

Abstract
Described are experiments that allow incorporation of cyanide cofactors and hydride substrate into active site models [NiFe]-hydrogenases (H2ases). Complexes of the type (CO)2(CN)2Fe(pdt)Ni(dxpe), (dxpe = dppe, 1; dxpe = dcpe, 2) bind the Lewis acid B(C6F5)3 (BArF3) to give the adducts (CO)2(CNBArF3)2Fe(pdt)Ni(dxpe), (1(BArF3)2, 2(BArF3)2). Upon decarbonylation using amine oxides, these adducts react with H2 to give hydrido derivatives Et4N[(CO)(CNBArF3)2Fe(H)(pdt)Ni(dxpe)], (dxpe = dppe, Et4N[H3(BArF3)2]; dxpe = dcpe, Et4N[H4(BArF3)2]). Crystallographic analysis shows that Et4N[H3(BArF3)2] generally resembles the active site of the enzyme in the reduced, hydride-containing states (Ni-C/R). The Fe-H…Ni center is unsymmetrical with rFe-H = 1.51(3) and rNi-H = 1.71(3) Å. Both crystallographic and 19F NMR analysis show that the CNBArF3− ligands occupy basal and apical sites. Unlike cationic Ni-Fe hydrides, [H3(BArF3)2]− and [H4(BArF3)2]− oxidize at mild potentials, near the Fc+/0 couple. Electrochemical measurements indicate that in the presence of base, [H3(BArF3)2]− catalyzes the oxidation of H2. NMR evidence indicates dihydrogen bonding between these anionic hydrides and ammonium salts, which is relevant to the mechanism of hydrogenogenesis. In the case of Et4N[H3(BArF3)2], strong acids such as HCl induce H2 release to give the chloride Et4N[(CO)(CNBArF3)2Fe(pdt)(Cl)Ni(dppe)].
Introduction
The hydrogenases (H2ases) are represented by three main families of enzymes defined by the metals at their active sites, the [FeFe]-, [Fe]-, and [NiFe]-H2ases.1 Other enzymes consume or produce hydrogen,2 but the [FeFe] and [NiFe]-H2ases are renown for their ability to catalyze the oxidation of H2 and reduction of protons at high rates and low overpotentials.3
The active sites of the hydrogenases are characterized by the presence of several remarkable cofactors. Crystallographic and spectroscopic analyses consistently point to the presence of Fe(CO)x(CN)y sites in both types of redox active hydrogenases.4 The diatomic cofactors are unusual in biology.5 As indicated by close contacts between otherwise nonbonded heteroatoms, the Fe-CN centers anchor the active sites to the protein by means of hydrogen bonding. For example in the [NiFe]-H2ase from D. fructosovorans one cyanide ligand hydrogen-bonds with serine 499 (N⋯N, N⋯O = 2.98, 2.88 Å, respectively). The second cyanide interacts with arginine 476, with N⋯N distances of 2.87 and 3.25 Å.6 Some of these amino acids can be mutated with retention of catalytic activity,7 which suggests that the N-terminus of the cyanide cofactors could be modified in models as well.
Motivated both by the novelty of the active site as well as possible practical applications, much effort has been invested into the structural and functional models of the [NiFe]-H2ase active site.1,8 Since 2009, several nickel-iron hydrides have been reported, but all of these models feature Fe(CO)3-x(PR3)x centers (x = 0, 1, 2).9-12 In contrast to these cationic models, the active site is expected to be anionic since six cofactors - two cyanide and four thiolate groups - are anionic.13 Incorporation of cyanide cofactors in models is avoided because the reactivity of the basic nitrogen site of cyanide.14 Structural modes for a CO-inhibited state of the [NiFe]-H2ases with cyanide cofactors have been reported, (CN)2(CO)2Fe(SR)2Ni(dppe) (1) (dppe = (C2H4(PPh2)2)15 and [(CN)2(CO)2Fe(SR)2Ni(S2CNEt2)]−.16 Both complexes feature Fe(CO)2(CN)2 centers bound to Ni(SR)2L2 sites. These complexes exhibit no biomimetic reactivity because of the coordinatively saturated nature of the Fe(CN)2(CO)2(SR)2 sites.
This paper describes a method for modifying Fe-CN centers to suppress their high basicity14 but retain the charge provided by the anionic cofactors. The approach entails attaching organoboranes to the Fe-CN groups. Although novel for metallobiochemistry, the interaction of boranes with metal cyanides is well known17 and is exploited in commercial hydrocyanation catalysis.18
In view of the inertness of cyanide-containing models for the [NiFe]-H2ases, attachment boranes to the Fe-CN centers could both labilize one CO ligand and suppress reactions of the Fe-CN centers, giving rise to reactive NiII-FeII centers capable of capturing a hydride substrate from dihydrogen.
Results and Discussion
N-Protected Active Site Models
Modeling efforts focused on the dicyano Ni-Fe dithiolate 1, which was prepared by combining NiCl2(dppe) and [Fe(CN)2(pdt)(CO)2]2−. Initial efforts to induce reactivity reminiscent of the H2ases focused on the decarbonylation of this species. The FT-IR spectrum of 1 exhibits a relatively high frequency νCO band at 2053 cm−1, suggesting that CO would be labile. UV-irradiation of a THF solution of 1 however afforded only insoluble solids, which are tentatively attributed to the formation of Fe-CN-M (M = Fe, Ni) linkages. Solutions of 1 were also found to be inert towards the decarbonylating agent Me3NO.
Next examined was the use of boranes to protect the Fe-CN centers while also electrophilically activating the iron center. A variety of triarylboranes were found to form adducts with 1, but this work focuses on B(C6F5)3 (BArF3). Adducts of this strong Lewis acid are often highly soluble in nonpolar solvents compatible with H2 activation experiments.19 Treatment of CH2Cl2 solutions of 1 with two equiv of BArF3 quantitatively afforded the 2:1 adduct (CO)2(CNBArF3)2Fe(pdt)Ni(dppe), 1(BArF3)2 (eq 1). The attachment of BArF3 strongly affects the νCN and νCO bands, resulting in shifts of about 100 and 50 cm−1, respectively (Table S1). The pattern of the νCO/CN bands is similar for the precursor and the adduct, consistent with retention of the cis-CO/trans-CN arrangement. 31P and 19F NMR spectra are also consistent with the proposed stereochemistry.
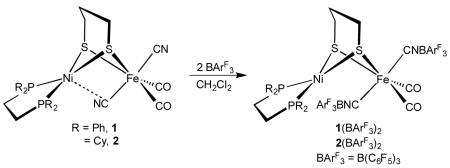 |
The structure of 1(BArF3)2 was verified by X-ray crystallography (Fig. 2). Owing to the steric bulk of the BArF3 groups, the Ni-Fe distance increased from 2.809 in 1 to 3.218 Å in the adduct.15 Similarly the Ni-S-Fe angles opened from 75.98 to 86.60°. The BArF3 substituents only subtly affect the diatomic ligands: the Fe-CN bonds are slightly elongated (Δavg = 0.03 Å) and the Fe-CO bond is slightly shortened (Δavg = −0.03 Å). The weak Ni⋯CN π-interaction (2.421 Å) apparent in 115 is absent in 1(BArF3)2.
Figure 2.
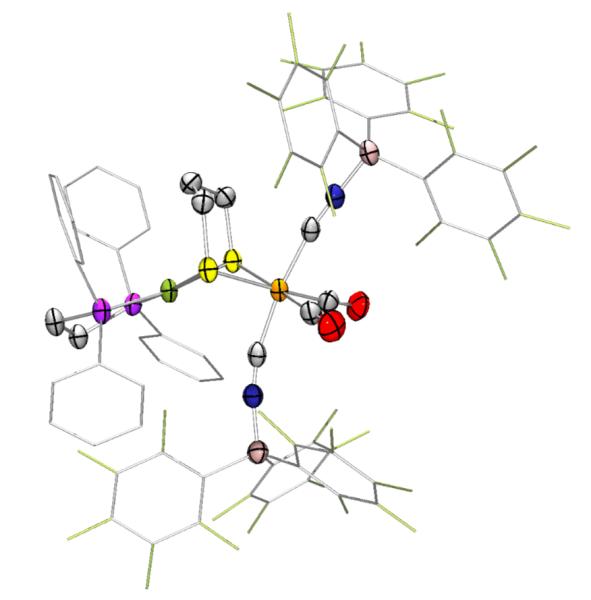
Structure of [(CO)2(CNBArF3)2Fe(pdt)Ni(dppe)], 1(BArF3)2 with ellipsoids shown at 50% probability and with H atoms omitted for clarity. Phenyl and pentafluorophenyl groups are deemphasized for clarity.
To examine the effects of the basicity of nickel, (CO)2(CN)2Fe(pdt)Ni(dcpe) (2), featuring the highly basic ligand dcpe (C2H4(P(C6H11)2)2), was also investigated. IR and 31P NMR spectroscopy shows that 2 exists as a mixture of isomers, consisting of both the cis-CO/trans-CN arrangement (as seen in 1) and the cis-CO/cis-CN arrangement. The decreased Lewis acidity of the Ni(dcpe) center, associated with the strong donor properties of dcpe,20 is proposed to weaken the Ni-NCFe interaction, giving rise to the second isomer. Much like 1, 2 was found to be unreactive toward Me3NO and decomposed in ambient light. Complexation of 2 with BArF3 gave a pair of isomeric adducts of the formula (CO)2(CNBArF3)2Fe(pdt)Ni(dcpe), 2(BArF3)2. Over the course of several hours, this mixture converted to the cis-CO/trans-CN arrangement seen for 1(BArF3)2.
The attachment of BArF3 to the cyanide ligands stabilizes isomers not observed in 1 and 2. As determined by 19F and 31P NMR spectroscopy, the cis-CO/trans-CN isomer of 1(BArF3)2 converted to the cis-CO/cis-CN isomer upon heating at 60 °C. Upon UV-irradiation (365 nm), 1(BArF3)2 converted to the trans-CO/cis-CN isomer as well as a small amount of the cis-CO/cis-CN isomer. Upon standing, trans-CO/cis-CN reverted to one of the two cis/cis isomers. Irradiation of the latter regenerated the trans-CO/cis-CN isomer (eq 2, showing only one of two cis/cis isomers). In no case was the cis-CO/trans-CN isomer of 1(BArF3)2 observed to reform, which is apparently a kinetically stabilized isomer.
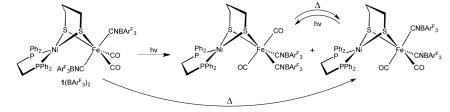 |
Irradiation of the cis-CO/trans-CN isomer of 2(BArF3)2 gave the trans-CO/cis-CN isomer. Unlike in 1(BArF3)2, the cis-CO/cis-CN isomer of 2(BArF3)2 was not found to reform. Iron carbonyl complexes with high frequency νCO bands are typically photolabile. Indeed photolysis of cis-CO/trans-CN 1(BArF3)2 under an atmosphere of 13CO gave complete exchange of CO while forming the trans-CO/cis-CN isomer. Furthermore, the photoisomerization of 1(BArF3)2 was found to be inhibited by CO. These results are consistent with a dissociative pathway for the photoisomerization of 1(BArF3)2 and 2(BArF3)2.
Hydride Derivatives
Being coordinatively saturated, the adducts 1(BArF3)2 and 2(BArF3)2 are unreactive toward H2. Addition of a decarbonylation agent, however, was found to induce rapid uptake of hydrogen (eq 3).
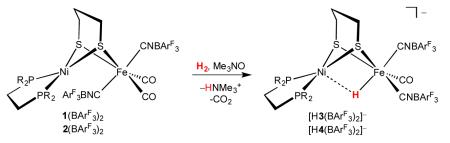 |
Thus, treatment of a CH2Cl2 solution of 1(BArF3)2 with 1.2 equiv of an amine oxide under an atmosphere of H2 afforded the hydride [(CO)(CNBArF3)2Fe(pdt)(H)Ni(dppe)]−, which was isolated as the salt Et4N[H3(BArF3)2]. The use of D2 in place of H2 gave Et4N[D3(BArF3)2], as verified by 1H and 2H NMR analyses. The decarbonylation agent Me3NO gave an initial mixture of isomers with the CNBArF3 ligands in dibasal (b/b) and apical-basel (a/b) arrangement in a 1:9.5 ratio. Upon standing in solution, the b/b isomer converted solely to the a/b isomer. Decarbonylation of 1(BArF3)2 with N-methylmorpholine-N-oxide (NMO) produced the b/b isomer as the major species (2.5:1 b/b:a/b ratio). Similarly, decarbonylation of 2(BArF3)2 under H2 produced the a/b isomer of Et4N[(CO)(CNBArF3)2Fe(H)(pdt)Ni(dcpe)] (Et4N[H4(BArF3)2]) when Me3NO was used as a decarbonylating agent with no b/b isomer observed.
The hydrides [H3(BArF3)2]− and [H4(BArF3)2]− were also produced using conventional hydride sources instead of H2. Thus, treatment of [1(BArF3)2] with amine oxides followed by Et4NBH4 gave Et4N[H3(BArF 3)2]. However, attempted synthesis of Et4N[H4(BArF3)2] by treatment of 2(BArF3)2 with amine oxides and Et4NBH4 produced more complicated results, yielding both monocarbonyl and dicarbonyl hydrides. Overall, H2 is a gentler, more selective reagent for generating hydrido complexes.
As observed in the enzymes, the IR spectra of [H3(BArF3)2]− and [H4(BArF3)2]− display two νCN and one νCO bands. The energy for νCO of [H4(BArF3)2]− is 11 cm−1 lower than that of [H3(BArF3)2]− indicating that changes at the Ni center (dppe vs dcpe) influences the Fe site. Values of νCO for [H3(BArF3)2]− and [H4(BArF3)2]− were found to be within the range observed for the Ni-R state of [NiFe]-H2ase (Table 1).
Table 1.
IR Bands for the CN-protected Species [H3(BArF3)2]− and [H4(BArF3)2]− as well as [NiFe]-H2ase Enzymes in the Ni-R State,21 and Other Ni-R Models.10
| Compound | νCN (cm−1) | νCO (cm−1) |
|---|---|---|
| Ni-R state, A. vinosum | 2072, 2059 | 1936 |
| Ni-R state, D. gigas | 2073, 2060 | 1940 |
| Ni-R state, D. fructosovorans | 2074, 2060 | 1938 |
| Ni-R state, D. vulgaris | 2074, 2061 | 1948 |
| [(CO)(CNBArF3)2Fe(H)(pdt)Ni(dppe)]−([H3(BArF3)2]−) | 2162, 2137 | 1963 |
| [(CO)(CNBArF3)2Fe(H)(pdt)Ni(dcpe)]− ([H4(BArF3)2]−) | 2158, 2131 | 1952 |
| [(CO)2(PPh3)Fe(H)(pdt)Ni(dppe)]+ | - | 2016, 1964 |
Structure of Et4N[H3(BArF3)2] and Comparisons with Enzyme Active Site
The structure of [H3(BArF3)2]− was confirmed by single crystal X-ray diffraction (Fig. 3). The hydride of [H3(BArF3)2]− was located in the difference Fourier map and refined isotropically. The Fe-H distance of 1.51(3) Å is shorter than the Ni-H bond (1.71(3) Å). In several respects, the structure of [H3(BArF3)2]− resembles the high resolution (1.4, 1.5 Å) structures of the hydride-containing states (Ni-C/R) of D. vulgaris (Table 2).22 The Fe⋯Ni distance of 2.5497(5) Å reasonably matches the values 2.59 and 2.57 Å found in these hydride-containing states. Reminiscent of the active site, the coordination geometry of the NiS2P2 center is distorted from square planar, with a twist angle between the NiS2 plane and NiP2 plane of 34.53°. Like the enzyme, iron is bound to one CO and two CN-derived ligands although the locations of the CO and the one CNBArF3− are interchanged relative to the enzyme.
Figure 3.
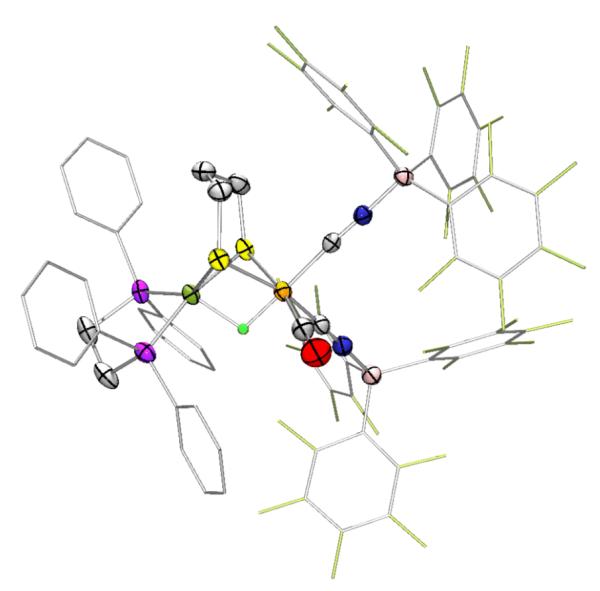
Structure of the anion in Et4N[(CO)(CNBArF3)2Fe(pdt)(H)Ni(dppe)], Et4N[H3(BArF3)2]) with ellipsoids shown at 50% probability and H atoms except for the bridging hydride omitted. Phenyl and pentafluorophenyl groups are deemphasized for clarity.
Table 2.
Key Distances (Å) and Angles (°) of [H3(BArF3)2]− and the Active Site of D. vulgaris Miyazaki F in the Reduced State(s).22
| [(CO)(CNBArF3)2Fe(pdt)(H) Ni(dppe)]− |
D. vulgaris Miyazaki F Ni-C/R (PDB: 1H2R) |
D. vulgaris Miyazaki F Ni-C/R (PDB: 1WUL) |
|
|---|---|---|---|
| Fe-Ni | 2.5497(5) | 2.59 | 2.57 |
| Fe-H | 1.51(3) | N/A | N/A |
| Ni-H | 1.71(3) | N/A | N/A |
| Fe-Capical | 1.889(3) (CNBArF3−) | 1.84 (CO) | 1.80 (CO) |
| Fe-Cbasal | 1.758(3) (CO), 1.866(2) (CNBArF3−) |
1.86 (CO), 2.21 (SO) | 1.99 (CN−), 2.03 (CO) |
| Fe-S | 2.3246(6), 2.2977(8) | 2.29, 2.36 | 2.26, 2.32 |
| Ni-S | 2.1952(8), 2.3046(8) | 2.43, 2.33 | 2.23, 2.52 |
| Ni-term-L | 2.1382(8), 2.1591(8) (L = PR3) |
2.32, 2.24 (L =SR−) |
2.13, 2.21 (L = SR−) |
| Cbasal-Fe-Cbasal | 94.0(1) | 89 | 93 |
| Cbasal-Fe-Capical | 90.6(1), 96.0(1) | 90, 98 | 90, 95 |
Reactivity of the Hydrido Complexes
The redox properties of [H3(BArF3)2]− and [H4(BArF3)2]− were assessed by cyclic voltammetry on CH2Cl2 solutions. These results underscore the influence of anionic donor ligands on the Fe site. Both hydrides oxidize at mild potentials: −0.08 V vs Fc0/+ (ipc/ipa = 0.43, scan rate = 0.1 V/s) for Et4N[H3(BArF3)2] and −0.10 V (ipc/ipa = 0.69, scan rate = 0.1 V/s) for Et4N[H4(BArF3)2] (Figs. S29, S30). The insensitivity of the [H3(BArF3)2]−/0 vs [H4(BArF3)2]−/0 couples to the diphosphine suggests that oxidation is localized on Fe. In the enzyme, oxidation of the Ni-R state is localized on Ni.
Reflecting their anionic character, both complexes reduce at more negative potentials in comparison with previously reported NiFe hydrides. A reversible reduction was observed for Et4N[H3(BArF3)2] at −1.99 V (Fig. S28) but no reduction was observed for Et4N[H4(BArF3)2] within the potential window of CH2Cl2 (< −2.5 V). Cationic NiFe hydrides, i.e., those based on Fe(CO)3-x(PR3)x centers, reduce at milder potentials (−1.33 to −1.56 V vs Fc0/+).10,11 Reduction potentials for those cationic Fe(II)Ni(II) hydrides are sensitive to substitution at Ni (dppe vs dcpe) and Fe (CO vs PR3).10,11
The acid base properties of [H3(BArF3)2]− and [H4(BArF3)2]− were examined, with limited success. Unlike cationic Ni-Fe hydrides, which exhibit pKaMeCN near 14, CH2Cl2 solution of [H3(BArF3)2]− did not undergo deprotonation upon treatment with the strong base DBU (pKaMeCN = 24.34). Solution of [H3(BArF3)2]− exhibit no exchange with D2O.
Et4N[H3(BArF3)2] was found to be an electrocatalyst for the oxidation of H2. When a solution was examined by cyclic voltammetry under 1 atm of H2, the current at −0.08 V, corresponding to the [H3(BArF3)2]−/0 couple, was found to increase upon addition of the base DBU (see Supporting Information).
Dihydrogen Bonding
The solution properties of [H3(BArF3)2]− are indicative of its hydridic character, consistent with its resemblance to the hydrogen-evolving state Ni-R. Specifically, the high field 1H NMR signal of Et N[H3(BArF 4 3)2] is sensitive to the presence of reagents that engage in dihydrogen bonding. For example, the addition of HNMe3[BArF24] (pKaMeCN = 17.61)23 shifts the hydride signal (δH) by about 2 ppm (Figure 4). The dependence of δH on the concentration of HNMe3+ is consistent with an equilibrium constant of 9.0±1.0 × 103 L/mol in CD2Cl2 (20 °C, eq 3). Dihydrogen bonding is also evident in the 1H NMR spectra when [H4(BArF3)2]− was titrated with [pyrrolidinium]BArF24 (pKaMeCN = 19.56).24 Although competitive with degradation of the complex, [H3(BArF3)2]− was found to undergo H/D scrambling with D2, forming HD. This process occurs only in the presence of HNMe3+.
Figure 4.

1H NMR chemical shift for Et4N[H3(BArF3)2] upon titration with HNMe3[BArF24] in CD2Cl2 (20 °C). Inset: 1H NMR (500 MHz) spectra of the titration of Et4N[H3(BArF3)2] with HNMe3BArF24 in CD2Cl2 (20 °C): a) 0, b) 1.0, c) 2.0, d) 3.0, and e) 4.0 equiv.
Although weak acids form dihydrogen bonds with [H3(BArF3)2]−, stronger acids, PhNH3+ (pKa = 10.62), induce release ~1 equiv of H2. Using HCl, the unsaturated product is trapped as the chloride adduct [Cl3(BArF3)2]− (eq 4). The complex [Cl3(BArF3)2]− was independently prepared by the reaction of 1(BArF3)2 with Me3NO and Et4NCl.
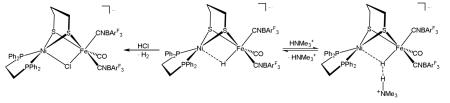 |
Conclusions
Borane protecting groups enable the preparation of the first models of the [NiFe]-H2ases with biomimetic ligation at Fe. The protecting groups suppress reactions at the Fe-CN centers, while retaining the anionic charge that is characteristic of these active sites.
Similar to work with the enzyme,21 models in this work were obtained in an inhibited state. Unlike the enzyme,25 the CO-inhibited NiFe models contain an extra CO ligand on Fe.26 Upon decarbonylation, the borane-protected Ni-Fe derivatives abstract hydride from H2. The generation of hydrides from H2 is unusual in H2ase models12,27 but is a key reaction of the enzyme. On the basis of structural and νCO data, these models compare well with the Ni-R state.28
The redox properties of the hydrides [H3(BArF3)2]− and [H4(BArF3)2)]− provide insights into the design of improved active site models. Because the basicities of dppe and dcpe differ greatly20 and the oxidations potentials differ by only 20 mV, these oxidations are assigned to the FeII/III couple. In contrast, oxidation of the Ni-R state to the Ni-C state is localized on nickel, i.e., a NiII-FeII/NiIII-FeII couple.29 These findings point to the limitations of diphosphines as mimics for the terminal ligands on the Ni site. As suggested with a recent model,30 the terminal thiolate ligands in the enzyme1 change their protonation state upon redox of the Ni center. Future models could benefit by including terminal ligands that have ionizable protons such as RSH.
Compared to cationic nickel-iron hydrides,10-12 the anionic models exhibit more biomimetic electrochemical and acid-base properties. Most importantly, anionic nickel-iron hydrides have hydridic character31 as indicated by NMR studies showing dihydrogen bonding, which leads to hydrogen evolution. On the basis of these results, a similar dihydrogen bond in the Ni-R state is proposed to precede H2 evolution (Figure 5).
Figure 5.
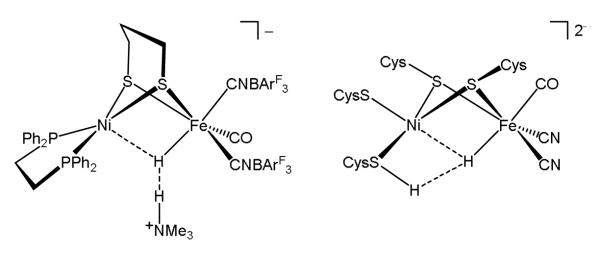
Dihydrogen bonding in [H3(BArF3)2]− and the analogous interaction proposed for Ni-R.
Bridging hydrides in related systems have protic character and do not engage in dihydrogen bonding.32 Dihydrogen bonding involving terminal iron hydrides is observed in models for the active site of the [FeFe]-H2ases.33 Thus it appears that the hydrides of [FeFe]- and [NiFe]-H2ases (in the Ni-R state) are similar in being hydridic.
Experimental Section
All procedures were carried out in a nitrogen filled MBraun glovebox or using standard Schlenk techniques. All solvents were degassed by sparging with nitrogen and dried by passage through activated alumina. IR spectra were recorded on a Perkin Elmer Spectrum 100. Cyclic voltammetry was performed under argon at room temperature using a CHI 630D potentiostat with glassy carbon working electrode, Pt wire counter electrode and the pseudo-reference electrode Ag wire, and with Fc internal standard. Production of H2 was quantified by gas chromatography on a column packed with 5 Å molecular sieves (carrier gas: Ar), using an Agilent 7820A instrument equipped with a thermal conductivity detector. The response factor for H2/CH4 was 3.8 under experimental conditions as established by calibration with standard H2-CH4 mixtures. NMR spectra were recorded on a Varian Unity 500 MHz NMR spectrometer. Signals for 19F and 31P NMR spectra were referenced using external standards of 1%CFCl3 in CDCl3 and 85% H3PO4, respectively. 1H NMR spectra were referenced to internal solvent signals. CD2Cl2 (Cambridge Isotope Laboratories) was dried over CaH2 before being vacuum distilled onto activated 4Å molecular sieves. B(C6F5)3 (Boulder Scientific) was sublimed under vacuum at 90 °C. Literature routes were followed for NiCl2(dppe),9 NiCl2(dcpe),11 Et4N[Fe(CO)4(CN)],34 K2pdt,16 and Et4N[FeBr(CO)3(CN)2].16
Revised Synthesis of (CO)2(CN)2Fe(pdt)Ni(dppe), 1
A solution of Et4N[FeBr(CO)3(CN)2] (0.900 g, 2.22 mmol) in MeOH (100 mL) was treated with a solution of K2pdt (0.410 g, 2.22 mmol) in MeOH (50 mL). The mixture was allowed to stir for 3 h at which time the solvent was removed under vacuum. NiCl2(dppe) (1.17 g, 2.22 mmol) was added along with 100 mL of MeCN and the mixture was stirred for 15 min. The mixture was filtered, and the solvent was removed from the filtrate under vacuum. The crude product was extracted into THF, and the crude product was precipitated from the filtered extract upon the addition of pentane. The crude product was dissolved in MeCN and loaded onto a neutral alumina column (Brockmann Level I). A faint red band was washed off the column with MeCN. The product eluted with MeOH as a red band. The solution was evaporated under vacuum, and the residue was extracted into THF. This solution was filtered through Celite, and product was precipitated from the filtrate with pentane. The product16 was collected by filtration and dried under vacuum. Yield: 0.958 g (59%).
(CO)2(CN)2Fe(pdt)Ni(dcpe), 2
A solution of Et4N[FeBr(CO)3(CN)2] (0.500 g, 1.23 mmol) in MeOH (20 mL) was treated with a solution of K2pdt (0.228 g, 1.23 mmol) in MeOH (20 mL). The mixture was let to stir for 3 h at which time the solvent was removed under vacuum. In the absence of light, the residue was extracted into 20 mL of MeCN, and the mixture was treated with a suspension of NiCl2(dcpe) (0.682 g, 1.23 mmol) in MeCN (20 mL). After stirring for 15 min, the mixture was filtered and the solvent was stripped from the filtrate. The crude product was extracted into 20 mL of THF, the mixture was filtered, and the product was precipitated from the filtrate upon the addition of pentane. Unlike 1, 2 did not require further purification by column chromatography. Yield: 0.423 g (45%). Anal. Calcd for C33H54FeN2NiO2P2S2 (found): C, 52.75 (52.34); H, 7.24 (7.59); N, 3.73 (3.99).
Data for cis-CO/trans-CN isomer: IR(CH2Cl2, cm−1): νCN = 2113, 2090; νCO = 2045, 1995. 31P NMR (CD2Cl2): δ 66.3 (s).
Dats for cis-CO/cis-CN isomer: IR (CH2Cl2, cm−1): νCN = 2120, νCO = 2033, 1982. 31P NMR (CD2Cl2): δ 67.8, 66.5 (AB quartet, JPP = 33 Hz).
(CO)2(CNBArF3)2Fe(pdt)Ni(dppe), 1(BArF3)2
A solution of (CO)2(CN)2Fe(pdt)Ni(dppe) (290 mg, 0.4 mmol) in 10 mL of CH2Cl2 was treated with a solution of BArF3 (410 mg, 0.8 mmol) in 30 mL of CH2Cl2 dropwise. The reaction caused a change in color from a red to an orange solution. The reaction solution was concentrated to 20 mL and layered with 100 mL of pentane followed by cooling to −30 °C. Yield: 570 mg (82%). Crystals suitable for X-ray diffraction were grown from a concentrated THF solution of 1(BArF3)2 layered with pentane and cooled at −30°C. IR (CH2Cl2, cm−1): νCN = 2194, 2166, νCO = 2088, 2050. 31P NMR (CD2Cl2): δ 56.4 (s). 19F NMR (CD2Cl2): δ −133.7 (d, o-F) −135.1 (d, o-F), −159.2 (t, p-F) −159.4 (t, p-F), −165.0 (dt, m-F) −165.6 (dt, m-F). Anal. Calcd for C69H30B2F30FeN2NiO2P2S2 (found): C, 47.32 (47.24); H, 1.73 (1.64); N, 1.60 (1.52).
(CO)2(CNBArF3)2Fe(pdt)Ni(dcpe), 2(BArF3)2
Analogously to the preparation of 1(BArF3)2, a solution of 2 (0.144 g, 0.0192 mmol) in CH2Cl2 (5 mL) was treated with a solution of BArF3 (0.196 g, 0.0383 mmol) resulting in a color change from red or orange. The solvent was removed under vacuum, redissolved in THF and crystallized by the addition of pentane followed by cooling to −30 °C. Yield: 0.253 g (74%).
Data for cis-CO/trans-CN isomer: IR (CH2Cl2, cm−1): νCN = 2190, 2149, νCO = 2085, 2046. 31P NMR (CD2Cl2): δ 69.8 (s). 19F NMR (CD2Cl2): −133.8 (d, o-F), −134.2 (d, o-F), −159.4 (t, p-F), −160.6 (t, p-F), −165.3 (t, m-F), −166.4 (t, m-F).
Data for cis-CO/cis-CN isomer: IR (CH2Cl2, cm−1): νCN = 2210; νCO = 2068, 2030. 31P NMR (CD2Cl2): δ 70.2, 69.5 (AB quartet, JPP = 30 Hz). 19F NMR (CD2Cl2): −133.1 (d, o-F), −134.9 (o-F), −158.7 (t, p-F), −159.3 (t, p-F), −165.0 (t, m-F), −165.5 (t, m-F).
Et4N[(CO)(CNBArF3)2Fe(pdt)(H)Ni(dppe)], Et4N[H3(BArF3)2]
In a thick-walled glass pressure tube, a solution of 1(BArF3)2 (150 mg, 0.0857 mmol) in CH2Cl2 (10 mL) was frozen by inserting the tube into liquid nitrogen. Under a flow of Ar, a buffer layer of CH2Cl (5 mL) was frozen on top of the frozen solution of 1(BArF3)2. A solution of Me3NO (7.7 mg, 0.103 mmol) in CH2Cl2 (2 mL) was frozen on top of the buffer layer. The head space was evacuated and refilled with 40 psig of H2 followed by thawing of the frozen solution. After stirring the mixture for 2 h, the solvent was removed under vacuum, and the residue was extracted into Et2O. The extract was filtered through Celite and analyzed by 19F NMR spectroscopy, which showed 60% of signals corresponded to product. A solution of this sample in 10 mL of CH2Cl2 was stirred with excess Et4NCl (142 mg, 0.857 mmol). The solvent was removed under vacuum, and the product was extracted into 10 mL of THF, which was then filtered through Celite and evaporated. The solid was dissolved in a minimal amount of THF and loaded onto a column of neutral alumina (Brockmann Level IV) packed with THF. A red band eluted with THF and was discarded. The remaining brown band was eluted off the column using CH2Cl2. The solution was concentrated to 5 mL, layered with 20 mL of pentane, and cooled to −30 °C yielding red crystals (44 mg, 28%). IR (CH2Cl2, cm−1): νCN = 2162, 2137, νCO = 1963. 1H NMR (CD2Cl2): δ −7.05 (t, JHP = 5.2 Hz). 31P{1H} NMR (CD2Cl2): δ −66.6 (s). 19F NMR (CD2Cl2): δ −134.4 (o–F), −162.5 −162.9 (t, p–F), −167.4 −167.6 (t, m–F). Anal. Calcd for C76H51B2F30FeN3NiOP2S2•0.3CH2Cl2 (found): C, 48.83 (48.81); H, 2.77 (2.43); N, 2.30 (2.24). Use of N-methylmorpholine-N-oxide in place of Me3NO favored the b/b isomer. Et4N[H3(BArF3)2] was prepared from 1(BArF3)2 and Me3NO using Et4NBH4 as the hydride source (see SI).
Et4N[(CO)(CNBArF3)2Fe(pdt)(H)Ni(dcpe)], Et4N[H4(BArF3)2]
This compound was prepared in a manner and yield similar to the preparation of Et4N[H3(BArF3)2]. IR (CH2Cl2, cm−1): νCN = 2158, 2132; νCO = 1952. 1H NMR (CD2Cl2): δ −6.56 (t, JHP = 3 Hz). 31P{1H} NMR (CD2Cl2): δ −85.1 (s). 19F NMR (CD2Cl2): δ −134.3 (d, o–F), −162.5 −163.0 (t, p–F), −167.3 −167.8 (t, m–F). Anal. Calcd for C76H75B2F30FeN3NiOP2S2 (found): C, 48.58 (48.83); H, 4.02 (4.00); N, 2.24 (2.35).
Et4N[(CO)(CNBArF3)2Fe(pdt)(Cl)Ni(dppe)], Et4N[Cl3(BArF3)2]
A solution of Et4N[H3(BArF3)2] (10 mg, 0.0054 mmol) in CD2Cl2 (0.7 mL) was treated with HCl (2M in Et2O, 6 μL, 2.2 equiv, 0.012 mmol) resulting the solution turning slightly lighter in color. 1H NMR confirmed the formation of H2. 31P NMR displayed a major species (~60%) as an AB quartet at δ–51.5 and −48.6 and 19F NMR displayed signals for two inequivalent BArF3 environments, both consistent with an apical/basal arrangement of CNBArF3− ligands. Assignment of this major species as Et4N[Cl3(BArF3)2] was confirmed by independent synthesis of Et4N[Cl3(BArF3)2] from 1(BArF3)2 and Et4NCl. Thus, a solution of 1(BArF3)2 (0.10 g, 0.057 mmol) in CH2Cl2 (10 mL) was treated with a solution of Me3NO (5.1 mg, 0.069 mmol) in CH2Cl2 (1 mL) followed by Et4NCl (9.5 mg, 0.057 mmol) in CH2Cl2 (5 mL). After the solution was allowed to stir for 15 min., solvent was removed under vacuum. The crude solid was dissolved in a minimal amount of THF and loaded onto a column of neutral alumina (Brockmann Level IV) eluting THF. The first red band was discarded, and the remaining brown band was eluted with CH2Cl2. The solution was concentrated to 5 mL, layered with 20 mL of pentane, and cooled to −30 °C precipitating an oil. Upon storing under vacuum, the oil converted to a solid (40 mg, 37%). IR (CH2Cl2, cm−1): νCN = 2181, 2149; νCO = 1996. 31P{1H} NMR (CD2Cl2): δ −51.5, −48.6 (AB quartet, JPP = 29 Hz). 19F NMR (CD2Cl2): δ −134.1, −134.6 (d, o–F); −161.9, −162.4 (t, p–F); −167.0, −167.2 (t, m–F). Anal. Calcd for C76H50B2ClF30FeN3NiOP2S2 (found): C, 48.33 (48.30); H, 2.67 (2.43); N, 2.22 (2.43).
Supplementary Material
Figure 1.
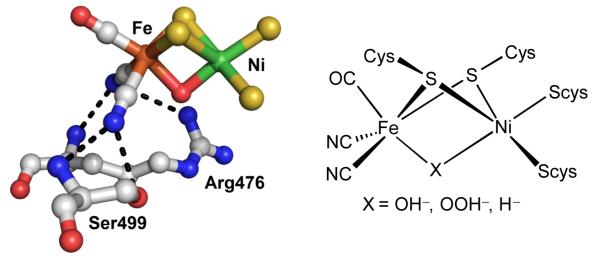
Left: Active site of [NiFe]-H2ase active site (Ni-B state) of D. fructosovorans with hydrogen-bonding interactions between Fe-CN and nearby residues (PDB 1YRQ). Right: Drawing of the active site of [NiFe]-H2ase showing various bridging ligands.
ACKNOWLEDGMENTS
This research was supported by NIH and the International Institute for Carbon Neutral Energy Research (WPI-I2CNER), sponsored by the World Premier International Research Center Initiative (WPI), MEXT, Japan. We thank Danielle Gray for assistance with the crystallography and Prof. Steven Zimmerman and Ying Li for assistance with the calculations of the binding constants.
Footnotes
Supporting Information 1H NMR, 19F NMR, 31P NMR, and IR spectra for new compounds. CIF files giving X-ray crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes The authors declare no competing financial interest.
REFERENCES
- (1).Tard C, Pickett CJ. Chem. Rev. 2009;109:2245. doi: 10.1021/cr800542q. [DOI] [PubMed] [Google Scholar]
- (2).Fontecilla-Camps JC, Amara P, Cavazza C, Nicolet Y, Volbeda A. Nature. 2009;460:814. doi: 10.1038/nature08299. [DOI] [PubMed] [Google Scholar]
- (3).Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Chem. Rev. 2007;107:4273. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]; Siegbahn PEM, Tye JW, Hall MB. Chem. Rev. 2007;107:4414. doi: 10.1021/cr050185y. [DOI] [PubMed] [Google Scholar]; Vincent KA, Parkin A, Armstrong FA. Chem. Rev. 2007;107:4366. doi: 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]
- (4).Ogata H, Lubitz W, Higuchi Y. Dalton Trans. 2009:7577. doi: 10.1039/b903840j. [DOI] [PubMed] [Google Scholar]; Ogata H, Kellers P, Lubitz W. J. Mol. Biol. 2010;402:428. doi: 10.1016/j.jmb.2010.07.041. [DOI] [PubMed] [Google Scholar]
- (5).Böck A, King PW, Blokesch M, Posewitz MC. Adv. Microb. Physiol. 2006;51:1. doi: 10.1016/s0065-2911(06)51001-x. [DOI] [PubMed] [Google Scholar]; Peters JW, Broderick JB. Annu. Rev. Biochem. 2012;81:429. doi: 10.1146/annurev-biochem-052610-094911. [DOI] [PubMed] [Google Scholar]
- (6).Volbeda A, Martin L, Cavazza C, Matho M, Faber BW, Roseboom W, Albracht SPJ, Garcin E, Rousset M, Fontecilla-Camps JC. JBIC, J. Biol. Inorg. Chem. 2005;10:239. doi: 10.1007/s00775-005-0632-x. [DOI] [PubMed] [Google Scholar]
- (7).De Lacey AL, Fernandez VM, Rousset M, Cavazza C, Hatchikian EC. J. Biol. Inorg. Chem. 2003;8:129. doi: 10.1007/s00775-002-0397-4. [DOI] [PubMed] [Google Scholar]
- (8).Ohki Y, Tatsumi K. Eur. J. Inorg. Chem. 2011;2011:973. [Google Scholar]; Gloaguen F, Rauchfuss TB. Chem. Soc. Rev. 2009;38:100. doi: 10.1039/b801796b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Barton BE, Whaley CM, Rauchfuss TB, Gray DL. J. Am. Chem. Soc. 2009;131:6942. doi: 10.1021/ja902570u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Barton BE, Rauchfuss TB. J. Am. Chem. Soc. 2010;132:14877. doi: 10.1021/ja105312p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Carroll ME, Barton BE, Gray DL, Mack AE, Rauchfuss TB. Inorg. Chem. 2011;50:9554. doi: 10.1021/ic2012759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ogo S, Ichikawa K, Kishima T, Matsumoto T, Nakai H, Kusaka K, Ohhara T. Science. 2013;339:682. doi: 10.1126/science.1231345. [DOI] [PubMed] [Google Scholar]
- (13).Armstrong FA. Science. 2013;339:658. doi: 10.1126/science.1233210. [DOI] [PubMed] [Google Scholar]
- (14).Jablonskyte A, Wright JA, Pickett CJ. Dalton Trans. 2010;39:3026. doi: 10.1039/b923191a. [DOI] [PubMed] [Google Scholar]
- (15).Jiang J, Maruani M, Solaimanzadeh J, Lo W, Koch SA, Millar M. Inorg. Chem. 2009;48:6359. doi: 10.1021/ic900929u. [DOI] [PubMed] [Google Scholar]
- (16).Li Z, Ohki Y, Tatsumi K. J. Am. Chem. Soc. 2005;127:8950. doi: 10.1021/ja051590+. [DOI] [PubMed] [Google Scholar]
- (17).Kristoff JS, Shriver DF. Inorg. Chem. 1973;12:1788. [Google Scholar]; Schelter EJ, Shatruk M, Heintz RA, Galan-Mascaros JR, Dunbar KR. Chem. Commun. 2005:1417. doi: 10.1039/b414262d. [DOI] [PubMed] [Google Scholar]
- (18).Brunkan NM, Brestensky DM, Jones WD. J. Am. Chem. Soc. 2004;126:3627. doi: 10.1021/ja037002e. [DOI] [PubMed] [Google Scholar]
- (19).Stephan DW. Dalton Trans. 2009:3129. doi: 10.1039/b819621d. [DOI] [PubMed] [Google Scholar]
- (20).Sowa JR, Jr., Zanotti V, Facchin G, Angelici RJ. J. Am. Chem. Soc. 1992;114:160. [Google Scholar]
- (21).De Lacey AL, Fernández VM, Rousset M, Cammack R. Chem. Rev. 2007;107:4304. doi: 10.1021/cr0501947. [DOI] [PubMed] [Google Scholar]
- (22).Ogata H, Hirota S, Nakahara A, Komori H, Shibata N, Kato T, Kano K, Higuchi Y. Structure. 2005;13:1635. doi: 10.1016/j.str.2005.07.018. [DOI] [PubMed] [Google Scholar]; Higuchi Y, Ogata H, Miki K, Yasuoka N, Yagi T. Structure. 1999;7:549. doi: 10.1016/s0969-2126(99)80071-9. [DOI] [PubMed] [Google Scholar]
- (23).Coetzee JF, Padmanabhan GR. J. Am. Chem. Soc. 1965;87:5005. [Google Scholar]
- (24).Kaljurand I, Kütt A, Sooväli L, Rodima T, Mäemets V, Leito I, Koppel IA. J. Org. Chem. 2005;70:1019. doi: 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- (25).Ogata H, Mizoguchi Y, Mizuno N, Miki K, Adachi S.-i., Yasuoka N, Yagi T, Yamauchi O, Hirota S, Higuchi Y. J. Am. Chem. Soc. 2002;124:11628. doi: 10.1021/ja012645k. [DOI] [PubMed] [Google Scholar]
- (26).Ohki Y, Yasumura K, Ando M, Shimokata S, Tatsumi K. Proc. Natl. Acad. Sci. U. S. A. 2010;107:3994. doi: 10.1073/pnas.0913399107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Matsumoto T, Kabe R, Nonaka K, Ando T, Yoon K-S, Nakai H, Ogo S. Inorg. Chem. 2011;50:8902. doi: 10.1021/ic200965t. [DOI] [PubMed] [Google Scholar]
- (27).Camara JM, Rauchfuss TB. Nature Chem. 2012;4:26. doi: 10.1038/nchem.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fichtner C, Laurich C, Bothe E, Lubitz W. Biochemistry. 2006;45:9706. doi: 10.1021/bi0602462. [DOI] [PubMed] [Google Scholar]
- (29).Lubitz W, Reijerse E, van Gastel M. Chem. Rev. 2007;107:4331. doi: 10.1021/cr050186q. [DOI] [PubMed] [Google Scholar]
- (30).Weber K, Krämer T, Shafaat HS, Weyhermuller T, Bill E, van Gastel M, Neese F, Lubitz W. J. Am. Chem. Soc. 2012;134:20745. doi: 10.1021/ja309563p. [DOI] [PubMed] [Google Scholar]
- (31).Raebiger JW, Miedaner A, Curtis CJ, Miller SM, Anderson OP, DuBois DL. J. Am. Chem. Soc. 2004;126:5502. doi: 10.1021/ja0395240. [DOI] [PubMed] [Google Scholar]; Rakowski DuBois M, DuBois DL. Acc. Chem. Res. 2009;42:1974. doi: 10.1021/ar900110c. [DOI] [PubMed] [Google Scholar]
- (32).Wang N, Wang M, Zhang T, Li P, Liu J, Sun L. Chem. Commun. 2008:5800. doi: 10.1039/b811352a. [DOI] [PubMed] [Google Scholar]
- (33).Carroll ME, Barton BE, Rauchfuss TB, Carroll PJ. J. Am. Chem. Soc. 2012;134:18843. doi: 10.1021/ja309216v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kayal A, Rauchfuss TB. Inorg. Chem. 2003;42:5046. doi: 10.1021/ic034455b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.