. MAP Kinase Phosphatases (MKPs, members of the dual specificity phosphatase, or DUSP, family) are a second family of phosphatases that regulate MAPK activity.11,34 However, unlike the KIM-PTPs, the DUSPs/MKPs have two structured domains: an N-terminal MAPK binding domain (MKBD) and a C-terminal dual specificity phosphatase domain [Fig. 1(B), right].11,34 In DUSPs/MKPs, the KIM is not a linear peptide but instead part of the structured MKBD domain35–37 and thus is much more constrained than that of e.g. MKKs or KIM-PTPs.
While the KIM is required for binding,38,39 multiple reports have shown that residues outside the KIM contribute to both binding and specificity.40,41 Recent crystal structures of MAPK:KIM-peptide complexes are leading to a precise understanding of how KIM sequences contribute to MAPK selectivity. In addition, new crystallographic and NMR studies are revealing how residues outside the canonical KIM motif interact with specific MAPKs and contribute further to MAPK selectivity and signaling pathway fidelity. Here, we focus on these recent studies, with an emphasis on the use of NMR spectroscopy, isothermal titration calorimetry (ITC) and small angle X-ray scattering (SAXS) to investigate these processes.
Crystallographic Studies of MAPKs with KIM Peptides and Regulatory/Substrate Proteins
The first crystal structures of a MAPK, p38, bound to a KIM peptide (MEF2A, a substrate, and MKK3b, an activating kinase) were described in 2002 by the Goldsmith laboratory.42 These ground-breaking structures revealed the initial insights into the molecular determinants that direct MAPK-KIM peptide interactions. Since then, an additional 29 MAPK:KIM-containing peptide or protein complexes have been determined of various MAPKs (ERK2, p38α, JNK1, JNK3, Fus3, ERK5) bound to KIM-containing peptides or proteins from activating kinases (MEK2,43,44 MKK5,45 MKK6,46 MKK3b,42,47 STE748), deactivating phosphatases (HePTP,44 Msg5,48 DUSP10/MKP535) and substrates/scaffolds (DCC,49 MEF2a,42 TAB1,50 JIP1,51,52 NFAT2,46 SAB,52 AFT2,52 Far1,48 PEA15,53 RSK1,43,46 MNK1,46 MK254,55). The available crystal structures of MAPK:KIM-containing peptide or protein complexes are listed in Table I and the overlays of the KIM peptides, along with their sequences, are shown in Fig. 2 (MAPK:peptide/MAPK:protein complexes overlaid using the C-terminal cores of the MAPK domains; C-terminal cores of the MAPK domains identified using procedures outlined in Page et al.56; the residues used for the alignments and the resulting RMSDs are listed in Table II). The MAPK KIM peptide binding pockets are labeled using the nomenclature in Garia et al.46 The KIM binding groove has four hydrophobic pockets: ΦA and ΦB (the first hydrophobic pockets identified), ΦL (L, lower; sometimes also referred to as ΦA-2) and ΦU (U, upper; also referred to as ΦH44). The KIM binding groove also has two electrostatic interaction sites (collectively also known as the CD site22): ΨU (U, upper) and ΨL (L, lower).
Table I.
Crystallographic Studies of MAPK Complexes
| MAPK complex | Orient | MAPK organism | P-loop visiblea | Res (Å) | PDB | Typeb | Ref |
|---|---|---|---|---|---|---|---|
| MAPK:peptide | |||||||
| ERK2 | |||||||
| ERK2:MKP360–76 | N→C | Rat | Partial | 2.50 | 2FYS | P | 82 |
| ERK2:MEK21–16 | N→C | Rat | 2.00 | n/a | K | 44 | |
| ERK2:HePTP16–31c | N→C | Rat | Y | 1.90 | 2GPH | P | 44 |
| ERK2:DCC1140–1166 | N→C | Rat | Partial | 1.95 | 3O71 | Su | 49 |
| ERK2:RSK1712–735c | C→N | Human | Partial | 2.40 | 3TEI | Su | 46 |
| ERK2:RSK1712–735c | C→N | Human | Partial | 2.30 | 4H3P | Su | 43 |
| ERK2:Synth–revD | C→N | Human | Y | 2.10 | 4FMQ | – | 46 |
| ERK2:MEK24–16c | N→C | Human | Y | 2.20 | 4H3Q | K | 43 |
| ERK2:MNK1434–451 | C→N | Human | Y | 1.55 | 2Y9Q | Su | 46 |
| p38α | |||||||
| p38α:MEF2A2–11 | N→C | Mouse | N | 2.30 | 1LEW | Su | 42 |
| p38α: MKK3b8–15c | N→C | Mouse | Partial | 2.30 | 1LEZ | K | 42 |
| p38α:MK2370–393 | C→N | Human | N | 2.00 | 2OKR | Su | 54 |
| pTpY–p38α:MKK3b8–18c | N→C | Mouse | N | 2.30 | 3P4K | K | 47 |
| p38α:MKK64–17 | N→C | Human | N | 1.95 | 2Y8O | K | 46 |
| p38α:TAB1395–415 | N→C | Mouse | N | 2.71 | 4KA3 | Su | 50 |
| JNK1 | |||||||
| JNK1:JIP1154–163 | N→C | Human | N | 2.35 | 1UKH | Sc | 51 |
| JNK1:NFAT4141–154 | N→C | Human | N | 1.33 | 2XRW | Su | 46 |
| JNK1:NFAT4141–154 | N→C | Human | N | 2.60 | 2XS0 | Su | 46 |
| JNK3 | |||||||
| JNK3:SAB341–350 | N→C | Human | Y | 2.08 | 4H3B | Su | 52 |
| JNK3:AFT248–55 | N→C | Human | Y | 3.00 | 4H36 | Su | 52 |
| JNK3:JIP1158–167 | N→C | Human | Partial | 1.99 | 4H39 | Sc | 52 |
| Fus3 | |||||||
| Fus3:STE79–20c | N→C | Yeast | Partial | 1.55 | 2B9H | K | 48 |
| Fus3:Msg525–38c | N→C | Yeast | Partial | 2.50 | 2B9I | P | 48 |
| Fus3:Far172–82c | N→C | Yeast | Partial | 2.30 | 2B9J | Su | 48 |
| MAPK:protein | |||||||
| ERK2 | |||||||
| ERK2:PEA–151–96 | Human | Partial | 1.80 | 4IZ7 | Sc | 53 | |
| T185E–ERK2:PEA–151–130 | C→N | Human | Y | 3.19 | 4IZ5 | Sc | 53 |
| pTpYERK2:PEA–151–96 | Human | Y | 1.93 | 4IZA | Sc | 53 | |
| ERK5 | |||||||
| ERK5:MKK52–126 | N→C | Human | Y | 2.60 | 4IC7 | K | 45 |
| p38α | |||||||
| p38α:MKP5/DUSP10139–288 | Mixed | Mouse | N | 2.71 | 3TG1 | P | 35 |
| p38α:MK22–406 | C→N | Human | N | 4.00 | 2ONL | Su | 54 |
| p38α:MK21–356 | C→N | Mouse | Partial | 2.70 | 2OZA | Su | 55 |
Partial indicates P–loops for which either the activation Thr or Tyr (or both) residue is visible, but other residues from the loop are still missing.
K, kinase, P, phosphatase, Su, substrate, Sc, scaffold.
Mutations in either the protein/peptide for optimal crystal formation.
Figure 2.
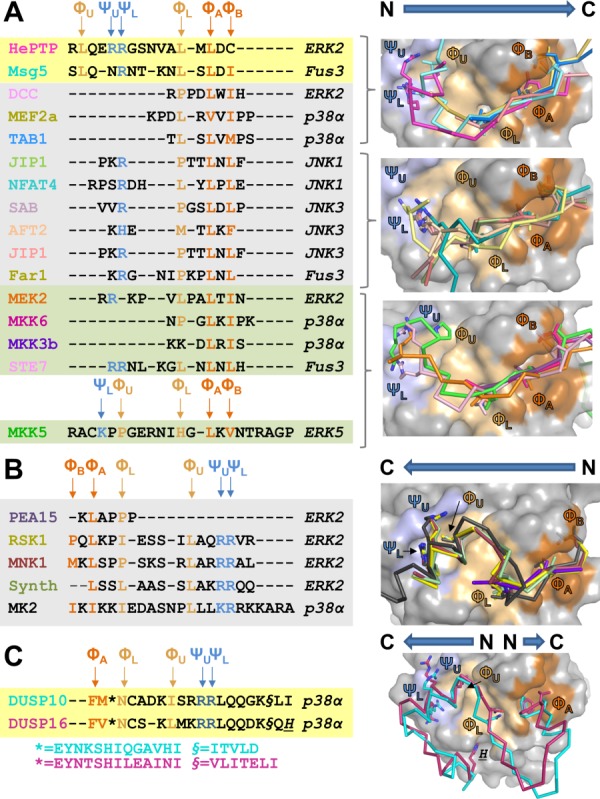
Structure-based sequence alignment of KIM peptides bound to MAPKs. (A) Left, structure-based sequence alignment of KIM peptides that bind the MAPK KIM binding pocket in the N→C direction; phosphatases on a yellow background, substrates/scaffolds on a grey background and kinases on a green background. Key interaction residues are colored. Positively charged amino acids that bind the electrostatic pocket are labeled ΨU and ΨL and colored blue; hydrophobic amino acids that bind the ΦA-X-ΦB binding groove are labeled ΦA and ΦB and colored orange; hydrophobic amino acids that bind the ΦL (also known as ΦA-2) and the ΦU (also known as ΦH) pockets are labeled and colored beige. The identities of the MAPKs present in the MAPK:KIMpeptide/protein complex structures are indicated to the right in italics. Right, MAPK:KIMpeptide/protein complexes superimposed using residues that optimally align the MAPK KIM docking grooves (Table II). MAPK surface is in grey, with the electrostatic binding pockets in light blue, the ΦA-X-ΦB binding groove in orange and the ΦL and ΦU binding pockets in beige. The bound KIM residues are shown as ribbons with the key interacting residues (ΨU, ΨL, ΦU, ΦL, ΦA, ΦB) shown as sticks. The ribbons are colored according to the MAPK interacting protein listed in A. B. Same as A, except KIM peptides bind in the C→N direction. C. Same as A, except bound KIM is part of a structured domain, the MAPK binding domain, and binds predominantly in the C→N direction. DUSP16 makes an additional interaction in a pocket below ΦL, which is indicated by H.
Table II.
RMSD of C-Terminal Core Domains Within and Between a MAPK Families
| MAPK | C-terminal core | Moving PDB | RMSD (Å2) | MAPK residues | ERK2 (3TEI) residues | RMSD (Å2) |
|---|---|---|---|---|---|---|
| ERK2 (3TEI) | 108–173 | 2FYSa | 1.04 | |||
| 190–201 | 2GPHa | 0.60 | ||||
| 207–254 | 3O71a | 0.59 | ||||
| 258–326 | 2Y9Q | 0.96 | ||||
| 4FMQ | 1.04 | |||||
| 4H3Q | 0.57 | |||||
| 4H3P | 0.58 | |||||
| 4IZ5 | 0.60 | |||||
| p38 (2Y80) | 109–117 | 1LEW | 0.30 | 109–113 | 108–112 | |
| 123–171 | 1LEZ | 0.40 | 123–169 | 122–168 | 0.65 | |
| 185–321 | 2OKR | 0.31 | 298–315 | 303–320 | ||
| 3P4K | 0.38 | |||||
| 4KA3 | 0.40 | |||||
| 3TGI | 1.10 | |||||
| JNK1 (2XRW) | 111–170 | 1UKH | 0.51 | 111–115 | 108–112 | |
| 199–250 | 2XS0 | 0.52 | 125–170 | 123–168 | 1.12 | |
| 267–281 | 311–328 | 303–320 | ||||
| 288–334 | ||||||
| JNK3 (4H39) | 149–207 | 4H3B | 0.48 | 149–153 | 108–112 | |
| 222–319 | 4H36 | 0.55 | 163–208 | 123–168 | 1.02 | |
| 326–371 | 349–366 | 303–320 | ||||
| FUS3 (2B9H) | 96–163 | 2B9I | 0.37 | 98–100 | 110–112 | |
| 180–319 | 2B9J | 0.24 | 111–156 | 123–168 | 0.46 | |
| 299–316 | 303–320 | |||||
| ERK5 (4IC7) | 140–143 | n/ab | n/ab | 140–143 | 108–111 | |
| 156–199 | 156–199 | 123–166 | 0.54 | |||
| 319–346 | 319–346 | 285–312 |
Mouse/rat ERK2 isoform; C-terminal core residues corresponding to the human isoform are 106–171, 188–199, 205–252, 256–324.
Only 1 ERK5 complex available.
Figure 2 readily illustrates that the mechanism of KIM engagement is not conserved among these interacting peptides. Some KIMs only interact with the hydrophobic pockets (i.e., MKK6), some only engage a subset of hydrophobic pockets (i.e., STE7, NFAT2, JIP1), while others occupy all pockets (RSK1, MNK1). In addition, while the structure of p38 bound to its substrate, the MAPKAP-2 (MK2) kinase was the first structure to show that p38 is capable of binding KIM peptides bi-directionally54,55 (that is, in a C-to-N, versus the more common N-to-C direction; Figs. 2(A,B)), a number of recent studies have shown that ERK is also capable of binding KIM peptides/proteins bi-directionally (i.e., RSK1 and PEA-15)46,53 and that DUSPs/MKPs, in which the KIM is not a linear peptide but instead part of the folded MKBD, actually bind MAPKs using a “mixed” directionality in which two separate sequences within the DUSP MKBD engage the KIM binding groove35,57 [Fig. 2(C); these “noncanonical” interactions are further discussed later]. So how do these interactions of KIM peptides contribute to MAPK selectivity? In a recent study by Garia et al., the authors showed that the charge and distance between the electrostatic and hydrophobic grooves facilitates the ability of KIM-motifs to discriminate between MAPKs.46 That is, KIM-motif residues that do not bind directly to the KIM-motif binding pocket contribute directly to the affinity and specificity of MAPK:KIMpeptide interactions. Furthermore, in the case of the MAPK-binding domains of DUSP10/MKP5 and DUSP16/MKP7, which share a core interaction motif, binding of additional structural elements of DUSP16/MKP7 lead to an extended interaction surface and thus stronger binding.57 These MAPK:KIM-peptide structures have not only revealed that the KIM binding pocket of p38, ERK, and potentially other MAPKs, is bi-directional binding competent but has also shown how both linear and structured domains engage the KIM binding groove and revealed the importance of intervening residues in the KIM in MAPK selectivity. Taken together, these structures have shown the MAPK KIM binding pocket is a very “adaptable” binding pocket that can bind a large variety of complementary sequences.
KIM Peptides Readily Discriminate Between JNK and p38/ERK but Discriminate Comparably Poorly Between p38 and ERK
Recent, systematic efforts to quantify the binding affinities of MAPK regulatory/substrate proteins for their respective MAPK has revealed that while the KIM-motifs from these proteins readily discriminate between JNK and p38/ERK, they discriminate comparably poorly between p38 and ERK. For example, in a recent study JNK-specific KIM peptides were only specific to JNK, with no detectable binding of these JNK-specific peptides by p38/ERK. In contrast, KIM peptides specific for either p38 (i.e., MKK3) or ERK (i.e., MEK1) exhibited discrimination factors of less than 4-fold between p38 and ERK.46 There was no detectable binding of these p38 and ERK specific peptides to JNK. Indeed, we have observed the same behavior of KIM peptides/proteins using ITC; i.e., the discrimination factor of DUSP16/MKP7 (a p38/JNK specific DUSP) for p38 versus ERK is only 0.7.57,58 Our unpublished ITC data shows that this behavior is consistent between the majority of p38 and ERK binding proteins.
Recent structures of MAPK:KIM-peptide complexes, compared to those previously determined, highlight the differences between the JNK, p38, and ERK KIM binding pockets, providing new insights into this selectivity. All four hydrophobic pockets (ΦA, ΦB, ΦL, ΦU) are present in ERK, p38, and FUS3 (Fig. 3) and each pocket is often, but not always, engaged by a single interacting KIM (e.g., compare FUS3:Msg5, where all four pockets of FUS3 are engaged by Msg5, with FUS3:Far1, where only three are engaged, Fig. 2). In contrast, only three hydrophobic pockets (ΦA, ΦB, and ΦL) are present in JNKs [Fig. 2(A)].46,51,52 Similarly, while both electrostatic pockets (ΨL, ΨU) are present and occupied by different KIM peptides in ERK, p38, and FUS3, only the ΨL pocket is engaged by KIM peptides that bind specifically to JNKs [Fig. 2(A)]. These differences between ERK/p38/FUS3 and the JNKs families arise primarily from two residue changes: ERK residues E81/D321 and corresponding JNK1 residues K83/E329 [compare Figs. 3(D,E)]. Namely, the ΨU interaction site does not exist in JNK1 because the negatively charged E81 residue in ERK is replaced by an oppositely charged K84 residue in JNKs and the longer side chain of E329 in JNKs, whose position is stabilized by K83, occludes the ΦU pocket. Nevertheless, other MAPK residues that define these pockets are largely but not perfectly conserved among the MAPKs, ensuring that the rest of the pocket stays intact (Fig. 3). Thus, these crystallographic studies are leading to a coherent understanding of how differences in both KIM peptide sequences and the residues in MAPKs that define the KIM peptide binding pockets contribute to MAPK selectively and, in turn, MAPK signal fidelity.
Figure 3.
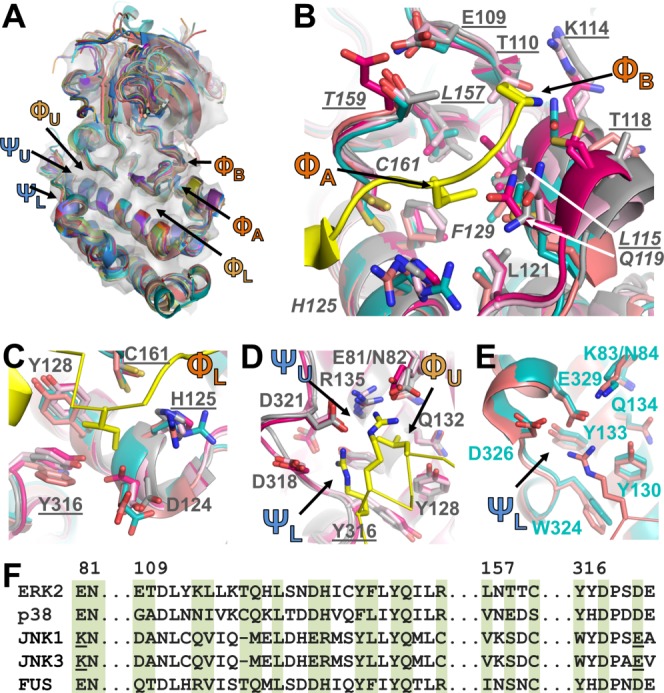
KIM binding pockets. (A) MAPKs listed in Table I aligned using the C-terminal cores described in Table II and displayed as cartoons; ERK2 shown as a transparent grey surface. Locations of KIM binding pockets labeled. (B) ΦA and ΦB binding pockets. ERK2 (grey; PDBID 3TEI), p38 (bright pink; PDBID 2Y8O), JNK1 (cyan, PDBID 2XRW), JNK3 (coral, PDBID 4H39), FUS3 (light pink, PDBID 2B9H); RSK1 KIM peptide in yellow (PDBID 3TEI). MAPK residues shown as sticks with ERK2 residues labeled; ΦA pocket residues underlined, ΦB pocket residues in italics. C. ΦL pocket, colored as in B. Residues that are also part of other KIM binding pockets underlined. D. ΦL, ΨU, ΨL pockets of ERK2, p38 and FUS3; colored as in B, labeled as in C. E. ΨL pocket of JNK1 and JNK3; colored as in B, labeled as in C, except JNK1 residues labeled. F. Sequence alignment of the MAPKs; residues that make up the KIM binding pockets highlighted in green (ERK2 residue numbers above alignment).
Biomolecular NMR Spectroscopy is Necessary to Understand the Regulation of MAPKs
While X-ray crystallography has provided essential insights into the regulation of MAPKs by their interacting proteins, over the last ∼10 years biomolecular NMR spectroscopy has also been used to complement and critically expand these efforts (Table III).57–61 This contribution was accelerated by new protein labeling techniques, new, more sensitive NMR spectrometers as well as novel NMR techniques.62 To overcome the broad line-widths that are characteristic of proteins ≥35 kDa, MAPKs must be expressed in D2O-based medium and TROSY versions of all 2D and 3D experiments must be recorded using high field NMR spectrometers (800–1000 MHz 1H Larmor frequency).62 Still, central regions of their spectrum exhibited overlapping peaks, thereby complicating resonance assignment. To overcome spectral overlap, single amino acid labeled (e.g. 15N-Leu, 15N-Tyr, 15N-Phe, or 15N-Val) samples can be used.63,64 Interestingly, the quality of the 2D [1H,15N] TROSY spectra of MAPKs are significantly different from one another, with often many fewer peaks detected than expected, despite their nearly identical 3D structures.58,61,65 Peaks in NMR spectra can be missing due to: (1) fast solvent (H2O) amide exchange on the NMR time scale, (2) lack of D2O back exchange after protein growth in D2O-based expression medium, especially for amino acids in the hydrophobic core of an enzyme, or (3) intermediate conformational exchange which broadens the peak line-widths beyond detection. The latter point seems to be especially critical for MAPKs and the differences in their NMR spectra (spectra quality of MAPKs tested by the authors is p38α>ERK2∼JNK1)58,61,65 are providing the first indications that dynamics likely plays a key role in their biological function. This supports the results of earlier H/D-MS studies from the Ahn laboratory that show the dynamics of key loops are affected be the interactions of MAPKs with peptides and ligands.66 A number of NMR studies focused on the DFG in/out loop equilibrium as well as the hinge region, showing that these regions change dynamics upon ligand (inhibitor) binding. 67,68
Table III.
Solution (NMR, SAXS, DXMS) Studies of MAPK Complexes
| MAPK complex | MAPK organism | Tech | BMRBa | Type | Ref |
|---|---|---|---|---|---|
| ERK2b | |||||
| pTpY-ERK2:ELK1311–327 | rat | DXMS | – | Su | 66 |
| pTpY-ERK2:ELK1391–399 | rat | DXMS | – | Su | 66 |
| ERK2:MKP3 | rat | DXMS | – | P | 66 |
| ERK2:STE72–19 | rat | NMR | 17748 | K | 65 |
| ERK2:ELK1311–327 | rat | NMR | 17748 | Su | 65 |
| ERK2:ETS1 | rat | NMR | 17748 | Su | 65 |
| ERK2:PEA-15 | rat | NMR | 17748 | Sc | 65 |
| ERK2:HePTP15–31 | rat | NMR | 17748 | P | 59 |
| ERK2:HePTP15–56 | rat | NMR | 17748 | P | 59 |
| ERK2:HePTP15–339 | rat | NMR | 17748 | P | 59 |
| ERK2:HePTP15–339 | rat | SAXS | – | P | 71 |
| pTpYERK2:HePTP15–339 | rat | SAXS | – | P | 71 |
| p38αc | |||||
| p38α:MKK3b15–32 | mouse | DXMS | – | K | 66 |
| p38α:MKK3b | mouse | NMR | 6468 | K | 47 |
| p38α:HePTP15–31 | human | NMR | 17471/6468 | P | 58 |
| p38α:HePTP15–56 | human | NMR | 17471/6468 | P | 58 |
| p38α:HePTP15–339 | human | NMR | 17471/6468/15680 | P | 58 |
| p38α:STEP214–229 | human | NMR | 17471/6468 | P | 60 |
| p38α:STEP214–256 | human | NMR | 17471/6468 | P | 60 |
| p38α:STEP214–539 | human | NMR | 17471/19046/6468 | P | 60 |
| p38α:PTPSL332–348 | human | NMR | 17471/6468 | P | 60 |
| p38α:PTPSL332–373 | human | NMR | 17471/6468 | P | 60 |
| p38α:PTPSL332–655 | human | NMR | 17471/6468 | P | 60 |
| p38α:STEP/PTPSLchimera | human | NMR | 17471/6468 | P | 60 |
| p38α:MKP5/DUSP10 | human | NMR | 17471/19330 | P | 57 |
| pTpYp38αd:MBP94–102 | human | NMR | 17940 | Su | 69 |
| p38α:HePTP15–339 | human | SAXS | – | P | 58 |
| p38α:STEP214–539 | human | SAXS | – | P | 60 |
| p38α:PTPSL332–655 | human | SAXS | – | P | 60 |
| p38α:STEP/PTPSLchimera | human | SAXS | – | P | 60 |
| p38α:MKP5/DUSP10 | human | SAXS | – | P | 57 |
The basis of any NMR spectroscopy analysis is the sequence-specific backbone assignment, which has been achieved for p38α47,58,61 (82%), activated pTpYp38α69 (56%) and ERK265 (65%) and include key MAPK regulatory elements, especially the KIM binding pockets (93%/95% for p38α/ERK2) the glycine-rich loops (100%/100% for p38α/ERK2) and the activation loops (83%/none for p38α/ERK2). Chemical shift perturbation (CSP) mapping experiments, in which the local environment of each HN/N pair of a protein (the MAPK, labeled, NMR active) are followed upon the addition of a peptide or protein (MAPK peptide/protein, unlabeled, NMR inactive), are subsequently used to identify MAPK residues that interact with MAPK regulatory/substrate proteins [Fig. 4(A), left]. These NMR experiments can be used to validate models of MAPK:MAPK regulatory/substrate protein interactions generated using alternative methods, such as molecular modeling, as was done for the pTpYERK2:ETS-1 complex.70 More importantly, the results of NMR CSP experiments can also be used to generate new models of MAPK:MAPK regulatory/substrate protein complexes. This procedure is illustrated in Fig. 4(A). Specifically, the first set of CSP titration studies are used to identify MAPK residues that interact directly with a particular MAPK regulatory/substrate protein. Reverse NMR CSP titration experiments are then performed to detect the interaction residues on the MAPK regulatory/substrate protein. These are similar to the CSP experiments performed on the MAPKs, except now the local environment of each HN/N pair of the MAPK regulatory/substrate protein (labeled, NMR active) is followed upon the addition of the MAPK (unlabeled, NMR inactive). This allows the complementary interaction site—that is, the MAPK regulatory/substrate protein residues that interact directly with the MAPK—to be determined. However, these experiments have one requirement: the sequence-specific backbone assignment of the MAPK regulatory/substrate protein must also be known. As these are often large proteins, this can be a time consuming, expensive, and even technical challenge. These NMR spectroscopy studies can subsequently be complemented by SAXS measurements, also performed in solution, to provide a model (“envelope”) of the MAPK:MAPK regulatory protein complex. SAXS data can then either be used in conjunction with NMR as well as other constraints to generate a structure (co-refinement) or it can be used to confirm a model that was generated using NMR constraints. Models for the MAPK:MAPK regulatory/substrate protein are then obtained using EROS (ensemble refinement of SAXS),71,72 EROS-NMR (ensemble refinement of SAXS in which NMR CSP are used as local energy constraints),58 HADDOCK57,73 or similar programs.
Figure 4.
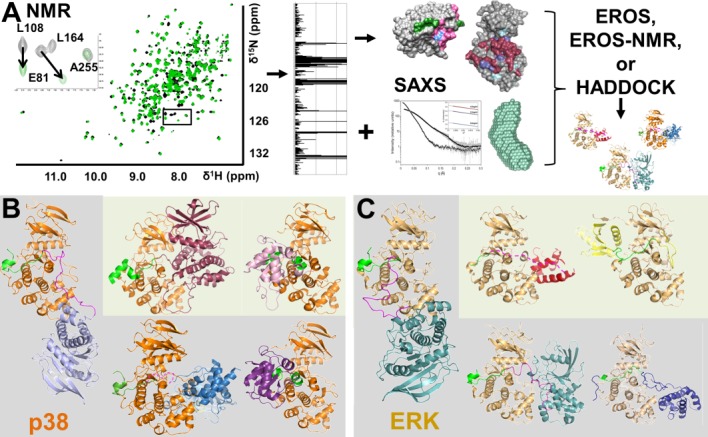
Interactions Outside the MAPK KIM Binding Groove. (A) Schematic illustration showing how NMR spectroscopy, using chemical shift perturbation (CSP) experiments, coupled with small angle X-ray scattering (SAXS) can be used to determine the structures of large (∼80 kDa) protein:protein complexes in solution. (B) p38:MAPK regulatory protein complexes. p38 shown in orange, the KIM residues of the MAPK regulatory proteins are shown in green and the KIS sequence of HePTP/STEP shown in pink. Structures determined using the procedures highlighted in A shown on a grey background while crystal structures are shown on a light green background (p38 in the same orientation in all figures). Left, p38:HePTP resting state complex (HePTP, light blue); upper middle, p38:MK2 (MK2, dark pink, PDBID 2OZA); upper right, p38:DUSP10 (DUSP10, pink, PDBID 3TG1); lower middle, p38:STEP (STEP, blue); lower right, p38:DUSP16 (DUSP16, purple). (C) ERK:MAPK regulatory protein complexes: ERK shown in beige while the KIM residues of the MAPK binding proteins are shown in green and the KIS sequence of HePTP shown in pink. ERK in the same orientation in all figures. Structures determined using the procedures highlighted in (A) shown on a grey background while crystal structures are shown in a light green background. Left, ERK:HePTP resting state (HePTP, light blue); upper middle, T185E-ERK:PEA-15 (PEA-15, red, PDBID 4IZ5); upper right, ERK5:MKK5 (MKK5, yellow; PDBID 4IC7); lower middle, pTpY-ERK:HePTPSTM (HePTPSTM, light blue); lower left, ERK:Ets-1 (Ets-1, dark blue).
Thus far, the interactions between MAPKs (p38, pTpY-p38 and ERK2) and MAPK scaffolds (Ets-1,65,70 PEA-1565), kinases (MKK3b,47 STE765), substrates (ELK1,65 MBP69), and phosphatases (HEPTP,58,59,71 STEP,60 PTPSL,60 DUSP16/MKP757) have been studied using these techniques and have allowed us to determine additional structures of full MAPK:MAPK regulatory protein complexes, which are providing new and unexpected insights into MAPK selectivity not only at the KIM binding pocket, but especially at binding pockets outside of it [Fig. 4(B), p38 complexes; Fig. 4(C), ERK2 complexes]. For example, the SAM domain of Ets-1 binds the FRS or FxF docking site on ERK2, which is located ∼15 Å away from the ERK2 KIM binding groove [Fig. 4(C), lower right].70,74 Similarly, previous studies showed that PEA-15 binds the ERK2 KIM binding groove in the non-canonical C->N direction [Fig. 2(B)].75 Subsequently, NMR CSP studies of Piserchio et al.,65 which probed the interaction of PEA-15 on ERK2, revealed that PEA-15 KIM binding is mediated primarily by the hydrophobic binding pockets of the KIM binding groove with very little interaction at the electrostatic (CD) sites. Both aspects of PEA-15 binding on ERK2 were subsequently confirmed by the PEA-15:ERK2 crystal structure Fig. 4(C), upper middle].53 Unique interactions outside the MAPK KIM binding groove have also been identified using these solution-based methods, especially the combination of NMR spectroscopy and SAXS. Specifically, NMR studies of p38 with the KIM containing family of PTPs have unequivocally demonstrated that while the KIM peptides of these proteins bind p38 via quite similar mechanisms, they interact with p38 outside of the KIM binding groove very differently explaining their different biological activities. These distinct interactions are described in detail in the next section.
Interactions Outside the KIM Binding Pocket using NMR Spectroscopy: the Differential Interactions of KIM-Containing PTPs with p38 and ERK
The KIM-PTPs are a small family of tyrosine-phosphatases that include HePTP, STEP and STEP-like PTP (PTPSL). Each phosphatase possesses a C-terminal catalytic domain (the PTP domain) and an N-terminal unstructured extension that contains the ∼15-amino-acid KIM. Despite their high sequence similarity, it has been shown that these three phosphatases regulate/deactivate p38 with different efficiencies.76 Although crystallographic data for p38/ERK and their interactions with various KIM peptides (including the ERK2:HePTP KIM-peptide complex44) provided key insights into KIM binding, a full understanding of the atomic-level interactions between this family of MAPK regulatory proteins both within and outside the KIM binding pockets has only recently been obtained, by combining NMR spectroscopy with SAXS and EROS/EROS-NMR/HADDOCK.58–60,71
These experiments showed that while there are similarities, there are also key differences in the mechanism of binding of HePTP, STEP, and PTPSL with p38.58,60 First, while the mode of KIM binding to p38 is largely conserved among the KIM-PTP family, with the KIMs from PTPSL/STEP and HePTP engaged residues within all four hydrophobic both electrostatic pockets of the MAPK KIM binding groove, they perturb it differently, especially in the ΦA and ΦB hydrophobic pockets. Differential engagement of these pockets in ERK2 by ERK-specific scaffolds (ETS, PAE-15) and substrates (Elk-1) have also been observed.65 Second, residue L108 from p38, which functions as a hinge connecting the p38 N- and C-terminal lobes, experiences CSPs with HePTP and PTPSL, but not STEP. The adjacent residue in ERK has also been shown to experience chemical shifts upon KIM peptide binding.65 Third, HePTP, but not PTPSL or STEP, also interacts with p38 via its kinase specificity sequence (KIS; the HePTP KIMKIS is composed of residues 15–56), resulting in the CSP of residues outside the p38 KIM binding groove. This observation, coupled with SAXS experiments that showed that the p38:HePTP complex is elongated in solution and reverse titrations that allowed us to unequivocally define which HePTP residues bind p38, led to the development of new computational procedure, ensemble refinement of SAXS (EROS)-NMR. This allowed the relative orientation of p38 and HePTP that best fits both the NMR CSP constrains and the experimental SAXS data to be determined [Fig. 4(A)].58 The ensemble model showed that the catalytic domain of HePTP is localized below the p38 KIM-binding groove and fluctuates in a fan-like motion below p38; explaining why the crystallization of this complex has proven intractable [Fig. 4(B)]. Notably, this structural arrangement of p38:HePTP was similar to the conformation determined for the ERK:HePTP complex using SAXS data and subsequent EROS refinement,71 although in this case the HePTP KIS did not engage ERK;59 that is, the HePTP catalytic domain was localized below the KIM binding groove [Fig. 4(C)]. By comparison, the structure of the trapped “catalytically active” state pTpY-ERK2:HePTPm complex (ERK2 phosphorylated on T183 and Y185, pTpY-ERK2, bound to a catalytically inactive mutant of HePTP, HePTPm) that was determined using similar procedures, revealed that the HePTP catalytic domain rotates by more than 65 Å in order to bind and dephosphorylate the ERK2 activation loop.71
Fourth, while the p38:HePTP and p38:PTPSL complexes are elongated in solution, the p38:STEP complex is compact. NMR experiments showed, unexpectedly, that STEP binding to p38 leads to multiple additional perturbed residues on p38 (these residues are not perturbed by HePTP or PTPSL), including D177/M179/V183/A190 (activation loop), L247/K248/S254/R256 (MAPK insert) and E328 (αI-αL16 loop).60 Thus, despite of the fact that the KIM sequences of PTPSL and STEP are identical, the proteins bind p38 very differently. Reverse NMR titration studies, which required the completion of the sequence-specific backbone assignment of STEP,63 coupled with NMR-constrained docking using HADDOCK led to a model of the full p38:STEP complex [Fig. 4(B)].60 This structure showed that the STEP catalytic domain binds p38 at the MAPK-specific insert, near the p38 activation loop, which was experimentally further confirmed by mutagenesis studies combined with ITC measurements. Critically, additional studies of the trapped “activated” pTpYp38:STEPm complex (STEP is catalytically inactive and thus unable to dephosphorylate dually phosphorylated p38) showed that the orientation of the STEP catalytic domain in the p38:STEP resting-state complex is not conducive to dephosphorylation of the tyrosine residue in the p38 phosphorylation loop and a significant rotation of the STEP catalytic domain is necessary to properly position the active site of STEP for catalysis. Together, these data provide a structural explanation for the increased dephosphorylation efficiency of both HePTP and PTPSL for p38 over STEP and, more importantly, provide atomic resolution evidence that residues outside the MAPK KIM binding pocket are important for regulatory protein binding and specificity.
KIM-Containing DUSPs Bind and Regulate MAPKs using a Structured KIM Domain that Binds MAPKs using a “Mixed” Directionality
DUSPs dephosphorylate both serine/threonine and tyrosine residues using an enzymatic mechanism conserved with that of tyrosine phosphatases in which a conserved catalytic cysteine residue (HCxxxxxR) functions as a nucleophile.34,77,78 However, the DUSP active site is shallow11,34—more similar to the depth of the active sites of serine/threonine specific phosphatases79–81—when directly compared with that of tyrosine phosphatases,38 which allows for phosphorylated serine/threonine and tyrosine residues as substrates. Twenty-five genes encode for DUSPs in the human genome, with DUSP24 and DUSP27 lacking enzymatic activity.12 Ten DUSPs contain a MAPK binding domain (MKBD) with a KIM interaction sequence that is required for a direct interaction with MAPKs. These 10 DUSP are also known as typical DUSPs or MKPs, which are commonly divided according to their cellular locations (nuclear, cytosolic or both) and their ability to recognize specific MAPKs.11,34
The domain architecture of typical DUSPs/MKPs is highly similar with a modestly conserved N-terminal MKBD domain and the more highly conserved C-terminal catalytic domain (DUSP8 and DUSP16/MKP7 also have a C-terminal PEST domain while DUSP10/MKP5 also has an N-terminal disintegrin domain).11 The lower sequence conservation of the MKBD likely contributes to the difference in MAPK substrate specificity. It is interesting to note that binding of the MKBD to MAPKs can further activate the phosphatase activity of the DUSP. However, a molecular basis for this activation has not been identified, although it was suggested that this could be due to an intramolecular interaction of the MKBD and the catalytic domain that changes upon MAPK binding.36
The MKBDs of DUSP6/MKP336 (NMR spectroscopy), DUSP10/MKP537 (X-ray crystallography) and DUSP16/MKP735 (X-ray crystallography) have been structurally characterized. All adopt a common mixed α/β-fold that is highly similar to that of sulfurtransferases (rhodaneses) and the DUSP Cdc25A. ∼5 α-helices fold around central β-sheet that itself consists of ∼5 parallel β-strands. The MKBD of DUSP10/MKP5 and DUSP16/MKP7 are highly similar,35,57 but there are larger structural differences in the MKBD from DUSP6,36 which also has the central 5-membered β-sheet but the surrounding α-helices and connecting loops adopt significantly different conformations.
It was hypothesized that the KIM sequences that contain conserved basic and hydrophobic residues, and are part of the well-folded MKBD of the typical DUSPs, engage the common complementary KIM-binding groove in MAPKs.41,44 This was only recently confirmed, when a crystal structure of the complex between the MKBD of DUSP10/MKP5 and p38 was reported35. Surprisingly and interestingly, this interaction is significantly different to the interaction of KIM-peptides from KIM-PTPs, MAPKKs, and other substrates/scaffolds. First, as predicted, it does not bind as a linear peptide, but instead maintains the MKBD fold when bound to p38. Second, it was immediately apparent that DUSP10/MKP5 engages the KIM-binding groove in a predominantly C-to-N terminal direction, versus the more typical N-to-C direction. Specifically, the N-terminal helix α2 of the DUSP10/MKP5 MKBD binds in the hydrophobic pocket and the more C-terminal helix α3 binds at the electrostatic (CD) site. Previous NMR spectroscopy analysis of the DUSP6 MKBD with ERK2 showed that the protein:protein interface was also mainly centered on residues belonging to the β3-α3 region of the DUSP6 MKBD.36 Most recently, we showed that while the interaction between the DUSP16/MKP7 MKBD with p38 also binds in a similar mostly C-to-N direction, it is also different from that of DUSP6 and DUSP10/MKP5, as it includes not only helix α2 and α3 (largely identical to the interaction of DUSP10/MKP5 MKBD) but also helix α4 [Figs. 2(C), 4(B)].57 Thus, with these additional structures, it is becoming apparent that while the overall interactions of DUSP/MKP MKBDs with MAPKs are somewhat structurally conserved, there is significant fine tuning, i.e. fewer and/or more additional interactions that likely alter the duration of MAPK activity and thus are a powerful regulatory component of the MAPK signaling pathways.
Conclusion
During the last few years, the structural biology of MAPKs and thus our understanding of the regulation of these essential enzymes has made significant progress—from the pioneering work of the Goldsmith laboratory and others to recent studies of the first structures of MAPKs:regulatory protein complexes. Some of these advancements have been propelled by the inclusion of solution techniques, especially biomolecular NMR spectroscopy and SAXS, as well as novel methods that combine all of the information to obtain accurate structural models. These studies are also showing the importance of protein dynamics for the regulation of MAPKs, both locally, e.g. gating of the active site by inhibitors, as well as globally. It will be very interesting to follow these new results during the next few years in order to see how these novel structural and dynamics data will lead to improved, specific drug design—it seems that MAPKs have their best years ahead.
Acknowledgments
The authors thank all current and previous members of the Page and Peti laboratory for their contribution to this ongoing research. The authors thank Drs. Natalie G. Ahn, Kevin N. Dalby, Ranajeet Ghose and Deborah K. Morrison for stimulating discussions about MAPKs and KSR1. We thank Dr. Gerhard Hummer for an exciting collaboration using EROS and EROS-NMR methods.
Glossary
- DUSP
dual specificity phosphatase
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- NMR
nuclear magnetic resonance
- PTP
protein tyrosine phosphatase
- SAXS
small angle X-Ray scattering.
References
- 1.Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci U S A. 1980;77:1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuo JF, Greengard P. Cyclic nucleotide-dependent protein kinases. IV. Widespread occurrence of adenosine 3′,5′-monophosphate-dependent protein kinase in various tissues and phyla of the animal kingdom. Proc Natl Acad Sci U S A. 1969;64:1349–1355. doi: 10.1073/pnas.64.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 4.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 5.Kim SC, Hahn JS, Min YH, Yoo NC, Ko YW, Lee WJ. Constitutive activation of extracellular signal-regulated kinase in human acute leukemias: combined role of activation of MEK, hyperexpression of extracellular signal-regulated kinase, and downregulation of a phosphatase, PAC1. Blood. 1999;93:3893–3899. [PubMed] [Google Scholar]
- 6.Boulton TG, Cobb MH. Identification of multiple extracellular signal-regulated kinases (ERKs) with antipeptide antibodies. Cell Regul. 1991;2:357–371. doi: 10.1091/mbc.2.5.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med. 2009;15:369–379. doi: 10.1016/j.molmed.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown MD, Sacks DB. Protein scaffolds in MAP kinase signalling. Cell Signal. 2009;21:462–469. doi: 10.1016/j.cellsig.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Imajo M, Tsuchiya Y, Nishida E. Regulatory mechanisms and functions of MAP kinase signaling pathways. IUBMB Life. 2006;58:312–317. doi: 10.1080/15216540600746393. [DOI] [PubMed] [Google Scholar]
- 10.Cobb MH, Xu S, Hepler JE, Hutchison M, Frost J, Robbins DJ. Regulation of the MAP kinase cascade. Cell Mol Biol Res. 1994;40:253–256. [PubMed] [Google Scholar]
- 11.Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26:3203–3213. doi: 10.1038/sj.onc.1210412. [DOI] [PubMed] [Google Scholar]
- 12.Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 13.McKay MM, Ritt DA, Morrison DK. Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci U S A. 2009;106:11022–11027. doi: 10.1073/pnas.0901590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koveal D, Schuh-Nuhfer N, Ritt D, Page R, Morrison DK, Peti W. A CC-SAM, for coiled coil-sterile alpha motif, domain targets the scaffold KSR-1 to specific sites in the plasma membrane. Sci Signal. 2012;5:ra94. doi: 10.1126/scisignal.2003289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 16.Chen RE, Thorner J. Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2007;1773:1311–1340. doi: 10.1016/j.bbamcr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Harkins PC, Ulevitch RJ, Han J, Cobb MH, Goldsmith EJ. The structure of mitogen-activated protein kinase p38 at 2.1-A resolution. Proc Natl Acad Sci U S A. 1997;94:2327–2332. doi: 10.1073/pnas.94.6.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature. 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- 19.Kallunki T, Su B, Tsigelny I, Sluss HK, Derijard B, Moore G, Davis R, Karin M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- 20.Bardwell AJ, Frankson E, Bardwell L. Selectivity of docking sites in MAPK kinases. J Biol Chem. 2009;284:13165–13173. doi: 10.1074/jbc.M900080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanoue T, Adachi M, Moriguchi T, Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol. 2000;2:110–116. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- 22.Tanoue T, Maeda R, Adachi M, Nishida E. Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J. 2001;20:466–479. doi: 10.1093/emboj/20.3.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu B, Stippec S, Robinson FL, Cobb MH. Hydrophobic as well as charged residues in both MEK1 and ERK2 are important for their proper docking. J Biol Chem. 2001;276:26509–26515. doi: 10.1074/jbc.M102769200. [DOI] [PubMed] [Google Scholar]
- 24.Bardwell L, Cook JG, Chang EC, Cairns BR, Thorner J. Signaling in the yeast pheromone response pathway: specific and high-affinity interaction of the mitogen-activated protein (MAP) kinases Kss1 and Fus3 with the upstream MAP kinase kinase Ste7. Mol Cell Biol. 1996;16:3637–3650. doi: 10.1128/mcb.16.7.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zanke B, Suzuki H, Kishihara K, Mizzen L, Minden M, Pawson A, Mak TW. Cloning and expression of an inducible lymphoid-specific, protein tyrosine phosphatase (HePTPase) Eur J Immunol. 1992;22:235–239. doi: 10.1002/eji.1830220134. [DOI] [PubMed] [Google Scholar]
- 26.Lombroso PJ, Murdoch G, Lerner M. Molecular characterization of a protein-tyrosine-phosphatase enriched in striatum. Proc Natl Acad Sci U S A. 1991;88:7242–7246. doi: 10.1073/pnas.88.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hendriks W, Schepens J, Brugman C, Zeeuwen P, Wieringa B. A novel receptor-type protein tyrosine phosphatase with a single catalytic domain is specifically expressed in mouse brain. Biochem J. 1995;305:499–504. doi: 10.1042/bj3050499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saxena M, Williams S, Brockdorff J, Gilman J, Mustelin T. Inhibition of T cell signaling by mitogen-activated protein kinase-targeted hematopoietic tyrosine phosphatase (HePTP) J Biol Chem. 1999;274:11693–11700. doi: 10.1074/jbc.274.17.11693. [DOI] [PubMed] [Google Scholar]
- 29.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hill JM, Vaidyanathan H, Ramos JW, Ginsberg MH, Werner MH. Recognition of ERK MAP kinase by PEA-15 reveals a common docking site within the death domain and death effector domain. EMBO J. 2002;21:6494–6504. doi: 10.1093/emboj/cdf641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith JA, Poteet-Smith CE, Malarkey K, Sturgill TW. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J Biol Chem. 1999;274:2893–2898. doi: 10.1074/jbc.274.5.2893. [DOI] [PubMed] [Google Scholar]
- 32.Lukas SM, Kroe RR, Wildeson J, Peet GW, Frego L, Davidson W, Ingraham RH, Pargellis CA, Labadia ME, Werneburg BG. Catalysis and function of the p38 alpha.MK2a signaling complex. Biochemistry. 2004;43:9950–9960. doi: 10.1021/bi049508v. [DOI] [PubMed] [Google Scholar]
- 33.Yang SH, Galanis A, Sharrocks AD. Targeting of p38 mitogen-activated protein kinases to MEF2 transcription factors. Mol Cell Biol. 1999;19:4028–4038. doi: 10.1128/mcb.19.6.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patterson KI, Brummer T, O'Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009;418:475–489. doi: 10.1042/bj20082234. [DOI] [PubMed] [Google Scholar]
- 35.Zhang YY, Wu JW, Wang ZX. A distinct interaction mode revealed by the crystal structure of the kinase p38alpha with the MAPK binding domain of the phosphatase MKP5. Sci Signal. 2011;4:ra88. doi: 10.1126/scisignal.2002241. [DOI] [PubMed] [Google Scholar]
- 36.Farooq A, Chaturvedi G, Mujtaba S, Plotnikova O, Zeng L, Dhalluin C, Ashton R, Zhou MM. Solution structure of ERK2 binding domain of MAPK phosphatase MKP-3: structural insights into MKP-3 activation by ERK2. Mol Cell. 2001;7:387–399. doi: 10.1016/s1097-2765(01)00186-1. [DOI] [PubMed] [Google Scholar]
- 37.Tao X, Tong L. Crystal structure of the MAP kinase binding domain and the catalytic domain of human MKP5. Protein Sci. 2007;16:880–886. doi: 10.1110/ps.062712807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Critton DA, Tortajada A, Stetson G, Peti W, Page R. Structural basis of substrate recognition by hematopoietic tyrosine phosphatase. Biochemistry. 2008;47:13336–13345. doi: 10.1021/bi801724n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saxena M, Williams S, Tasken K, Mustelin T. Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase. Nat Cell Biol. 1999;1:305–311. doi: 10.1038/13024. [DOI] [PubMed] [Google Scholar]
- 40.Munoz JJ, Tarrega C, Blanco-Aparicio C, Pulido R. Differential interaction of the tyrosine phosphatases PTP-SL, STEP and HePTP with the mitogen-activated protein kinases ERK1/2 and p38alpha is determined by a kinase specificity sequence and influenced by reducing agents. Biochem J. 2003;372:193–201. doi: 10.1042/BJ20021941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tarrega C, Blanco-Aparicio C, Munoz JJ, Pulido R. Two clusters of residues at the docking groove of mitogen-activated protein kinases differentially mediate their functional interaction with the tyrosine phosphatases PTP-SL and STEP. J Biol Chem. 2002;277:2629–2636. doi: 10.1074/jbc.M108874200. [DOI] [PubMed] [Google Scholar]
- 42.Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol Cell. 2002;9:1241–1249. doi: 10.1016/s1097-2765(02)00525-7. [DOI] [PubMed] [Google Scholar]
- 43.Gogl G, Toro I, Remenyi A. Protein-peptide complex crystallization: a case study on the ERK2 mitogen-activated protein kinase. Acta Cryst. 2013;D69:486–489. doi: 10.1107/S0907444912051062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou T, Sun L, Humphreys J, Goldsmith EJ. Docking interactions induce exposure of activation loop in the MAP kinase ERK2. Structure. 2006;14:1011–1019. doi: 10.1016/j.str.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 45.Glatz G, Gogl G, Alexa A, Remenyi A. Structural mechanism for the specific assembly and activation of the extracellular signal regulated kinase 5 (ERK5) module. J Biol Chem. 2013;288:8596–8609. doi: 10.1074/jbc.M113.452235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garai A, Zeke A, Gogl G, Toro I, Fordos F, Blankenburg H, Barkai T, Varga J, Alexa A, Emig D, Albrecht M, Remenyi A. Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci Signal. 2012;5:ra74. doi: 10.1126/scisignal.2003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akella R, Min X, Wu Q, Gardner KH, Goldsmith EJ. The third conformation of p38alpha MAP kinase observed in phosphorylated p38alpha and in solution. Structure. 2010;18:1571–1578. doi: 10.1016/j.str.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 48.Remenyi A, Good MC, Bhattacharyya RP, Lim WA. The role of docking interactions in mediating signaling input, output, and discrimination in the yeast MAPK network. Mol Cell. 2005;20:951–962. doi: 10.1016/j.molcel.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 49.Ma W, Shang Y, Wei Z, Wen W, Wang W, Zhang M. Phosphorylation of DCC by ERK2 is facilitated by direct docking of the receptor P1 domain to the kinase. Structure. 2010;18:1502–1511. doi: 10.1016/j.str.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 50.Xin F, Wu J. Crystal structure of the p38alpha MAP kinase in complex with a docking peptide from TAB1. Sci China Life Sci. 2013;56:653–660. doi: 10.1007/s11427-013-4494-0. [DOI] [PubMed] [Google Scholar]
- 51.Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, Lee JI, Park SY, Kim JH, Hwang KY, Hyun YL, Jeon YH, Ro S, Cho JM, Lee TG, Yang CH. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 2004;23:2185–2195. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laughlin JD, Nwachukwu JC, Figuera-Losada M, Cherry L, Nettles KW, LoGrasso PV. Structural mechanisms of allostery and autoinhibition in JNK family kinases. Structure. 2012;20:2174–2184. doi: 10.1016/j.str.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mace PD, Wallez Y, Egger MF, Dobaczewska MK, Robinson H, Pasquale EB, Riedl SJ. Structure of ERK2 bound to PEA-15 reveals a mechanism for rapid release of activated MAPK. Nature Commun. 2013;4:1681. doi: 10.1038/ncomms2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.ter Haar E, Prabhakar P, Liu X, Lepre C. Crystal structure of the p38 alpha-MAPKAP kinase 2 heterodimer. J Biol Chem. 2007;282:9733–9739. doi: 10.1074/jbc.M611165200. [DOI] [PubMed] [Google Scholar]
- 55.White A, Pargellis CA, Studts JM, Werneburg BG, Farmer BT., 2nd Molecular basis of MAPK-activated protein kinase 2:p38 assembly. Proc Natl Acad Sci U S A. 2007;104:6353–6358. doi: 10.1073/pnas.0701679104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Page R, Lindberg U, Schutt CE. Domain motions in actin. J Mol Biol. 1998;280:463–474. doi: 10.1006/jmbi.1998.1879. [DOI] [PubMed] [Google Scholar]
- 57.Kumar GS, Zettl H, Page R, Peti W. Structural basis for the regulation of the MAP kinase p38alpha by the dual specificity phosphatase 16 MAP kinase binding domain in solution. J Biol Chem. 2013;288:28347–28356. doi: 10.1074/jbc.M113.499178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Francis DM, Koveal D, Rozycki B, Hummer G, Page R, Peti W. Structural basis of p38α regulation by hematopoietic tyrosine phosphatase. Nat Chem Biol. 2011;7:916–924. doi: 10.1038/nchembio.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piserchio A, Francis DM, Koveal D, Dalby KN, Page R, Peti W, Ghose R. Docking interactions of hematopoietic tyrosine phosphatase with MAP kinases ERK2 and p38alpha. Biochemistry. 2012;51:8047–8049. doi: 10.1021/bi3012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Francis DM, Kumar GS, Koveal D, Tortajada A, Page R, Peti W. The differential regulation of p38alpha by the neuronal kinase interaction motif protein tyrosine phosphatases, a detailed molecular study. Structure. 2013;21:1612–1623. doi: 10.1016/j.str.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vogtherr M, Saxena K, Grimme S, Betz M, Schieborr U, Pescatore B, Langer T, Schwalbe H. NMR backbone assignment of the mitogen-activated protein (MAP) kinase p38. J Biomol NMR. 2005;32:175. doi: 10.1007/s10858-005-2449-x. [DOI] [PubMed] [Google Scholar]
- 62.Barrett PJ, Chen J, Cho MK, Kim JH, Lu Z, Mathew S, Peng D, Song Y, Van Horn WD, Zhuang T, Sonnichsen FD, Sanders CR. The quiet renaissance of protein nuclear magnetic resonance. Biochemistry. 2013;52:1303–1320. doi: 10.1021/bi4000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Francis DM, Page R, Peti W. Sequence-specific backbone H, C and N assignments of the 34 kDa catalytic domain of PTPN5 (STEP) Biomol NMR Assign. 2013 doi: 10.1007/s12104-013-9480-8. in press. [DOI] [PubMed] [Google Scholar]
- 64.Piserchio A, Dalby KN, Ghose R. Assignment of backbone resonances in a eukaryotic protein kinase - ERK2 as a representative example. Methods Mol Biol. 2012;831:359–368. doi: 10.1007/978-1-61779-480-3_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Piserchio A, Warthaka M, Devkota AK, Kaoud TS, Lee S, Abramczyk O, Ren P, Dalby KN, Ghose R. Solution NMR insights into docking interactions involving inactive ERK2. Biochemistry. 2011;50:3660–3672. doi: 10.1021/bi2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- 67.Vogtherr M, Saxena K, Hoelder S, Grimme S, Betz M, Schieborr U, Pescatore B, Robin M, Delarbre L, Langer T, Wendt KU, Schwalbe H. NMR characterization of kinase p38 dynamics in free and ligand-bound forms. Angew Chem. 2006;45:993–997. doi: 10.1002/anie.200502770. [DOI] [PubMed] [Google Scholar]
- 68.Honndorf VS, Coudevylle N, Laufer S, Becker S, Griesinger C. Dynamics in the p38alpha MAP kinase-SB203580 complex observed by liquid-state NMR spectroscopy. Angew Chem. 2008;47:3548–3551. doi: 10.1002/anie.200705614. [DOI] [PubMed] [Google Scholar]
- 69.Nielsen G, Schwalbe H. NMR spectroscopic investigations of the activated p38alpha mitogen-activated protein kinase. Chembiochem. 2011;12:2599–2607. doi: 10.1002/cbic.201100527. [DOI] [PubMed] [Google Scholar]
- 70.Lee S, Warthaka M, Yan C, Kaoud TS, Piserchio A, Ghose R, Ren P, Dalby KN. A model of a MAPK*substrate complex in an active conformation: a computational and experimental approach. PLoS One. 2011;6:e18594. doi: 10.1371/journal.pone.0018594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Francis DM, Rozycki B, Tortajada A, Hummer G, Peti W, Page R. Resting and active states of the ERK2:HePTP complex. J Am Chem Soc. 2011;133:17138–17141. doi: 10.1021/ja2075136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rozycki B, Kim YC, Hummer G. SAXS ensemble refinement of ESCRT-III CHMP3 conformational transitions. Structure. 2011;19:109–116. doi: 10.1016/j.str.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Vries SJ, van Dijk AD, Krzeminski M, van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AM. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- 74.Callaway K, Waas WF, Rainey MA, Ren P, Dalby KN. Phosphorylation of the transcription factor Ets-1 by ERK2: rapid dissociation of ADP and phospho-Ets-1. Biochemistry. 2010;49:3619–3630. doi: 10.1021/bi100199q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Callaway K, Rainey MA, Dalby KN. Quantifying ERK2-protein interactions by fluorescence anisotropy: PEA-15 inhibits ERK2 by blocking the binding of DEJL domains. Biochim Biophys Acta. 2005;1754:316–323. doi: 10.1016/j.bbapap.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 76.Zhang YY, Wu JW, Wang ZX. Mitogen-activated protein kinase (MAPK) phosphatase 3-mediated cross-talk between MAPKs ERK2 and p38alpha. J Biol Chem. 2011;286:16150–16162. doi: 10.1074/jbc.M110.203786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang CY, Tan TH. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012;2:24. doi: 10.1186/2045-3701-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ragusa MJ, Dancheck B, Critton DA, Nairn AC, Page R, Peti W. Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat Struct Mol Biol. 2010;17:459–464. doi: 10.1038/nsmb.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grigoriu S, Bond R, Cossio P, Chen JA, Ly N, Hummer G, Page R, Cyert MS, Peti W. The molecular mechanism of substrate engagement and immunosuppressant inhibition of calcineurin. PLoS Biol. 2013;11:e1001492. doi: 10.1371/journal.pbio.1001492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.O'Connell N, Nichols SR, Heroes E, Beullens M, Bollen M, Peti W, Page R. The molecular basis for substrate specificity of the nuclear NIPP1:PP1 holoenzyme. Structure. 2012;20:1746–1756. doi: 10.1016/j.str.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu S, Sun JP, Zhou B, Zhang ZY. Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proc Natl Acad Sci U S A. 2006;103:5326–5331. doi: 10.1073/pnas.0510506103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeeves M, McClelland DM, Barr AJ, Overduin M. Sequence-specific 1H, 13C and 15N backbone resonance assignments of the 34 kDa catalytic domain of human PTPN7. Biomol NMR Assign. 2008;2:101–103. doi: 10.1007/s12104-008-9095-7. [DOI] [PubMed] [Google Scholar]