

Abstract
Substantial advances have been made in modeling protein association mechanisms and in calculating association rate constants (ka). We now have a clear understanding of the physical factors underlying the wide range of experimental ka values. Half of the association problem, where ka is limited by diffusion, is perhaps solved, and for the other half, where conformational changes become rate-limiting, a number of promising methods are being developed for ka calculations. Notably, the binding kinetics of disordered proteins are receiving growing attention, with “dock-and-coalescence” emerging as a general mechanism. Progress too has been made in the modeling of protein association kinetics under conditions mimicking the heterogeneous, crowded environments of cells, an endeavor that should ultimately lead to a better understanding of cellular functions.
Introduction
The association of proteins with small molecules and other macromolecules constitute the key steps of all cellular functions. In addition to the structures of the complexes formed and their binding affinities, association mechanisms and rate constants uniquely contribute to the characterization of cellular functions. Association processes are often under kinetic, rather than thermodynamic, control [1], e.g., when several macromolecules compete for the same binding site [2] or when a protein is faced with alternative pathways [3]. Understanding the physical principles governing association mechanisms and rate constants and furthermore, realistically modeling them, are thus of paramount importance.
In many cases, convincing arguments can be made that rapid association, not just high affinity, is essential for biological function. For example, some bacteria produce enzymes (e.g., barnase in Bacillus amyloliquefaciens and nuclease colicins in Escherichia coli; Fig. 1) that are exported to rapidly kill off competitor cells by cleaving their nucleic acids. However, these enzymes can also potentially cleave the host cell’s own nucleic acids. This ill fate is prevented by co-producing inhibitors (i.e., barstar and immunity proteins), and rapid enzyme-inhibitor association here is obviously crucial to ensure the self-defense of the host cell.
Fig. 1.
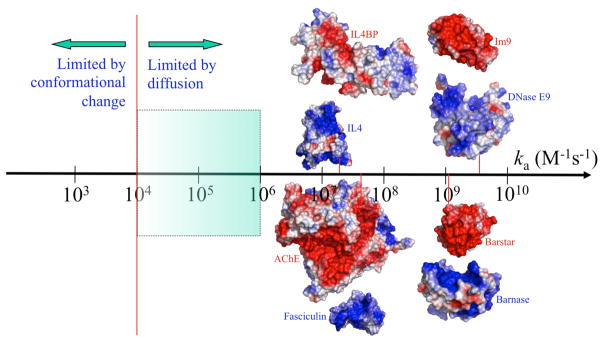
The wide spectrum of experimental values for the rate constants of protein-protein association. The conformational change-limited regime and diffusion-limited regime are generally separated around ka = 104 M−1s−1 (indicated by a vertical line). In the diffusion-limited regime, if there is no long-range force to bias the diffusion, ka usually falls between 104 and 106 M−1s−1 (shaded region). Proteins that associate with ka > 107 M−1s−1 invariably feature strongly complementary electrostatic surfaces (as shown for four complexes; blue and red represent positive and negative electrostatic potentials, respectively); ka values for the four systems measured at ionic strengths of 150, 50, 15, and 25 mM (from left to right) are indicated by short lines touching the ka axis.
Various cellular functions rely on altering association (ka) and dissociation (kd) rate constants. Translation initiation, putatively the rate-limiting and most highly regulated phase of bacterial protein synthesis, provides a good example. Crucial to translation initiation is the binding of the initiator tRNA (i.e., fMet-tRNAfMet) to the ribosome 30S subunit [2]. This binding, in the absence of initiation factors (IF1-3), has the same low ka and low kd as the binding of noncognate, elongator tRNAs [4•]. Pre-binding of IF1-3 to the 30S subunit results in increases in ka of as much as 400-fold for the initiator tRNA but by only ~10-fold for noncognate tRNAs, as well as significant, uniform increases in kd for all the tRNAs. The resulting disparity in ka is essential for the accuracy of initiator tRNA selection.
Post-translational modifications such as phosphorylation present a different mechanism for altering ka. A recent study [5] found that response levels in cellular signaling correlated with the phosphorylation-dependent binding affinities of a linker peptide on a kinase for the SH2 domain of an effector protein. However, as the phosphorylation in this case affected ka but not kd, a similar correlation can be argued if ka, instead of the association constant (i.e., Ka = ka/kd), is used. Even if Ka was the determinant for the cellular function in this study, it remains important to point out that the increases in Ka upon phosphorylation were achieved through increases in ka, as opposed to decreases in kd. The latter might not be a viable option for signaling proteins, as the complexes formed with their targets generally have to be short-lived. It has been proposed that structural disorder allows signaling proteins to bypass the requirement for maintaining high ka, without sacrificing specificity [6].
The question of whether Ka or ka is the better predictor of cellular response in signaling was specifically addressed by Kiel and Serrano [7]. They introduced c-Raf mutations that were expected to change either only Ka (by changing kd) or only ka (by compensatory changes in kd) for association with Ras. The effects of these mutations on MAP kinase signaling correlated better with predicted ka than with predicted Ka. While signaling networks may not generally be dictated by single protein-protein association steps, this study does highlight the importance of considering association kinetics, not just equilibrium, in predicting the outcome of signaling.
The association constant is determined by the end states of the binding process, namely the unbound state, in which the two subunits separately reside in the solvent environment, and the bound state, in which the subunits form specific, noncovalent interactions. In contrast, to determine the association rate constant one has to consider the whole binding process. This process involves both overall translational and rotational diffusion of the molecules, which brings the binding sites on the two molecules into proximity, and internal conformational changes, which allow the molecules to achieve their native stereospecific fit. There is a long history of deriving theoretical results for ka by formulating the diffusional and internal motions of simple molecular models [1] and in developing algorithms for computing ka through simulating the motions of realistically represented protein molecules [8–12].
The past few years have seen major progress in modeling protein association mechanisms and in calculating association rate constants. In highlighting this progress below, we will focus on association between protein molecules, but will occasionally include examples where the subunits are nucleic acids, small molecules, and multi-component complexes.
An overview on the wide spectrum of protein association rate constants
The observed ka values for protein-protein association range from ~1 to 1010 M–1s–1 (Fig. 1). It is now clear that this 10 orders of magnitude spectrum involves two different rate-limiting mechanisms [13,14]. The binding process between two proteins, A and B, can be modeled by the reaction scheme [6,13]
| (1) |
where A · B denotes an initial complex in which at least a portion of the binding site on A forms near-native contact with the cognate portion of the binding site on B, and C denotes the final, native complex. The formation of the initial complex predominantly involves the translational and rotational diffusion of the molecules, whereas the subsequent step predominantly involves the conformational changes of the molecules. The overall rate constants for association and dissociation are
| (2) |
| (3) |
Both the diffusional step and the conformational step can be rate-limiting for association. The former regime occurs if kc ≫ kD− (e.g., when structural differences are small between the bound and unbound states). Then ka ≈ kD Recent calculations for over 100 protein-protein complexes found that, in the absence of any biasing force in the diffusional step, diffusion would set an upper bound of 104 to 106 M−1s−1 for ka [15••]. Electrostatic attraction can bias the diffusional step and push up kD by 3 or more orders of magnitude. Indeed, proteins that associate with rate constant higher than 107 M−1s−1 always have highly complementary electrostatic surfaces (Fig. 1).
When large-scale structural differences exist between the bound and unbound states, the conformational step can become rate-limiting. In this regime, ka ≈(kD/ kD−) kc kD/kD− is the association constant for forming the initial complex and probably has a lower bound of 102 to 103 M−1, similar to those for low-affinity protein-protein complexes. kc could be compared to the rate of transition from a folding intermediate to the native state of a protein, which can be as low as 10−2 to 10−3 s−1 [16,17]. Therefore, a lower bound of 1 M−1s−1 is expected when association is rate-limited by conformational changes.
The diffusion-limited regime
In general, ka values higher than 104 M−1s−1 can be proposed to be diffusion-limited. Most ka calculations have been restricted to this regime [11]. Here, the initial complex can be placed very close to the native complex, and its precise location can be determined by sampling near the bound state in the 6-dimensional translational and rotational space [18]. This and other developments culminated in the transient-complex theory for protein association [13]. The transient complex specifically refers to the initial complex in the diffusion-limited regime, in which the two proteins have near-native separation and relative orientation but have yet to form most of the stereospecific native contacts. The association rate constant is predicted as
| (4) |
where ka0 is the “basal” rate constant, i.e., the rate constant at which the two proteins reach the transient complex by free diffusion, and the Boltzmann factor captures the rate enhancement by inter-protein electrostatic attraction. Applications to structurally similar proteins show that different degrees of electrostatic complementarity across the binding interface can produce orders of magnitude disparities in ka [19]. Differences in sizes and shapes of the interface can also contribute to variations in ka by modulating the basal rate constant, which is dictated by the extent of the orientational restraints between the subunits in the transient complex [15••,20••]. The orientational restraints themselves may result in preferred pathways for reaching the transient complex [21]. Pre-binding of a third protein near the interface can modify the placement of the transient complex, potentially contributing to a dramatic increase in ka [22].
Another important development is the automated implementation of the transient-complex theory, resulting in the TransComp web server (http://pipe.sc.fsu.edu/transcomp/) for ka prediction [15••]. To illustrate the level of accuracy of the server predictions a comparison to experimental results for 49 diverse protein-protein complexes is shown in Fig. 2. In an intriguing application, extensive TransComp calculations suggested that a conserved cationic surface on the kinesin motor domain enhances the association rate with, and contributes to, the directional movement on microtubules [23•].
Fig. 2.

Level of accuracy of TransComp ka predictions. (a) Comparison of predicted and experimental ka values for 49 protein-protein complexes. (b) Structures of 10 of the 49 complexes, illustrating the structural diversity. Adapted from Qin et al. [15••].
Recently machine-learning protocols have also been employed to predict kinetic parameters from atomic coordinates [24,25]. The benefit of these approaches is that they too are able to identify, from diverse training sets, which structural and energetic features are the more powerful predictors, with solvent mediated hydrogen bonding and surface complementarity identified as being particularly important for the prediction of ka [25].
Structural and dynamic differences of proteins result in a variety of association mechanisms [15••,20••]. The association of many proteins that fall in the diffusion-limited regime can be viewed as rigid docking of the subunits accompanied by fast local conformational adjustments (Fig. 3a). In other cases the docking of one subunit may require a larger scale breathing motion of the other subunit to open the binding site (Fig. 3b). As long as the breathing motion is fast on the timescale of the diffusion approach between the subunits, the overall association process is still diffusion-limited [26].
Fig. 3.

Three different mechanisms of protein association, illustrated by three proteins binding to G-actin (shown as pink surface) [20••]. (a) Profilin reaches the transient complex (not shown) with G-actin by diffusion and then quickly forms the native contacts all at once to produce the native complex. (b) Vitamin-D binding protein must undergo breathing motion to transiently widen the opening between its domains 1 and 3 to allow G-actin to enter. (c) The Wiskott-Aldrich syndrome protein has a disordered actin-regulatory region. The binding of this regulatory region to G-actin follows the dock-and-coalesce mechanism: the WH2 segment (in blue) docks to the cleft between subdomains 1 and 3 of G-actin, allowing the remaining segment (in green) to sample conformations and coalesce to its cognate subsite in the cleft between subdomains 2 and 4 of G-actin.
Modeling conformational changes during association
When conformational changes are slow on the timescale of the diffusion approach between the subunits, they become rate-limiting. In this regime, internal motions have to be explicitly modeled during the association. A few such simulation studies, on binding of small molecules to proteins, have been published over the years [27,28]. Using algorithms originally designed for calculating the association rate constants of rigid molecules [8,9] to calculate the association of flexible molecules presents the formidable challenge of having to simulate internal motions over the length of time required for achieving successful association.
Recently a new algorithm was developed [29], based on breaking the association process into two problems confined to either the outer region, where the molecules can be modeled as rigid, or the inner region, which covers the binding site but is small enough such that modeling of internal motions can be affordable. The algorithm has been tested on the binding of small molecules to proteins [Qin, Greives, & Zhou, to be published]. Another interesting approach is based on long, extensive molecular dynamics simulations, from which a Markov state model of the binding process was constructed [30•]. Application of either approach has yet to be made to study protein-protein association.
Association of disordered proteins with structured targets
Intrinsically disordered proteins (IDPs) represent an extreme form of molecular flexibility [31,32]. Their coupled folding and binding processes have increasingly been investigated experimentally and computationally [33,34••]. When bound to structured targets, they often form open structures with extended interaction surfaces [35]. It is unlikely that an IDP would form its extensive interactions with its target all at once, as the resulting severe orientational restraints would lead to extremely low ka [6,15••]. Rather, different segments of the IDP have to form contacts with their cognate subsites at different times. A likely scenario, consistent with molecular simulations [36–39], is the sequential formation of these contacts [15••,20••]. In such a “dock-and coalescence” mechanism (Fig. 3c), one particular segment of the IDP first docks to its cognate subsite on the target, thereby allowing the remaining segments to explore conformational space and coalesce around their cognate subsites.
In the simplest case, the docking step is rate-limiting for overall association. Then mutations, either on the docking segment of the IDP or on its cognate subsite, that perturb the docking step would significantly affect the overall ka, whereas those perturbing the coalescing step or even deletion of the entire coalescing segment would not. Such telltale signs have indeed been observed experimentally [40–43•]. Molecular simulations have suggested that the flexibility of IDPs helps reduce the free energy barrier for the coalescing step [39,44], potentially making the docking step rate-limiting. Importantly, for IDPs that bind to their targets with a rate-limiting docking step, the TransComp method can again be used to predict the association rate constants, by studying just the binding of the docking segments with their cognate subsites [15••]. TransComp calculations have now produced ka values that are in quantitative agreement with experimental results for a variety of IDPs, suggesting that dock-and-coalesce may be a general mechanism for IDPs binding to their structured targets, often with docking as the rate-limiting step [15••,20••,34••].
Compared to structured proteins, IDPs achieve similar levels of specificity for their targets, but with the specific interactions more widely distributed over space. Therefore during dissociation these interactions do not have to be broken all at once, leading to an increase in kd. In this way IDPs can form highly specific and yet short-lived complexes with their targets, fulfilling the twin requirements for signaling proteins [6].
Influence of cellular environments on association kinetics
Understanding cellular functions ultimately requires the modeling of protein association processes not in dilute solutions but in solutions mimicking the crowded, heterogeneous cellular environments. It is now recognized that such environments can significantly affect both thermodynamic and kinetic properties of protein association [45]. A reasonable approach to model association kinetics in a crowded solution is by accounting for the effects of the crowder molecules on the interaction energetics and the motional dynamics of the reactant molecules [46]. This approach has been validated by simulations of a simple system in which both reactant and crowder molecules are modeled as hard spheres [47].
For protein association that is rate-limited by the conformational step, the dominant contribution of crowding can be captured by its effect on the free energy of the transition state for the conformational step (relative to the unbound state). If this transition state is structurally similar to the native complex, then the effect of crowding on free energy barrier is close to that on the binding free energy. An efficient method has been developed for calculating the latter quantity [48].
For protein association in the diffusion-limited regime, crowding exerts its dominant effect on the diffusional approach of the subunits toward the transient complex. The presence of crowder molecules slows down the relative diffusion of the associating subunits but also produces an effective interaction energy between them [47]. The transient-complex theory has been modified to account for these effects and predict the association rate constant under crowding as [49•]
| (5) |
where γ is the factor by which the relative diffusion constant is slowed down by the crowders and is the crowding-induced interaction energy between the subunits in the transient complex. Hard-core repulsion from the crowder molecules can lead to apparent attraction between the subunits (i.e., ). Then and would have opposing consequences on the association rate, leaving a nearly null net effect. Therefore despite the presence of crowders the association would appear unimpeded. This prediction is confirmed by recent kinetic experiments under in vitro crowding and in living cells [50,51•].
Prospects
Over recent years major progress has been made in modeling protein association mechanisms and in calculating association rate constants. For half of the association problem, where ka is higher than 104 M−1s−1 and limited by diffusion, the determinants for ka are now well understood and the calculations of ka are quite accurate. For the other half, where conformational changes become rate-limiting, promising methods are being developed and it will be interesting to see how practical and predictive when these are ready for application to study protein-protein association.
Quantitative modeling of protein association kinetics can provide answers to open questions concerning essential cellular functions. For example, how do initiation factors achieve the substantial rate increase for the binding of the initiator tRNA to the ribosome 30S subunit? Interference with this rate increase may potentially be a new mechanism of antibiotic action. More broadly, modeling of association mechanisms may uncover intermediates along kinetic pathways, which could form a new class of targets for drug design [52].
An area where the progress in ka calculations reported here could have immediate impact is the modeling of signaling networks. One of the more popular approaches to modeling such networks dynamically is to construct a set of rate equations to monitor key cellular events over time [53]; for example, the nucleocytoplasmic shuttling of proteins associated with the TGF-β pathway [54], and the modulation of disease-associated pathways due to protein missense mutations [7,55]. While some of the kinetic parameters for such equations have been or can be experimentally determined, others may need to be predicted. It is here that the accurate prediction of association rate constants from bound or even unbound protein structures can be extremely valuable.
Highlights.
Protein association rate constants are essential for cellular functions.
Half of the problem of modeling association kinetics has a solution.
Binding of disordered proteins appears to follow a dock-and-coalesce mechanism.
Crowded cellular environments influence protein association and cellular functions.
Acknowledgments
This work was supported in part by Grants GM58187 and GM88187 from the National Institutes of Health (to HXZ) and funding from Cancer Research UK (to PAB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhou HX. Rate theories for biologists. Q Rev Biophys. 2010;43:219–293. doi: 10.1017/S0033583510000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Milon P, Rodnina MV. Kinetic control of translation initiation in bacteria. Crit Rev Biochem Mol Biol. 2012;47:334–348. doi: 10.3109/10409238.2012.678284. [DOI] [PubMed] [Google Scholar]
- 3.Zhao H, Beckett D. Kinetic partitioning between alternative protein-protein interactions controls a transcriptional switch. J Mol Biol. 2008;380:223–236. doi: 10.1016/j.jmb.2008.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4•.Antoun A, Pavlov MY, Lovmar M, Ehrenberg M. How initiation factors maximize the accuracy of tRNA selection in initiation of bacterial protein synthesis. Mol Cell. 2006;23:183–193. doi: 10.1016/j.molcel.2006.05.030. experimental study demonstrating that initiation factors accelerate the binding of the initiation tRNA to the ribosome 30S subunit, and this acceleration is essential for the accuracy of translation. [DOI] [PubMed] [Google Scholar]
- 5.Chen CH, Martin VA, Gorenstein NM, Geahlen RL, Post CB. Two closely spaced tyrosines regulate NFAT signaling in B cells via Syk association with Vav. Mol Cell Biol. 2011;31:2984–2996. doi: 10.1128/MCB.05043-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou HX. Intrinsic disorder: signaling via highly specific but short-lived association. Trends Biochem Sci. 2012;37:43–48. doi: 10.1016/j.tibs.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiel C, Serrano L. Cell type-specific importance of ras-c-raf complex association rate constants for MAPK signaling. Sci Signal. 2009;2:ra38. doi: 10.1126/scisignal.2000397. [DOI] [PubMed] [Google Scholar]
- 8.Northrup SH, Allison SA, Mccammon JA. Brownian dynamics simulation of diffusion-influenced bimolecular reactions. J Chem Phys. 1984;80:1517–1526. [Google Scholar]
- 9.Zhou HX. Kinetics of diffusion-influenced reactions studied by Brownian dynamics. J Phys Chem. 1990;94:8794–8800. [Google Scholar]
- 10.Northrup SH, Erickson HP. Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc Natl Acad Sci U S A. 1992;89:3338–3342. doi: 10.1073/pnas.89.8.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabdoulline RR, Wade RC. Biomolecular diffusional association. Curr Opin Struct Biol. 2002;12:204–213. doi: 10.1016/s0959-440x(02)00311-1. [DOI] [PubMed] [Google Scholar]
- 12.Frembgen-Kesner T, Elcock AH. Absolute protein-protein association rate constants from flexible, coarse-grained Brownian dynamics simulations: the role of intermolecular hydrodynamic interactions in barnase-barstar association. Biophys J. 2010;99:L75–77. doi: 10.1016/j.bpj.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alsallaq R, Zhou HX. Electrostatic rate enhancement and transient complex of protein-protein association. Proteins. 2008;71:320–335. doi: 10.1002/prot.21679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schreiber G, Haran G, Zhou HX. Fundamental aspects of protein-protein association kinetics. Chem Rev. 2009;109:839–860. doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15••.Qin S, Pang X, Zhou HX. Automated prediction of protein association rate constants. Structure. 2011;19:1744–1751. doi: 10.1016/j.str.2011.10.015. presenting an automated implementation of the TransComp method for ka calculations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldberg ME, Semisotnov GV, Friguet B, Kuwajima K, Ptitsyn OB, Sugai S. An early immunoreactive folding intermediate of the tryptophan synthease beta 2 subunit is a ‘molten globule’. FEBS Lett. 1990;263:51–56. doi: 10.1016/0014-5793(90)80703-l. [DOI] [PubMed] [Google Scholar]
- 17.Finke JM, Jennings PA. Interleukin-1 beta folding between pH 5 and 7: experimental evidence for three-state folding behavior and robust transition state positions late in folding. Biochemistry. 2002;41:15056–15067. doi: 10.1021/bi026197k. [DOI] [PubMed] [Google Scholar]
- 18.Alsallaq R, Zhou HX. Energy landscape and transition state of protein-protein association. Biophys J. 2007;92:1486–1502. doi: 10.1529/biophysj.106.096024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pang X, Qin S, Zhou HX. Rationalizing 5000-fold differences in receptor-binding rate constants of four cytokines. Biophys J. 2011;101:1175–1183. doi: 10.1016/j.bpj.2011.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20••.Pang X, Zhou KH, Qin S, Zhou HX. Prediction and dissection of widely-varying association rate constants of actin-binding proteins. PLoS Comput Biol. 2012;8:e1002696. doi: 10.1371/journal.pcbi.1002696. illustrating three different mechanisms of protein association. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mirzaei H, Beglov D, Paschalidis IC, Vajda S, Vakili P, Kozakov D. Rigid body energy minimization on manifolds for molecular docking. J Chem Theory Comput. 2012;8:4374–4380. doi: 10.1021/ct300272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qin SB, Zhou HX. Dissection of the high rate constant for the binding of a ribotoxin to the ribosome. Proc Natl Acad Sci U S A. 2009;106:6974–6979. doi: 10.1073/pnas.0900291106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23•.Grant BJ, Gheorghe DM, Zheng W, Alonso M, Huber G, Dlugosz M, McCammon JA, Cross RA. Electrostatically biased binding of kinesin to microtubules. PLoS Biol. 2011;9:e1001207. doi: 10.1371/journal.pbio.1001207. role of conserved cationic surface on kinesin in directional movement on microtubulin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bai H, Yang K, Yu D, Zhang C, Chen F, Lai L. Predicting kinetic constants of protein-protein interactions based on structural properties. Proteins. 2011;79:720–734. doi: 10.1002/prot.22904. [DOI] [PubMed] [Google Scholar]
- 25.Moal IH, Bates PA. Kinetic rate constant prediction supports the conformational selection mechanism of protein binding. PLoS Comput Biol. 2012;8:e1002351. doi: 10.1371/journal.pcbi.1002351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou HX, Wlodek ST, McCammon JA. Conformation gating as a mechanism for enzyme specificity. Proc Natl Acad Sci U S A. 1998;95:9280–9283. doi: 10.1073/pnas.95.16.9280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wade RC, Luty BA, Demchuk E, Madura JD, Davis ME, Briggs JM, McCammon JA. Simulation of enzyme-substrate encounter with gated active sites. Nat Struct Biol. 1994;1:65–69. doi: 10.1038/nsb0194-65. [DOI] [PubMed] [Google Scholar]
- 28.Kang M, Roberts C, Cheng YH, Chang CEA. Gating and intermolecular interactions in ligand-protein association: coarse-grained modeling of HIV-1 protease. J Chem Theory Comput. 2011;7:3438–3446. doi: 10.1021/ct2004885. [DOI] [PubMed] [Google Scholar]
- 29.Greives N, Zhou HX. BDflex: a method for efficient treatment of molecular flexibility in calculating protein-ligand binding rate constants from brownian dynamics simulations. J Chem Phys. 2012;137:135105. doi: 10.1063/1.4756913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30•.Buch I, Giorgino T, De Fabritiis G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc Natl Acad Sci U S A. 2011;108:10184–10189. doi: 10.1073/pnas.1103547108. method for treating protein flexibility in modeling ligand-binding kinetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 32.Janin J, Sternberg MJ. Protein flexibility, not disorder, is intrinsic to molecular recognition. F1000 Biol Rep. 2013;5:2. doi: 10.3410/B5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34••.Zhou HX, Pang XD, Lu C. Rate constants and mechanisms of intrinsically disordered proteins binding to structured targets. Phys Chem Chem Phys. 2012;14:10466–10476. doi: 10.1039/c2cp41196b. suggesting dock-and-coalesce as a general mechanism for the binding of intrinsically disordered proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gunasekaran K, Tsai CJ, Kumar S, Zanuy D, Nussinov R. Extended disordered proteins: targeting function with less scaffold. Trends Biochem Sci. 2003;28:81–85. doi: 10.1016/S0968-0004(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 36.Ahmad M, Gu W, Helms V. Mechanism of fast peptide recognition by SH3 domains. Angew Chem Int Ed Engl. 2008;47:7626–7630. doi: 10.1002/anie.200801856. [DOI] [PubMed] [Google Scholar]
- 37.Turjanski AG, Gutkind JS, Best RB, Hummer G. Binding-induced folding of a natively unstructured transcription factor. PLoS Comput Biol. 2008;4:e1000060. doi: 10.1371/journal.pcbi.1000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J. Intrinsically disordered p53 extreme C-terminus binds to S100B(betabeta) through “fly-casting”. J Am Chem Soc. 2009;131:2088–2089. doi: 10.1021/ja809547p. [DOI] [PubMed] [Google Scholar]
- 39.De Sancho D, Best RB. Modulation of an IDP binding mechanism and rates by helix propensity and non-native interactions: association of HIF1alpha with CBP. Mol Biosyst. 2012;8:256–267. doi: 10.1039/c1mb05252g. [DOI] [PubMed] [Google Scholar]
- 40.Stone SR, Dennis S, Hofsteenge J. Quantitative evaluation of the contribution of ionic interactions to the formation of the thrombin-hirudin complex. Biochemistry. 1989;28:6857–6863. doi: 10.1021/bi00443a012. [DOI] [PubMed] [Google Scholar]
- 41.Betz A, Hofsteenge J, Stone SR. Interaction of the N-terminal region of hirudin with the active-site cleft of thrombin. Biochemistry. 1992;31:4557–4562. doi: 10.1021/bi00134a004. [DOI] [PubMed] [Google Scholar]
- 42.Myles T, Le Bonniec BF, Betz A, Stone SR. Electrostatic steering and ionic tethering in the formation of thrombin-hirudin complexes: the role of the thrombin anion-binding exosite-I. Biochemistry. 2001;40:4972–4979. doi: 10.1021/bi0023549. [DOI] [PubMed] [Google Scholar]
- 43•.Lacy ER, Filippov I, Lewis WS, Otieno S, Xiao L, Weiss S, Hengst L, Kriwacki RW. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat Struct Mol Biol. 2004;11:358–364. doi: 10.1038/nsmb746. experimental study demonstrating a sequential binding mechanism for an intrinsically disordered protein. [DOI] [PubMed] [Google Scholar]
- 44.Huang Y, Liu Z. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the “fly-casting” mechanism. J Mol Biol. 2009;393:1143–1159. doi: 10.1016/j.jmb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 45.Zhou HX, Rivas G, Minton AP. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou HX, Qin S. Simulation and modeling of crowding effects on the thermodynamic and kinetic properties of proteins with atomic details. Biophys Rev. 2013 doi: 10.1007/s12551-013-0101-7. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou HX, Szabo A. Comparison between Molecular Dynamics Simulations and the Smoluchowski Theory of Reactions in a Hard Sphere Liquid. J Chem Phys. 1991;95:5948–5952. [Google Scholar]
- 48.Qin S, Zhou HX. Atomistic modeling of macromolecular crowding predicts modest increases in protein folding and binding stability. Biophys J. 2009;97:12–19. doi: 10.1016/j.bpj.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49•.Qin S, Cai L, Zhou HX. A method for computing association rate constants of atomistically represented proteins under macromolecular crowding. Phys Biol. 2012;9:066008. doi: 10.1088/1478-3975/9/6/066008. a method for calculating ka in crowded cellular environments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phillip Y, Harel M, Khait R, Qin S, Zhou HX, Schreiber G. Contrasting factors on the kinetic path to protein complex formation diminish the effects of crowding agents. Biophys J. 2012;103:1011–1019. doi: 10.1016/j.bpj.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51•.Phillip Y, Kiss V, Schreiber G. Protein-binding dynamics imaged in a living cell. Proc Natl Acad Sci U S A. 2012;109:1461–1466. doi: 10.1073/pnas.1112171109. ka measurement in living cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tzeng H-S, Kalodimos CG. Allosteric inhibition through suppression of transient conformational states. Nat Chem Biol. 2013 doi: 10.1038/nchembio.1250. in press. [DOI] [PubMed] [Google Scholar]
- 53.Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol. 2003;15:221–231. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- 54.Schmierer B, Tournier AL, Bates PA, Hill CS. Mathematical modeling identifies Smad nucleocytoplasmic shuttling as a dynamic signal-interpreting system. Proc Natl Acad Sci U S A. 2008;105:6608–6613. doi: 10.1073/pnas.0710134105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng TM, Goehring L, Jeffery L, Lu YE, Hayles J, Novak B, Bates PA. A structural systems biology approach for quantifying the systemic consequences of missense mutations in proteins. PLoS Comput Biol. 2012;8:e1002738. doi: 10.1371/journal.pcbi.1002738. [DOI] [PMC free article] [PubMed] [Google Scholar]