

Abstract
Growth factor deprivation is a physiological mechanism to regulate cell death. We utilize an interleukin-2 (IL-2)-dependent murine T-cell line to identify proteins that interact with Bad upon IL-2 stimulation or deprivation. Using the yeast two-hybrid system, glutathione S-transferase (GST) fusion proteins and co-immunoprecipitation techniques, we found that Bad interacts with protein phosphatase 1α (PP1α). Serine phosphorylation of Bad is induced by IL-2 and its dephosphorylation correlates with appearance of apoptosis. IL-2 deprivation induces Bad dephosphorylation, suggesting the involvement of a serine phosphatase. A serine/threonine phosphatase activity, sensitive to the phosphatase inhibitor okadaic acid, was detected in Bad immunoprecipitates from IL-2-stimulated cells, increasing after IL-2 deprivation. This enzymatic activity also dephosphorylates in vivo 32P-labeled Bad. Treatment of cells with okadaic acid blocks Bad dephosphorylation and prevents cell death. Finally, Ras activation controls the catalytic activity of PP1α. These results strongly suggest that Bad is an in vitro and in vivo substrate for PP1α phosphatase and that IL-2 deprivation-induced apoptosis may operate by regulating Bad phosphorylation through PP1α phosphatase, whose enzymatic activity is regulated by Ras.
Keywords: apoptosis/Bad/interleukin-2/protein phosphatase PP1α/Ras
Introduction
Hemopoietic cell lines are dependent on the presence of appropriate cytokine(s) for their continued growth and survival. In the absence of cytokine, cells cease to proliferate, and undergo programmed cell death or apoptosis. The Bcl-2 protein family can be divided into two groups based on their ability to promote or inhibit apoptosis; those that promote apoptosis include Bad, Bax, Bak and Bik, whereas Bcl-2, Bcl-x and Mcl-1 are inhibitors (Cory 1995; Adams and Cory, 1998; Chao and Korsmeyer, 1998). The similarity between family members varies widely, although all members possess at least one of four motifs termed the BH domain (Cory, 1995; Chao and Korsmeyer 1998). Bcl-2 family proteins form homo- and heterodimers; the balance between specific homo- and heterodimers is thought to be critical to the maintenance of cell survival or the induction of apoptosis (Steller, 1995; Chinnaiyan and Dixit, 1996; Jacobson, 1997; Reed, 1997). Whereas up- or down-regulation of these proteins may account for the survival of certain cell types in response to extracellular stimuli, it is also possible that survival factors use protein kinases or phosphatases to alter the ability of apoptotic proteins to promote cell survival or death.
The Bad protein is a divergent Bcl-2 family member belonging to a subset that shares identity only in the BH3 domain (Yang et al., 1995; Zha et al., 1997). Deletion mapping and site-directed mutagenesis have indicated that the BH3 domain of Bad is critical for its heterodimerization with Bcl-x or Bcl-2 and for its death agonist activity (Kelekar et al., 1997; Ottilie et al., 1997). Bad lacks the hydrophobic C-terminal sequence that, in the case of Bcl-2, is required for its targeting to mitochondria (Nguyen et al., 1993).
Some growth factors, such as interleukin 3 (IL-3), tumor necrosis factor (TNF), nerve growth factor (NGF) and granulocyte–macrophage colony-stimulating factor (GM-CSF), induce Bad phosphorylation at Ser112 and Ser136 (Zha et al., 1996; Datta et al., 1997; del Peso et al., 1997; Scheid and Duronio, 1998; Craddock et al., 1999; Pastorino et al., 1999), resulting in its association with the 14-3-3 protein. This abolishes its interaction with Bcl-x or Bcl-2, allowing these latter proteins to promote cell survival (Zha et al., 1996; Hsu et al., 1997). Consistent with this model, mutation of the phosphorylation sites on Bad disrupts its ability to promote cell survival (Zha et al., 1996).
The serine/threonine phosphatases are usually classified as type 1 (PP1) or type 2 (PP2), depending on their substrate specificity and sensitivity to inhibitors. Type 2 phosphatases are subdivided into major classes, including PP2A, calcium-dependent calcineurin (PP2B) and magnesium-dependent PP2C (Ingebritsen and Cohen, 1983). It has been shown recently that there is cross-talk between PP1 and PP2A and Ras signaling, suggesting that these phosphatases act downstream or in parallel to Ras (Baharians and Schönthal, 1999; Rajesh et al., 1999; Sieburth et al., 1999). PP1 represents a family of holoenzymes generated by specific interactions between catalytic subunits and a wide variety of regulatory or anchoring proteins involved in targeting, as well as in controlling phosphatase activity (Hubbard and Cohen, 1993; Faux and Scott, 1996). The catalytic subunit of PP1 (PP1c) is a 38 kDa protein that is highly conserved in evolution. Four isoforms of the enzyme, α, β, γ1 and γ2, encoded by three genes (γ1 and γ2 result from alternative splicing) are differentially expressed in mammals (Takizawa et al., 1994). Protein sequence variations among these isoforms are observed mainly in the C-termini (Sasaki et al., 1990), which play a regulatory role in catalytic activity, as demonstrated by proteolysis (Martin et al., 1991) and phosphorylation studies. PP1 is a major eukaryotic protein serine/threonine phosphatase that regulates diverse cellular processes such as cell cycle progression, proliferation, protein synthesis, muscle contraction, carbohydrate metabolism, transcription, cytokinesis and neuronal signaling (Shenolikar and Nairn, 1991; Shenolikar, 1994; Wera and Hemmings, 1995; He et al., 1997; Cheng et al., 2000). In this study, we report that PP1α interacts in vitro and in vivo with the pro-apoptotic molecule Bad. Our data indicate that IL-2 induces serine phosphorylation of Bad, which is a substrate for PP1α phosphatase. Ras activation controls the enzymatic activity of PP1α, leading to Bad dephosphorylation and, consequently, to apoptosis. The functional relevance of PP1α-mediated Bad dephosphorylation is discussed in the context of apoptosis.
Results
To identify proteins that interact with Bad in IL-2-stimulated or -deprived cells, Bad was used as bait in the two-hybrid method to screen cDNA libraries made from IL-2-stimulated or -deprived TS1αβ cells. The nucleotide sequence of one specific clone isolated showed a 100% match to a partial cDNA encoding the proximal N-terminal region of the catalytic PP1α subunit. The full-length cDNA was cloned in pGAD and tested for interaction with Bad. The interaction between the two-hybrid proteins is indicated by the induction of LacZ expression (Figure 1A); neither Bad nor PP1α alone restores LacZ expression. Ras–Raf interaction was used as a positive control.



Fig. 1. Interaction of Bad and PP1α phosphatase. (A) Interaction of Bad and the catalytic subunit of PP1α phosphatase in the two-hybrid system. The S.cerevisiae L40 reporter strain was co-transformed with the plasmids indicated. The interaction between the two-hybrid proteins is indicated by induction of LacZ expression (blue patches). There are two patches for each strain and each patch represents an independent transformant. L40 carrying Ras and Raf was used as positive control. DB, fusion with the DNA-binding domain of Lex10. AD, fusion with the activation domain of Gal4. (B) Reciprocal co-immunoprecipitation of Bad and PP1α. Cytoplasmic lysates from IL-2-stimulated (5 ng/ml) or -deprived cells were immunoprecipitated with anti-Bad antibody, transferred to nitrocellulose and immunoblotted with anti-PP1α and anti-Bad antibody, the latter as internal control. Similarly, PP1α was immunoprecipitated from cytoplasmic lysates from IL-2-stimulated or -deprived cells and immunoblotted with anti-Bad or anti-PP1α antibody, the latter as internal control. Protein bands were detected using the ECL system. Molecular weights of the corresponding proteins are shown. Similar results were obtained in three independent experiments. (C and D) Expression of (C) GST–Bad or (D) GST–PP1α fusion proteins was induced with or without IPTG, and proteins were isolated by affinity chromatography with glutathione–agarose beads and incubated with or without (lane E) cytoplasmic lysates from IL-2-stimulated or -deprived cells. After washing, eluted proteins were resolved by SDS–PAGE, transferred to nitrocellulose and blotted with anti-PP1α or anti-Bad antibody. Protein bands were detected using the ECL system. Molecular weight markers of the corresponding proteins are indicated.
To validate the results obtained using the yeast two-hybrid system, the interaction of Bad and PP1α in intact cells was studied by co-immunoprecipitation using Bad- and PP1α-specific antibodies. We performed reciprocal co-immunoprecipitation experiments of cytoplasmic proteins under IL-2 stimulation or deprivation conditions (Figure 1B). High Bad levels are detected by Western blot analysis of anti-PP1α immunoprecipitates of IL-2-stimulated cells. The amount of Bad detected decreased slightly in 12 h IL-2-deprived cells, with low levels detectable after 24 h of IL-2 deprivation. The PP1α protein level did not diminish, as shown by probing the membrane with anti-PP1α antibody. In reciprocal experiments, high PP1α levels were detected in anti-Bad immunoprecipitates from IL-2-stimulated or 12 h IL-2-deprived cells, decreasing after 24 h of IL-2 deprivation (Figure 1B). Reprobing the membrane with anti-Bad antibody showed that there were similar amounts of Bad in IL-2-stimulated or -deprived cells. These results show that PP1α phosphatase-complexed Bad is recovered in TS1αβ cells, confirming the results obtained in the two-hybrid system.
The Bad–PP1α interaction was also studied in in vitro binding experiments. Bad was produced as a glutathione S-transferase (GST) fusion protein and purified on glutathione–agarose beads. Buffer or cytoplasmic lysates from IL-2-stimulated or -deprived cells were incubated with GST–Bad and, after several washing steps, proteins were resolved by SDS–PAGE and the blot was developed with anti-PP1α or anti-Bad antibody. PP1α from IL-2-stimulated or -deprived cells interacts with the GST–Bad fusion protein (Figure 1C), whereas there is no interaction in the absence of TS1αβ extracts (Figure 1C, lane E). In reciprocal experiments, GST–PP1α was purified on glutathione–agarose beads; after washing, proteins were resolved by SDS–PAGE and the blot was developed as above (Figure 1D). Bad from IL-2-stimulated or -deprived cells interacted with GST–PP1α fusion protein; no interaction was observed in the absence of TS1αβ extracts (lane E).
Following IL-2 deprivation, TS1αβ cells ceased to proliferate and undergo apoptosis, as detected by annexin staining. As early as 4 h after IL-2 deprivation, ∼14% of cells were apoptotic, reaching ∼44% at 24 h, whereas control cells maintained in the presence of IL-2 showed no apoptosis (Figure 2A). To study the effect of IL-2 deprivation in Bad and PP1α expression, TS1αβ cells were stimulated or deprived of IL-2 for different periods of time, and total Bad and PP1α protein expression was analyzed by Western blotting. Expression of Bad and PP1α was observed in control IL-2-stimulated cells and was similar throughout the starvation period analyzed (Figure 2B). It is interesting to note that, although similar levels of Bad and PP1α expression were detected after IL-2-deprivation, starvation induced a strong increase in apoptosis after lymphokine deprivation (Figure 2A). Previous studies indicated that IL-3 induces Bad phosphorylation at Ser112 and Ser136 and, in the absence of phosphorylation at these sites, Bad induces cell death (Datta et al., 1997). We found that IL-2 induces Bad phosphorylation at the same serine residues as IL-3 (Figure 2C). In the absence of IL-2, which correlates with apoptotic cell death (Figure 2A), Bad phosphorylation was not detected.



Fig. 2. Effect of IL-2 deprivation on Bad and PP1α. (A) Cells were cultured in the presence or absence or IL-2 and harvested at different times. To analyze apoptosis, cells were stained with annexin and propidium iodide and analyzed using fluorescence flow cytometry. Region G corresponds to apoptotic cells. The percentage of apoptosis in each sample is superimposed. Similar results were obtained in three independent experiments. (B) TS1αβ cells were IL-2-stimulated or -deprived for the times indicated, then lysed. Protein extracts were separated by SDS–PAGE, transferred to nitrocellulose and probed with anti-Bad, anti-PP1α and pan-Ras antibodies, the latter as internal control. Protein bands were detected using the ECL system. Molecular weights of the corresponding proteins are shown. Similar results were obtained in three independent experiments. (C) Cytoplasmic lysates from IL-2-stimulated or -deprived cells were immunoprecipitated with anti-Bad antibody, transferred to nitrocellulose and blotted with phospho-Bad Ser112, Ser136 or anti-Bad antibody, the latter as internal control of protein loading. Protein bands were detected using ECL. Molecular weights of the corresponding proteins are shown.
Given that (i) Bad phosphorylation at serine residues regulates its apoptotic function, (ii) Bad–PP1α association is detected by immunoprecipitation and (iii) PP1α is a serine/threonine phosphatase, we explored the possibility that phosphorylated Bad may be a PP1α substrate. Figure 3A shows phosphatase activity in Bad immunoprecipitates of IL-2-stimulated or -deprived cells. The phosphatase activity in the immunoprecipitates was measured using 32P-labeled phosphorylase a as substrate. Phosphatase activity was detected in Bad immunoprecipitates of IL-2-stimulated cells, increasing 12 and 24 h after IL-2 deprivation. No phosphatase activity was detected in anti-Jun immunoprecipitates of IL-2-stimulated or -deprived cells. This result suggests that IL-2 deprivation induces phosphatase activation in a time-dependent manner.

Fig. 3. Estimation of serine/threonine phosphatase activity in Bad immunoprecipitates. (A) Phosphatase activity was estimated in Bad immunoprecipitates from IL-2-stimulated or -deprived cells using [32P]phosphorylase a as substrate (open bars). As a negative control, phosphatase activity was estimated in c-Jun immunoprecipitates (black bars). SD is shown for n = 3. Phosphatase activity is represented as a percentage of the maximal activity detected in control IL-2-stimulated cells. (B) Different concentrations of okadaic acid (OA) were added to Bad immunoprecipitates from IL-2-stimulated or 24 h-deprived cells. The reaction was as above. SD is shown for n = 3. Open bars, IL-2 deprivation; shaded bars, IL-2 stimulation. Phosphatase activity is represented as the percentage of maximal activity in untreated immunoprecipitates. (C) Different concentrations of OA were added to the supernatant of Bad immunoprecipitates from IL-2-stimulated cells. The reaction was as in (A). SD is shown for n = 3. Phosphatase activity is represented as the percentage of maximal activity in untreated supernatants. (D) Total extracts (lane T) or cytoplasmic lysates from IL-2-stimulated or -deprived cells immunoprecipitated with Bad were transferred to nitrocellulose and immunoblotted with PP1α. The blot was reprobed with anti-PP2A, anti-PP2B (calcineurin) and Bad antibody. Protein bands were detected using the ECL system. Molecular weights of the corresponding proteins are shown. Similar results were obtained in two independent experiments.
PP1 and PP2A are abundant serine/threonine-specific protein phosphatases expressed in mammalian cells (Kremmer et al., 1997). PP2A enzymatic activity is reported to be more potently inhibited by okadaic acid (OA) than that of PP1, exhibiting an IC50 of 1 nM, compared with an IC50 of 10–15 nM for PP1 (Cohen et al., 1989, 1990). Controlled concentrations of OA can selectively inhibit PP2A, with little or no effect on PP1 (Favre et al., 1997). To confirm that PP1α is the active phosphatase associated with Bad, different OA concentrations were added to Bad immunoprecipitates from IL-2-stimulated or -deprived cells, after which phosphatase activity was assayed using phosphorylase a as substrate. OA concentrations that inhibit PP2A activity (10–9 M) had no effect on the Bad-associated phosphatase activity in vitro (expressed as a percentage of maximal activity in untreated cells; Figure 3B). Addition of 10–8 M OA to Bad immunoprecipitates results in ∼40% inhibition of phosphatase activity, which is almost undetectable after addition of 10–6 M OA (Figure 3B). The in vitro effect of OA on phosphatase activity was comparable in Bad immunoprecipitates from IL-2-stimulated and -deprived cells (data not shown). The selective effect of OA, together with the fact that PP1α interacts with Bad, suggests that the phosphatase activity observed in Bad immunoprecipitates is likely to be the result of PP1α phosphatase. The effect of OA on phosphatase activity was also estimated in supernatants of Bad immunoprecipitates. The OA concentration that had no effect on phosphatase activity in Bad immunoprecipitates (10–9 M; Figure 3B) shows ∼50% inhibition in the supernatants (Figure 3C). Because of the low OA concentration, this result suggests that we are able to detect PP2A and PP1 activity in the supernatant.
We performed immunoprecipitation and Western blot analyses to confirm that PP1α, and not PP2A or PP2B (calcineurin), was responsible for the enzymatic activity detected in Bad immunoprecipitates. PP1α was detected in total extracts from control cells (Figure 3D, lane T). Comparable PP1α levels were detected by Western blot analysis of anti-Bad immunoprecipitates from control IL-2-stimulated cells or cells deprived of IL-2 for 12 h; the levels decreased after 24 h of deprivation. Reprobing the membrane with anti-PP2A or anti-PP2B antibody showed PP2A and PP2B expression only in total extracts from control cells (lane T). These findings suggest that PP1α is the OA-sensitive phosphatase responsible for the phosphatase activity detected in Bad immunoprecipitates.
To determine whether PP1α is the serine/threonine phosphatase specifically involved in Bad dephosphorylation, cells were treated with two concentrations of OA. Treatment with 1 µM OA, a concentration that inhibits both PP1 and PP2A activities, in the absence of IL-2, prevented Bad dephosphorylation at Ser112 and Ser136 (Figure 4A). No changes in total Bad or PP1α levels were observed after OA treatment of the cells, suggesting that OA does not affect protein expression. In contrast, treatment of IL-2-deprived cells with 50 nM OA did not prevent Bad dephosphorylation at Ser112 and Ser136 (Figure 4B), although this OA concentration induces degradation of phosphorylated IκB (Figure 4C). Expression of Bad and Ras was not affected by treatment of cells with 50 nM OA. Since IκB degradation is controlled by PP2A (Sontag et al., 1997), our results suggest that PP2A is not involved in Bad dephosphorylation and that PP1α dephosphorylates Bad in an OA-sensitive manner. As IL-2 deprivation in TS1αβ cells correlates with Bad dephosphorylation and apoptosis, and as OA treatment is able to block Bad dephosphorylation, we hypothesized that inhibition of phosphatase activity by OA in IL-2-deprived cells may prevent apoptosis. IL-2-deprived cells treated with 1 µM OA for 6 h showed a significant reduction in the fraction of apoptotic cells compared with untreated cells (Figure 4D).



Fig. 4. Effect of IL-2 and OA on Ser112 and Ser136 phosphorylation of Bad. (A) Cells were treated with or without 1 µM OA in the absence of IL-2 for different periods of time. Cytoplasmic lysates were immunoprecipitated with anti-Bad antibody and transferred to nitrocellulose. The membrane was probed with phospho-Bad Ser136, Ser112, anti-Bad and anti-PP1α antibody, the last to verify that OA treatment in vivo does not affect PP1α expression. Molecular weights of the corresponding proteins are shown. Protein bands were detected using ECL Plus. (B) Cells were treated with or without 50 nM OA in the absence of IL-2 for different periods of time. Cytoplasmic lysates were treated as in (A). The membrane was probed with phospho-Bad Ser112, Ser136 and anti-Bad antibody, the latter as internal control of protein loading. Molecular weights of the corresponding proteins are shown; protein bands were detected using ECL Plus. (C) Cells were treated with or without 50 nM OA in the absence of IL-2 for different periods of time, then lysed. Protein extracts were separated by SDS–PAGE, transferred to nitrocellulose and probed with anti-IκB and pan-Ras antibodies, the latter as an internal control of protein loading. Molecular weights of the corresponding proteins are shown. Protein bands were detected using ECL. (D) Cells were treated for 6 h with or without 1 µM OA in the presence or absence of IL-2. Cells were washed, stained with annexin and propidium iodide, then analyzed by flow cytometry. Region G of the fluorescence scale represents apoptosis. The percentage of apoptotic cells in each sample is superimposed.
To confirm conclusively that phosphorylated Bad is a PP1α substrate, Bad was labeled in vivo with [32P]orthophosphate and immunoprecipitated; the 32P- labeled Bad–PP1α complexes were assayed for the ability of PP1α to dephosphorylate 32P-labeled Bad (Figure 5A). PP1α dephosphorylates in vitro immunoprecipitated 32P-labeled Bad and no phosphatase activity was observed in c-Jun immunoprecipitates of 32P-labeled cells (Figure 5A). Bad phosphorylation was measured by autoradiography (Figure 5B), and Western blot analysis confirmed the presence of PP1α (Figure 5B). To confirm that PP1α is the phosphatase that dephosphorylates Bad, we performed an in vitro assay of Bad dephosphorylation using purified recombinant PP1α. GST–Bad fusion protein was incubated with or without recombinant PP1α phosphatase. Proteins were separated by SDS–PAGE, and blotted and developed with anti-Bad Ser112 and Ser136 antibody. In vitro, purified PP1α was able to dephosphorylate GST–Bad on Ser112 and Ser136 (Figure 6). As an internal control for protein loading, the blot was reprobed with anti-PP1α and anti-Bad antibody. This result suggests that Bad is a substrate for PP1α phosphatase.
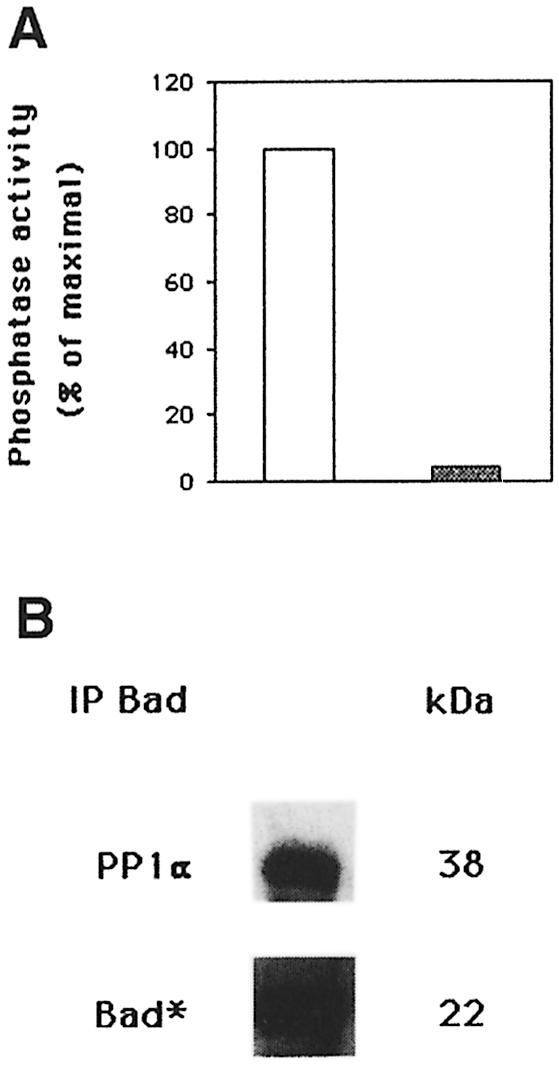
Fig. 5. PP1α dephosphorylates 32P-labeled Bad. (A) Cells were labeled for 2 h with [32P]orthophosphate (100 µCi/ml) in phosphate-free Dulbecco’s modified Eagle’s medium (DMEM) with IL-2. Cells were lysed, Bad immunoprecipitated and phosphatase activity estimated by dephosphorylation of 32P-labeled Bad (open bar). As a negative control, phosphatase activity was estimated in c-Jun immunoprecipitates of 32P-labeled cells (shaded bar). (B) Immunoblot of the membrane with anti-PP1α antibody is shown in the upper panel. Quantification by autoradiography of 32P incorporation into Bad is shown in the lower panel.

Fig. 6. In vitro dephosphorylation of GST–Bad. GST–Bad fusion protein was incubated for 30 min at 30°C with recombinant PP1α phosphatase. Proteins were separated by SDS–PAGE, transferred to nitrocellulose, blocked and incubated with anti-Bad Ser112 and Ser136. Membranes were reprobed with anti-PP1α and anti-Bad antibody. Protein bands were detected using the ECL system. Molecular weights of the corresponding proteins are shown. Similar results were obtained in two independent experiments.
Recently, a direct correlation between Ras activation and PP1/PP2A activation has been shown (Baharians and Schönthal, 1999; Rajesh et al., 1999; Sieburth et al., 1999). In addition, we have shown that Ras activation leads to apoptotic cell death upon IL-2 deprivation, which is prevented by expression of the dominant-negative Ras mutant N17 (Gómez et al., 1998). It was of interest to analyze whether Ras activation could be involved in PP1α activation in IL-2-deprived cells. For this purpose, the mutant N17 or pHook3 vector (mock transfectants) was transiently transfected in cells that were maintained in the presence or absence of IL-2. Cells were selected using magnetic beads and analyzed for PP1 activity (Figure 7A). The proportion of apoptotic cells was also estimated (Figure 7B). Figure 7A shows phosphatase activity in Bad immunoprecipitates of mock or Ras N17 IL-2-stimulated or -deprived transfected cells. The phosphatase activity in the immunoprecipitates was measured using 32P-labeled phosphorylase a as substrate. Phosphatase activity was detected in Bad immunoprecipitates of IL-2-stimulated or -deprived mock transfected cells. Interestingly, PP1 activity was strongly inhibited in IL-2-deprived Ras N17 transfected cells. Non-transfected cells were used as a control. Figure 7A also shows that Ras expression in Ras N17 transfected cells is greater than that in mock transfectants. This result suggests that IL-2 deprivation-induced PP1 activity is mediated by Ras activation.


Fig. 7. Overexpression of Ras N17 induces PP1α activity inhibition and prevents apoptosis. (A) Cells were transfected with pHook3 (mock transfectants) or Ras N17 and pHook3 by the DEAE–dextran method and maintained for 24 h in the presence (open bars) or absence (shaded bars) of IL-2. Transfected cells were selected using magnetic beads and phosphatase activity was estimated in Bad immunoprecipitates using [32P]phosphorylase a as substrate. Untransfected cells were used as control. Similar results were obtained in two independent experiments. Phosphatase activity is represented as the percentage of maximal activity in untransfected cells. Expression of the transiently transfected Ras N17 was confirmed by direct comparison of Ras protein levels in mock and transfected cells. (B) Cells were treated as in (A) and then washed, stained with annexin and propidium iodide and analyzed by flow cytometry. Region G of the fluorescence scale represents apoptosis. The percentage of apoptotic cells in each sample is superimposed.
The effect of Ras inhibition was also analyzed in apoptosis. Cells were transfected with pHook3 (mock transfectants) or pHook3 and Ras N17. Transfected cells were isolated using magnetic beads and the percentage of apoptotic cells was estimated by annexin staining (Figure 7B). When cells were transfected with Ras N17 and maintained in the absence of IL-2, the fraction of apoptotic cells was much lower than that in mock transfectants. The number of apoptotic cells remained low when either transfectant was maintained in the presence of IL-2. Taken together, our results strongly suggest that Bad is a substrate for PP1α phosphatase, the enzymatic activity of which correlates with IL-2 deprivation-induced apoptosis and Ras activation.
Discussion
We were searching for Bad-interacting proteins that might induce apoptosis. Using the yeast two-hybrid approach, we show that Bad interacts with PP1α, the catalytic subunit of the serine/threonine phosphatase PP1. These results were confirmed by in vitro binding of cellular extracts to purified fusion proteins containing Bad or PP1α. Under both conditions, we detected Bad–PP1α interaction. We corroborated the results by co-immunoprecipitation experiments in which we recovered Bad or PP1α from cytoplasmic lysates.
Using the yeast two-hybrid system, Durfee et al. (1993) cloned a gene encoding PP1α2 phosphatase, an alternatively spliced isoform of PP1α that interacts with Rb. This suggests a potential role for PP1 in the M–G1 phases of the cell cycle. PP1α has also been implicated in the control of cell proliferation and mitosis, and in the mitotic dephosphorylation of Rb (Axton et al., 1990; Kinoshita et al., 1990; Alberts et al., 1993; Ludlow et al., 1993; Mumby and Walter, 1993). The pattern of changes in PP1 activity observed during the cell cycle is consistent with the idea that phosphatase may function as a cell cycle regulator. Finally, cyclin-dependent protein kinases phosphorylate a consensus sequence present in the C-terminus of all PP1 isotypes, inhibiting their activity (Dohadwala et al., 1994; Yamano et al., 1994).
PP1α phosphatase activity in Bad immunoprecipitates increased with the starvation period, which corresponds to an increase in the percentage of apoptotic cells, suggesting a correlation between apoptosis and PP1α phosphatase activity. This enzymatic activity in Bad immunoprecipitates was inhibited using OA concentrations that specifically block PP1 and PP2A activity (Cohen et al., 1989, 1990) and by inhibition of Ras activity. Western blot experiments confirmed that PP2A and PP2B were not observed in Bad–PP1α complexes. Bad is dephosphorylated after 6 h of IL-2 deprivation, indicating the effect of a phosphatase. Inhibition of Bad dephosphorylation may occur through the inhibition of PP1α phosphatase by OA. Treatment of the cells with low OA concentrations had no effect on Bad phosphorylation, but allowed IκB degradation, a process controlled by PP2A (Sun et al., 1995; Sontag et al., 1997). OA alone can prevent cell death and Bad dephosphorylation in IL-2-deprived cells, suggesting that, at least in part, Bad phosphorylation is important for cell survival, since IL-2 stimulation induces Bad phosphorylation. Prolonged exposure of cells to OA would also presumably inhibit other critical PP1α-regulated processes. Ras inhibition also prevents cell death through down-modulation of PP1 activity. Consistent with a direct role for PP1α in Bad dephosphorylation, this phosphatase efficiently dephosphorylates in vitro 32P-labeled Bad in vivo. In addition, recombinant PP1α is able to dephosphorylate GST–Bad fusion protein in vitro. Phosphorylated Bad results in cytoplasmic Bad sequestration by 14-3-3 protein, preventing binding to Bcl-x, thus promoting cell survival (Datta et al., 1997; del Peso et al., 1997). PP1α was associated with Bad even when cells had been deprived of IL-2 for 24 h, by which time no serine phosphorylation was detected. The interaction observed under starvation conditions may account for the absence of Bad phosphorylation after IL-2 deprivation. In fact, the PP1α activity observed in Bad immunoprecipitates increased with the starvation period and could be prevented by expression of a dominant-negative Ras mutant.
Serine/threonine phosphatases appear to be critical molecules in the control of apoptosis and proliferation. We and others have shown that Ca2+-dependent phosphatase, calcineurin, is involved in the control of Bcl-2 expression (Gómez et al., 1998; Srivastaba et al., 1999). Furthermore, IL-3 deprivation-induced apoptosis involves PP2A-mediated Bcl-2 dephosphorylation (Deng et al., 1998). It has been shown that calcineurin dephosphorylates Bad (Wang et al., 1999). This study does not contradict our results, since the authors used a growth factor-independent cell model in which they overexpressed Bad and/or calcineurin and, significantly, signaling through the IL-2 receptor does not mobilize intracellular Ca2+. Alternatively, binding of either PP1α or calcineurin to Bad may have different functional consequences and points out the potential versatility of Bad in interacting with and regulating other components involved in apoptosis.
Activated kinases, such as Akt, MAPK/Erk, Rsk, PAK and PKA, probably induce Bad phosphorylation, partially inhibiting or masking PP1α phosphatase activity (Datta et al., 1997; Bonni et al., 1999; Harada et al, 1999; Scheid et al., 1999; Schurmann et al., 2000). A balance between kinase and phosphatase activity may exist in IL-2-stimulated cells; when cells are deprived of lymphokine, kinase activity is down-regulated, shifting the equilibrium to the phosphatase activity. Alternatively, kinase activity may be constant under IL-2 deprivation conditions, suggesting that the reduced amount of phospho-Bad is a result of active dephosphorylation of Bad rather than of kinase suppression. The in vitro and in vivo studies strongly suggest that PP1α dephosphorylates Bad, although we do not exclude the possibility that other phosphatases can remove activating phosphates in Bad. In addition, we can not rule out the possibility that other phosphorylated proteins may be a substrate for PP1α phosphatase. Our results are the first evidence showing that Bad is a substrate for PP1α phosphatase and the Bad–PP1α interaction provides a mechanism for controlling the phosphorylation state. Consequently, the activity of this pro-apoptotic molecule links PP1α phosphatase to the apoptotic machinery. The association between Bad and PP1α may function to maintain the appropriate physiological level of phosphorylation required for cell growth or apoptosis. Bad phosphorylation can be considered in dynamic terms, with tight regulation of phosphorylation by the concerted action of kinases and PP1α phosphatase activity. We have shown a specific regulatory role for PP1α in Ras-mediated signal transduction. We propose that Ras activation influences the catalytic activity of PP1α, leading to Bad dephosphorylation and, consequently, to apoptosis. Our findings help to advance understanding of the mechanisms that regulate Bad dephosphorylation, which is required for its pro-apoptotic function.
Materials and methods
Cells and cultures
TS1αβ is a murine T-cell line stably transfected with the α and β chains of the human IL-2 receptor (Pitton et al., 1993); it can be propagated independently in the presence of IL-2, IL-4 or IL-9. Cells were cultured in RPMI-1640 (BioWhittaker, Walkersville, MD) supplemented with 5% heat-inactivated fetal calf serum (FCS) (Gibco-BRL, Gaithersburg, MD), 2 mM glutamine, 10 mM HEPES, 0.55 mM arginine, 0.24 mM asparagine, 50 µM 2-mercaptoethanol (2-ME) and 5 ng/ml recombinant IL-2 (rIL-2).
Lymphokines, antibodies, reagents and plasmids
Human rIL-2 was provided by Roussel Uclaf (Paris, France). Okadaic acid was from Calbiochem (La Jolla, CA) or Sigma (St Louis, MO). Anti-Bad antibody was from Calbiochem or Transduction Laboratories (Lexington, KY). Anti-phospho-Bad antibodies were from New England BioLabs (Beverly, MA) and anti-protein phosphatase 1 (PP1) antibodies from UBI (Lake Placid, NY), Calbiochem or Transduction Laboratories. Polyclonal PP2A antibody was generated at Pierre et Marie Curie University. Anti-PP2B (calcineurin) antibody was from Transduction Laboratories. Recombinant PP1α protein was from Calbiochem and annexin V–FITC from Immunotech (Marseille, France). Peroxidase-conjugated goat anti-rabbit, -mouse, or -guinea pig antibody was from Dako (Glostrup, Denmark). ECL, ECL Plus and [32P]orthophosphate were from Amersham (Amersham, UK). NP-40 was from Boehringer-Mannheim (Mannheim, Germany). Capture-Tec pHook3 kit was from Invitrogen (San Diego, CA). Glutathione–agarose beads and protease inhibitor cocktail were from Sigma. PP1α cDNA was kindly provided by Dr I.Sánchez (Institute of Microbiology, Salamanca, Spain). Bad and PP1α cloned into the pLex 10 or pGAD10 vectors were provided by Dr A.Germani (Hôpital Cochin, Paris, France).
cDNA libraries and the two-hybrid screen
Two cDNA libraries from IL-2-stimulated or -deprived TS1αβ cells from polyadenylated RNA were constructed in fusion with the Gal4 activation domain in pGAD10 (Bartel et al., 1993). Bad, cloned into the pLex10 vector, was used as a bait to screen the cDNA libraries in the Saccharomyces cerevisiae L40 strain (MATa, trp1, leu2, his3, LYS::lexA–His3, URA3::lexA–lacZ) using standard procedures (Bartel and Fields, 1997).
Sequence analysis
Sequencing of cDNA inserts from positive clones of the two-hybrid screening was performed on both strands with an automatic sequencer (Applied Biosystems 373A, Foster City, CA). Sequences were compared using the FASTA program.
Preparation of GST fusion proteins
PP1α and Bad were inserted into pGEX-4T1 or pEBG vectors, respectively (Pharmacia Biotech and New England BioLabs). Fusion protein expression was induced with or without isopropyl-β-d-thiogalactopyranoside (IPTG), respectively; proteins were then isolated from lysates by affinity chromatography with glutathione–agarose beads. For purity controls, proteins were eluted from agarose beads in SDS sample buffer, separated in 10% polyacrylamide gels and stained with Coomassie Blue.
Immunoprecipitation and Western blotting
Cells (1 × 107) were IL-2-stimulated or -deprived and lysed for 20 min at 4°C in lysis buffer (50 mM Tris–HCl pH 8, 1% NP-40, 137 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 10% glycerol, 1 mM orthovanadate and protease inhibitor cocktail). Lysates were immunoprecipitated with the appropriate antibody and protein A–Sepharose was added for 1 h at 4°C. After washing, immunoprecipitates were separated by SDS–PAGE. Alternatively, cells were lysed in Laemmli sample buffer and protein extracts separated by SDS–PAGE, transferred to nitrocellulose, blocked with 5% non-fat dry milk in Tris-buffered saline (TBS; 20 mM Tris–HCl pH 7.5, 150 mM NaCl) and incubated with primary antibody in TBS–0.5% non-fat dry milk. The membrane was washed with 0.05% Tween-20 in TBS and incubated with peroxidase-conjugated second antibody. After washing, proteins were developed using the enhanced chemiluminescence (ECL) or ECL Plus system. When stripping was required, membranes were incubated with 62.5 mM Tris–HCl pH 6.8, 2% SDS and 0.1 M 2-ME for 1 h at 55°C and washed extensively with TBS buffer before reblocking and probing.
In vitro phosphatase assay
Cells (1 × 107) were lysed in lysis buffer then supernatants were immunoprecipitated (2 h, 20°C) with anti-Bad antibody and incubated with protein A–Sepharose beads (45 min, 18°C). Immunoprecipitates were washed with phosphatase buffer [50 mM Tris–HCl pH 7.5, 0.1% 2-ME, 0.1 mM EDTA, 1 mg/ml bovine serum albumin (BSA)] and mixed with [32P]phosphorylase a, diluted in phosphatase buffer supplemented with 75 mM caffeine. The reaction was incubated at 30°C for 40 min, terminated by adding 200 µl of 20% trichloroacetic acid (TCA) and centrifuged. A total of 185 µl was used to estimate the generation of free phosphate liberated from [32P]phosphorylase a. Supernatants from anti-Bad immunoprecipitates were also used to estimate phosphatase activity. For phosphorylase phosphatase activity inhibition, OA at different concentrations was added to the reaction mixture. In some cases, control IL-2-stimulated cells were labeled with [32P]orthophosphate (100 µCi/ml) in phosphate-free medium (2 h, 37°C). Cells were lysed and supernatants immunoprecipitated with anti-Bad antibody, followed by protein A–Sepharose beads. Immunoprecipitates were diluted in phosphate buffer and incubated at 30°C for 40 min. Phosphatase activity was determined by measuring the generation of free phosphate released from 32P-labeled Bad.
GST–Bad fusion protein was incubated with recombinant PP1α protein phosphatase in P buffer [50 mM Tris–HCl pH 7, 5 mM dithiothreitol (DTT), 200 µM MnCl2, 100 µM EDTA and 200 µg/ml BSA] for 30 min at 30°C. Proteins were separated by SDS–PAGE, transfered to nitrocellulose, blocked and incubated with primary antibody. Membrane was washed and incubated with peroxidase-conjugated second antibody. Proteins were developed using the ECL system.
Cell cycle analysis by annexin staining
A total of 2.5 × 105 cells were washed with ice-cold PBS, diluted in ice-cold binding buffer and stained with annexin and propidium iodide (PI). Samples were maintained on ice for 10 min in the dark and then analyzed using an Epics XL flow cytometer (Coulter, Miami, FL).
Transient transfection
Transfections were performed using the DEAE–dextran method. Cells in exponential growth were washed in TS buffer (25 mM Tris–HCl pH 7.4, 137 mM NaCl, 5 mM KCl, 0.7 mM CaCl2, 0.5 mM MgCl2 and 0.6 mM Na2HPO4). A total of 5 µg of Ras N17 and pHook3, 750 µl of TS buffer and 750 µl of freshly prepared DEAE–dextran (1 mg/ml) in TS buffer were mixed successively with cells and incubated for 20 min at room temperature. After incubation, 13 ml of RPMI-1640 with 5% FCS were added. Cells were incubated at 37°C for 1 h, centrifuged and resuspended in 12 ml of RPMI-1640–5% FCS alone or supplemented with 5 ng/ml IL-2. The pHook3 vector drives the expression of a hapten-specific single chain antibody (sFv) on the surface of transfected cells. Cells expressing the sFv were isolated from the culture by binding to hapten-coated (pHox) magnetic beads and then analyzed.
Acknowledgments
Acknowledgements
We would like to thank Dr I.Sánchez (Salamanca, Spain) and Dr A.Germani (Paris, France) for gifts of reagents used in this study and C.Mark for editorial assistance. This work was partially supported by grants from the Association pour la Recherche sur le Cancer (ARC) 9294 and 9449 and from the Dirección General de Investigación y Ciencia (DGIyC). The Department of Immunology and Oncology was founded and is supported by the Spanish Research Council (CSIC) and Pharmacia–Upjohn.
References
- Adams J.M. and Cory,S. (1998) The Bcl-2 protein family: arbiters of cell survival. Science, 281, 1322–1326. [DOI] [PubMed] [Google Scholar]
- Alberts A.S., Thorburn,A.M., Shenolikar,S., Mumby,M.C. and Feramisco,J.R. (1993) Regulation of cell cycle progression and nuclear affinity of the Rb protein by protein phosphatases. Proc. Natl Acad. Sci. USA, 90, 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axton J.M., Dombradi,V., Cohen,P.T. and Glover,D.M. (1990) One of the protein phosphatase 1 isoenzymes in Drosophila is essential for mitosis. Cell, 63, 33–46. [DOI] [PubMed] [Google Scholar]
- Baharians Z. and Schönthal,A.H. (1999) Reduction of H-Ras induced cellular transformation by elevated expression of protein phosphatase type 2A. Mol. Carcinog., 24, 246–254. [DOI] [PubMed] [Google Scholar]
- Bartel P.L. and Fields,S. (1997) The Yeast Two-Hybrid System. Oxford University Press, New York, NY. [Google Scholar]
- Bartel P.L., Chien,C.T., Sterglanz,R. and Fields,S. (1993) Using the two-hybrid system to detect protein–protein interaction. In Cellular Interactions in Development: A Practical Approach. Oxford University Press, Oxford, UK, pp. 153–179. [Google Scholar]
- Bonni A., Brunet,A., Liu,M.Z. and Greenberg,M.E. (1999) Cell survival promoted by the Ras–MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science, 286, 1358–1362. [DOI] [PubMed] [Google Scholar]
- Chao D.T. and Korsmeyer,S.J. (1998) Bcl-2 family: regulators of cell death. Annu. Rev. Immunol., 16, 395–410. [DOI] [PubMed] [Google Scholar]
- Cheng A., Dean,N.M. and Honkanen,R.E. (2000) Serine/threonine protein phosphatase type 1γ is required for the completion of cytokinesis in human A549 lung carcinoma cells. J. Biol. Chem., 275, 1846–1854. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan A.M. and Dixit,V.M. (1996) The cell-death machine. Curr. Biol., 6, 555–562. [DOI] [PubMed] [Google Scholar]
- Cohen P., Klumpp,S. and Schelling,D.L. (1989) An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett., 250, 596–600. [DOI] [PubMed] [Google Scholar]
- Cohen P., Holmes,C.F. and Tsukitani,Y. (1990) Okadaic acid: a new probe for the study of cellular regulation. Trends Biochem. Sci., 15, 98–102. [DOI] [PubMed] [Google Scholar]
- Cory S. (1995) Regulation of lymphocyte survival by the Bcl-2 gene family. Annu. Rev. Immunol., 13, 513–543. [DOI] [PubMed] [Google Scholar]
- Craddock B.L., Orchiston,E.A., Hinton,H. and Welham,M.J. (1999) Dissociation of apoptosis from proliferation, PKB activation and Bad phosphorylation in IL-3-mediated PI3 kinase signaling. J. Biol. Chem., 274, 10633–10640. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Dukek,H., Tao,X., Masters,S., Fu,H., Gotoh,Y. and Greenberg,M.E. (1997) Akt phosphorylation of Bad couples survival signals to the cell-intrinsic death machinery. Cell, 91, 231–241. [DOI] [PubMed] [Google Scholar]
- del Peso L., Garcia,M.G., Page,C., Herrera,R. and Nuñez,G. (1997) Interleukin-3-induced phosphorylation of Bad through the protein kinase Akt. Science, 278, 687–689. [DOI] [PubMed] [Google Scholar]
- Deng X., Ito,T., Carr,B., Mumby,M. and Stratford,M.J. (1998) Reversible phosphorylation of Bcl-2 following IL-3 or Bryostatin 1 is mediated by direct interaction with protein phosphatase 2A. J. Biol. Chem., 273, 34157–34163. [DOI] [PubMed] [Google Scholar]
- Dohadwala M., Cruz e Silva,E.F., Hall,F.L., Willians,R.T., Carbonaro,O.A., Naiva,A.E., Greengard,P. and Berndt,N. (1994) Phosphorylation and inactivation of protein phosphatase 1 by cyclin-dependent kinases. Proc. Natl Acad. Sci. USA, 91, 6408–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durfee T., Becherer,K., Chen,P.L., Yeh,S.H., Yang,Y., Kilburn,A.E., Lee,W.H. and Elledge,S.J. (1993) The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev., 7, 555–569. [DOI] [PubMed] [Google Scholar]
- Faux M.C. and Scott,J.D. (1996) More on targeting with protein phosphorylation: conferring specificity by location. Trends Biochem. Sci., 21, 312–315. [PubMed] [Google Scholar]
- Favre B., Turowski,P. and Hemmings,B.A. (1997) Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin, okadaic acid and tautomycin. J. Biol. Chem., 272, 13856–13863. [DOI] [PubMed] [Google Scholar]
- Gómez J., Martínez-A,C., González,A., García,A. and Rebollo,A. (1998) The Bcl-2 gene is differentially regulated by IL-2 and IL-4: role of the transcription factor NF-AT. Oncogene, 17, 1235–1243. [DOI] [PubMed] [Google Scholar]
- Harada H., Becknell,B., Wilm,M., Mann,M., Huang,L.J., Taylor,S.J., Scott,J.D. and Korsmeyer,S.J. (1999) Phosphorylation and inactivation of Bad by mitochondria-anchored protein kinase A. Mol. Cell, 3, 413–422. [DOI] [PubMed] [Google Scholar]
- He B., Gross,M. and Roizman,B. (1997) The γ protein of herpes simplex virus complexes with PP1α to dephosphorylate the α subunit of eukaryotic TIF2 and preclude the shut off of protein synthesis by double strand RNA-activated protein kinase. Proc. Natl Acad. Sci. USA, 94, 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S.Y., Kaipai,L., Zhu,L. and Hsueh,A.J. (1997) Interference of Bad-induced apoptosis in mammalian cells by 14-3-3 isoforms and P11. Mol. Endocrinol., 11, 1858–1867. [DOI] [PubMed] [Google Scholar]
- Hubbard M.J. and Cohen,P. (1993) On target with new mechanism for the regulation of protein phosphorylation. Trends Biochem. Sci., 18, 172–177. [DOI] [PubMed] [Google Scholar]
- Ingebritsen T.S. and Cohen,P. (1983) The protein phosphatases involved in cellular regulation. Classification and substrate specificities. Eur. J. Biochem., 132, 255–261. [DOI] [PubMed] [Google Scholar]
- Jacobson M.D. (1997) Apoptosis: Bcl-2-related proteins get connected. Curr. Biol., 7, 277–281. [DOI] [PubMed] [Google Scholar]
- Kelekar A., Chang,B.S., Harlan,J.E., Fesik,S.S. and Thompson,C.B. (1997) Bad is a BH3-containing protein that forms an inactivating dimer with Bcl-x. Mol. Cell. Biol., 17, 7040–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita N., Ohkura,H. and Yanagida,M. (1990) Distinct essential roles of type 1 and 2A protein phosphatases in the control of the fission yeast cell division cycle. Cell, 63, 405–415. [DOI] [PubMed] [Google Scholar]
- Kremmer E., Ohst,K., Kiefer,J. and Walter,G. (1997) Separation of PP2A core enzyme and holoenzyme with monoclonal antibodies against the regulatory A subunit: abundant expression of both forms in T cells. Mol. Cell. Biol., 17, 1692–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow J.W., Glendening,C.L., Livingston,D.M. and DeCaprio,J.A. (1993) Specific enzymatic dephosphorylation of the retinoblastoma protein. Mol. Cell. Biol., 13, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B.L., Shriner,C.L. and Brautigan,D.L. (1991) Modulation of type I protein phosphatase by synthetic peptides corresponding to the carboxyl terminus. FEBS Lett., 285, 6–10. [DOI] [PubMed] [Google Scholar]
- Mumby M.C. and Walter,G.W. (1993) Protein serine/threonine phosphatases: structure, regulation and functions in cell growth. Physiol. Rev., 73, 673–699. [DOI] [PubMed] [Google Scholar]
- Nguyen M., Millar,D.G., Yong,V.W., Korsmeyer,S.J. and Shore,G.C. (1993) Targeting of Bcl-2 to the mitochondrial outer membrane by a COOH-terminal signal anchor sequence. J. Biol. Chem., 268, 25265–25268. [PubMed] [Google Scholar]
- Ottilie S. et al. (1997) Dimerization properties of human Bad. J. Biol. Chem., 272, 30866–30872. [DOI] [PubMed] [Google Scholar]
- Pastorino J.G., Tafani,M. and Farber,J.L. (1999) TNF induces phosphorylation and translocation of Bad through a PI3 kinase-dependent pathway. J. Biol. Chem., 274, 19411–19416. [DOI] [PubMed] [Google Scholar]
- Pitton C., Rebollo,A., Van Snick,J., Theze,J. and Garcia,A. (1993) High affinity and intermediate affinity forms of the human IL-2 receptor expressed in an IL-9-dependent murine T cell line deliver proliferative signals via differences in their transduction pathways. Cytokine, 5, 362–371. [DOI] [PubMed] [Google Scholar]
- Rajesh D., Schell,K. and Verma,A.K. (1999) Ras mutation, irrespective of cell type and p53 status, determines a cell’s destiny to undergo apoptosis by okadaic acid, an inhibitor of protein phosphatase 1 and 2A. Mol. Pharmacol., 56, 515–525. [DOI] [PubMed] [Google Scholar]
- Reed J.C. (1997) Double identity for proteins of the Bcl-2 family. Nature, 387, 773–776. [DOI] [PubMed] [Google Scholar]
- Sasaki K., Shima,H., Kitagawa,Y., Irino,S., Sugimura,T. and Nagao,M. (1990) Identification of members of the PP1 gene family in the rat and enhanced expression of PP1α gene in rat hepatocellular carcinoma. Jpn. J. Cancer Res., 81, 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid M.P. and Duronio,V. (1998) Dissociation of cytokine-induced phosphorylation of Bad and activation of PKB/Akt: involvement of MEK upstream of Bad phosphorylation. Proc. Natl Acad. Sci. USA, 95, 7439–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid M.P., Schubert,K.M. and Duronio,V. (1999) Regulation of Bad phosphorylation and association with Bcl-x by the MAPK/Erk kinase. J. Biol. Chem., 274, 31108–31113. [DOI] [PubMed] [Google Scholar]
- Schurmann A., Mooney,A.F., Sanders,L.C., Sells,M.A., Wang,H.G., Reed,J.C. and Bokoch,G.M. (2000) p21-activated kinase 1 phosphorylates the death agonist Bad and protects cells from apoptosis. Mol. Cell. Biol., 20, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenolikar S. (1994) Protein serine/threonine phosphatases, new avenues for cell regulation. Annu. Rev. Cell. Biol., 10, 55–86. [DOI] [PubMed] [Google Scholar]
- Shenolikar S. and Nairn,A.C. (1991) Protein phosphatases: recent progress. Adv. Second Messenger Phosphoprotein Res., 23, 1–121. [PubMed] [Google Scholar]
- Sieburth D.S., Sundaram,M., Howard,R.M. and Han,M. (1999) A PP2A regulatory subunit positively regulates Ras-mediated signaling during C.elegans vulval induction. Genes Dev., 13, 2562–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E., Sontag,J.M. and García,A. (1997) Protein phosphatase 2A is a critical regulator of PKC ζ signaling targeted by SV40 small t to promote cell growth and NF-κB activation. EMBO J., 16, 5662–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastaba R.K., Sasaki,C.Y., Hardwick,J.M. and Logo,D.L. (1999) Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T cells (NFAT)-induced Fas ligand transcription. J. Exp. Med., 190, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steller H. (1995) Mechanisms and genes of cellular suicide. Science, 267, 1445–1449. [DOI] [PubMed] [Google Scholar]
- Sun S.C., Maggirwar,S.B. and Harhaj,E. (1995) Activation of NF-κB by phosphatase inhibitors involves the phosphorylation of IκB at phosphatase 2A sensitive sites. J. Biol. Chem., 270, 18347–18351. [DOI] [PubMed] [Google Scholar]
- Takizawa N., Mizuno,Y., Ito,Y. and Kikuchi,K. (1994) Tissue distribution of isoforms of type I PP1 in mouse tissues and its diabetic alterations. J. Biochem., 116, 411–415. [DOI] [PubMed] [Google Scholar]
- Wang H.G. et al. (1999) Ca++-induced apoptosis through calcineurin dephosphorylation of Bad. Science, 284, 339–343. [DOI] [PubMed] [Google Scholar]
- Wera S. and Hemmings,B.A. (1995) Serine/threonine protein phosphatases. Biochem. J., 311, 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano H., Ishii,K. and Yanagida,M. (1994) Phosphorylation of dis2 protein phosphatase at the C-terninal cdc2 consensus and its potential role in cell cycle regulation. EMBO J., 13, 5310–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E., Zha,J., Jockel,J., Boise,L.H., Thompson,C.B. and Korsmeyer,S.J. (1995) Bad: a heterodimeric partner for Bcl-x and Bcl-2 displaces Bax and promotes cell death. Cell, 80, 285–291. [DOI] [PubMed] [Google Scholar]
- Zha J., Harada,H., Yang,E., Jocker,J. and Korsmeyer,S. (1996) Serine phosphorylation of death agonist Bad in response to survival factor results in binding to 14-3-3 not Bcl-x. Cell, 87, 619–628. [DOI] [PubMed] [Google Scholar]
- Zha J., Harada,H., Osipov,K., Jockel,J., Waksman,G. and Korsmeyer,S.J. (1997) BH3 domain of Bad is required for heterodimerization with Bcl-x and pro-apoptotic activity. J. Biol. Chem., 272, 24101–24104. [DOI] [PubMed] [Google Scholar]