

Abstract
Background
It is during embryogenesis that the plant body plan is established and the meristems responsible for all post-embryonic growth are specified. The molecular mechanisms governing conifer embryogenesis are still largely unknown. Their elucidation may contribute valuable information to clarify if the distinct features of embryo development in angiosperms and gymnosperms result from differential gene regulation. To address this issue, we have performed the first transcriptomic analysis of zygotic embryo development in a conifer species (Pinus pinaster) focusing our study in particular on regulatory genes playing important roles during plant embryo development, namely epigenetic regulators and transcription factors.
Results
Microarray analysis of P. pinaster zygotic embryogenesis was performed at five periods of embryo development from early developing to mature embryos. Our results show that most changes in transcript levels occurred in the first and the last embryo stage-to-stage transitions, namely early to pre-cotyledonary embryo and cotyledonary to mature embryo. An analysis of functional categories for genes that were differentially expressed through embryogenesis highlighted several epigenetic regulation mechanisms. While putative orthologs of transcripts associated with mechanisms that target transposable elements and repetitive sequences were strongly expressed in early embryogenesis, PRC2-mediated repression of genes seemed more relevant during late embryogenesis. On the other hand, functions related to sRNA pathways appeared differentially regulated across all stages of embryo development with a prevalence of miRNA functions in mid to late embryogenesis. Identification of putative transcription factor genes differentially regulated between consecutive embryo stages was strongly suggestive of the relevance of auxin responses and regulation of auxin carriers during early embryogenesis. Such responses could be involved in establishing embryo patterning. Later in development, transcripts with homology to genes acting on modulation of auxin flow and determination of adaxial-abaxial polarity were up-regulated, as were putative orthologs of genes required for meristem formation and function as well as establishment of organ boundaries. Comparative analysis with A. thaliana embryogenesis also highlighted genes involved in auxin-mediated responses, as well as epigenetic regulation, indicating highly correlated transcript profiles between the two species.
Conclusions
This is the first report of a time-course transcriptomic analysis of zygotic embryogenesis in a conifer. Taken together our results show that epigenetic regulation and transcriptional control related to auxin transport and response are critical during early to mid stages of pine embryogenesis and that important events during embryogenesis seem to be coordinated by putative orthologs of major developmental regulators in angiosperms.
Keywords: Conifer embryogenesis, Epigenetics, Gymnosperm, Transcriptomics, Transcription factor
Background
Embryogenesis is a crucial period in the life cycle of most plant species. Molecular aspects of reproductive biology and embryo development have been widely studied in model angiosperms, which diverged from the gymnosperms more than 300 million years ago to follow a distinct evolutionary pathway within the Spermatophyta [1]. As such, striking differences are visible during reproduction and embryogenesis, of which the double fertilization in angiosperms versus single fertilization in gymnosperms is a major example. Important differences observed during conifer embryo development also include: (1) nuclear duplication without cytokinesis during proembryogeny instead of the initial asymmetric cell division commonly observed in the zygote of angiosperms; (2) the frequent occurrence of polyembryony; (3) the differentiation of tube cells during early embryo development; and (4) the formation of multiple cotyledons during late embryo development [2]. These differences imply that differences in the molecular regulation of embryo development must exist between the two groups of plants. Pinus represents the largest genus within the coniferous family Pinaceae, and among the gymnosperms is also the most widespread genus of trees in the northern hemisphere. A substantial amount of information on the repertoire of transcribed genes in several pine species is available in a variety of databases [3,4]. However, most of the transcriptomics studies using this information to date have focused on stress resistance/tolerance and wood development [5-8]. The most comprehensive study of transcript profiling in Pinus embryos has been conducted in P. taeda, where approximately 68,700 ESTs have been generated from somatic and zygotic embryos [9]. The authors suggested that differences between the embryo developmental pathways in angiosperms and gymnosperms are primarily the result of differential control of spatial and temporal gene expression along with the expression of unique proteins. However, the expression dynamics of genes transcribed at different stages of embryo development was not studied. In A. thaliana, Spencer et al. [10] concluded that in terms of overall transcriptional profiles of several embryo stages, temporal expression differences were more significant than spatial differences. Very recently, differential gene expression during somatic embryogenesis of Norway spruce (P. Abies) has been probed using a microarray of P. taeda sequences, which allowed identification of molecular events regulating putative processes associated with pattern formation and differentiation [11]. Most studies in conifers up to now have relied on the use of somatic embryos but, while somatic embryogenesis is a useful experimental model system for studying embryology in conifers, it is also recognized that the conditions provided during in vitro culture, such as the provision of synthetic auxins, can have an impact on transcript profiles [12]. Most developmental responses to auxin appear to be mediated through changes in gene expression and external application of auxin cause profound effects in plant growth and development [13]. Moreover, abnormal morphology has been reported for somatic embryos of P. pinaster, which are routinely induced on auxin-containing medium [14]. Although zygotic embryogenesis is the model against which somatic embryogenesis is typically compared, zygotic embryo development has rarely been studied because the isolation of zygotic embryos from immature conifer seeds is technically challenging, especially at early stages of embryo development [15].
In the present study, we have characterized the transcriptome of P. pinaster zygotic embryos isolated at different developmental stages, from early embryogenesis to embryo maturation, using a loblolly pine cDNA microarray containing approximately 25,000 unique cDNAs [7], with the aim of identifying transcripts and biological processes associated with specific developmental stages, and emphasizing early embryo development. To our knowledge, this is the first genome-wide study of transcript profiles across zygotic embryogenesis in pines. Our approach uncovered major regulatory genes with putative roles in epigenetic and transcriptional control of key developmental processes. Comparative transcriptomic analyses against an A. thaliana embryogenesis model [16] further highlighted these regulatory functions.
Results
Microarray analysis of the Pinus pinaster developing embryo transcriptome
Transcript level dynamics during P. pinaster zygotic embryogenesis were analyzed using the PtGen2 cDNA microarray (GPL11184) for hybridization of samples representing five sequential periods of embryo development. Based on previous studies in which maritime pine embryo development was monitored [15,17], we isolated dominant zygotic embryos at five time points representing consecutive stages of embryo development grouped as early, pre-cotyledonary, early cotyledonary, cotyledonary and mature embryos (Figure 1). The PtGen2 microarray contains 25 848 (26 496 total features minus buffer blanks and duplicate spots) amplimers of cDNA clones derived from thirty-six cDNA libraries constructed exclusively from loblolly pine (P. taeda) root and needle tissue; no embryonic tissue was utilized in its construction [7,18]. The use of this array for cross-species hybridization with target samples from diverse Pinus species, including P. pinaster, has been previously demonstrated [7,18]. In fact, loblolly pine cDNA arrays have been successfully used also for gene expression analysis in other conifer genera [11,19]. In total, 30 microarray slides were used in our study (Figure 1A). Box plots of the expression values pre- and post-normalization confirmed that the data were successfully normalized (Additional file 1). The quality of the microarray datasets was demonstrated by verifying reproducibility among replicates by hierarchical cluster analysis using Pearson correlation and average linkage (Figure 1B). Samples harvested at the same time point clustered together and separated from samples of other time periods, with the exception of Day15. Technical replicates always clustered together and their correlation values ranged from 0.7 to 0.85. Overall, a close relationship between technical replicates and among samples harvested at the same time point (biological replicates) was observed, whereas samples from distant time points showed greater variability.
Figure 1.

Microarray hybridization. (A) Staging system (T0 to T7) used for Pinus pinaster zygotic embryo development [15], showing how samples were divided into five developmental groups/time-points representing early embryogenesis (T0 to T2), pre- cotyledonary (T3 and T4), early cotyledonary (T4B), late embryogenesis (T5) and mature embryo (T7). Bar: T0 and T1 = 300 μm; T2, T3 and T4 = 400 μm; T4B = 800 μm; T5, T6 and T5 = 0.1 cm. Three biological replicates of each sample harvested on Day 0 to Day 25 and two technical replicates were used for hybridization with the reference sample, which consisted of a pool containing equal amounts of total RNA from all five time points. (B) Cluster analysis of the thirty replicates generated using MeV with Pearson correlation and average linkage.
In order to improve subsequent annotation, P. taeda ESTs corresponding to the 3′ and 5′ ends of cDNAs spotted on the PtGen2 array were used in a BLASTX search against the SustainPineDB, which contains non-duplicated set of transcripts for P. pinaster (Unigenes). A total of 10 922 spots were aligned to the same unigene using either the 3′ or the 5′ end sequences. Another 6911 spots for which only a single end sequence was available (the 3′end in most of cases) aligned with a single unigene. There were 3105 spots that aligned to different unigenes depending on whether the 3′ or 5′ end sequence was used, and these were consequently associated with 6210 unigenes. In all cases, duplicated spots aligned to the same unigene, and in a few cases different clones aligned to the same unigene. In total, 20 938 spots were aligned with 14 996 different unigenes from SustainPineDB (Additional file 2). By contrast, no significant alignment was found in SustainPineDB for 5294 of the microarray spots. In such cases, the P. taeda 3′ EST sequence, when available, was used during annotation. Alternatively, the 5′ EST sequence was used. The 14 996 recovered unigenes plus the 5294 P. taeda EST end sequences were annotated by comparison against the NCBI protein database (nr) using BLASTX (E-value < 1e-10). Orthologs were found for 13 280 unigenes and 3482 cDNA clones (Additional file 3). Identity (matches/alignment length) distribution peak was at 75%. Of the top hits, 47.4% corresponded to Picea sitchensis, 10.3% to Vitis vinifera and 3.4% to Ricinus communis. The remaining hits did not correspond to more than 2.5% for any one species (Additional file 4). Gene Ontology terms (http://www.geneontology.org/) [20] were subsequently associated with 12 659 unigenes and EST sequences using Blast2GO (Additional file 3). The annotated GO terms ranked from level 2 to 11, and were concentrated around level 7. Most of the sequences that could not be annotated were shorter than 1 kb (Additional file 4). We also used BLASTX (E-value < 1e-10) to look at A. thaliana orthologous proteins corresponding to each of the P. pinaster unigenes. A total of 13 265 unigenes aligned with 7732 A. thaliana proteins, thus providing Plant Ontology, pathway and gene family annotations based on TAIR mappings (http://www.arabidopsis.org) (Additional file 3).
Time-course analysis of gene functional categories during embryo development
To get an overview of the processes and functions significantly associated with different stages of zygotic embryo development we first performed a functional assessment of expressed transcripts using the regression model in maSigFun for the analysis of time-course microarray experiments. In this analysis, functional categories whose genes significantly changed transcript levels with the same pattern over time indicate a high level of co-expression within them and a relationship to the embryogenic process [21]. Differential expression (FDR < 0.01) was noted in 103 functional categories that clustered into nine profiles (Figure 2). Some profiles showed similar trends in expression at the same developmental stages. For example, profiles 1, 6 and 7 were up-regulated during early embryogenesis, while profiles 3, 4 and 5 were down-regulated during the same period. However, the profiles all showed different fold-changes that distinguished them from each other at these stages. The genes assigned to each category based on their ontological annotations are described in Additional file 3. The number of categories within each profile ranged from 2 to 29. In addition to GO biological processes, functions and cell components, the identified functional categories included 18 EC numbers, six pathways, one gene family (cytoskeleton in profile 1), and seven plant ontology terms. However, it should be noted that EC numbers were redundant to GO terms in all cases.
Figure 2.

Clustering of functional categories that showed similar expression profiles during P. pinaster zygotic embryogenesis. Functional categories include gene ontologies, plant ontologies, enzyme codes, pathways, families and structures annotations. The expression of a category is represented by the median expression values of the genes annotated within that category. Dots indicate the distribution (mean and quartiles) of the median values for the categories included in each profile/cluster. Functional categories are listed in the inset.
Amino acid transport and metabolism, as well as nucleotide metabolism, were among the most prevalent functional categories up-regulated during early embryogenesis, but showed a gradual decrease at subsequent stages of development (profiles 1 and 6). Enzymes at important branch points between carbon and nitrogen metabolism, such as glutamate dehydrogenase, or involved in the biosynthesis of central metabolites for carbon and energy metabolism, such as acetyl coenzyme A, or in glutathione metabolism, such as glutathione thiolesterase, were also prevalent in these transcript profiles. Functions related to GDP- mannose metabolism, particularly GDP-mannose 4,6-dehydratase activity, which is associated with GDP-L-fucose biosynthesis, were also up-regulated during early embryogenesis. Cytoskeleton gene family members were up-regulated in early embryogenesis, as were pathways for the biosynthesis of cell wall components, such as dTDP/UDP-L-rhamnose (Figure 2).
Profile 9, in which functional categories are highly expressed during early to mid- embryogenesis, but drastically down-regulated towards the mature embryo (Day25) was of particular interest. All co-expressed categories identified in this profile appeared to be associated with mechanisms of epigenetic regulation, including maintenance of chromatin silencing, regulation of histone acetylation or methylation, and regulation of DNA methylation. In contrast, processes mediated by small RNAs appeared prevalent as a subset of co-expressed categories in profile 3, with an expression trend that increased towards late embryogenesis. Included in this profile were miRNA metabolic process, siRNA and miRNA binding, and gene silencing by miRNA. A high number of co-expressed categories followed a similar trend, including functions related to purine catabolic recycling activities involving xanthine dehydrogenase and xanthine oxidase enzymes, and the guanosine nucleotide degradation pathway. DNA replication and repair processes, which are indicative of a high DNA replication rate, were also identified in this profile together with biosynthesis of structural components, such as phytyl diphosphate. In profile 2, which differs from profile 3 by its sustained increase in expression through the mature embryo stage, processes related to fatty acid metabolism were clearly over-represented, but other mechanisms, such as those related to polarized growth, chromatin organization and fine regulation of cytokinin, were also present.
Transcriptional profile analysis during embryo development
For the analysis of individual genes, differentially transcribed sequences were extracted using maSigPro [22]. The method first adjusts a global regression model to identify sequences that are differentially transcribed with respect to time, after which a variable selection strategy is applied to study differences between groups and find significantly different profiles. A total of 3081 spots were differentially transcribed during embryo development in P. pinaster (FDR < 0.001; Additional file 5). Of these, 384 spots were associated to two unigenes, 2210 spots were associated to one unigene, and 487 spots could not be associated to any unigene in SustainpineDB and the EST was used instead. However, some spots mapped to the same unigene and the number of unique unigenes was 2633, therefore the total number of differentially transcribed sequences considered in our analyses was 3120 (2633 unigenes and 487 ESTs). Orthologs were found in the NCBI nr database for 2814 of these sequences, while 2161 had orthologs in the A. thaliana TAIR10 database. We were able to associate GO terms to 2485 of these sequences (Additional file 5).
Based on their transcript levels across the five developmental time points, the 3081 spots could be grouped in 6 clusters, formed by 796, 631, 812, 555, 125 and 162 spots, respectively. The spots mapped to 837, 646, 846, 594, 134 and 170 unigenes and ESTs, respectively, to sum up 3227 differentially transcribed sequences (Figure 3). Some of the unigenes that mapped to multiple spots fell into more than one cluster. The six transcript profiles could be further grouped by pattern into up (clusters 1 and 3), down (clusters 2 and 4), up-down-up (cluster 5), and down-up-down (cluster 6) clusters describing changes from early embryogenesis to the mature embryo. We found that 1683 (52.2%) transcripts were up-regulated while 1240 (38.4%) were down-regulated across the embryo development time course. The remaining 170 (5.3%) and 134 (4.2%) transcripts were either up- or down-regulated, respectively, during the intermediate time points (clusters 5 and 6), which correspond to the pre-and early-cotyledonary stages of development.
Figure 3.

Clustering of differentially expressed genes according to their expression profiles during P. pinaster zygotic embryogenesis, and representative gene ontology terms in each cluster. Transcripts having similar expression profiles, which were differentially expressed in time according to maSigPro analysis, were clustered together, and a representative median expression profile was inferred from the expression of all the genes in each cluster. For each cluster, the number of transcripts in each gene ontology term is indicated in a bar graph.
Changes in transcript profiles during developmental transitions that included the most extreme time points, Day0 → Day5 and to Day15 → Day25, were more evident and involved a larger number of genes than changes at the intermediate time points, especially Day11 → Day15. This is not particularly surprising considering the shorter time-window between Day11 and Day15, as well as the close developmental proximity between early-cotyledonary and cotyledonary embryos, which differ mainly regarding the enlargement of pre-formed organs.
We evaluated the biological process GO term distribution in each cluster (Figure 3). Terms were joined in a superior level when the number of transcripts was smaller than 20. The analysis of GO terms showed that the metabolic process oxidation-reduction is over- represented in most clusters, followed by response to stress. The exceptions were clusters 5 and 6, which included relatively few annotated genes. However, certain GO terms were identified only in specific clusters, suggesting their association with defined periods of embryo development. For example, anatomical structure development (which included organ and shoot morphogenesis, and photomorphogenesis) and multicellular organismal development (includes embryo development) were both associated with early embryogenesis (cluster 4). Post-embryonic development, developmental process involved in reproduction, and reproductive structure development (which included seed, fruit, embryo and flower development) were constrained to clusters 1 and 3, which represented transcripts accumulating mostly from Day11-15 up to the mature embryo. Organ development transcripts (which included post-embryonic organ, shoot and root development, and leaf senescence) were also present in clusters 1 and 3, but also cluster 6. Although the more generic processes cellular response to stimulus, response to chemical, and stress or abiotic stimulus were present in all clusters, response to hormone stimulus was associated only with mid- and late embryogenesis stages (clusters 1 and 6), while response to inorganic substance was exclusive to late embryogenesis (cluster 1). Eighty-three (83) differentially expressed transcripts were only found in gymnosperms (annotated in Additional file 5). These corresponded with 53 unigenes and 26 ESTs, as several transcripts were associated with more than one unigene. On average, about 3% of the sequences in each cluster corresponded to gymnosperm-specific sequences, and they were equally distributed between early and late embryogenic stages. Few of these sequences showed similarity to NCBI accessions.
Differential transcript profiles associated to epigenetic regulation
We focused our analysis of differentially transcribed genes on the identification of putative master regulators that might drive expression of the embryo transcriptome, with an emphasis on epigenetic regulators and transcription factors that could potentially impact development. A list of 24 selected genes whose annotations suggest involvement in epigenetic regulation is presented in Table 1. Several associated with chromatin remodelling, including histone post-translational modifications, DNA methylation, and regulation of small RNA biogenesis and processing, were up-regulated at different stages of embryo development. However, 71% of the transcripts in Table 1 were found in clusters 1 and 4, which display opposite patterns of transcription during early and late embryo development.
Table 1.
Differentially transcribed genes implicated in epigenetic regulation
| Cluster | Gene ID | Pp unigene | At Locus | E-value | Annotation |
|---|---|---|---|---|---|
| 1 |
6.2.9.22 |
732 |
AT1G08460 |
2E-141 |
Histone deacetylase 8 (HDA8) |
| 1 |
3.1.21.21 |
1009 |
AT3G44680 |
0 |
Histone deacetylase 9 (HDA9) |
| 1 |
9.3.15.18 |
25311 |
AT1G09700 |
2E-43 |
dsRNA-binding protein 1 (DRB1), HYPONASTIC LEAVES 1 (HYL1) |
| 1 |
2.2.5.12 |
38 |
AT1G48410 |
0 |
ARGONAUTE 1 (AGO1) |
| 1 |
9.4.14.10 |
9118 |
AT1G48410 |
0 |
ARGONAUTE 1 (AGO1) |
| 1 |
1.4.19.11 |
5048 |
AT5G21150 |
3E-109 |
ARGONAUTE 9 (AGO9) |
| 1 |
7.2.18.7 |
3877 |
AT2G23380 |
2E-151 |
CURLY LEAF (CLF), INCURVATA 1 (ICU1), SDG1, SET1 |
| 1 |
1.2.3.11 |
401 |
AT5G22750 |
2E-155 |
DNA/RNA helicase protein RAD5 |
| 1 |
2.2.17.8 |
62665 |
AT3G20550 |
6E-84 |
SMAD/FHA domain-containing protein DAWDLE, DDL |
| 1 |
9.3.17.23 |
15368 |
AT5G14170 |
1E-164 |
SWIB/MDM2 domain superfamily protein CHC1 |
| 1 |
4.1.9.18 |
60482 |
AT5G14170 |
1E-164 |
SWIB/MDM2 domain superfamily protein CHC1 |
| 2 |
10.2.21.21 |
28419 |
AT1G01040 |
0 |
ABNORMAL SUSPENSOR 1 (ASU1), CARPEL FACTORY (CAF), DCL1, |
| |
|
|
|
|
DICER-LIKE1 (DCL1), EMBRYO DEFECTIVE 60 / 76 (EMB60 / 76), |
| |
|
|
|
|
SHORT INTEGUMENTS 1 (SIN1), SUSPENSOR 1 (SUS1) |
| 3 |
10.2.13.22 |
1158 |
AT1G77300 |
5E-27 |
H3-K4 specific histone methyltransferases, ASH1 HOMOLOG 2 (ASHH2) |
| 3 |
4.4.11.7 |
441, 7804 |
AT4G38040 |
7E-150 |
Exostosin family protein |
| 3 |
8.4.10.21 |
7514 |
AT3G17590 |
3E-75 |
Transcription regulatory protein SNF5 homologue, BUSHY GROWTH (BSH) |
| 4 |
5.3.8.1 |
17585 |
AT5G03740 |
4E-19 |
Histone deacetylase 2C (HD2C) |
| 4 |
7.1.7.5 |
18705 |
AT1G77540 |
5E-30 |
H3/H4 histone acyl-CoA N-acyltransferase |
| 4 |
1.4.19.18 |
25013 |
AT2G47210 |
4E-124 |
myb-like transcription factor family protein |
| 4 |
1.3.3.10 |
2926 |
AT5G66750 |
0 |
CHROMATIN REMODELLING 1 (CHR1), DECREASED DNA |
| |
|
|
|
|
METHYLATION 1 (DDM1), SOMNIFEROUS 1 (SOM1) |
| 4 |
8.2.9.24 |
4432 |
AT4G16280 |
9E-73 |
RNA-mediated chromating silencing protein, FLOWERING TIME CONTROL |
| |
|
|
|
|
PROTEIN FCA |
| 4 |
6.4.2.10 |
5264 |
AT3G57300 |
1E-157 |
INO80 ortholog |
| 5 |
8.1.4.5 |
739, 26140 |
AT1G57820 |
0 |
Zinc C3HC4-type RING finger protein, ORTH2, VARIANT IN |
| |
|
|
|
|
METHYLATION 1 (VIM1) |
| 6 |
11.2.17.19 |
19820 |
AT5G26040 |
5E-136 |
Histone deacetylase 2 (HDA2) |
| 6 | 2.3.12.22 | 3694 | AT5G04940 | 5E-123 | SU(VAR)3-9 homolog 1 (SUVH1) |
Four putative histone deacetylase (HDA) genes were identified among the differentially expressed transcripts. While two of them, HDA8 and HDA9, showed increasing transcription from early embryogenesis up to the cotyledonary embryo stage (cluster 1), HD2C, which belongs to a class found only in plants [23], showed an opposite transcript profile (cluster 4). The fourth, HDA2, was up-regulated in mid-embryogenesis especially at the early cotyledonary stage (cluster 6).
Genes related to the methylation of histones were also identified with different transcript profiles. A putative ortholog of the A. thaliana polycomb-group (Pc-G) gene CURLY LEAF (CLF), part of the Polycomb Repressive Complex 2 (PRC2), was up-regulated in mid- to late embryogenesis (cluster 1). A putative ortholog of ASH1 HOMOLOG 2 gene (ASHH2), which encodes a protein with histone lysine N-methyltransferase activity implicated in the regulation of gene expression via H3K36 trimethylation [24], showed increasing transcription towards maturation (cluster 3). Finally, a putative SU(VAR)3-9 homolog 1 (SUVH1) was specifically up-regulated in early cotyledonary embryos (cluster 6). Several putative orthologs to SWI2/SNF2 chromatin-remodelling ATPases that modulate the accessibility of genomic regions to the transcriptional machinery [25] were differentially expressed. Two of them were found in cluster 1, namely CHC1 and RAD5, while a third one, BUSHY GROWTH (BSH), which is a putative ortholog of yeast SNF5 (subunit of the SWI/SNF chromatin remodelling complex), was up-regulated in late embryogenesis (cluster 3). Cluster 4 contained a differentially expressed ortholog of INO80 which is a member of the SNF2 superfamily of ATPases.
Transcripts for genes related to DNA methylation, DECREASE IN DNA METHYLATION 1 (DDM1) and DNA methyltransferase 1-associated protein (DMAP1), were up-regulated during early embryogenesis through the pre-cotyledonary embryo stage (cluster 4). A putative ortholog of VARIANT IN METHYLATION 1 (ORTH2/VIM1) was found in cluster 5.
Finally, several transcripts with homology to known regulators of small RNA biogenesis, processing, and function, were also differentially regulated during embryo development. Transcripts encoding a putative DAWDLE (DDL) were identified in cluster 1. Putative orthologs of HYPONASTIC LEAVES1 (HYL1) and DICER-LIKE1 (DCL1) were found in cluster 1 and 2, respectively. Additionally, two putative ARGONAUTE transcripts, AGO1 and AGO9, were found in cluster 1. With an opposite transcript profile, we also identified a FLOWERING LOCUS CA (FCA) putative ortholog in cluster 4.
Transcription factors involved in consecutive embryo stage-to-stage transitions
Transcripts displaying a fold-difference ≥ 2 between consecutive embryo developmental stages were examined to identify genes that might be relevant for a specific period of development (Additional file 6). Consistent with the time-course analyses of transcript profiles and functional categories, the most dramatic changes in expression were noticed in the transition from Day0 → Day5, where 173 transcripts were specifically down-regulated and 78 transcripts were up-regulated, and in the transition from Day15 → Day25, where 280 and 139 transcripts were specifically up and down-regulated, respectively (Figure 4). Only four transcripts were specifically differentially regulated (up-regulated) in Day11 → Day15 period (Figure 4), and no genes were up-regulated simultaneously in Day0 → Day5, Day5 → Day11 and Day11 → Day15 transitions, suggesting major differences between the transcriptomes of early and late stage embryos.
Figure 4.
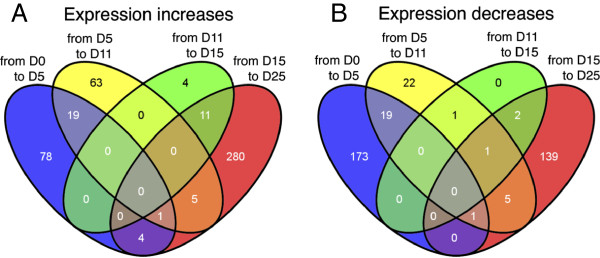
Venn diagrams of genes regulated between two consecutive stages. The number of genes showing a fold-change >2 between consecutive stages is shown. For each transition, genes showing increasing (A) or decreasing (B) expression between consecutive stages are represented in different diagrams.
Focusing on genes involved in transcriptional regulation, we identified transcripts annotated as likely transcription factors (TFs) in Table 2. Among the 23 TFs identified, about 2/3 were up-regulated in a specific developmental transition, with Day15 → Day25 showing the highest number of differentially expressed TFs. The bHLH, followed by the NAC and MYB transcription factor families, were most represented in our analyses. The best characterized putative TF found to be up-regulated in early embryogenesis (Day0 → Day5) was KANADI 2 (KAN2), a member of the GARP transcription factor family. Up-regulation during early and mid embryogenesis was also observed for a putative ortholog of AINTEGUMENTA (ANT) (Table 2), while a putative YABBY2 transcript seemed to be important in the transition from early cotyledonary to cotyledonary stage. Putative orthologs for three NAC transcripts, namely NAM/NARS2, ARABIDOPSIS NAC DOMAIN CONTAINING PROTEIN 75 (ANAC075) and ATAF1, were up-regulated in the Day15 → D25 transition (Table 2), while a putative bHLH transcript, LEUCINE RESISTANT 3 (ILR3), was strongly up-regulated in Day15 → Day25. Worth noting was the strong down-regulation from Day0 → Day5 (20.9 fold) of a putative AUXIN RESPONSE FACTOR, ARF16, which may serve to underscore the relevance of auxin response mechanisms in the early stage embryos. In this early stage, a putative ortholog coding for a bZIP TF, FLORAL TRANSITION AT THE MERISTEM 3 (FTM3), and a putative WRKY28 were found to be down-regulated. TFs that were specifically down-regulated in the Day15 → Day25 transition included members of the bHLH superfamily, such as a putative FAMA homologue. Additionally, a homologue of the PLATZ family of plant-specific TFs, and HAP5B, whose roles are poorly or not yet characterized were also down-regulated at this period.
Table 2.
Transcription factor genes showing a transcript level fold-change ≥ ; 2 between consecutive stages of embryo development
| Cl | Gene ID | Pp unigene | At Locus | E-value | Annotation | Up-regulated | FC | Down regulated | FC |
|---|---|---|---|---|---|---|---|---|---|
| 1 |
6.3.22.5 |
20408 |
AT4G38620 |
7E-72 |
R2R3 MYB protein 4 (MYB4) |
D0 → D5 |
2.7 |
- |
- |
| D5 → D11 |
2.4 |
|
|
||||||
| 1 |
12.1.13.17 |
7865 |
AT3G54390 |
2E-45 |
Sequence-specific DNA binding protein |
D0 → D5 |
2.2 |
- |
- |
| 1 |
6.4.7.23 |
1862 |
AT1G68920 |
8E-56 |
Basic helix-loop-helix (bHLH) DNA-binding superfamily protein |
D0 → D5 |
2.1 |
- |
- |
| 6 |
8.2.2.4 |
1030 |
AT1G32240 |
8E-41 |
KANADI family transcription factor 2 (KAN2) |
D0 → D5 |
2.1 |
- |
- |
| 1 |
6.1.20.1 |
27680 |
AT4G37750 |
2E-96 |
AINTEGUMENTA (ANT) |
D5 → D11 |
2.4 |
- |
- |
| 3 |
10.3.2.2 |
2388 |
AT1G08465 |
1E-41 |
YABBY family protein 2 (YAB2) |
D11 → D15 |
2.2 |
- |
- |
| 3 |
3.3.10.21 |
57615 |
AT1G12540 |
3E-13 |
Basic helix-loop-helix (bHLH) DNA-binding superfamily protein |
D11 → D15 |
2.0 |
- |
- |
| D15 → D25 |
4.2 |
|
|
||||||
| 3 |
1.1.20.3 |
37794 |
AT3G49950 |
5E-46 |
GRAS family transcription factor |
D15 → D25 |
2.0 |
- |
- |
| 3 |
2.3.14.1 |
25514 |
AT1G52880 |
4E-53 |
ARABIDOPSIS NAC DOMAIN CONTAINING PROTEIN 18 (ANAC018), NO APICAL MERISTEM/SEED MORPHOLOGY 2 (NAM/NARS2) |
D15 → D25 |
2.3 |
- |
- |
| 3 |
2.4.19.23 |
17360 |
AT1G22640 |
1E-150 |
MYB-type transcription factor 3 (MYB3) |
D15 → D25 |
2.3 |
- |
- |
| 3 |
2.4.7.21 |
2808 |
AT4G29230 |
7E-98 |
NAC-domain protein 75 (ANAC075) |
D15 → D25 |
2.4 |
- |
- |
| 3 |
3.1.19.21 |
12873 |
AT5G47390 |
4E-71 |
MYB-like transcription factor |
D15 → D25 |
2.0 |
- |
- |
| 3 |
3.2.1.2 |
16071 |
AT1G01720 |
7E-75 |
NAC-domain protein 2 (ANAC002), ATAF1 |
D15 → D25 |
2.7 |
- |
- |
| 3 |
4.4.11.10 |
270 |
AT3G20910 |
6E-31 |
Nuclear factor Y subunit A9 (NF-YA9) |
D15 → D25 |
2.0 |
- |
- |
| 3 |
5.2.18.9 |
16530 |
AT5G54680 |
1E-54 |
Basic helix-loop-helix (bHLH) 105 (BHLH105), LEUCINE RESISTANT 3 (ILR3) |
D15 → D25 |
4.1 |
- |
- |
| 2 |
1.3.17.22 |
8657 |
AT2G18160 |
2E-14 |
Basic leucine-zipper 2 (BZIP2), FLORAL TRANSITION AT THE MERISTEM 3 (FTM3) |
- |
- |
D0 → D5 |
3.3 |
| 2 |
5.1.1.2 |
37241 |
AT4G18170 |
2E-27 |
WRKY transcription factor 28 (WRKY28) |
- |
- |
D0 → D5 |
2.1 |
| 5 |
5.3.10.21 |
2451 |
AT4G30080 |
3E-151 |
Auxin response factor 16 (ARF16) |
- |
- |
D0 → D5 |
20.9 |
| 6 |
1.4.9.14 |
22607 |
AT3G24140 |
1E-51 |
bHLH superfamily protein FAMA |
- |
- |
D15 → D25 |
2.2 |
| 4 |
10.3.12.3 |
17529 |
AT2G12646 |
3E-67 |
PLATZ transcription factor family protein |
- |
- |
D15 → D25 |
3.0 |
| 6 |
3.3.17.13 |
7237 |
AT1G65620 |
1E-56 |
ASYMMETRIC LEAVES 2 (AS2) |
- |
- |
D15 → D25 |
3.2 |
| 4 |
4.3.3.12 |
4970 |
AT1G56170 |
4E-56 |
Nuclear factor Y subunit C2 (NF-YC2), HAP5B |
- |
- |
D15 → D25 |
2.7 |
| 4 |
4.4.11.2 |
18019 |
AT3G19500 |
2E-31 |
bHLH DNA-binding superfamily protein |
- |
- |
D15 → D25 |
2.5 |
| 1 | 6.4.7.23 | 1862 | AT1G68920 | 8E-56 | bHLH DNA-binding superfamily protein | - | - | D15 → D25 | 2.1 |
Comparative time-course analysis of differentially expressed transcripts during embryogenesis in P. pinaster versus A. thaliana
We compared the transcript profiles for genes involved in embryo development in A. thaliana and P. pinaster in order to search for correlations between the levels of putative orthologous transcripts. For each developmental stage that we considered as equivalent between species, genes were plotted in a scatter graph using the A. thaliana and P. pinaster expression values as coordinates (Additional file 7). Linear regression analysis demonstrated that the highest correlations were in the first (Day0 and globular embryos) and last (Day25 and mature embryos) stages, while the lowest correlations were in the third stage (Day11 and torpedo embryos).
To compare transcript profiles between the two species, the fold-changes in transcript levels at each stage versus the average value throughout embryogenesis in each species was quantified for each data series. The transcript profiles for 224 genes in P. pinaster and A. thaliana had a Pearson correlation higher than 0.90 (Additional file 8). An enrichment analysis using Agrigo [26] of the 206 genes that had GO annotations in the A. thaliana TAIR10 database revealed that seven biological functions were significantly over- represented (FDR < 0.001; Additional file 9), specifically catabolic process, cellular component organization, developmental process, multicellular organismal process and several metabolic process categories.
Among the genes with highly correlated transcript profiles between both species we found: (i) six EMBRYO DEFECTIVE genes, namely GNOM / EMB30 (AT1G13980), S- ADENOSYL-L-HOMOCYSTEIN HYDROLASE 1 / EMB1395 (AT4G13940), SECY HOMOLOG 2 / EMB2289 (AT2G31530), EMB2296 (AT2G18020), RIBOSOMAL PROTEIN 1 / EMB2207 (AT2G18020) and CYTOKINESIS DEFECTIVE 1 / EMB101 (AT2G39770); (ii) several differentially expressed genes within the developmental process category that have been previously shown to affect embryo development in A. thaliana via transcription and regulatory processes, such as SKP1-Cullin/CDC53-F-box (also LEAF CURLING RESPONSIVENSS, LCR) and ETHYLENE-INSENSITIVE 2 (EIN2; AT5G03280); and (iii) two relevant epigenetic regulators, MULTICOPY SUPPRESSOR OF IRA 1 (MSI1; AT5G58230) and UBIQUITIN CARRIER PROTEIN 2 (UBC2; AT2G02760).
Validation of microarray data
To validate the microarray expression data, a set of ten differentially transcribed genes putatively involved in epigenetic and transcriptional regulation (Tables 1 and 2) were analysed using RT-qPCR. Selected genes showed different transcription profiles during zygotic embryo development. Five genes belonged to cluster 1, two genes to cluster 6, and one gene to each of the clusters 2, 3 and 4 (Figure 3). Four of the genes (Unigenes 1030, 7237, 27680 and 17529) were transcription factors. Microarray green/red intensity ratios (M values) and RT-qPCR transcription levels were obtained for each gene (Additional file 10). The magnitudes of both data series were equalized or normalized calculating the fold-change between each time point and the average value, then log2 transformed as shown in Figure 5. Correlation between microarray and RT-qPCR data was demonstrated by high Pearson correlation coefficients that ranged from 0.69 (Unigene 25311) to 0.99 (Unigene 17529).
Figure 5.

Validation of microarray transcript profiles. Fold-changes for selected transcripts obtained by microarray analysis and RT-qPCR are shown for each developmental time point. Insets represent the profiles based on microarray M-values intensity ratios (triangles) or RT-qPCR relative expression normalized values (squares).
Discussion
Zygotic embryo isolation is a major challenge in most plant species, especially in the early stages of embryo development [16]. Our method allowed for rapid separation of embryos by stage and verification of structural integrity before freezing, which enabled collection of sufficient amounts of tissue for RNA isolation. In this way, we were able to perform the first genome-wide transcript profiling study that covers a wide time-window of zygotic embryo development in a conifer species.
Many genes required for embryo development are not embryo-specific as their basal functions are needed throughout the plant’s life cycle [27]. Le et al. [28] reported that out of 16,000 genes active throughout A. thaliana seed development, only 289 are seed-specific. However, significant quantitative changes in gene activity occur across specific developmental stages. In fact, each stage of seed development has a characteristic gene set that is either specific or up-regulated with respect to genes active at other stages [28]. We found that oxidation-reduction metabolic processes were over-represented in most clusters, which is indicative of high metabolic activity taking place in the developing embryos. Maintenance of cellular redox homeostasis by redox metabolites, such as glutathione, likely plays an important role in the context of embryo development. In fact, glutathione metabolism was highlighted in our analyses of differentially expressed functional categories (glutathione thiolesterase activity) and by the high number of transcripts putatively encoding glutathione transferases. These observations would appear in line with previous reports that the abundance of glutathione in proliferating cells is critical for shoot and root meristem development through roles in auxin transport and signalling [29]. Moreover, it has been shown that manipulation of glutathione metabolism during in vitro embryogenesis can affect embryo yield and quality, i.e. an environment with elevated levels of reduced glutathione results in increased numbers of immature embryos during somatic embryogenesis induction, while a more oxidized environment promotes embryo development [30].
Conifers are a major group within the gymnosperms, and are interesting subjects for studying embryogenesis due to distinguishing characteristics that when compared to angiosperms may reveal unique genes and gene networks that could further illuminate plant embryo development and its evolutionary implications. In our analyses, we found that approximately 3% of the differentially regulated transcripts appeared unique to gymnosperms with most putatively coding for unknown or uncharacterized proteins. While these unknown proteins might play important roles in conifer embryo development, still it appears that embryogenesis is mainly accomplished by the coordinated activities of a similar set of transcripts in both angiosperms and gymnosperms, as has been previously suggested [9]. The features that differentiate embryo development in these two plant groups probably result primarily from differential gene regulation. Thus, we have focused our study on the analysis of regulatory mechanisms of gene expression that play important roles during plant development, namely epigenetic control and transcriptional control by transcription factors.
Epigenetic regulation pathways across embryo development
Covalent modification of histones, DNA methylation, chromatin-remodelling enzymes and small RNAs, among other factors, play a central role in gene expression by modulating access to DNA and defining distinct chromatin states that ultimately determine selective readout of the genomic sequence [31].
Maintenance of chromatin silencing, specific histone post-translational modifications, and regulation of DNA methylation and transposition, appeared as co-regulated functions during early embryogenesis in our time-course analysis of functional categories across pine embryogenesis. In early embryogenesis, co-regulated functional categories as well as the identified differentially regulated transcripts, pointed to the importance of gene silencing mechanisms related to the control of transposable elements (TE). In fact, DNA methylation and heterochromatin maintenance were highlighted by both the analysis of co-regulated functional categories and the up-regulation of a putative DDM1, which is a key regulator of heterochromatic formation in A. thaliana required for TE-specific DNA methylation [32]. An identical transcript profile was observed for a putative FCA, recently implicated in the regulation of RNA sequences related to transposons, retrotransposons, and dispersed repeats that are normally silenced by the RNA-directed DNA methylation pathway [33]. Also, a putative ORTH2/VIM1, a mediator of DNA methylation status implicated in the establishment/maintenance of chromatin structure during cell division [34], was up-regulated during early embryogenesis in pine, although it showed a distinct transcript profile across embryo development being down-regulated specifically at mid embryogenesis.
DCL1[35], a homologue of which was up-regulated in Day0 samples, has been recently suggested to play a role in TAS-derived small interfering RNA-triggered DNA methylation by directly processing TAS gene transcripts [36]. DCL1 is required for cell differentiation events as early as the eight-cell stage A. thaliana embryos [37], where it participates in early embryonic patterning. Through its action on miRNA biogenesis, DCL1 prevents the accumulation of miRNA targets that promote differentiation during later stages of embryogenesis, namely transcription factors [37].
Evidence for negative regulation of histone H4 acetylation together with positive regulation of H3K9 methylation found in the time-course analysis of functional categories suggests a trend towards transcription repression during early embryogenesis. Specifically, H3K9 trimethylation, but not H3K9 methylation or dimethylation, is found in highly expressed genes [38]. In contrast, hyperacetylated histones are generally associated with gene activation, whereas hypoacetylated histones are related to gene repression [23]. Our results suggest that different groups of genes are being targeted for epigenetic regulation during embryogenesis through the action of different classes of deacetylases, e.g. a putative HD2C in early embryogenesis versus putative HDA8 and HDA9 in late embryogenesis. Inhibition of histone deacetylases (HDACs) in A. thaliana is known to affect embryo development as well as the expression of seed-associated genes, including transcription factors [39,40]. In another conifer, Norway spruce, Uddenberg et al. [41] reported a similar behaviour, suggesting a connection between changes in acetylation patterns and the levels of embryogenesis-related gene expression.
A homologue of SUVH1, which encodes a H3 lysine-9 specific histone-lysine N- methyltransferase, was up-regulated specifically in early cotyledonary embryos (cluster 6), suggesting a possible role in the organization of transcriptionally repressive chromatin [42]. Altogether these results suggest that different well-known mechanisms of gene silencing are active during early embryo development in pine.
From the mid to late embryogenesis stages, large chromatin remodelling events also seem to take place in conifer embryos as suggested by significant increases in transcription of several apparent chromatin-remodelling ATPases, such as putative CHC1, RAD5 and BSH. Another gene up-regulated in mid- to late embryogenesis (cluster 1) was a homolog of CLF, a member of the polycomb-group (Pc-G) proteins, which regulate many developmental processes in plants and animals by repressing gene expression in a cell-specific manner via trimethylation of histone H3 lysine 27 (H3K27me3) [43]. CLF was initially characterized as a suppressor of floral homeotic genes [44], but other genes such as CUP-SHAPED COTYLEDON 2 (CUC2) and PIN1 have been described as Pc-G target genes [45,46]. CUC genes, for example, which are regulated by PIN1, are crucial for the establishment of the embryonal shoot apical meristem and the formation of two separated cotyledons by presumably preventing cell proliferation and cotyledonary outgrowth in the intercotyledonary regions [47]. The Picea abies PaNAC01, an orthologue of CUC, is also regulated by polar auxin transport and it is associated with differentiation of the shoot apical meristem and formation of separated cotyledons [48]. Consistent with these findings, loss of H3K27me3 in Arabidopsis was shown to be associated with misregulation of genes involved in auxin responses [49]. It seems likely that in our results, the observed differential regulation of CLF and other chromatin-remodelling genes is related to the regulation of some of these processes.
In what concerns small RNA pathways, several transcripts with homology to known regulators of small RNA biogenesis, processing, and function were differentially expressed during pine embryo development, including a putative DDL, mostly up-regulated from mid to late embryogenesis, that could act in the biogenesis of miRNAs and endogenous siRNAs. DDL does not affect transcription of miRNAs directly but acts through other proteins, such as DCL, by facilitating their access or recognition of pri-miRNAs [50]. The processing of miRNAs from longer primary transcripts (pri-miRNAs) requires the activity of several proteins, including DCL1 and the double-stranded RNA-binding protein, HYPONASTIC LEAVES1 (HYL1) [35,51,52], for which a putative ortholog was found up-regulated in the same developmental periods as the putative DDL.
Argonaute (AGO) proteins are part of the RNA-induced silencing complex (RISC) that bind small RNAs and cause gene silencing. Two putative AGO transcripts similar to AGO1 and AGO9, were highly represented in the late pine embryo transcriptome. AGO9 belongs to a phylogenetic clade in which all members recognize 24-nucleotide small interfering RNAs and act to silence TEs and other repetitive sequences at the transcriptional level [53]. In contrast, orthologs of AGO1 are primarily mediators of miRNA activities [54,55], although AGO1 is also involved in the production of RDR6-dependent siRNAs [56,57]. In summary, different gene silencing mechanisms seem to be more active in opposite stages of pine embryo development. In early embryogenesis, mechanisms that target TE and repetitive sequences appear dominant, while PRC2-mediated repression of genes involved in specific developmental processes, such as formation of cotyledons, seems to be more relevant during late embryogenesis. In contrast, genes associated with sRNA pathways were found to be differentially regulated across all stages of embryo development.
Transcriptional regulators and auxin-mediated events
When chromatin structure allows expressed TFs to gain access to their binding sites, these proteins play a master role in the regulation of gene expression. A putative ortholog of ARF16 showed a dramatic decrease in transcription from the early embryo to precotyledonary stage of development, suggesting it could play a major role in early pine embryogenesis. ARFs are key regulators of auxin-modulated gene expression [58] that activate or repress target genes by binding to the promoters of early auxin response genes. ARF16 and ARF10 were shown to repress WOX5 transcription and restrict it to the A. thaliana root quiescent center [59] where it is required to maintain pluripotent columella stem cells [60]. Interestingly, in addition to being expressed in early embryogenesis, the putative ARF16 transcript profile in pine (cluster 5) also showed an increase from early cotyledonary to mature embryos, which is a profile similar to the one described for ARF16 in A. thaliana embryos [61]. Another gene up-regulated in early pine embryos was a putative ortholog of AUXIN RESISTANT1 (AUX1), which encodes an auxin influx carrier. Together with ATP Binding Cassette subfamily B (ABCB) transporters and PIN proteins, AUX1 carriers are primary coordinators of polar auxin transport. A homolog of N-MYC DOWNREGULATED-LIKE 1 (NDL1), which plays a role in modulating auxin transport, possibly by regulating auxin transport carrier proteins like PIN2 and AUX1 [62], was also up-regulated in early stage pine embryos.
If these gene products serve conserved functions in gymnosperm embryogenesis, then the interplay between auxin and transcription factors with defined spatial and temporal expression patterns is critical for the establishment of the pine embryo patterning. For example, the putative ARF16 expression pattern in pine embryos seems to be consistent with what has been described for ARF16 in A. thaliana, where it is involved in establishment of apical-basal patterning by participating in initiation of the root apical meristem formation in an early stage of embryogenesis.
KANADI protein (KAN2) plays an important part in early angiosperm embryogenesis, presumably by modulating the flow of auxin through regulating polar expression of PIN proteins [63]. Our studies suggest a KAN2 homolog may be important in the transition from early stage pine embryogenesis to the precotyledonary embryo stage. Interestingly, differential regulation of a homolog of PIN3 (Additional file 6), an auxin efflux carrier, and of a homolog of GNOM (cluster 4, Additional file 5), important for the recycling of PIN proteins between endosomal compartments and the plasma membrane [64], observed during the same period of development is in agreement with such a role for the putative KAN2. At the heart stage of A. thaliana embryogenesis, KAN2 as well as KAN1 and KAN3, display a similar expression pattern in the abaxial basal portion of emerging cotyledon primordial [63]. The same authors proposed that pattern formation along the central–peripheral axis results from interplay between auxin and the KANADI and Class III HD-Zip transcription factors.
Eshed et al [65] proposed that initial asymmetric leaf development, regulated primarily by mutual antagonism between KANADI and PHABULOSA (PHB)-like genes, is translated into polar YABBY expression, which subsequently contributes both to abaxial cell fate and abaxial/adaxial juxtaposition-mediated lamina expansion. A putative YABBY gene family member, YABBY2, was significantly up-regulated in pine from early cotyledonary stage onwards, which could be consistent with involvement in abaxial cell fate determination and leaf lamina growth along the abaxial–adaxial boundary [66,67]. Another transcript, a putative AS2/LOB, encoding a plant-specific protein involved in the determination of adaxial-abaxial polarity in leaf primordia [68-70] and repression of class 1 KNOTTED-like homeobox (KNOX) genes [71], was down-regulated in the Day15 → Day25 transition. Pine genes likely involved in the formation and function of apical meristems were also differentially expressed, including a putative NARS2/NAM, which was significantly up- regulated in late pine embryogenesis. NARS2/NAM has been implicated both in the formation of organ boundaries and embryonic shoot apical meristem. For example, the A. thaliana CUC1[72] and CUC2[73] as well as the petunia NO APICAL MERISTEM (NAM)[74], are expressed in boundaries between floral organ primordia and in the boundary between the cotyledons. However, mutations in CUC1, CUC2, and NAM also affect the initiation of the shoot apical meristem [72-75]. AINTEGUMENTA (ANT) has been implicated in the regulation of shoot apical meristem function [76]. A putative pine ANT homolog may be important for the transition from pre-cotyledonary to early cotyledonary embryos, as suggested from its up-regulation during this period. Plants containing mutations in ANT genes exhibit increased sensitivity to disruptions in polar auxin transport [77], suggesting that its role during embryogenesis might be related to auxins.
Correlation between profiles of putative orthologs between A. thaliana and P. pinaster embryogenesis
By matching embryo stages between two distantly related species, comparative analyses between the model angiosperm A. thaliana and P. pinaster lead us to conclude that several important processes in embryo development are conserved between angiosperms and gymnosperms. In fact, a few genes known to be essential for A. thaliana embryo development, including EMBRYO DEFECTIVE genes, and respective putative pine orthologs were highlighted by our analyses. One of these genes, GNOM (EMB30), plays a pivotal role in early A. thaliana embryogenesis being required for embryo patterning [78]. Its function in endosomal recycling of PIN1 (and PIN2) to the basal plasma membrane is fundamental for apical-basal polarity establishment during embryogenesis. Through tissue-specific expression of GNOM in gnom mutant embryos, Wolters et al. [78] showed that both apical and basal embryo organization depend on GNOM provascular expression, and proposed that GNOM-dependent PIN relocalization (cell-autonomous) and sink-driven auxin transport (non cell-autonomous) trigger the initiation of auxin transport routes in embryonic pattern formation. Additional relevant genes involved in auxin-mediated responses were identified in this study by comparative analysis. For example, LCR has been shown to negatively regulate several auxin-responsive genes during leaf development [79], and EIN2 is involved, together with AUX1 and GNOM, in establishing a concentration gradient of auxin in the root tip important for root-hair positioning within the epidermal layer [80].
Finally, there were two relevant epigenetic regulators whose transcript levels decreased over time in highly similar fashion, suggesting equivalent roles in developing A. thaliana and P. pinaster embryos: MSI1 and UBC2. MSI1 is a core protein of the plant PRC2 complex required for normal seed development [81]. MSI1 is needed to maintain the correct temporal and organ-specific expression of homeotic genes, including AGAMOUS and APETALA2, as well as to establish epigenetic marks, such as H3K4 di-methylation and H3K9 acetylation, in SOC1 chromatin [82-84]. UBC2 is involved in the regulation of ubiquitination of histone H2B and control of flowering time in A. thaliana through modulation of FLOWERING LOCUS C/ MADS AFFECTING FLOWERING chromatin [85].
Conclusions
Taken together, our results indicate that characteristic transcriptional changes are associated with each developmental period, as has been previously observed for A. thaliana embryogenesis [10,28], and suggest that major events during embryogenesis are orchestrated by putative orthologs of regulators of these processes in angiosperms. Our analyses were limited to some extent because the stages of embryo development used in this study did not cover certain periods where major events that distinguish angiosperm and gymnosperm embryogenesis take place, such as the first zygote divisions after fertilization and polyembryony. Thus, further work should focus on the earliest stages of pine zygotic embryo development, before dominant embryos are identified. An expanded knowledge of conifer genomes will also be essential to further understand the molecular basis for characteristic features of embryogenesis in gymnosperms. Additional insight into the molecular regulation of embryo development in these species will have great utility for the improvement of conifers and their vegetative propagation through somatic embryogenesis.
Methods
Plant material
Immature female cones from Pinus pinaster Ait. were randomly collected from open- pollinated trees growing in a clonal orchard established by grafting between 1970 and 1975 at Escaroupim National Forest, Portugal (longitude 8°44’W, latitude 39°4’N). Cone harvesting was performed at five different time points from July to August 2007, selecting from 3–5 trees at each date. Immediately after cone harvest on each date seeds were removed from the cones and opened to expose the megagametophytes. These were dissected using fine forceps and a scalpel under a stereomicroscope to excise the dominant embryo. Isolated embryos were quickly evaluated for developmental stage and immediately frozen in liquid N2 into different pools depending on stage. The five time points were designated as Day0 for the first harvesting date and Day5, 11, 15 and 25 according to the day of harvest after Day0. Following a previously described embryo staging system [15], Day0 included embryos in developmental stages T0, T1 and T2 (early embryo stages), Day5 included stages T3 and T4 (pre-cotyledonary embryos), Day11 included stage T4B (early cotyledonary embryos), Day15 included stage T5 (cotyledonary embryos) and Day25 included stage T7 embryos (mature embryos) (Figure 1A). Several separate pools, each containing 30–60 zygotic embryos for each stage, were prepared for each time point and kept in separate tubes to serve as biological replicates in subsequent analyses.
RNA isolation and amplification
Total RNA was extracted from each pool of frozen zygotic embryos using the RNeasyPlant Mini Kit (Qiagen, Valencia CA, USA) incorporating the on-column DNase I digestion with the RNase-Free DNase Set (Qiagen) in the isolation procedure, according to manufacturer’s instructions. RNA concentration was determined spectrophotometrically using a NanoDrop 1000 (Thermo Scientific, Waltham, Massachusetts, USA) and agarose gel electrophoresis was used to ascertain RNA quality and integrity.
After checking RNA integrity and to rule out any possibility of DNA contamination before proceeding to RNA amplification, samples were subject to an additional DNase digestion treatment immediately before amplification using TURBO DNase (Ambion, Foster City CA, USA) according to manufacturer’s instructions. RNA amplification was performed using the Amino Allyl MessageAmp II aRNA Amplification Kit (Ambion). In brief, the reaction consisted of a reverse transcription step that generated a first-strand cDNA containing a T7 promoter sequence during a 2 h reaction at 42°C, followed by second-strand cDNA synthesis for 2 h at 16°C. After cDNA purification, in vitro transcription for 12–14 h at 37°C was used to generate antisense amplified RNA. A total of 100 ng total RNA was amplified in two sequential rounds of amplification where amino allyl UTP nucleotide incorporation was performed only during the second round of amplification, per the manufacturer’s instructions.
Microarray experiment and signal acquisition
The PtGen2 cDNA microarray used in this study has been previously described [7] and additional information regarding the array can be obtained from NCBI’s Gene Expression Omnibus (GEO) under the Platform accession GPL11184. Experimental samples consisted of amplified RNA from zygotic embryos at each of the five time points shown in Figure 1A. Three biological replicates were analyzed for each time point and two technical replicates were conducted for each sample. The hybridization reference sample consisted of a pool containing equal amounts of total RNA from all five time points investigated. Fluorescent labelling, hybridization, pre- and post-hybridization washes, scanning, and data processing were performed according to Lorenz et al. [86], except that 2 μg of Cy-labeledamplified RNA was used to hybridize with the array after fragmentation using Fragmentation Reagent (Ambion, Cat. #AM8740). Microarrays were scanned using a ProScanArrayTM confocal scanner (Perkin Elmer, Waltham, MA) equipped with 532 nm and 635 nm lasers. Raw fluorescence data were processed using ImaGene (Bio-Discovery Inc., El Segundo, CA, USA).
Low-quality raw data and blank controls were filtered as previously described [7,87,88].
Microarray data normalization and annotation
Filtered raw data was analyzed using the Bioconductor Limma package [89]. Data was normalized within array (Print-tip loess) and between arrays (Cyclic Loess) according to Smyth and Speed [90], and intensity ratios were transformed into log2 values. No background correction was applied and duplicated probes were treated independently during normalization.
Samples were clustered using the MeV program [91]. To incorporate updated transcript information for P. pinaster, the PtGen2 cDNA microarray (GPL11184) was re-annotated against sequences in the P. pinaster database, SustainPineDB (Version 2; http://www.scbi.uma.es/sustainpine), which contains 92 478 unique transcripts from maritime pine. The EST sequences representing both the 3′ and 5′ ends of the cDNA clones used to generate the array [5] were aligned using BLASTX (E-value 1e-10) to identify P. pinaster transcripts corresponding to each spot.
Transcript profiles and functional category analysis
P. pinaster unigenes or ESTs were used as BLASTX queries to identify homologous sequences (E-value < 1e-10) in the NCBI protein (nr) or A. thaliana TAIR10 databases. Corresponding gene ontologies, enzymatic code numbers, plant ontology, pathway and gene family terms were collected using Blast2GO [92], which was also used for the enrichment analysis.
Differentially transcribed genes were identified from data for the different time points and replicates using the Bioconductor maSigPro package [22]. We defined a quadratic regression model to identify differentially transcribed genes across the five time points based on the premise that this model should have sufficient power to analyze a reduced number of time points [22]. A false discovery rate (FDR) of 0.01 was applied to identify significantly differentially transcribed genes. The maSigPro stepwise regression was adjusted to use the “two.ways.backward” method, with alfa 0.01 and an R2 cut-off value fit of 0.7. The use of high R2 gene models has been proposed for capturing biological meaningful expression trends and profile differences [22]. Significantly differentially transcribed genes were clustered using a hierarchical approach based on correlation distances. Unigenes not present in flowering plants (or other more distant taxa), but present in gymnosperms, were annotated as specific to gymnosperms. To make this identification, the complete list of proteins in the NCBI proteins (nr) database belonging to gymnosperm taxa (Tax IDs 58021, 3312, 58020 and 58022) was generated (GI list) and downloaded from NCBI Entrez database (104709 proteins). We used the BLASTX options negative_gilist or gilist, to respectively exclude or restrict comparisons to gymnosperms. Functional assessment was performed using maSigFun, which fits the regression model for groups of genes annotated to a similar functional category [21]. In this case, the groupings were based on gene ontology terms, enzyme code numbers, plant ontology terms, gene families and pathways, and a selection of other categories for which we had significant models.
Intensity values from the time-course microarray analysis of A. thaliana embryogenesis were available from the work of Xiang et al. (http://www2.bri.nrc.ca/plantembryo/) [16] and were normalized to obtain the expression values by subtracting a constant background value, as described by the authors. For the comparative analysis performed in our study, the A. thaliana globular, heart, torpedo, bent and mature embryo stages were considered equivalent to the Day0, Day5, Day11, Day15 and Day25 embryo samples from P. pinaster, respectively. For each gene, the fold-change between the transcript level values at each stage versus the average of the values in all the stages for each species was calculated. Since the P. pinaster unigene putative homologues in A. thaliana had been previously identified, transcript profiles were compared using Pearson correlation.
Validation of microarray data using real-time RT-qPCR
RT-qPCR was used for independent verification of the transcript profiles of a sub-set of genes identified from the microarray data. Primers were designed with Primer3 program (http://primer3.sourceforge.net/) to have a Tm of 58°C, bind the gene conserved regions and amplify around 100 bp (Additional file 10). Reverse transcription was performed using cDNA synthesized from 1.25 μg of total RNA using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche Diagnostics, USA) with the Random Hexamer primers according to the manufacturer’s instructions. Subsequent quantitative PCR (qPCR) was performed in a Roche LightCycler 480 system following standard cycling conditions. The SYBR Green I Master kit (Roche diagnostics, USA) was used to prepare the qPCR reaction mixtures, each one containing 1.8 μl of cDNA and 400 nM of each primer in a total volume of 20 μl. DNA contamination was discarded by the absence of signal after qPCR amplification of RNA samples with the UBIQUITIN primers. Negative controls (no template) were included in all the runs and the presence of only one specific peak was checked in the melting curve (dissociation curve). A pool containing equal amounts of total RNA from all five time points was included as a calibrator and positive control. Two biological replicates of each sample and three technical replicates per biological replicate were analyzed. The efficiency of each pair of primers was calculated based on the regression analysis of the PCR reactions kinetics by program LinReg 11.3 [93], and the Ct values by Roche Lightcycler software (Additional file 10). The relative amount of each transcript was calculated by the delta-delta-Ct method [94] using UBIQUITIN as reference gene, which has been probed reliable as endogenous control during P. pinaster somatic embryogenesis [95].
Availability of supporting data
The data set supporting the results of this article is available in the NCBI GEO database, accession number GSE32551 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32551).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MS was involved in the experimental design, sample collection and preparation, microarray hybridization, interpretation of the results and writing of the manuscript. JVB performed the bioinformatic analyses, participated in the interpretation of results and preparation of the manuscript. WWL supervised sample labelling, microarray hybridizations and took part in the critical review of the manuscript. ASR contributed for the validation of microarray data. RA contributed to the experimental design and provided inputs for interpretation of results. JFDD contributed materials and facilities and was involved in the critical review of the manuscript. CM conceived and supervised the project, participated in the interpretation of results, wrote and edited the manuscript. All authors have read and approved the final manuscript.
Supplementary Material
Microarray data normalization and replicate clustering. Box plot of the distribution of the red and green intensity ratio (M) from the thirty hybridized chip arrays before (A) and after (B) normalization.
Microarray intensity values for the 30 slides after normalization and correspondence between spot, EST accession number and Pinus pinaster unigene.
Functional annotation of the Pinus pinaster unigenes. Annotation was based on the homologous in NCBI and Arabidopsis databases, including gene putative prediction, gene ontology, pathways and families.
Quality assessment of the gene annotation. (A) Similitude values of the BLASTX alignments. (B) Species with the best BLASTX alignment of each query sequence. (C) Number of gene ontology terms per sequence for each group of terms. (D) Relation between number of gene ontology terms and length of the query sequence.
Functional annotation, specificity, clustering results and statistical analysis of the differentially expressed transcripts.
Clustering, fold-change and functional annotation of the differentially expressed transcripts in consecutive embryo stage-to-stage transitions.
Correlation of gene expression during A. thaliana and P. pinaster embryogenesis. The A. thaliana globular, heart, torpedo, bent and mature embryo stages [16] were considered equivalent to the Day0, Day5, Day11, Day15 and Day25 embryo samples of P. pinaster, respectively. For each sampling time, genes were plotted in a scatter graph using the A. thaliana (Y-axis) and P. pinaster (X-axis) expression values as coordinates.
Correlation of the expression profiles between A. thaliana and P. pinaster along five embryogenesis stages. Enrichment analysis values of the gene ontologies over/under-represented in the common subset of highly co-regulated transcripts.
Enrichment analysis of the genes with a highly similar expression profile in A. thaliana and P. pinaster.
Primers, efficiencies, Ct values and normalization of the target genes used for the data validation by qRT-PCR.
Contributor Information
José J de Vega-Bartol, Email: devegabartol@itqb.unl.pt.
Marta Simões, Email: simoesmh@gmail.com.
W Walter Lorenz, Email: wlorenz@uga.edu.
Andreia S Rodrigues, Email: sofiasantos@itqb.unl.pt.
Rob Alba, Email: robert.m.alba@monsanto.com.
Jeffrey F D Dean, Email: jeffdean@uga.edu.
Célia M Miguel, Email: cmiguel@itqb.unl.pt.
Acknowledgements
FCT is acknowledged for financial support through grants SFRH/BD/32037/2006 (to M. Simões) and SFRH/BD/79779/2011 (to A.S. Rodrigues), projects PTDC/AGR- GLP/102877/2008, P-KBBE/AGR-GPL/0001/2009 and PEst-OE/EQB/LA0004/2011. Part of this work was also supported by Fundação Luso-Americana para o Desenvolvimento (FLAD). Alexandre Aguiar and Isabel Carrasquinho from Instituto Nacional de Investigação Agrária e Veterinária (INIAV) are acknowledged for making plant material available.
References
- Smith SA, Beaulieu JM, Donoghue MJ. An uncorrelated relaxed-clock analysis suggests an earlier origin for flowering plants. Proc Natl Acad Sci USA. 2010;107:5897–5902. doi: 10.1073/pnas.1001225107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CG. Conifer reproductive biology. NY: Springer; 2009. [Google Scholar]
- Fernandez-Pozo N, Canales J, Guerrero-Fernandez D, Villalobos DP, Diaz-Moreno SM, Bautista R, Flores-Monterroso A, Guevara MA, Perdiguero P, Collada C, Cervera MT, Soto A, Ordas R, Cantón FR, Avila C, Cánovas FM, Claros MG. EuroPineDB: a high- coverage Web database for maritime pine transcriptome. BMC Genomics. 2011;12:366. doi: 10.1186/1471-2164-12-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz WW, Ayyampalayam S, Bordeaux JM, Howe GT, Jermstad KD, Neale DB, Rogers DL, Dean JFD. Conifer DBMagic: a database housing multiple de novo transcriptome assemblies for 12 diverse conifer species. Tree Genet Genomes. 2012;8:1477–1485. doi: 10.1007/s11295-012-0547-y. [DOI] [Google Scholar]
- Lorenz WW, Sun F, Liang C, Kolychev D, Wang H, Zhao X, Cordonnier-Pratt M-M, Pratt LH, Dean JFD. Water stress-responsive genes in loblolly pine (Pinus taeda) roots dentified by analyses of expressed sequence tag libraries. Tree Physiol. 2006;26:1–16. doi: 10.1093/treephys/26.1.1. [DOI] [PubMed] [Google Scholar]
- Canales J, Flores-Monterrosso A, Rueda-López M, Avila C, Cánovas FM. Identification of genes regulated by ammonium availability in the roots of maritime pine trees. Amino Acids. 2010;39:991–1001. doi: 10.1007/s00726-010-0483-9. [DOI] [PubMed] [Google Scholar]
- Lorenz WW, Alba R, Yu Y-S, Bordeaux JM, Simões M, Dean JFD. Microarray analysis and scale-free gene networks identify candidate regulators in drought-stressed roots of loblolly pine (P. taeda L.) BMC Genomics. 2011;12:264. doi: 10.1186/1471-2164-12-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalobos DP, Diaz-Moreno SM, Said E-SS, Cañas RA, Osuna D, Van Kerckhoven SHE, Bautista R, Claros MG, Cánovas FM, Cantón FR. Reprogramming of gene expression during compression wood formation in pine: coordinated modulation of S- adenosylmethionine, lignin and lignan related genes. BMC Plant Biol. 2012;12:100. doi: 10.1186/1471-2229-12-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairney J, Zheng L, Cowels A, Hsiao J, Zismann V, Liu J, Ouyang S, Thibaud-Nissen F, Hamilton J, Childs K, Pullman GS, Zhang Y, Oh T, Buell CR. Expressed sequence tags from loblolly pine embryos reveal similarities with angiosperm embryogenesis. Plant Mol Biol. 2006;62:485–501. doi: 10.1007/s11103-006-9035-9. [DOI] [PubMed] [Google Scholar]
- Spencer MWB, Grene R, Lindsey K. Transcriptional profiling of the Arabidopsis embryo. Plant Physiol. 2007;143:924–940. doi: 10.1104/pp.106.087668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestman D, Larsson E, Uddenberg D, Cairney J, Clapham D, Sundberg E, Arnold S. Important processes during differentiation and early development of somatic embryos of Norway spruce as revealed by changes in global gene expression. Tree Genet Genomes. 2011;7:347–362. doi: 10.1007/s11295-010-0336-4. [DOI] [Google Scholar]
- Lara-Chavez A, Egertsdotter U, Flinn BS. Comparison of gene expression markers during zygotic and somatic embryogenesis in pine. Vitro Cell Dev Biol-Plant. 2012;48:341–354. [Google Scholar]
- Zhao Y. Auxin biosynthesis and its role in plant development. Annu Rev Plant Biol. 2010;61:49–64. doi: 10.1146/annurev-arplant-042809-112308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marum L, Loureiro J, Rodriguez E, Santos C, Oliveira MM, Miguel C. Flow cytometric and morphological analyses of Pinus pinaster somatic embryogenesis. J Biotechnol. 2009;143:288–295. doi: 10.1016/j.jbiotec.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Gonçalves S, Cairney J, Oliveira M, Miguel C. Identification of differentially expressed genes during embryogenesis in maritime pine (Pinus pinaster) Silva Lusitana. 2005;13(2):203–216. [Google Scholar]
- Xiang D, Venglat P, Tibiche C, Yang H, Risseeuw E, Cao Y, Babic V, Cloutier M, Keller W, Wang E, Selvaraj G, Datla R. Genome-wide analysis reveals gene expression and metabolic network dynamics during embryo development in Arabidopsis. Plant Physiol. 2011;156:346–356. doi: 10.1104/pp.110.171702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel C, Gonçalves S, Tereso S, Marum L, Maroco J, Margarida Oliveira M. Somatic embryogenesis from 20 open-pollinated families of Portuguese plus trees of maritime pine. Plant Cell Tiss Organ Cult. 2004;76:121–130. [Google Scholar]
- Lorenz WW, Simões M, Miguel C, Dean JFD. Analysis of gene expression changes in Pinus species using a loblolly pine cDNA microarray. Quebec city: IUFRO-CITA 2008 Joint Conference; 2008. [Google Scholar]
- Van Zyl L, Arnold Von S, Bozhkov P, Chen Y, Egertsdotter U, Mackay J, Sederoff RR, Shen J, Zelena L, Clapham DH. Heterologous array analysis in pinaceae: hybridization of Pinus taeda cDNA arrays with cDNA from needles and embryogenic cultures of P. taeda, P. sylvestris or Picea abies. Comparative and Functional Genomics. 2002;3:306–318. doi: 10.1002/cfg.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nueda MJ, Sebastián P, Tarazona S, García-García F, Dopazo J, Ferrer A, Conesa A. Functional assessment of time course microarray data. BMC Bioinforma. 2009;6(10):S9. doi: 10.1186/1471-2105-10-S6-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Nueda MJ, Ferrer A, Talon M. MaSigPro: a method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics. 2006;22:1096–1102. doi: 10.1093/bioinformatics/btl056. [DOI] [PubMed] [Google Scholar]
- Hollender C, Liu Z. Histone deacetylase genes in Arabidopsis development. J Integr Plant Biol. 2008;50:875–885. doi: 10.1111/j.1744-7909.2008.00704.x. [DOI] [PubMed] [Google Scholar]
- Grini PE, Thorstensen T, Alm V, Vizcay-Barrena G, Windju SS, Jørstad TS, Wilson ZA, Aalen RB. The ASH1 HOMOLOG 2 (ASHH2) histone H3 methyltransferase is required for ovule and anther development in Arabidopsis. PLoS ONE. 2009;4:e7817. doi: 10.1371/journal.pone.0007817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev mBiochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Du Z, Zhou X, Ling Y, Zhang Z, Su Z. AgriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38:W64. doi: 10.1093/nar/gkq310. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzafrir I, Pena-Muralla R, Dickerman A, Berg M, Rogers R, Hutchens S, Sweeney TC, McElver J, Aux G, Patton D, Meinke D. Identification of genes required for embryo development in Arabidopsis. Plant Physiol. 2004;135:1206–1220. doi: 10.1104/pp.104.045179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le BH, Cheng C, Bui AQ, Wagmaister JA, Henry KF, Pelletier J, Kwong L, Belmonte M, Kirkbride R, Horvath S, Drews GN, Fischer RL, Okamuro JK, Harada JJ, Goldberg RB. Inaugural article: global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc Natl Acad Sci USA. 2010;107:8063–8070. doi: 10.1073/pnas.1003530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashandy T, Guilleminot J, Vernoux T, Caparros-Ruiz D, Ljung K, Meyer Y, Reichheld J-P. Interplay between the NADP-linked thioredoxin and glutathione systems in Arabidopsis auxin signaling. Plant Cell. 2010;22:376–391. doi: 10.1105/tpc.109.071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasolla C. Glutathione redox regulation of in vitro embryogenesis. Plant Physiol Biochem. 2010;48:319–327. doi: 10.1016/j.plaphy.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Tsukahara S, Kobayashi A, Kawabe A, Mathieu O, Miura A, Kakutani T. Bursts of retrotransposition reproduced in Arabidopsis. Nature. 2009;461:423–426. doi: 10.1038/nature08351. [DOI] [PubMed] [Google Scholar]
- Bäurle I, Smith L, Baulcombe DC, Dean C. Widespread role for the flowering-time regulators FCA and FPA in RNA-mediated chromatin silencing. Science. 2007;318:109–112. doi: 10.1126/science.1146565. [DOI] [PubMed] [Google Scholar]
- Liu S, Yu Y, Ruan Y, Meyer D, Wolff M, Xu L, Wang N, Steinmetz A, Shen W-H. Plant SET- and RING-associated domain proteins in heterochromatinization. Plant J. 2007;52:914–926. doi: 10.1111/j.1365-313X.2007.03286.x. [DOI] [PubMed] [Google Scholar]
- Kurihara Y, Takashi Y, Watanabe Y. The interaction between DCL1 and HYL1 is important for efficient and precise processing of pri-miRNA in plant microRNA biogenesis. RNA. 2006;12:206–212. doi: 10.1261/rna.2146906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Mao L, Qi Y. Roles of dicer-like and argonaute proteins in TAS-derived small interfering RNA-triggered DNA methylation. Plant Physiol. 2012;160:990–999. doi: 10.1104/pp.112.200279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodine MD, Bartel DP. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev. 2010;24:2678–2692. doi: 10.1101/gad.1986710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudier F, Ahmed I, Bérard C, Sarazin A, Mary-Huard T, Cortijo S, Bouyer D, Caillieux E, Duvernois-Berthet E, Al-Shikhley L, Giraut L, Després B, Drevensek S, Barneche F, Dèrozier S, Brunaud V, Aubourg S, Schnittger A, Bowler C, Martin-Magniette M-L, Robin S, Caboche M, Colot V. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 2011;30:1928–1938. doi: 10.1038/emboj.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai HH, Tai GCC, Beardmore T. Dynamic histone acetylation of late embryonic genes during seed germination. Plant Mol Biol. 2005;59:909–925. doi: 10.1007/s11103-005-2081-x. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Kikuchi A, Kamada H. The Arabidopsis histone deacetylases HDA6 and HDA19 contribute to the repression of embryonic properties after germination. Plant Physiol. 2008;146:149–161. doi: 10.1104/pp.107.111674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddenberg D, Valladares S, Abrahamsson M, Sundström JF, Sundås-Larsson A, Arnold Von S. Embryogenic potential and expression of embryogenesis-related genes in conifers are affected by treatment with a histone deacetylase inhibitor. Planta. 2011;234:527–539. doi: 10.1007/s00425-011-1418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas-Mollano JA, Lao NT, Kavanagh TA. Intron-regulated expression of SUVH3, an Arabidopsis Su(var)3-9 homologue. J Exp Bot. 2006;57:3301–3311. doi: 10.1093/jxb/erl093. [DOI] [PubMed] [Google Scholar]
- Schmitges FW, Prusty AB, Faty M, Stützer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, Bunker RD, Wirth U, Bouwmeester T, Bauer A, Ly-Hartig N, Zhao K, Chan H, Gu J, Gut H, Fischle W, Müller J, Thomä NH. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell. 2011;42:330–341. doi: 10.1016/j.molcel.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Goodrich J, Puangsomlee P, Martin M, Long D, Meyerowitz EM, Coupland G. A polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature. 1997;386:44–51. doi: 10.1038/386044a0. [DOI] [PubMed] [Google Scholar]
- Nikovics K, Blein T, Peaucelle A, Ishida T, Morin H, Aida M, Laufs P. The balancebetween the MIR164A and CUC2 genes controls leaf margin serration in Arabidopsis. Plant Cell. 2006;18:2929–2945. doi: 10.1105/tpc.106.045617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhorn J, Reimer JJ, Leuz I, Göbel U, Huettel B, Farrona S, Turck F. Development- related PcG target in the apex 4 controls leaf margin architecture in Arabidopsis thaliana. Development. 2012;139:2566–2575. doi: 10.1242/dev.078618. [DOI] [PubMed] [Google Scholar]
- Breuil-Broyer S, Morel P, de Almeida-Engler J, Coustham V, Negrutiu I, Trehin C. High-resolution boundary analysis during Arabidopsis thaliana flower development. Plant J. 2004;38:182–192. doi: 10.1111/j.1365-313X.2004.02026.x. [DOI] [PubMed] [Google Scholar]
- Larsson E, Sitbon F, Arnold Von S. Differential regulation of Knotted1-like genes during establishment of the shoot apical meristem in Norway spruce (picea abies) Plant Cell Rep. 2012;31:1053–1060. doi: 10.1007/s00299-011-1224-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafos M, Kroll P, Hohenstatt ML, Thorpe FL, Clarenz O, Schubert D. Dynamic regulation of H3K27 trimethylation during Arabidopsis differentiation. PLoS Genet. 2011;7:e1002040. doi: 10.1371/journal.pgen.1002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Bi L, Zheng B, Ji L, Chevalier D, Agarwal M, Ramachandran V, Li W, Lagrange T, Walker JC, Chen X. The FHA domain proteins DAWDLE in Arabidopsis and SNIP1 in humans act in small RNA biogenesis. Proc Natl Acad Sci USA. 2008;105:10073–10078. doi: 10.1073/pnas.0804218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraguri A, Itoh R, Kondo N, Nomura Y, Aizawa D, Murai Y, Koiwa H, Seki M, Shinozaki K, Fukuhara T. Specific interactions between dicer-like proteins and HYL1/DRB-family dsRNA-binding proteins in Arabidopsis thaliana. Plant Mol Biol. 2005;57:173–188. doi: 10.1007/s11103-004-6853-5. [DOI] [PubMed] [Google Scholar]
- Dong Z, Han M-H, Fedoroff N. The RNA-binding proteins HYL1 and SE promote accurate in vitro processing of pri-miRNA by DCL1. Proc Natl Acad Sci USA. 2008;105:9970–9975. doi: 10.1073/pnas.0803356105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen C, Vaucheret H, Brodersen P. Lessons on RNA silencing mechanisms in plants from eukaryotic argonaute structures. Genes Dev. 2004;18:1187–1197. doi: 10.1101/gad.1201404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaucheret H, Vazquez F, Crété P, Bartel DP. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004;18:1187–1197. doi: 10.1101/gad.1201404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumberger N, Baulcombe DC. Arabidopsis ARGONAUTE1 is an RNA slicer that selectively recruits microRNAs and short interfering RNAs. Proc Natl Acad Sci USA. 2005;102:11928–11933. doi: 10.1073/pnas.0505461102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-M, Chen L-T, Patel K, Li Y-H, Baulcombe DC, Wu S-H. 22-Nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc Natl Acad Sci USA. 2010;107(34):15269–15274. doi: 10.1073/pnas.1001738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuperus JT, Montgomery TA, Fahlgren N, Burke RT, Townsend T, Sullivan CM, Carrington JC. Identification of MIR390a precursor processing-defective mutants in Arabidopsis by direct genome sequencing. Proc Natl Acad Sci USA. 2010;107:466–471. doi: 10.1073/pnas.0913203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscum E, Reed JW. Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol Biol. 2002;49:387–400. doi: 10.1023/A:1015255030047. [DOI] [PubMed] [Google Scholar]
- Ding Z, Friml J. Auxin regulates distal stem cell differentiation in Arabidopsis roots. Proc Natl Acad Sci USA. 2010;107:12046–12051. doi: 10.1073/pnas.1000672107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar AK, Luijten M, Miyashima S, Lenhard M, Hashimoto T, Nakajima K, Scheres B, Heidstra R, Laux T. Conserved factors regulate signalling in Arabidopsis thaliana shoot and root stem cell organizers. Nature. 2007;446:811–814. doi: 10.1038/nature05703. [DOI] [PubMed] [Google Scholar]
- Haecker A, Gross-Hardt R, Geiges B, Sarkar A, Breuninger H, Herrmann M, Laux T. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development. 2004;131:657–668. doi: 10.1242/dev.00963. [DOI] [PubMed] [Google Scholar]
- Mudgil Y, Uhrig JF, Zhou J, Temple B, Jiang K, Jones AM. Arabidopsis N-MYC DOWNREGULATED-LIKE1, a positive regulator of auxin transport in a G protein- mediated pathway. Plant Cell. 2009;21:3591–3609. doi: 10.1105/tpc.109.065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhaki A, Bowman JL. KANADI and class III HD-Zip gene families regulate embryo patterning and modulate auxin flow during embryogenesis in Arabidopsis. Plant Cell. 2007;19:495–508. doi: 10.1105/tpc.106.047472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geldner N, Anders N, Wolters H, Keicher J, Kornberger W, Muller P, Delbarre A, Ueda T, Nakano A, Jürgens G. The Arabidopsis GNOM ARF-GEF mediates endosomal recycling, auxin transport, and auxin-dependent plant growth. Cell. 2003;112:219–1032. doi: 10.1016/S0092-8674(03)00003-5. [DOI] [PubMed] [Google Scholar]
- Eshed Y, Izhaki A, Baum SF, Floyd SK, Bowman JL. Asymmetric leaf development and blade expansion in Arabidopsis are mediated by KANADI and YABBY activities. Development. 2004;131:2997–3006. doi: 10.1242/dev.01186. [DOI] [PubMed] [Google Scholar]
- Siegfried KR, Eshed Y, Baum SF, Otsuga D, Drews GN, Bowman JL. Members of the YABBY gene family specify abaxial cell fate in Arabidopsis. Development. 1999;1038(126):4117–4128. doi: 10.1242/dev.126.18.4117. [DOI] [PubMed] [Google Scholar]
- Sarojam R, Sappl PG, Goldshmidt A, Efroni I, Floyd SK, Eshed Y, Bowman JL. Differentiating Arabidopsis shoots from leaves by combined YABBY activities. Plant Cell. 2010;22:2113–2130. doi: 10.1105/tpc.110.075853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Xu Y, Dong A, Sun Y, Pi L, Xu Y, Huang H. Novel as1 and as2 defects in leaf adaxial-abaxial polarity reveal the requirement for ASYMMETRIC LEAVES1 and 2 and ERECTA functions in specifying leaf adaxial identity. Development. 2003;130:4097–4107.1046. doi: 10.1242/dev.00622. [DOI] [PubMed] [Google Scholar]
- Lin W-C, Shuai B, Springer PS. The Arabidopsis LATERAL ORGAN BOUNDARIES-domain gene ASYMMETRIC LEAVES2 functions in the repression of KNOX gene expression and in adaxial-abaxial patterning. Plant Cell. 2003;15:2241–1049. doi: 10.1105/tpc.014969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno Y, Ishikawa T, Watanabe K, Terakura S, Iwakawa H, Okada K, Machida C, Machida Y. Histone deacetylases and ASYMMETRIC LEAVES2 are involved in the establishment of polarity in leaves of Arabidopsis. Plant Cell. 2007;19:445–457. doi: 10.1105/tpc.106.042325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semiarti E, Ueno Y, Tsukaya H, Iwakawa H, Machida C, Machida Y. The ASYMMETRIC LEAVES2 gene of Arabidopsis thaliana regulates formation of a symmetric lamina, establishment of venation and repression of meristem-related homeobox genes in leaves. Development. 2001;128:1771–1783. doi: 10.1242/dev.128.10.1771. [DOI] [PubMed] [Google Scholar]
- Takada S, Hibara K, Ishida T, Tasaka M. The CUP-SHAPED COTYLEDON1 gene of Arabidopsis regulates shoot apical meristem formation. Development. 2001;1059(128):1127–1135. doi: 10.1242/dev.128.7.1127. [DOI] [PubMed] [Google Scholar]
- Aida M, Ishida T, Fukaki H, Fujisawa H, Tasaka M. Genes involved in organ separation in Arabidopsis: an analysis of the cup-shaped cotyledon mutant. Plant Cell. 1997;9:841–857. doi: 10.1105/tpc.9.6.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souer E, van Houwelingen A, Kloos D, Mol J, Koes R. The no apical meristem gene of Petunia is required for pattern formation in embryos and flowers and is expressed at meristem and primordia boundaries. Cell. 1996;85:159–170. doi: 10.1016/S0092-8674(00)81093-4. [DOI] [PubMed] [Google Scholar]
- Duval M, Hsieh T-F, Kim SY, Thomas TL. Molecular characterization of AtNAM: a member of the Arabidopsis NAC domain superfamily. Plant Mol Biol. 2002;50:237–1068. doi: 10.1023/A:1016028530943. [DOI] [PubMed] [Google Scholar]
- Mudunkothge JS, Krizek BA. Three Arabidopsis AIL/PLT genes act in combination to regulate shoot apical meristem function. Plant J. 2012;71:108–121. doi: 10.1111/j.1365-313X.2012.04975.x. [DOI] [PubMed] [Google Scholar]
- Krizek BA. Auxin regulation of Arabidopsis flower development involves members of the AINTEGUMENTA-LIKE/PLETHORA (AIL/PLT) family. J Exp Bot. 2011;62:3311–3319. doi: 10.1093/jxb/err127. [DOI] [PubMed] [Google Scholar]
- Wolters H, Anders N, Geldner N, Gavidia R, Jürgens G. Coordination of apical and basal embryo development revealed by tissue-specific GNOM functions. Development. 2011;138:117–126. doi: 10.1242/dev.059147. [DOI] [PubMed] [Google Scholar]
- Song JB, Huang SQ, Dalmay T, Yang ZM. Regulation of leaf morphology by microRNA394 and its target LEAF CURLING RESPONSIVENESS. Plant Cell Physiol. 2012;53:1283–1294. doi: 10.1093/pcp/pcs080. [DOI] [PubMed] [Google Scholar]
- Fischer U, Ikeda Y, Ljung K, Serralbo O, Singh M, Heidstra R, Palme K, Scheres B, Grebe M. Vectorial information for Arabidopsis planar polarity is mediated by combined AUX1, EIN2, and GNOM activity. Curr Biol. 2006;16:2143–2149. doi: 10.1016/j.cub.2006.08.091. [DOI] [PubMed] [Google Scholar]
- Köhler C, Hennig L, Bouveret R, Gheyselinck J, Grossniklaus U, Gruissem W. Arabidopsis MSI1 is a component of the MEA/FIE polycomb group complex and required for seed development. EMBO J. 2003;22:4804–4814. doi: 10.1093/emboj/cdg444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig L, Taranto P, Walser M, Schönrock N, Gruissem W. Arabidopsis MSI1 is required for epigenetic maintenance of reproductive development. Development. 2003;1088(130):2555–2565. doi: 10.1242/dev.00470. [DOI] [PubMed] [Google Scholar]
- Exner V, Taranto P, Schönrock N, Gruissem W, Hennig L. Chromatin assembly factor CAF-1 is required for cellular differentiation during plant development. Development. 2006;133:4163–4172. doi: 10.1242/dev.02599. [DOI] [PubMed] [Google Scholar]
- Bouveret R, Schönrock N, Gruissem W, Hennig L. Regulation of flowering time by Arabidopsis MSI1. Development. 2006;133:1693–1702. doi: 10.1242/dev.02340. [DOI] [PubMed] [Google Scholar]
- Cao Y, Dai Y, Cui S, Ma L. Histone H2B monoubiquitination in the chromatin of FLOWERING LOCUS C regulates flowering time in Arabidopsis. Plant Cell. 2008;20:2586–2602. doi: 10.1105/tpc.108.062760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz WW, Yu Y-S, Simões M, Dean JFD. Processing the loblolly pine PtGen2 cDNA microarray. J Vis Exp. 2009;25:e1182. doi: 10.3791/1182. doi:10.3791/1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ghosh S, Guo SW. Quantitative quality control in microarray image processing and data acquisition. Nucleic Acids Res. 2001;29:E75–5. doi: 10.1093/nar/29.15.e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alba R, Fei Z, Payton P, Liu Y, Moore SL, Debbie P, Cohn J, D’Ascenzo M, Gordon JS, Rose JKC, Martin G, Tanksley SD, Bouzayen M, Jahn MM, Giovannoni J. ESTs, cDNA microarrays, and gene expression profiling: tools for dissecting plant physiology and development. Plant J. 2004;39:697–714. doi: 10.1111/j.1365-313X.2004.02178.x. [DOI] [PubMed] [Google Scholar]
- Smyth GK. In: Bioinformatics and computational biology solutions using R and bioconductor. Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editor. NY: Springer; 2005. Limma: linear models for microarray data; pp. 397–420. [Google Scholar]
- Smyth GK, Speed T. Normalization of cDNA microarray data. Methods. 2003;1109(31):265–273. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 Microarray software suite. Meth Enzymol. 2006;1112(411):134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Ruijter J, Ramakers C, Hoogaars W, Karlen Y, Bakker O, Van Den Hoff M, Moorman A. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37(6):e45. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M. A new mathematical model for relative quantification in real-time RT- PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vega-Bartol JJ, Santos RR, Simões M, Miguel CM. Normalizing gene expression by quantitative PCR during somatic embryogenesis in two representative conifer species: Pinus pinaster and Picea abies. Plant Cell Rep. 2013;32:715–729. doi: 10.1007/s00299-013-1407-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Microarray data normalization and replicate clustering. Box plot of the distribution of the red and green intensity ratio (M) from the thirty hybridized chip arrays before (A) and after (B) normalization.
Microarray intensity values for the 30 slides after normalization and correspondence between spot, EST accession number and Pinus pinaster unigene.
Functional annotation of the Pinus pinaster unigenes. Annotation was based on the homologous in NCBI and Arabidopsis databases, including gene putative prediction, gene ontology, pathways and families.
Quality assessment of the gene annotation. (A) Similitude values of the BLASTX alignments. (B) Species with the best BLASTX alignment of each query sequence. (C) Number of gene ontology terms per sequence for each group of terms. (D) Relation between number of gene ontology terms and length of the query sequence.
Functional annotation, specificity, clustering results and statistical analysis of the differentially expressed transcripts.
Clustering, fold-change and functional annotation of the differentially expressed transcripts in consecutive embryo stage-to-stage transitions.
Correlation of gene expression during A. thaliana and P. pinaster embryogenesis. The A. thaliana globular, heart, torpedo, bent and mature embryo stages [16] were considered equivalent to the Day0, Day5, Day11, Day15 and Day25 embryo samples of P. pinaster, respectively. For each sampling time, genes were plotted in a scatter graph using the A. thaliana (Y-axis) and P. pinaster (X-axis) expression values as coordinates.
Correlation of the expression profiles between A. thaliana and P. pinaster along five embryogenesis stages. Enrichment analysis values of the gene ontologies over/under-represented in the common subset of highly co-regulated transcripts.
Enrichment analysis of the genes with a highly similar expression profile in A. thaliana and P. pinaster.
Primers, efficiencies, Ct values and normalization of the target genes used for the data validation by qRT-PCR.