

Abstract
Microglial cells aggregate around amyloid plaques in Alzheimer's disease, but, despite their therapeutic potential, various aspects of their reactive kinetics and role in plaque pathogenesis remain hypothetical. Through use of in vivo imaging and quantitative morphological measures in transgenic mice, we demonstrate that local resident microglia rapidly react to plaque formation by extending processes and subsequently migrating toward plaques, in which individual transformed microglia somata remain spatially stable for weeks. The number of plaque-associated microglia increased at a rate of almost three per plaque per month, independent of plaque volume. Larger plaques were surrounded by larger microglia, and a subset of plaques changed in size over time, with an increase or decrease related to the volume of associated microglia. Far from adopting a more static role, plaque-associated microglia retained rapid process and membrane movement at the plaque/glia interface. Microglia internalized systemically injected amyloid-binding dye at a much higher rate in the vicinity of plaques. These results indicate a role for microglia in plaque maintenance and provide a model with multiple targets for therapeutic intervention.
Keywords: amyloid, presenilin, microglia, multiphoton imaging, Alzheimer's disease, inflammation
Introduction
A defining feature of Alzheimer's disease (AD) is the cerebral accumulation of amyloid. Because a significant amyloid load is already present at the time of earliest clinical symptoms (Lopresti et al., 2005), a therapeutic approach that addresses the deleterious effects of existing amyloid deposits is essential (Schenk, 2002). Although the production of β-amyloid (Aβ), the fibrillary component of amyloid plaques, is thought to be primarily neuronal (Goedert, 1987; Calhoun et al., 1999), it is the microglial cells that react to amyloid as to other brain injuries, and microglia have been shown to have the ability to phagocytose Aβ, at least in culture (Bard et al., 2000; Rogers et al., 2002; D'Andrea et al., 2004; Frackowiak et al., 2005). Indeed, the most successful of the amyloid clearing therapies, immunization, has been indirectly linked to microglial activation (Hardy and Selkoe, 2002; Wilcock et al., 2004; Barger, 2005).
It has long been recognized that microglia are associated with cerebral amyloid deposits (Itagaki et al., 1989; Wegiel and Wisniewski, 1990; Stalder et al., 1999), but other basic questions about their role in AD pathophysiology remain controversial (Morgan et al., 2005). Hypotheses with respect to the potential role of microglia in plaque turnover and clearance are paralleled by those focusing on the potential negative inflammatory effects mediated by microglia (Monsonego and Weiner, 2003; Morgan et al., 2005; Town et al., 2005; Wyss-Coray, 2006). In vivo imaging of amyloid over time has demonstrated long-term stability of amyloid deposits (Christie et al., 2001) and existing amyloid deposits remain even when overproduction of Aβ is interrupted using a tet-off system (Jankowsky et al., 2005). Recent evidence has also highlighted the role of infiltrating over resident cells, hypothesizing their importance for restricting plaque growth (Simard et al., 2006). Together, these results seem to indicate that, although microglia are capable of amyloid clearance, additional factors are necessary for induction of effective phagocytic activity in vivo and that the nature of their response is heterogeneous.
We have used repeated in vivo imaging with time delays varying from minutes to weeks, to study the dynamic interaction of microglia and amyloid. A double-transgenic mouse expressing mutant amyloid precursor protein (APP) and presenilin 1 (PS1) (Radde et al., 2006) was crossed with mice overexpressing green fluorescent protein (GFP) in microglia via an Iba-1 promoter (Hirasawa et al., 2005). As shown previously (Davalos et al., 2005; Nimmerjahn et al., 2005), in control mice, microglia processes are remarkably motile, surveying a defined area of their environment and expanding previous reports, also exhibit substantial migration in baseline conditions. We compare this with plaque-bearing APPPS1 mice, show the time course of various alterations in normal microglial function, and provide direct evidence for ongoing uptake of Aβ. These data provide a model involving multiple previously hypothetical microglia-related pathways, illustrate the roles of resident microglia, and highlight potential sites for therapeutic intervention.
Materials and Methods
Mice.
Hemizygous Iba-1–GFP mice (Hirasawa et al., 2005) were crossed with hemizygous APPPS1 mice that express both the APP and PS1 transgenes through coinjection of the constructs (Radde et al., 2006), and, for this study, GFP+ littermates were analyzed either with (APPPS1+) or without (APPPS1−) the double Alzheimer's-associated mutations [Thy-1 promoter (Andra et al., 1996); APPKM670/671NL and PS1L166P]. Both lines were generated on a C57BL/6 background, and both male and female mice were used in this study. Mice were group housed in pathogen-free conditions until surgery, after which they were singly housed. All procedures were in accordance with an animal protocol approved by the University of Tübingen and the government of Baden-Württemberg.
Surgery.
One week before imaging, a rectangular cranial window was installed under isoflurane anesthesia. Briefly, animals were secured on a modified stereotax while placed on a warming pad. The skin and the periosteum were removed to expose the skull. Lines forming a rectangle (from the midline ±2 mm and at approximately −0.5 and −4.5 mm from bregma) were gently drilled onto the skull surface. Using a pair of angled forceps, the circumscribed portion of the skull was then gently removed, being careful not to disturb or puncture the dura. A matching, custom-cut glass coverslip (thickness, 130–170 μm) was held with a micromanipulator and slowly lowered until it was parallel to the edge of the skull. Once correctly positioned, the coverslip was sealed to the skull with light-curing dental cement (Flowline; Heraeus Kulzer, Hanau, Germany). A 2-mm-high wall made of dental cement was built up around the coverslip to create a chamber for water immersion. In addition, a dental-cement screw receptacle was created to accept a metal rod for securing the mouse during imaging. The vast majority of mice so operated had stable windows for at least several months, with very few instances of obscuring dural thickening.
Imaging.
Based on pilot studies of the onset and decay of signal, 2 h before each imaging session, all mice for the quantitative studies (including APPPS1−) were injected intraperitoneally with 10 mg/kg methoxy-X04 (Klunk et al., 2002) (5 mg/ml in 50% DMSO; 50% NaCl, pH 12). Although the rapidity of amyloid labeling is somewhat slower with intraperitoneal versus intravenous injections, our preliminary work indicated that these injections led to reduced cardiovascular side effects and improved feasibility/consistency for multiple injections (10 per mouse in many cases). To facilitate repeated imaging of the same sites in the quantitative studies, Texas Red dextran (70,000 Da molecular weight; 12.5 mg/ml in sterile PBS; Invitrogen, Carlsbad, CA) was injected intravenously immediately before imaging, providing a fluorescent angiogram (Spires et al., 2005). Before each imaging session, mice were anesthetized with isoflurane (induction at 5% and immediately reduced and held constant at 1%), and their skull was held stable within a custom-built circular stereotaxic apparatus via a tiltable and rotatable metal rod screwed into the aforementioned screw receptacle. The window was then cleaned with EtOH, and the mouse/apparatus was secured under the microscope within a custom-built bridge connected to a motorized x–y stage (Luigs & Neumann, Ratingen, Germany). Under the guidance of a video camera (Pixelfly; pco imaging, Kelheim, Germany) using a 10× objective under ambient illumination, custom software recorded the location of two recognizable brain landmarks based on the window borders and underlying blood vessels, allowing precise relocation of multiple regions across imaging sessions. All imagery was taken using a 40× water immersion lens (0.8 numerical aperture, U-V-I 0/D; Leica Microsystems, Bensheim, Germany). A Leica DMLFS microscope attached to a Spectra Physics (San Jose, CA) Mai-Tai laser (tunable 770–990 nm) provided multiphoton excitation at 910 nm, and detection was via two non-descanned detectors (R6357 P.M.T.; Hamamatsu, Bridgewater, NJ) located close to the objective lens. The two channels were detected simultaneously, split by an FITC/tetramethylrhodamine isothiocyanate filter (reflection short pass, 560; bandpass, 525/50; bandpass, 610/75). Pilot studies determined the minimal excitation necessary to detect microglia and their processes, without causing detectable microglial activation, even after multiple xyzt scans per day. All subsequent imaging used a protocol with substantially less excitation time. GFP, the Congo Red derivative methoxy-X04, and Texas Red were all detected in two imaging channels, with the emission curve of methoxy-X04 leading to a 50/50 split between channels. For the quantitative studies, imagery was subsequently divided into three channels via a custom algorithm (as displayed in Fig. 1 A), the validity of which was tested by changing the excitation wavelength to 810 nm, where at the 910 nm optimized detection settings, no GFP was visible. As described in Figure 1, two distinct imaging protocols were used in different mice to study microglial process dynamics (50 μm Z-stacks every 9 min, 10 sessions; n = 3, APPPS1+; n = 4, APPPS1−) and plaque development/microglial migration (100 μm Z-stacks, two rounds of five sessions over 3 d separated by 1 month; n = 4, APPPS1+; n = 2, APPPS1−). An average of five different xyz locations (starting depth, 35–70 μm) could be consistently imaged across all sessions for each mouse. Additional mice were used for in vivo/histological mapping (n = 2) and as non-methoxy-injected controls (n = 2). In all subjects, the imaged location was selected in a semirandom manner (e.g., no bias attributable to existing plaque size or other characteristics), with the only exclusion criteria being the presence of a large vessel on the cortical surface. No microglial activation was observed because of imaging, as evidenced by subsequent overview scans of the imaged area versus surrounding non-imaged area and by postmortem histology of the imaged regions.
Figure 1.
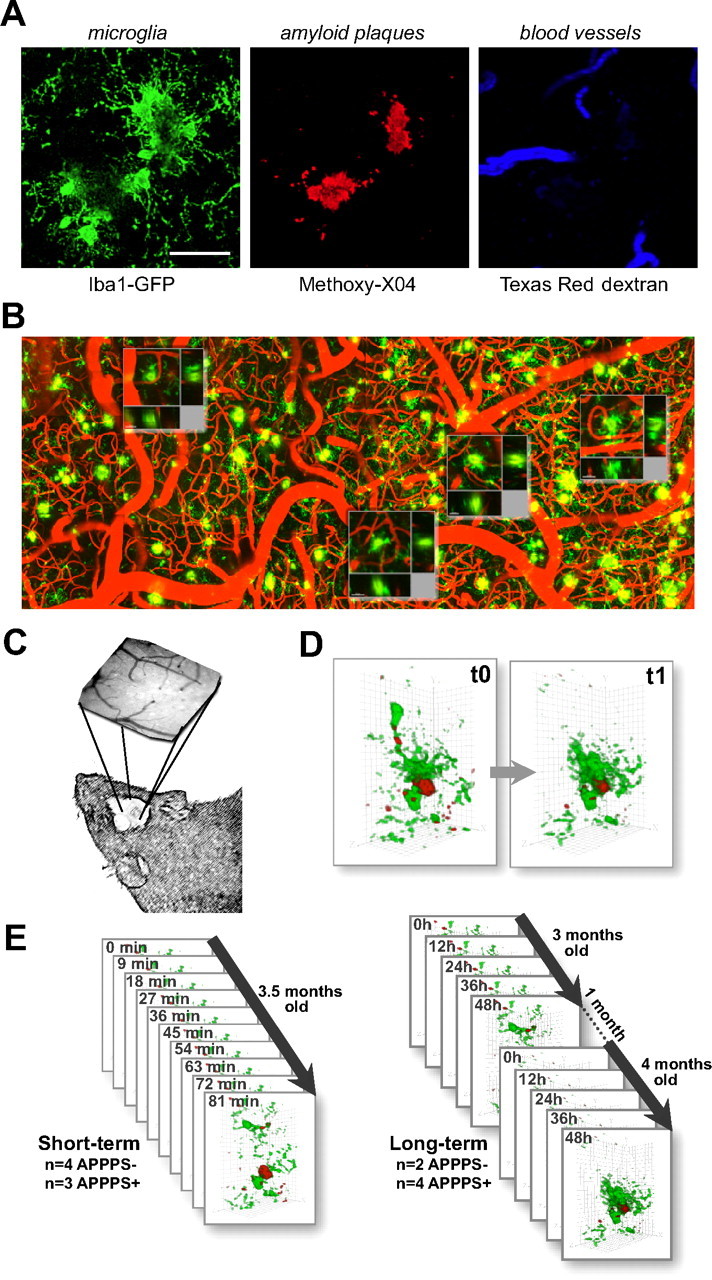
In vivo imaging methods and time course for quantification. A, Simultaneous imaging of three fluorescent signals was performed: green, microglia (through Iba-1-driven GFP expression); red, amyloid deposits [after systemic injection of methoxy-X04 (Klunk et al., 2002)]; and blue, blood vessel labeling as a spatial reference (through systemic injection of Texas Red-coupled dextran). B, An overview image before channel separation of blood vessels (red), microglia (green), and amyloid (yellow) spanning 1.8 mm, which allowed for matching individual plaques and microglia with subsequent histological sections. The location of Z projections from 4 of the 10 high-magnification image stacks from this mouse are shown as insets. The rightmost location is shown with histological analysis in Figure 6 (flipped vertically). C, A coordinate system was devised for consistent imaging of the same volumes using ambient illumination of blood vessels on the brain surface. D, Volume rendering of microglia/plaque interaction over time, in this case illustrating microglial migration. E, Image stacks were taken in multiple locations over time in each mouse (mean, 5 volumes per mouse for both APPPS1+ and APPPS1−), using two different protocols: “short term” for study of the dynamics of individual microglia and “long term” for study of plaque development and microglia migration/volume changes. Scale bar (in A), 50 μm. Images are from a maximum Z projection with a depth of 50 μm.
Histology.
Immediately after the last imaging session, mice were transcardially perfused with ice-cold 4% paraformaldehyde in PBS, and the brains were removed, postfixed overnight in the same fixative, cryoprotected in increasing concentrations of glycerol (10–20%) in phosphate buffer, frozen in 2-propanol (Merck, Darmstadt, Germany), and stored at −80°C until sectioning on a freezing-sliding microtome (40 μm sections at a plane parallel to the window orientation). Sections were either observed after washing without additional manipulation or were immunostained using antibodies to visualize microglia (rat polyclonal Iba-1; Waco, Richmond, VA), Aβ [mouse monoclonal β1 or rabbit polyclonal NT12 (Paganetti et al., 1996; Sturchler-Pierrat et al., 1997; Pfeifer et al., 2002); kindly provided by P. Paganetti and M. Staufenbiel, Novartis Pharma, Basel, Switzerland], astrocytes (rabbit anti-GFAP; Dako, High Wycombe, UK), and/or lysosomes [mouse monoclonal lamp-1 (lysosome-associated membrane protein 1) (BD Biosciences, San Diego, CA) and rat monoclonal lamp-1 (Abcam, Cambridge, UK)]. Visualization was via the appropriate fluorescent-coupled secondary antibodies (Alexa 568/594/633; Invitrogen) or via biotinylated antibodies, an avidin–biotin reaction and Vector SG Blue. Verification of the specificity of the Aβ and lysosomal labeling was done using Fab-fragment-specific secondary antibodies (Invitrogen) and/or by preblocking Fc receptors with Mouse BD Fc Block (rat monoclonal CD16/32; BD Biosciences PharMingen). Images of entire serial sections were captured using the Mosaic X software feature of Axiovision (Zeiss, Jena, Germany), and subsequent high-magnification imaging was done with a Zeiss LSM 510 Meta confocal microscope, which allowed measurement of emission spectra via lambda scanning and flexible definition of excitation parameters and detection wavelengths to avoid crosstalk between fluorescent channels.
Data analysis.
To ensure accurate analysis of slightly varying volumes across imaging sessions, after deconvolution with AutoQuantX (AutoQuant Imaging, Troy, NY), all data were registered using an automated algorithm within Amira software (Mercury Computer Systems, Berlin, Germany). Custom software then eliminated voxels that were not present in all imaging sessions. In practice, the change in imaged angle was rarely >5°, and thus the vast majority of imaged voxels were present in all datasets. The first of the five datasets from each month in the long-term imaging protocol had higher GFP intensity and were not used for quantitative analyses. Several different custom tools were developed using NIH ImageJ to analyze changes in microglia and plaque size and location, as well as microglia process movement. The location of each microglia soma or plaque center was recorded in three dimensions by a trained observer [and the three-dimensional (3D) change in this location over time was used for migration calculation], and the total number of voxels within each of these stacks over a predetermined threshold was automatically counted. The average plaque or microglia volume was then calculated by dividing the total number of voxels by the number of counted elements. Although visualization of the full extent of ramified processes was apparent in both APPPS1− and APPPS1+ mice, we cannot exclude the possibility that the observed increase in microglial volume is attributable in part to an increased expression of GFP via the Iba-1 promoter. Process movement and length was measured by tracing processes in three dimensions, which for movement was aided by color coding of the imagery according to subtraction with the adjacent time points (see Fig. 4). Polarity for the microglia near plaques was measured by dividing processes into two categories, split by a line through the center of the soma, at an angle tangent to the nearest plaque perimeter. Methoxy-X04 particles colocalized within microglia were manually counted in three dimensions. When differentiating microglia based on relation to amyloid, “on” were microglia with soma contacting the plaque surface (GFP-positive voxels directly neighboring or colocalized with methoxy-positive voxels), and “near” were those with soma within 50 μm from the plaque surface. Many of the figures, movies, and qualitative visualizations were done with the assistance of Imaris software (Bitplane, Zurich, Switzerland). The quantitative data follow individual plaques, microglia, or imaged volumes over time, and thus the respective statistics use these as independent observations. This analysis accurately captures the desired variability in amyloid extent between heterogeneneous locations within an individual subject (n = 4–6 locations per subject, with plaque-containing subvolumes ranging from n = 3–18 per location). Total numbers are given for each comparison, and no additional exclusion criteria were applied. Data reported are mean ± SEM, and reported significance is based on ANOVA (in some cases repeated measures), followed by Fisher's PLSD for categorical variables, regression (linear/logarithmic) for continuous variables, or, in the case of multivariate analyses of continuous variables, a standard least squares regression model.
Figure 4.

Microglia process dynamics and length are altered in relation to amyloid. A, Two time points, separated by 9 min, are superimposed with voxels that were present in the first but not the second (retraction) indicated in purple, and in the second but not the first (extension) indicated in yellow, with all unchanged voxels in white. Process retractions and extensions in microglia from APPPS1− mice typically occur isotropically distributed throughout their ramified tree structure. In APPPS1+ mice, polarized microglia (arrow) and macrophagic microglia (arrowhead) “near” or “on” amyloid plaques no longer survey their environment in a distributed, isotropic manner. Inset, Right panel, Process movement (in this case, extension) was measured with lines drawn in three dimensions between the start and end of new processes. Scale bar, 50 μm. B, Significant differences in the total process length per cell were seen for the different categories, with microglia near plaques exhibiting increased process length, and those immediately “on” plaques with substantially reduced length (main group effect, F (1,22) = 22.2; p < 0.01 for all comparisons). Both types of plaque-associated microglia also showed significant polarity (indicated for the “near” microglia by diagonal shading for processes oriented toward plaques and solid for processes oriented in the opposite direction), with microglia “on” plaques only having processes oriented away from the plaque surface. C, Although morphology between the microglia types was substantially altered, the overall extent of process movement was similar (analyzed microglia: APPPS1−, n = 10; APPPS1+“near”, n = 10; APPPS1+“on”, n = 6), with only the microglia “on” amyloid exhibiting a slight but significant change in speed of process extension/retraction (μm/time point − 9 min).
Results
Visualization of microglia and amyloid over time
Simultaneous imaging of microglia, amyloid, and blood vessels was performed as described in Materials and Methods (Fig. 1). Imaging parameters were first optimized to ensure that the amount of laser power used did not result in any detectable microglia reaction or photodynamic damage, even with multiple sessions of scanning the same volume. Mice used in the quantitative studies were divided into two groups imaged at different timescales. The first was imaged at brief intervals (minutes) to study process dynamics and subcellular events, and the other was imaged over a longer time period (days to 1 month) to study plaque formation and microglial migration, number, and size (Fig. 2 A). A total of 39 amyloid-containing volumes from four APPPS1+ mice and 33 volumes from two APPPS− mice were imaged at 3 months of age and again at 4 months, over a total of 10 sessions (“long-term” mice). A coordinate system was devised whereby multiple separate image stacks could be precisely located across imaging sessions, and 3D alignment of the subsequent image stacks based on blood vessels was performed to correct for slight changes in stack placement or orientation. Thus, image sequences of disparate timescales could be reliably generated for quantitative analyses.
Figure 2.
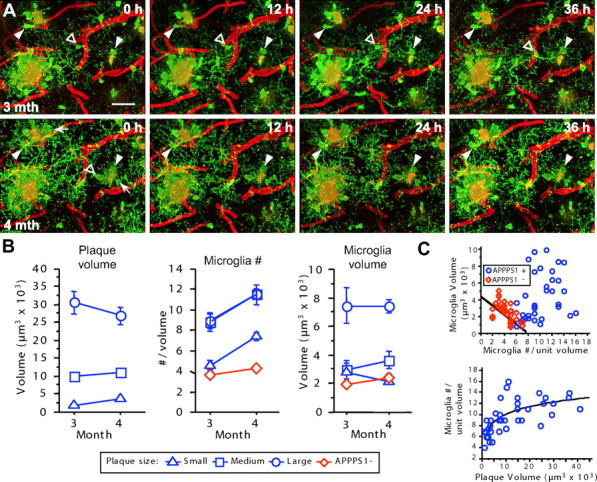
Existing amyloid deposits are stable over time despite the microglial response. A, Long-term imaging of the same volume is shown with microglia in green, amyloid in yellow, and blood vessels in red (non-channel separated). Individual microglia surrounding amyloid remain stable over periods of 1 month (arrowheads), but new microglia are also evident (arrows). Microglial processes in each 12 h time interval have new spatial locations, with the exception of some of the largest processes, often also tied to amyloid. An example is also apparent of a resting microglia (open arrowhead) that first sends processes and subsequently migrates to contact a plaque. All images represent Z projections of 30 μm. Scale bar, 50 μm for all images. B, Quantification of mean plaque volume and microglia number and volume over time. Overall plaque volume remained stable over 1 month but differed between size categories. The number of microglia in each volume increased significantly over 1 month, independent of plaque volume. Although larger plaques were surrounded by larger microglia, the average microglia size did not increase over time. C, Size and volume of microglia are inter-related, and the upper limit of the microglial number increase around plaques is compensated by an increase in individual microglial volume. In APPPS1− mice (red diamonds), the number of microglia and the volume of individual microglia in any volume showed an inverse relationship (R 2 = 0.35; p < 0.001). The number of microglia surrounding plaques increased in relation to plaque volume, reaching an apparent ceiling at a plaque size of ∼15,000 μm3 (logarithmic regression, R 2 = 0.51; p < 0.0001). Values for each mouse represent the average of the long-term imaging sessions of the second month (similar results were obtained regardless of which month's values were used for this analysis).
Microglia number and volume increase
Microglia morphology was quantified over multiple sessions for a period of 1 month (Fig. 2). As a first analysis, imaged volumes were categorized according to their plaque size (radius: small < 10 μm < medium < 15 μm < large; n = 18, 9, 12 respectively), which revealed a change in the size of individual plaques over 1 month (Fig. 2 B) (repeated-measures ANOVA, size × month interaction, F (2,36) = 15.5; p < 0.001]. Small plaques exhibited an average size increase of 84% (F (1,17) = 19.1; p < 0.001), and large plaques decreased in size by 12% (F (1,11) = 9.6; p < 0.05). From 3 to 4 months of age, the number of microglia in volumes containing plaques increased by an average of 2.7 microglia (Fig. 2 B) (repeated-measures ANOVA; F (1,38) = 78.5; p < 0.001). This increase was independent of the size of the amyloid deposit (repeated-measures ANOVA, month × plaque size, interaction, F (2,36) = 0.1; p = 0.95). Microglial number also increased slightly in APPPS− mice but at a significantly lower rate than in the plaque-bearing APPPS1+ mice (Fig. 2 B) (repeated-measures ANOVA, month × transgene interaction, F (1,70) = 29.1; p < 0.001). The number of microglia surrounding an individual plaque reached an upper limit in relation to plaque size (Fig. 2 B,C).
The average volume of plaque-associated microglia was substantially higher than the volume of resting microglia in APPPS1− mice (Fig. 2 B) (221%; F (1,70) = 12.1; p < 0.001). A strong positive relationship was also found between plaque volume and the average volume of associated microglia (R 2 = 0.77; p < 0.001). Microglia surrounding medium plaques were only 35% larger than those around small plaques, but the large plaques had substantially larger microglia than the medium plaques (225%). Together with the data above, microglia volume thus differentiates the large from medium plaques, which had equivalent numbers of microglia. Interestingly, in the control mice, an inverse relationship was seen between the number of microglia in any given volume and the average microglia volume (Fig. 2 C), suggesting that regionally, the neuropil may be surveyed by many smaller or fewer larger microglia. Microglia volume was similar in fixed tissue immunostained against Iba-1 (data not shown), indicating that increases were not simply a result of increased GFP expression.
Microglial migration
Extending previous reports that studied shorter time intervals (Davalos et al., 2005; Nimmerjahn et al., 2005), we were able to observe substantial movement of microglia somata over time in both the APPPS1+ and APPPS1− mice (Figs. 2 A, 3). In APPPS+ mice, approximately half of the microglia adjacent to plaques (within 50 μm) subsequently migrated such that their somata were immediately adjacent to the plaque periphery (over a period of 24–48 h), whereas the somata of the other half remained distant (10–50 μm) from the plaque periphery with their processes, nevertheless maintaining a connection to the amyloid surface. Previously resting microglia with symmetrical ramified processes thus first became polarized and, on reaching the plaque, had enlarged somas and reduced ramification (Figs. 2 A, 3 B). Despite microglia movement toward the plaques, an overall decrease in microglial migration was observed in APPPS1+ mice compared with APPPS1− mice (Fig. 3 C) (F (1,352) = 15.9; p < 0.001) primarily because the vast majority of the more numerous microglia in the APPS1+ mice (Radde et al., 2006) remained spatially stable around plaques. Plaque-associated microglial process extension and migration left formerly surveyed areas of the neuropil without detectable microglial coverage, which, over time, in some cases, was followed by influx of new/surrounding microglial cells and processes. Although we cannot completely exclude the possibility of microglia cell death, disappearance of a plaque-associated microglia over the 1 month observation period was rare and would also be explained by a microglia leaving the imaged volume. In APPPS1− control mice, 48% of microglia migrated a distance >5 μm (up to 38 μm) over a 1 month period, whereas the percentage was only 29% in APPPS+ mice (Fig. 3 D).
Figure 3.
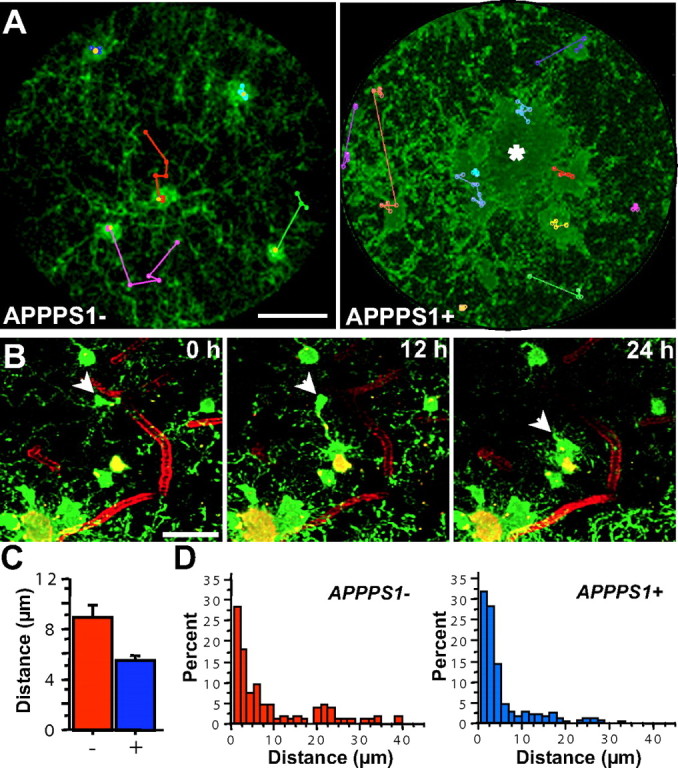
Microglia migrate in control mice and also in plaque-bearing mice, unless indefinitely attached to amyloid. A, Z projections (100 μm thick) of microglia in APPPS1− or APPPS1+ mice with the movement history of individual microglia superimposed in colored “tracks” (actual distance moved was calculated via manual identification of the soma location in three dimensions at various time points). The displayed session is the fifth (first session from the second month; yellow marker). The center of the amyloid deposit in the APPPS1+ mouse is indicated by an asterisk. Scale bar, 20 μm. B, A plaque-associated microglia (arrow) first sends processes toward the plaque (12 h), and subsequently migrates until the soma is in contact with the amyloid (24 h). Scale bar is 50 μm and images represent Z projections of 25 μm. C, The average movement of microglial soma over 1 month is significantly decreased in the plaque mice (APPPS1+, n = 248; APPPS1−, n = 106). D, Shown is a histogram of migration for individual microglia in APPPS1− mice (red) and APPPS1+ mice (blue). In both groups, the majority of microglia soma remained spatially stable over 1 month, but a subpopulation exhibits significant migration, up to 40 μm.
Alterations in fine morphology and process motility
Overall, microglia in the vicinity of plaques had 48% higher average process length than microglia from APPPS1− mice, whereas the microglia “on” plaques only had 27% of the process length compared with microglia from APPPS1− mice (Fig. 4). The plaque-associated glia also became significantly polarized with the “near” microglia having 62% of their processes in the direction of plaques (F (1,8) = 10.9; p < 0.05), and the “on” microglia exhibiting no fine processes in the plaque direction.
Similar to a previous report (Nimmerjahn et al., 2005), microglia in control mice rapidly surveyed their environment, sending processes extending in many directions symmetrically from their cell soma (Fig. 4) (supplemental Movie S1, available at www.jneurosci.org as supplemental material), at an average rate of 0.83 μm/min, with extension and retraction at equivalent rates. In plaque-bearing APPPS1+ mice, processes extended rapidly from the cell soma to contact amyloid within hours (Fig. 2 A), such that the surface area of plaques was covered by microglial processes. Speed of process movement, however, was surprisingly similar regardless of the association of microglia with amyloid (Fig. 4), with extensions and retractions equivalent between groups and only the remaining processes of the “on”-plaque microglia exhibiting a small but significant difference in speed (20%; main group effect, F (2,49) = 10.3; p < 0.001 for main effect and all group post hoc tests) compared with microglia in APPPS1− mice.
Fluctuation in plaque size
As mentioned above (Fig. 2), smaller amyloid deposits increased in size, whereas some of the larger plaques decreased. To examine the variability in plaque volume across imaging sessions and potentially exclude effects attributable to labeling/imaging, plaque volume change was screened by viewing the four imaging sessions of each month as independent measures and only plotting those with a significant change (p < 0.05) over 1 month (Fig. 5 E) (n = 13; 33%). Compared with the majority of plaques that maintained a constant size, the growing plaques had little initial microglia coverage (average total volume for the first month), and, conversely, the plaques that decreased in volume had the most microglial coverage (Fig. 5 A,C, insets) (Fig. 5 F, R 2 = 0.36; F (1,37) = 20.43; p < 0.001). The relationship between microglia and plaque volumes became stronger when only including the plaques with statistically consistent differences over 1 month (R 2 = 0.63; F (1,11) = 18.35; p < 0.01) and also held true for the change in microglia volume over 1 month (R 2 = 0.37; F (1,11) = 6.4; p < 0.05). That both the microglia volume of the first month and the change in microglia volume over the following month predict plaque volume change indicates that we have captured an ongoing process (e.g., the microglia volume has already increased to a predictive level and continues to do so). To ask whether the related variable, microglia number, also influenced a change in plaque size, a standard least squares analysis was performed with total microglia volume, number, and their interaction as factors. Although this statistical model slightly improved the prediction of the plaque volume change (R 2 = 0.41; F (3,35) = 8.19; p < 0.001), total volume was the only statistically significant individual factor (F = 6.44; p < 0.05).
Figure 5.
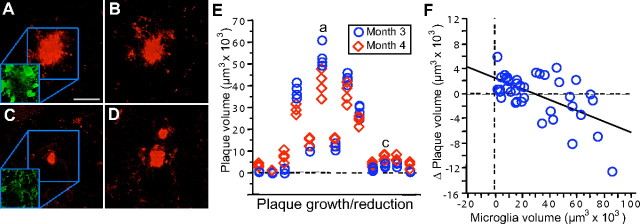
Amyloid plaque volume changes over time. A–D, Although the majority of plaques did not change in size over 1 month, examples of a reduction in plaque volume (A, B) and increase in plaque volume (C, D) were evident. Insets illustrate the contrasting microglia coverage from the shrinking plaque (A) versus a growing plaque (B). E, The individual volume measurements for the 13 of 39 plaques that had consistent volume change over multiple imaging sessions. Measurements from the four independent imaging sessions at 3 months of age are shown as blue circles, and data from the following month are shown as red diamonds. Only plaques with a significant difference between months are shown, and the plaques from A–D are indicated. F, The change in plaque volume over 1 month is related to microglia volume (total volume of all microglia/imaged volume, first month). Plaques that increased in volume over 1 month tended to have less microglia coverage, whereas those that decreased in volume had the most microglia. Scale bar: (in A) A–D, 25 μm. Figures are maximum Z projections of 25 μm.
The pooled monthly analysis provided a robust comparison for long-term size changes, but the shorter-term imaging protocols indicated more rapid alterations in amyloid fibril size and morphology [in both minutes (supplemental Movies S2, S3, available at www.jneurosci.org as supplemental material) and hours (variability in plaque volume evident in Fig. 5 E)]. Thus, rather than method variability such as in the labeling efficiency of methoxy-X04, it could be that the plaque size itself fluctuated even over a 12 h time span. Indeed, predictive analysis between microglia volume and the extent of plaque volume change (SD), indicated a strong relationship (R 2 = 0.53; F (1,37) = 42.3; p < 0.001). Although this statistic is influenced by the correlation in absolute sizes, when plaque size was included as a factor, microglia volume had equivalent predictive value [standard least squares model (R 2 = 0.57; F (3,35) = 15.24; p < 0.001); independent factors plaque volume (F = 2.6; p = 0.12) and total microglia volume (F = 2.8; p = 0.10)]. Together, the evidence thus suggests an ongoing dynamic homeostasis between amyloid and microglia, with modest growth in the subset of regions having low microglia reaction and, in the smaller subset of regions with the most microglial reaction, removal of existing amyloid. In particular, the nature of the microglia response was important, with increases in number eclipsed by the importance of an increase in microglial volume.
Microglia uptake of injected dye
The systemically injected amyloid-binding dye methoxy-X04 showed the expected high-contrast labeling of amyloid plaques (Klunk et al., 2002), but we also noticed a punctate fluorescence in the neuropil (Fig. 6 A) that was not present in mice before injection of the dye (Fig. 6 E). The vast majority of these puncta colocalized with GFP, and additional 3D analysis indicated that the puncta were within GFP+ microglia (Fig. 6 B). Although the puncta were also present within microglia in methoxy-injected APPPS1− mice, the density of puncta was markedly higher in APPPS1+ mice, particularly in the vicinity of amyloid plaques (Fig. 6 E) (main group effect F (5,318) = 116.8; all post hoc tests for the two plaque-associated groups, p < 0.001 compared with each of the other groups). Subsequent high-magnification imaging of individual particles over time revealed both movement of the particles within the cytoplasm and, less frequently, the disappearance of individual particles (Fig. 6 A) (supplemental Movie S3, available at www.jneurosci.org as supplemental material). Higher-power imaging of the plaque–glial interface also revealed rapid membrane movement (supplemental Movies S2, S3, available at www.jneurosci.org as supplemental material). To further study particle localization and whether they represent a nonspecific dye uptake or uptake of Aβ, we performed detailed postmortem morphological analyses.
Figure 6.
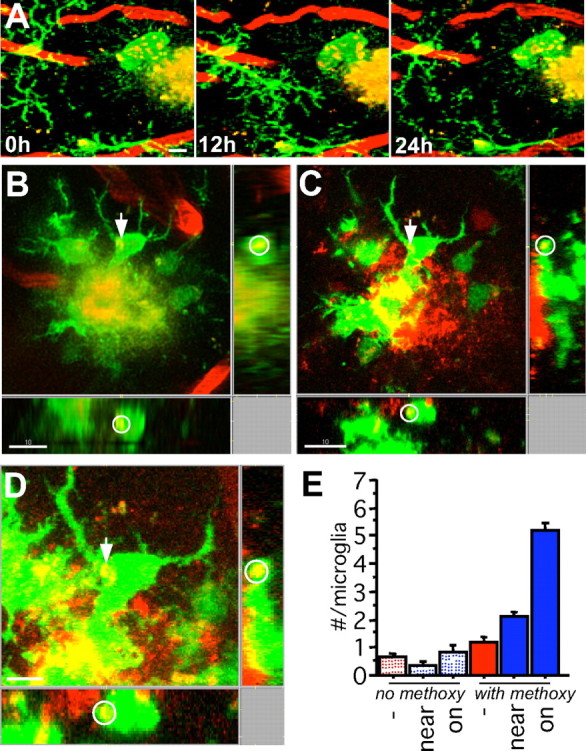
Uptake of Aβ by plaque-associated microglia. A, A macrophagic plaque-associated cell exhibits many internalized fluorescent particles that move within the cell soma over time (maximum Z projection, 35 μm). B–D, In vivo (B) and histological (C, D) analyses of the same plaque, microglia, and internalized puncta (B; red, blood vessels; green, microglia; yellow, methoxy X-04) confirm the localization and, via immunostaining, reveals colocalization with Aβ (C, D; red, Aβ via NT-12/Alexa 568, green, GFP+ microglia). The same punctum is indicated by an arrow and can be visualized as within the microglia in the orthogonal x and y projections (indicated by a circle). E, Quantification of puncta colocalized with microglia in vivo in uninjected mice versus those injected with methoxy-X-04 indicates a dramatic increase in the transgenic mice, particularly in the vicinity of amyloid plaques [number of microglia analyzed: no methoxy, 59/49/26; with methoxy, 62/57/71 (APPPS1−/near/on)]. Scale bars: A–C, 10 μm; D, 5 μm.
To match individual locations, an overview volume was imaged with overlapping tiles, followed by higher-resolution imaging of individual microglia and plaques within this volume (Fig. 1 B). Immediately after imaging, tissue was fixed, serial sectioned for 3D reconstruction, and immunostained with antibodies against Aβ. Subsequent confocal microscopy images revealed similar microglial morphology to that imaged in vivo and surrounding plaques, the spots of dye uptake imaged in vivo also were positive for Aβ (Fig. 6 B–D). Examination of this colocalization throughout the tissue revealed much reduced Aβ labeling within microglia in areas distant from plaques and no such labeling in APPPS1− mice. Although examples could be found in which fine amyloid fibrils emanated from the plaque core and were interdigitated with microglial processes as shown previously (Stalder et al., 2001), the observed puncta had a round form much larger than, and not continuous with, amyloid fibrils. Indeed, they were most often found within the microglia soma opposite to the plaque–glia interface, moved within the cell soma independently of the plaque itself (supplemental Movie S3, available at www.jneurosci.org as supplemental material), and appeared and disappeared over time.
The internalized particles were then further characterized as to subcellular compartment. Immunostaining for the lysosomal marker lamp-1 revealed small and large puncta throughout the neuropil and occasional loci of staining near blood vessels (Fig. 7). Larger and more frequent lamp-1 immunostaining was observed in the vicinity of plaques, and many of these puncta colocalized with GFP+ microglia (Fig. 7). This observation was specific to microglia, because no such increase or plaque-associated colocalization was observed in astrocytes (Fig. 7). Because we were concerned about nonspecific labeling of lysosomes and microglia generally, parallel tests to verify specificity were conducted with Fab-fragment secondary antibodies and/or a mouse Fc fragment preblock.
Figure 7.
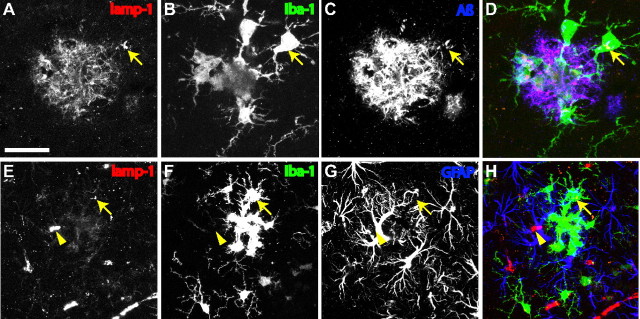
Aβ is localized specifically within microglial lysosomes. A–D, lamp-1-positive lysosomes (A) colocalize with Aβ immunostaining (C) within GFP+ microglia (B), in the vicinity of amyloid plaques (C). Arrows indicate the most prominent example of a large Aβ-positive microglial lysosome, also apparent in the three-channel overlay (D). E–H, No internalization of Aβ or plaque-associated increase in lysosome labeling is observed for astrocytes. Arrow again indicates larger lysosomes found within a plaque-associated microglia. Although lysosomes can be found within astrocytes, no specific association with plaques is observed. Colocalization is observed however in the immunostaining of blood vessels (arrowhead), surrounded by labeling of astrocytic end feet. Images in both A–D and E–H represent maximum Z projections of 8 μm. Scale bar: (in A) A–D, 20 μm; E–H, 40 μm.
Discussion
Microglial cells undoubtedly have multiple roles in the brain (Town et al., 2005), and therefore their role in Alzheimer's disease pathogenesis has remained enigmatic. The present study thus sought to satisfy several goals. The first was to study the basic biology and kinetics of the microglial response to amyloid. Although several previous studies of amyloid-associated microglia have documented their static morphology, until individual cells are followed over time with quantitative measures, the origin, extent, and the time course of microglial transformation could not have been known. Second, previous reports have shown that plaque formation occurs at a very rapid rate and individual plaques subsequently remain relatively stable (Christie et al., 2001), but the role of microglia in the “maintenance” of this process has not been studied. By following individual plaques and microglia over various time intervals, our results support the idea that the initial rapid resident microglia reaction is followed by an ongoing heterogeneous response, not only a case of microglia providing a static physical barrier but rather a dynamic process of homeostasis. That plaque-associated microglia remain very motile at the plaque–glial interface and exhibit signs of uptake of the Aβ peptide, provides evidence that they can inhibit additional fibrillization and plaque growth. New cells continue to be added over time, and their volume predicts the fluctuation in plaque size. Finally, by comprehensively studying various aspects of the microglial response, we sought to provide a benchmark for therapeutic studies with simultaneous insight into mechanism and efficacy.
Similar to recent reports (Davalos et al., 2005; Nimmerjahn et al., 2005), we found that microglia in normal mice are rapidly and continuously surveying their environment. By imaging over a longer time course, we were also able to demonstrate that the positioning of microglia continues to evolve in the adult brain, with a subset of glia migrating over relatively large distances. Although the potential role of invading cells has been highlighted recently (Simard et al., 2006), our results indicate that the time course of this response (days) is inconsistent with a role in halting the initial rapid plaque growth and that process outgrowth by resident cells is a more likely candidate. By following the spatial location and various morphological measures over time, we were also able to demonstrate that, once microglia become attached to amyloid, their somata remain spatially stable, that individual microglia increase in volume, and that additional microglia [via either infiltration (Stalder et al., 2005) or cell division (Bondolfi et al., 2002)] supplement the reaction of existing resident microglia. Whether the ramified existing microglia that we observed at the first imaging session truly represent resident microglia or represent a population that has recently infiltrated the brain requires more detailed study. The increase in number of microglia [2.7 per imaged volume; assuming neocortical volume of ∼50 mm3 (Bondolfi et al., 2002)], would yield a total increase of 250,000 per month, similar to the 400,000 per month previously reported via stereological analysis on fixed tissue (Radde et al., 2006). Although rigorous quantification of microglia cell division is still lacking, the relative infrequency of invading cells (Stalder et al., 2005; El Khoury et al., 2007) would suggest that the majority of the new microglia are derived by proliferation.
The inverse relationship between average microglia volume and the number of microglia in normal mice indicates a process ensuring neuropil coverage, perhaps actively via cell–cell contact. This was also apparent after plaque formation with the overall volume or surface area of plaque coverage being conserved. Average microglia volume and number predicted the change in plaque volume, with overall microglial volume having the strongest predictive value. The increase in microglia number and volume around plaques appeared to be independently regulated. Although we can only speculate about governing mechanisms, the limited number of “available” microglia in any given volume could lead these to compensate with a greater volumetric increase as larger plaques form. Whether this process leads to a continued heightened state of activation, with increased ability to remove amyloid fibrils, is an important area for additional study. Although molecular mechanisms will clearly be critical, it seems that basic issues of time (e.g., the initial reaction being primarily the resident microglia) and space also play a significant role.
Previous research has highlighted the intimate relationship of amyloid fibrils and microglia membranes, without finding any evidence for the presence of fibrillar amyloid within microglia proper (Stalder et al., 1999, 2001). One might view our finding of Aβ-positive particles within microglia as contradictory to these findings, but several lines of evidence indicate that differing methodology has allowed visualization of an additional phenomena. First, our results primarily stem from the in vivo observation of an increase in an internalized fluorescent molecule (systemically injected methoxy-X04). This observation alone is a clear indication that microglia in the vicinity of plaques are more actively performing endo/pino/phagocytosis or are more capable of the same. Second, the subcellular localization of the observed particles within the soma opposite the plaque and their spheroid morphology are wholly different. Fibrils extending from the plaque to create such a morphology would have to exhibit bulbous endings rather than the well characterized tapering fine fibrils. Finally, study of these particles over time revealed both turnover and movement within the cell soma, in directions independent of the plaque itself. Postmortem histological analysis did demonstrate instances of the previously reported interdigitation of fibrils within microglia, at locations different from the internalized fluorescence, confirming the independence of these two observations in vivo. The definitive answers as to whether these particles represent uptake of dyes bound to soluble Aβ or fibrillary amyloid and whether methoxy-bound amyloid is processed differently require more detailed study.
Given the unexpectedly active relationship between microglia and existing amyloid deposits, what might the role of microglia be in the growth kinetics of amyloid deposits? It is clear from this and other studies (Bacskai and Hyman, 2002) that plaques form at a rapid rate, one that makes direct observation of the first fibrillization phase difficult. However, even when observed shortly after initial plaque formation, in the majority of cases, the entirety of the amyloid surface area is covered by microglia processes. The plaques that had less microglial coverage were the ones that tended to increase in volume over time. Microglia are known to surround sites of injury, as a putative mechanism to “seal” the injury site. Another interpretation of our results in relation to plaques is that this is a chronic process with the potential for continued fibrillization via the presence of local soluble Aβ. Our observations indicate that microglia can continuously take up Aβ, perhaps through an active process or simply as a side effect of internalization of other local debris. Regardless of mechanism, this would lead to a lower concentration at that interface and thus prevent fibril extension. Alternatively, Aβ protofibrils could be forming at the end of existing deposits, and the then-activated microglia are able to phagocytose and reprocess these fibrils (which as individual small fibrils are beyond the resolving power of even the electron microscope).
These results demonstrate a dramatic, rapid microglia response to amyloid plaques, which, without additional manipulation, is ultimately inefficient in clearing existing deposits. The description of the system as a whole (with changes in processes, migration, individual microglial volume, numbers surrounding individual plaques, and amount of phagocytic activity) extends previous hypotheses with respect to microglia biology and their involvement in plaque pathogenesis. Furthermore, such studies provide clues to additional therapeutic targets. Why for instance does the number of surrounding microglia reach an upper limit, and can that be altered by changing migratory or cell-cycle capabilities or by enabling increased infiltration from the periphery? The ongoing phagocytic activity, uptake of labeled amyloid, and potential reprocessing of amyloid indicate that microglia are capable of Aβ clearance in vivo, but what factors are necessary to lead to clearance of existing dense deposits? Given the observation that microglia volume predicts their ability to reduce plaque size, what independent factors can be used to enhance this process? Answers to these questions and identification of the molecular factors involved in recruitment, activation, and uptake will ultimately provide the mechanistic clues necessary for development of additional therapeutic targets for amyloid clearance.
Footnotes
This work was supported in part by the following: Deutsche Forschungsgemeinschaft Grant CA 477-1/2 and Alzheimer Association Grant IIRG-05-13464 (M.E.C.); National Institutes of Health Grants AG020226 and AG0011039 (W.E.K.); and German National Genome Network Grant NGFN2 and German Competence Network in Degenerative Dementias (Bundesministerium für Bildung und Forschung Grant 01GI0705) (M.J.). Neuroptix holds a license agreement with the University of Pittsburgh based on the X-compound technology described in this manuscript. Drs. Mathis and Klunk have a financial interest in this license agreement. We thank Ana Fulgencio, Bettina Wegenast-Braun, Claudia Schäfer, and Jörg Odenthal for experimental support, Matthias Staufenbiel and Paolo Paganetti for providing the anti-Aβ antibodies, Carlos Portera-Cailliau and Thomas Deller for critical comments on this manuscript, and Cornelius Schwarz for surgical support and expertise.
References
- Andra K, Abramowski D, Duke M, Probst A, Wiederhold KH, Burki K, Goedert M, Sommer B, Staufenbiel M. Expression of APP in transgenic mice: a comparison of neuron-specific promoters. Neurobiol Aging. 1996;17:183–190. doi: 10.1016/0197-4580(95)02066-7. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Hyman BT. Alzheimer's disease: what multiphoton microscopy teaches us. The Neuroscientist. 2002;8:386–390. doi: 10.1177/107385802236963. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Barger SW. Vascular consequences of passive Abeta immunization for Alzheimer's disease. Is avoidance of “malactivation” of microglia enough? J Neuroinflammation. 2005;2:2. doi: 10.1186/1742-2094-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci. 2002;22:515–522. doi: 10.1523/JNEUROSCI.22-02-00515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, Hyman BT. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J Neurosci. 2001;21:858–864. doi: 10.1523/JNEUROSCI.21-03-00858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging. 2004;25:675–683. doi: 10.1016/j.neurobiolaging.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Frackowiak J, Potempska A, LeVine H, Haske T, Dickson D, Mazur-Kolecka B. Extracellular deposits of A beta produced in cultures of Alzheimer disease brain vascular smooth muscle cells. J Neuropathol Exp Neurol. 2005;64:82–90. doi: 10.1093/jnen/64.1.82. [DOI] [PubMed] [Google Scholar]
- Goedert M. Neuronal localization of amyloid beta protein precursor mRNA in normal human brain and in Alzheimer's disease. EMBO J. 1987;6:3627–3632. doi: 10.1002/j.1460-2075.1987.tb02694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hirasawa T, Ohsawa K, Imai Y, Ondo Y, Akazawa C, Uchino S, Kohsaka S. Visualization of microglia in living tissues using Iba1-EGFP transgenic mice. J Neurosci Res. 2005;81:357–362. doi: 10.1002/jnr.20480. [DOI] [PubMed] [Google Scholar]
- Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Gonzales V, Savonenko AV, Wen JC, Jenkins NA, Copeland NG, Younkin LH, Lester HA, Younkin SG, Borchelt DR. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med. 2005;2:e355. doi: 10.1371/journal.pmed.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, Meltzer CC, Schimmel K, Tsopelas ND, DeKosky ST, Price JC. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med. 2005;46:1959–1972. [PubMed] [Google Scholar]
- Monsonego A, Weiner HL. Immunotherapeutic approaches to Alzheimer's disease. Science. 2003;302:834–838. doi: 10.1126/science.1088469. [DOI] [PubMed] [Google Scholar]
- Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–753. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Paganetti PA, Lis M, Klafki HW, Staufenbiel M. Amyloid precursor protein truncated at any of the gamma-secretase sites is not cleaved to beta-amyloid. J Neurosci Res. 1996;46:283–293. doi: 10.1002/(SICI)1097-4547(19961101)46:3<283::AID-JNR1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Käser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Hölscher C, Mathews PM, Jucker M. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Strohmeyer R, Kovelowski CJ, Li R. Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia. 2002;40:260–269. doi: 10.1002/glia.10153. [DOI] [PubMed] [Google Scholar]
- Schenk D. Amyloid-beta immunotherapy for Alzheimer's disease: the end of the beginning. Nat Rev Neurosci. 2002;3:824–828. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder M, Deller T, Staufenbiel M, Jucker M. 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging. 2001;22:427–434. doi: 10.1016/s0197-4580(01)00209-3. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Wisniewski HM. The complex of microglial cells and amyloid star in three-dimensional reconstruction. Acta Neuropathol (Berl) 1990;81:116–124. doi: 10.1007/BF00334499. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]