

Abstract
Folate deficiency and resultant increased homocysteine levels have been linked experimentally and epidemiologically with neurodegenerative conditions like stroke and dementia. Moreover, folate deficiency has been implicated in the pathogenesis of psychiatric disorders, most notably depression. We hypothesized that the pathogenic mechanisms include uracil misincorporation and, therefore, analyzed the effects of folate deficiency in mice lacking uracil DNA glycosylase (Ung−/−) versus wild-type controls. Folate depletion increased nuclear mutation rates in Ung−/− embryonic fibroblasts, and conferred death of cultured Ung−/− hippocampal neurons. Feeding animals a folate-deficient diet (FD) for 3 months induced degeneration of CA3 pyramidal neurons in Ung−/− but not Ung+/+ mice along with decreased hippocampal expression of brain-derived neurotrophic factor protein and decreased brain levels of antioxidant glutathione. Furthermore, FD induced cognitive deficits and mood alterations such as anxious and despair-like behaviors that were aggravated in Ung−/− mice. Independent of Ung genotype, FD increased plasma homocysteine levels, altered brain monoamine metabolism, and inhibited adult hippocampal neurogenesis. These results indicate that impaired uracil repair is involved in neurodegeneration and neuropsychiatric dysfunction induced by experimental folate deficiency.
Keywords: neurodegeneration, memory impairment, despair, folate deficiency, base excision repair, neurogenesis
Introduction
Folate deficiency is associated with elevated levels of homocysteine, cerebrovascular and neurological diseases, and mood disorders (D'Anci and Rosenberg, 2004). The importance of folate in the nervous system was initially demonstrated in studies that established a greatly increased risk of neurodevelopmental disorders in the offspring of folate-deficient pregnant women (Smithells et al., 1976; Laurence et al., 1981; Blom et al., 2006). In the adult, epidemiological studies have linked lack of folate to neurodegenerative and neuropsychiatric diseases, including stroke, Parkinson's disease, dementia, and depression (Reynolds, 2002; He et al., 2004; Irizarry et al., 2005; Lamberti et al., 2005). In 1962, Victor Herbert reported insomnia, irritability, fatigue, and memory impairment as prominent symptoms of his self-inflicted folate deficiency, all of which were amenable to folate supplementation (Herbert, 1962).
The mechanisms by which chronic folate deficiency adversely affects CNS function are incompletely understood. Folic acid plays an essential role in one-carbon metabolism: it is required both in the remethylation of homocysteine to methionine and in the synthesis of S-adenosyl-methionine, the principal biological methyl donor in numerous methylation reactions. Reduced DNA methylation during folate deficiency results in altered gene expression and thereby may disrupt genome integrity (Bergman and Mostoslavsky, 1998). Dietary folate also has a major impact on homocysteine levels, which may exert direct neurotoxic and pro-oxidative actions (Lipton et al., 1997; Kruman et al., 2000; Ho et al., 2003) with an inverse relationship between plasma folate and homocysteine concentrations (Selhub et al., 1993).
Importantly, the methylation of deoxyuridine monophosphate (dUMP) to thymidylate (TMP) requires folic acid. Under conditions of folate depletion, a block in the methylation of dUMP to TMP increases intracellular deoxyuridine triphosphate (dUTP) by several orders of magnitude while concomitantly deoxythymidine triphosphate (dTTP) levels drop (Goulian et al., 1980b). Furthermore, a deficiency in folate has been reported to lead to an increase in genome instability (Beetstra et al., 2005), possibly the result of uracil misincorporation into DNA in proliferating cells and the subsequent formation of A:U mismatches. In addition, spontaneous hydrolytic deamination of cytosine both in proliferating and in differentiated cells also results in erroneous uracil residues in the form of U:G mispairs (Barnes and Lindahl, 2004). Base excision repair replaces these uracil bases (Friedberg et al., 2006). Uracil-DNA N-glycosylase (UNG) is the most widely distributed glycosylase that removes uracil from single- as well as double-stranded DNA (Kavli et al., 2002). We have previously demonstrated that UNG is of major importance for brain tissue repair after transient brain ischemia (Endres et al., 2004, 2005).
Here, we tested the hypothesis that neuronal dysfunction induced by chronic folate deficiency is aggravated in the absence of efficient uracil excision repair. We demonstrate significant alterations on the molecular, neurochemical, histological, and behavioral levels with distinct interactions between Ung genotype and folate deficiency.
Materials and Methods
Cell culture
Primary neuronal cultures of cerebral cortex or hippocampus were obtained from mouse embryos [embryonic day 16 (E16) to E17] as described previously (Harms et al., 2000). Briefly, cortices and hippocampi were dissected, trypsinized, dissociated, and plated in 24-well plates precoated with poly-l-lysine (0.5% w/v in PBS) and collagen (0.03% w/v) at a density of 200,000 cells/cm2. Cultures were kept at 36.5°C and 5% CO2 and were fed beginning from 4 d in vitro (DIV4) with cultivation medium (starter medium without glutamate) by replacing half of the medium twice a week. DMSO was used as the vehicle for methotrexate treatment (final concentration of 0.003% in all cultures), which was begun on DIV8. Additional cultures were exposed to 100 μm homocysteine (HC; Sigma-Aldrich) beginning on DIV8. Folate (FA; Sigma-Aldrich) was added to the cultivation medium, resulting in an increase of folate concentrations by 2.5- and 10-fold. Baseline concentration was 4 mg/L (∼10 μm), and cultures were supplemented with 25 and 100 μm FA. Staining with propidium iodide (PI) was performed for assessment of cell viability (1 μg/ml for 20 min at 37°C). Phase-contrast pictures from randomly chosen high-power fields from different cultures were taken and counted by a naive researcher as dead (PI-positive) or viable. Approximately 6000 neurons per condition were counted within five randomly taken high-power fields from three independent experiments. Aliquots of the medium were collected for analysis of lactate dehydrogenase (LDH) activity as described previously (Harms et al., 2000). Additionally, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, which measures the amount of blue formazan produced by viable mitochondria from MTT (Sigma-Aldrich), was performed at 72 h.
Mice, diets, and measurement of plasma homocysteine/methionine
The generation of mice deficient in Ung has been described previously (Endres et al., 2004). Folate deficiency was induced as described previously (Endres et al., 2005). For selective intestinal decontamination, experimental diets (Altromin control diet C1000 and Altromin special diet C1027 lacking folic acid) were supplemented with 1% succinylsulfathiazole (Sigma-Aldrich). All animals were kept on a 12 h light/dark schedule with ad libitum access to their respective diet and water. All applicable local and federal regulations for animal welfare were followed. The experimental protocol was approved by local authorities (Landesamt für Arbeitsschutz, Gesundheitsschutz und technische Sicherheit Berlin). Serum homocysteine levels were determined by HPLC using a homocysteine reagent kit for HPLC in serum (Chromosystems). Methionine was determined on the LC 3000 amino acid analyzer (Eppendorf-Biotronik).
Immunohistochemistry, quantification, and imaging
After the experiments were completed, animals were deeply anesthetized with ketamine and perfused transcardially with 4% paraformaldehyde in 0.1 m phosphate buffer. Tissue was essentially processed as described previously (Katchanov et al., 2001; Kronenberg et al., 2005). Briefly, after fixation in 4% paraformaldehyde in 0.1 m phosphate buffer over 48 h, brains were transferred to 30% sucrose for dehydration. Brains were cut in the coronal plane in 40-μm-thick sections and cryoprotected. Sections were stained free floating with all antibodies diluted in Tris-buffered saline containing 3% donkey serum and 0.1% Triton X-100. Primary antibodies were applied in the following concentrations: anti-bromodeoxyuridine (BrdU; rat, 1:500; Harlan Seralab), anti-S100β (rabbit, 1:2500; Swant), anti-NeuN (mouse, 1:100; Millipore Bioscience Research Reagents), anti-doublecortin (DCX; goat, 1:200; Santa Cruz Biotechnologies), and anti-calretinin (rabbit, 1:250; Swant).
Immunohistochemistry.
Immunohistochemistry followed the peroxidase method with biotinylated secondary antibodies (all 1:500; Jackson ImmunoResearch Laboratories), ABC Elite reagent (Vector Laboratories), and diaminobenzidine (Sigma) as chromogen.
Immunofluorescence.
For immunofluorescence, FITC-, RhodX-, or Cy5-conjugated secondary antibodies were all used at a concentration of 1:250. Fluorescent sections were coverslipped in polyvinyl alcohol with diazabicyclooctane as antifading agent.
Cell counts.
Cell counts were determined in one-in-six series of sections covering the entire hippocampus in its rostrocaudal extension with the peroxidase/diaminobenzidine method. The optical fractionator principle was modified in that only cells in the uppermost focal plane (at 40× magnification) were excluded to avoid oversampling (Kronenberg et al., 2003). For phenotypic analysis of BrdU-labeled cells, 50 randomly selected cells per animal were analyzed for costaining with mature neuronal marker NeuN or astrocytic marker S100β using a spectral confocal microscope (TCS SP2; Leica). Appropriate gain and black level settings were determined on control slices stained with secondary antibodies alone.
The procedure for quantification of pyramidal neurons in the hippocampal cornu ammonis (CA) was essentially identical to those described previously (Furukawa et al., 1997). Briefly, pyramidal cells in hematoxylin and eosin (H&E)-stained brain sections of CA1 and CA2/3 were sampled and numerical density calculated by dividing the number of neurons counted by the reference space. Pyramidal cells of CA1 were distinguished from those of CA2/3 by their smaller size and by the fact that they are more tightly packed (Kruman et al., 2002). Similarly, neuronal density was measured in deep frontoparietal cortical layers (V, VI) as described previously (Yanamoto et al., 2005) (see Fig. 2 D). Numerical density was also evaluated in the basolateral complex of the amygdala, which is easily identified by internal landmarks (i.e., amygdalar capsule, external capsule) (see Fig. 2 E) and consists of two major nuclei, the lateral nucleus and the basolateral nucleus (Millhouse and DeOlmos, 1983). Glial cells were excluded from the counts on the basis of size and cytological characteristics (von Bohlen und Halbach and Unsicker, 2002).
Figure 2.
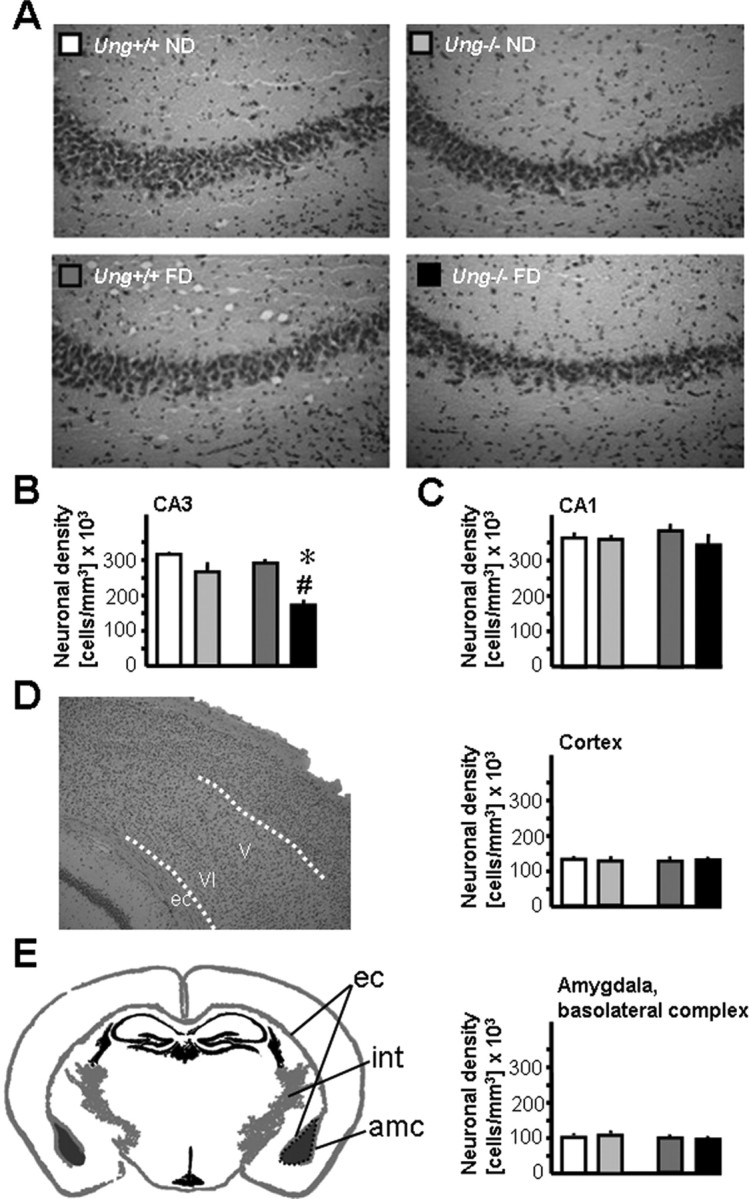
Increased susceptibility of Ung−/− mice to neurodegeneration after chronic folate depletion in vivo. A, Representative images of 10-μm-thick H&E-stained sections illustrating reduced neuronal density in CA3 of Ung−/− animals as a consequence of chronic folate deficiency (bottom right). B, Density of CA3 pyramidal neurons; ANOVA for genotype: F (1,16) = 22.8, p < 0.0005; treatment: F (1,16) = 11.3, p < 0.005; interaction: F (1,16) = 3.8, p = 0.068; 1-β = 0.44. C–E, Similar neuronal densities in other brain regions. C, Hippocampal subfield CA1. D, Deep cortical layers of frontoparietal cortex. E, Basolateral complex of amygdala. ec, External capsule; int, internal capsule; amc, amygdalar capsule. *p < 0.05, ND versus FD; # p < 0.05, Ung+/+ versus Ung−/−.
Quantification of reduced glutathione levels
Frozen tissue samples were homogenized in 10 mm EDTA buffer, pH 7.0, using a VirTis VirSonic Ultrasonic Cell Disruptor 100. After centrifugation at 14,000 rpm, at 4°C for 15 min, supernatants were aliquoted for measurement of glutathione (GSH) levels, and the remaining cellular pellet was retained for analysis of uracil misincorporation (see below). Pellets were stored at −80°C until subsequent extractions were performed. The protein concentration in each sample was determined using a Bio-Rad protein assay kit (Bio-Rad).
GSH levels were essentially quantified as described previously (Cardozo-Pelaez et al., 2000). Briefly, protein was precipitated in 6.5% sulfosalicylic acid, and nonprotein thiol content was quantified after centrifugation at 10,000 rpm, at 4°C for 10 min. Supernatants were added to a reaction mixture containing 2-nitro-5-thiobenzoic acid as chromogen. Absorbance was measured at 412 nm using a SpectraMax 340 96-well plate reader (Molecular Devices). GSH content was determined by comparison with standards of known amounts of GSH. Data were expressed as nanomoles of GSH per milligram of protein.
HPLC analysis of 2′-deoxyuridine
2′-Deoxyuridine (2-dU) levels were quantified from remaining nuclear pellets by preparing and resolving purified DNA into deoxynucleoside components as described previously (Bolin et al., 2006). The amount of 2-dU and 2′-deoxyguanosine (2-dG) was calculated by comparing the peak area of 2-dU and 2-dG obtained from 150 μl of injected enzymatic hydrolysate of DNA sample to a calibration curve for both nucleosides. HPLC analysis was based on a modification of a previously published method (Hailer et al., 2005). Nucleosides were separated using a reverse-phase C18 Omnisphere 4.6 × 250 mm with a 5 μm particle size column (VARIAN) and a water:acetonitrile mobile phase at a flow rate of 1 ml/min with an increasing acetonitrile gradient of 0.5% per minute using a Model 600S Solvent Delivery Module (Waters). 2-dU and 2-dG were detected by a Model 996 Photodiode Array Detector (Waters). Nucleoside identity was corroborated by comparison of retention time and absorbance spectrum of samples and pure standards. Peak areas were monitored at λ260 for 2-dU, and λ290 for 2-dG with the data recorded, stored, and analyzed on a PC Pentium computer using Millenium32 Software version 3.05.01 (Waters). Data were expressed as nanograms of 2-dU per micrograms of total DNA. Total DNA (in micrograms) was calculated based on the relationship that 1 μg of DNA contains 0.648 nmol of 2-dG (Cardozo-Pelaez et al., 1998).
Quantification of cerebral neurotransmitter and BDNF levels
Hippocampus, striatum, frontal cortex, parietal cortex, amygdala, and motor and somatosensory cortex were dissected on a cold plate (−16°C) according to Franklin and Paxinos (1997). Samples were weighed and stored at −80°C. Frozen tissue samples were homogenized by ultrasonication in 20–50× vol of deionized water at 4°C. Immediately after sonication, an aliquot of the homogenate (200–300 μl) was added to an equal volume of 0.2 N perchloric acid and centrifuged at 25,000 × g for 10 min at 4°C. The supernatant was used for the measurement of 5-HT, 5-hydroxyindoleacetic acid (5-HIAA), and norepinephrine (NA). For the determination of BDNF levels as described below, another aliquot of the homogenate was added to an equal volume of lysing buffer containing 0.1 m Tris-HCl, pH 7.0, 0.4 m NaCl, 0.1% NaN3, and a variety of protease inhibitors as contained in Protease Inhibitor Tablets (“complete”; purchased from Roche Diagnostics) and was stored at −80°C until analysis. 5-HT and 5-HIAA were analyzed as described previously (Sperk, 1982) using HPLC with electrochemical detection. NA was measured by HPLC with electrochemical detection after extraction to alumina according to a previously published protocol (Felice et al., 1978) with minor modifications (Sperk et al., 1981).
Endogenous levels of BDNF were measured in the rethawed homogenates using commercial ELISA kits in principle according to the manufacturer's instructions (Promega) but adapted to a fluorometric technique as described in detail previously (Hellweg et al., 2003). BDNF content was expressed as equivalents of recombinant human BDNF. The detection limit of the assay was 1 pg/ml. Determinations of recovery and specific and unspecific neurotrophin binding (the latter against mouse IgG1 obtained from MOPC 21) involved quadruplicate fluorescence determinations for each tissue sample. Using this improved fluorometric ELISA, it was feasible to quantify BDNF in brain tissue with a minimal wet weight of ∼5 pg (Hellweg et al., 2003, 2006). BDNF levels were expressed as picograms per milligram of tissue (wet weight).
Big Blue transgenic mouse mutation detection assay
Analyses were performed in mice after 3 months of folate depletion and in mouse embryonic fibroblasts (MEFs). Primary MEFs were cultured in folate-deficient, high-glucose DMEM (Invitrogen) supplemented with 10% FBS, Glutamax, and the antibiotics penicillin and streptomycin (Invitrogen). High-molecular-weight genomic DNA was isolated and mutations within the cII gene were scored essentially as we have described previously (Endres et al., 2004).
Behavioral tests
Behavioral tests were performed in principle as described previously (Winter et al., 2004, 2005). All behavioral analyses were performed during the dark (i.e., active) phase of a 12:12 h light–dark cycle.
Spontaneous activity.
Spontaneous activity was assessed essentially as described previously (Winter et al., 2005). Briefly, animals were individually placed in single cages (30 × 20 × 15 cm) evenly distributed across rows and columns for each group in a “checkerboard” manner. Animals from different groups were placed in alternating order to control for a position effect. Time, speed, and distance of spontaneous activity was measured overnight over a period of 8 h. Dim, indirect illumination in a neutral setting of a soundproof chamber provided uniform and sufficient light for automated detection of movements using PC-based VideoMot System software (TSE Systems). Here, the sampling rate was set at 1 Hz, and activity was defined as a movement of the center of gravity of the mouse >0.4 cm.
Morris water maze.
The Morris water maze task was used to assess spatial learning and memory. Experiments were adapted from previously published protocols with minor modifications (Harker and Whishaw, 2002; Winter et al., 2004). A 160-cm-diameter, 60.5-cm-high swimming pool with cues present at fixed positions was filled to a depth of 29 cm with 21°C opaque water. A clear Plexiglas platform (10 × 10 cm) was submerged at a fixed position with the top located 0.5 cm beneath the surface of the water in the center of one of the four quadrants. Swimming performance (e.g., speed, latency, distance) was tracked with a computer-based system (TSE Systems). The experiment consisted of a place task (acquisition period) with 3 trials/d for 8 consecutive days and a probe trial (spatial probe) on day 9. Each trial consisted of maximally 90 s starting from one of the three remaining quadrants with the face of the animal facing the wall. If after 90 s an animal did not reach the platform, it was guided to the platform. After reaching the platform, animals were allowed to remain there for 30 s. The intertrial interval was 1 min, and mice were quickly dried with a towel and placed under a heating lamp at exactly 37°C between each trial to avoid hypothermia. In the place task, the latencies, path lengths, and swim speeds for a single day were averaged to come up with a daily mean. For the probe trial, the platform was removed, and mice were allowed to swim for 90 s.
Elevated plus maze.
The elevated plus maze apparatus was made of black Plexiglas and consisted of two opposite open arms (30 × 5 × 0.25 cm) and two enclosed arms (30 × 5 × 15 cm) with side and end walls. The arms extend from a central platform (5 × 5 cm) and are elevated 50 cm above the floor standing on a tripod. Open-arm exploration was encouraged by testing under dim evenly distributed indirect light. At the beginning of the test, each mouse was placed in the center of the maze facing one of the closed arms. Time spent on the open arms was used as an index of anxiety (Winter et al., 2005).
Porsolt's forced-swim test.
The modified version of Porsolt's forced-swim test was performed at the end of behavioral testing as described previously (Winter et al., 2005). Briefly, mice were placed individually in glass cylinders (15 × 21 cm) filled with water (22°C) up to a height of 8.5 cm. Behaviors were monitored for a period of 300 s using a time sampling technique for subsequent analysis. Climbing behavior consisted of upward-directed movements of the forepaws along the side of the cylinder. Immobility was defined as motionless floating in the water with no additional activity other than that required to keep the head above the water.
Statistical analysis
Experiments were performed in a blinded manner. Values are presented as mean ± SEM. Statistical comparisons were performed by two-way ANOVA with level of significance set at 0.05 and two-tailed p values. Tukey post hoc testing was performed where appropriate. Water maze performance in the place task was analyzed by ANOVA with repeated measurements.
Results
Neurodegeneration induced by folate deficiency
Folate depletion induces cell death in Ung−/− neurons
The effect of folate depletion was analyzed in primary neocortical and hippocampal Ung−/− and Ung+/+ neurons (Fig. 1 A–D). To do so, cells were exposed to the dihydrofolate-reductase inhibitor methotrexate (MTX). Cell death was quantified by measurement of LDH release into the medium, by counting healthy neurons in vitro, as well as by measuring MTT as a metabolic activity marker. There was only borderline vulnerability to MTX in Ung+/+ neurons, whereas MTX treatment resulted in major cell death of cortical and, even more so, hippocampal Ung−/− neurons. Notably, a significant genotype × treatment interaction for LDH release, percentage of viable neurons, and MTT metabolism was evident in both cortical and hippocampal cultures (for statistical analysis, see Fig. 1).
Figure 1.

Increased cell death in Ung−/− neurons to methotrexate and homocysteine: reversal by folic acid supplementation. Neocortical (CTX) and hippocampal (HIP) neurons were exposed to the dihydrofolate-reductase inhibitor methotrexate (MTX) or vehicle. A, Increase in LDH activity relative to vehicle-treated sister cultures. Two-way ANOVA (72 h) for genotype: F (1,150) = 9.8, p < 0.001 in CTX and F (1,70) = 292.3, p < 0.001 in HIP; treatment: F (1,150) = 27.3, p < 0.001 in CTX and F (1,70) = 69.1, p < 0.001 in HIP; genotype × treatment interaction: F (1,150) = 5.2, p < 0.001 in CTX and F (1,70) = 28.2, p < 0.001 in HIP. B, MTT was measured as a metabolic activity marker and presented as loss of MTT metabolism relative to vehicle-treated sister cultures. ANOVA for genotype: F (1,70) = 0.6, p = 0.5 in CTX and F (1,70) = 305.5, p < 0.001 in HIP; treatment: F (1,70) = 11.3, p < 0.001 in CTX and F (1,70) = 101.4, p < 0.001 in HIP; interaction: F (1,70) = 4.0, p = 0.006 in CTX and F (1,70) = 42.2, p < 0.001 in HIP. C, D, Viable versus damaged neurons were identified by phase-contrast microscopy and propidium iodide counterstaining. Viable neurons are presented as a percentage of all neurons/high-power field. ANOVA for genotype: F (1,40) = 33.1, p < 0.001 in CTX and F (1,40) = 232, p < 0.001 in HIP; treatment: F (1,40) = 35.7, p < 0.001 in CTX and F (1,40) = 56.9, p < 0.001 in HIP. Interaction: F (1,40) = 9.8, p < 0.001 in CTX and F (1,40) = 27, p < 0.001 in HIP. E–G, CTX and HIP neurons were exposed to either HC plus vehicle or HC plus FA. Basal FA concentration in Neurobasal medium is 4 mg/L (∼10 μm). E, Increase in LDH activity relative to vehicle-treated sister cultures. Two-way ANOVA (72 h) for factor genotype: F (1,28) = 8.2, p = 0.008 in CTX and F (1,28) = 29.3, p < 0.001 in HIP; treatment: F (1,28) = 3.3, p = 0.017 in CTX and F (1,28) = 13.4, p < 0.001 in HIP; genotype × treatment interaction: F (1,28) = 0.711, p = 0.7 in CTX and F (1,28) = 3.1, p = 0.026 in HIP. F, G, Viable versus damaged neurons were identified by phase-contrast microscopy and propidium iodide counterstaining. Viable neurons are presented as a percentage of all neurons/high-power field. ANOVA for genotype: F (1,32) = 0.7, p = 0.40 in CTX and F (1,32) = 39, p < 0.001 in HIP; treatment: F (1,32) = 1.5, p = 0.25 in CTX and F (1,32) = 38.1, p < 0.001 in HIP. Interaction: F (1,32) = 0.5, p = 0.7 in CTX and F (1,32) = 10.4, p < 0.001 in HIP. Scale bars: D, G, 50 μm. A–F, # p < 0.05 for neocortical and *p < 0.05 for hippocampal neurons, Ung−/− versus Ung+/+ within the same MTX, HC, or HC plus FA treatment condition. E, F, + p < 0.05 versus corresponding HC without FA. Baseline LDH release and MTT metabolism are given in supplemental Tables 2A (for A, B) and 2B (for E) (available at www.jneurosci.org as supplemental material). All experiments were performed at least in triplicate.
Increased sensitivity of Ung−/− hippocampal neurons to homocysteine is alleviated by increasing concentration of folate in culture
Hyperhomocysteinemia mediates many of the major metabolic sequelae of low-folate status. Here, we tested how increasing folate concentrations above control levels in culture would impact the sensitivity of Ung−/− and Ung+/+ neurons to HC neurotoxicity. There was only borderline vulnerability to 100 μm HC in Ung+/+ cortical and hippocampal neurons. In contrast, hippocampal Ung−/− neurons proved highly susceptible to homocysteine. Importantly, increasing the concentration of folate in the cultivation medium above baseline levels largely reversed this effect (Fig. 1 E–G).
Chronic folate deficiency induces selective loss of pyramidal cells in CA3 in Ung−/− mice
To test the effects of chronic folate deficiency in vivo, Ung−/− and littermate Ung+/+ mice of 12 ± 2 months of age were subjected to a diet deficient in folate (FD) versus regular diet (ND) for 3 months (Endres et al., 2005). Ung−/− animals have been described in detail previously (Endres et al., 2004, 2005). Briefly, Ung−/− mice develop normally, show no overt phenotype, gain weight at a rate equal to that of wild-type mice, and have normal life spans. The effectiveness of FD was ascertained by measurement of serum homocysteine levels. Regardless of genotype, there was a significant increase in homocysteine levels (in micromoles per liter) as an effect of treatment (Ung+/+ ND, 8.5 ± 2.4; Ung−/− ND, 9.6 ± 1.4; Ung+/+ FD, 67 ± 29.9; Ung−/− FD, 49.9 ± 20.1). Methionine levels did not differ across groups (data not shown). Body weight (in grams) after 3 months of folate-deficient diet was also similar across groups (Ung+/+ ND, 31.9 ± 1.5; Ung−/− ND, 31.2 ± 2; Ung+/+ FD, 30.2 ± 1.1; Ung−/− FD, 31.6 ± 0.8).
CA3 pyramidal neurons are particularly vulnerable to DNA damage and excitotoxicity (Nadler et al., 1980; Furukawa et al., 1997; Kruman et al., 2002). We therefore quantified neuronal density in CA3. Examination of H&E-stained sections of hippocampus revealed massive degeneration of CA3 pyramidal neurons in Ung−/− mice that had been maintained on the folate-deficient diet (Fig. 2 A,B). However, the effects on CA3 pyramidal cell density of either folate deficiency or Ung−/− genotype alone remained subthreshold. Two-way ANOVA and Tukey post hoc testing demonstrated that CA3 cell loss was significantly increased in folate-deficient Ung−/− mice compared with the other experimental groups (Fig. 2 B). Loss of CA3 neurons was selective as demonstrated by stereology-based assessment of neuronal densities in CA1 (Fig. 2 C), deep layers (V, VI) of frontoparietal cortex (Fig. 2 D), and basolateral complex of amygdala (Fig. 2 E), which did not yield significant differences among the four experimental groups.
Effect of folate deficiency on brain 2-dU levels in Ung−/− versus Ung+/+ mice
Previous studies have indicated a critical role for base excision repair of misincorporated uracil in neuronal survival (Endres et al., 2004, 2005; Kruman et al., 2004). Misincorporation of 2′-dU into DNA was quantified in homogenates of whole cerebral hemispheres. 2′-dU levels were significantly elevated in Ung−/− compared with Ung+/+ mice and were further increased by folate deficiency (Fig. 3 A).
Figure 3.

Biochemical consequences of folate deficiency and Ung genotype. A, Uracil misincorporation in DNA; ANOVA for genotype: F (1,29) = 5.6, p = 0.02. B, BDNF protein levels in hippocampus; ANOVA for interaction: F (1,31) = 16.6, p < 0.0005. C, GSH levels in whole cerebral hemispheres; ANOVA for interaction: F (1,20) = 5.6, p = 0.03. D, GSH levels in cerebellum; ANOVA for interaction: F (1,20) = 20.4, p = 0.0002. *p < 0.05, ND versus FD; # p < 0.05, Ung+/+ versus Ung−/−; + p < 0.05 for interaction; two-way ANOVA and Tukey post hoc; n = 5–9 animals per group.
Folate deficiency decreases hippocampal BDNF levels in Ung−/− mice
BDNF signaling promotes neuronal survival (Thoenen, 1991; Soriano et al., 2006), and decreased hippocampal BDNF expression has been linked to depressive behaviors in mice (Ridder et al., 2005). Here, BDNF expression was measured at the protein level. We observed a significant genotype × treatment interaction (F (1,31) = 16.6; p < 0.0005) with reduced hippocampal BDNF levels in Ung−/− mice receiving folate-deficient chow but increased BDNF expression in folate-deficient wild-type mice and Ung−/− mice receiving the control diet (Fig. 3 B). Elevated amygdalar BDNF levels have been linked specifically to increased anxiety (Govindarajan et al., 2006). Here, BDNF levels in amygdala (in picograms per milligram of wet weight) were significantly increased as an effect of Ung−/− genotype (Ung+/+ ND, 41.7 ± 2.8; Ung−/− ND, 62.5 ± 6.1; Ung+/+ FD, 53.1 ± 4.6; Ung−/− FD, 61.7 ± 7.6).
Folate deficiency decreases reduced-form GSH levels in Ung−/− mice
GSH is the major cellular buffer against reactive oxygen species (ROS). Loss of mitochondrial GSH and the subsequent increase in ROS production are associated with induction of apoptosis (Tan et al., 1998; Armstrong et al., 2002). GSH levels decrease after severe brain damage [e.g., after ischemia/reperfusion (Park et al., 2000)], whereas “compensatory” GSH increases may indicate milder chronic oxidative stress (Shea et al., 2004; Tchantchou et al., 2005). Here, chronic folate deficiency decreased GSH levels (in nanomoles per milligram of protein) in whole brain hemispheres and profoundly in cerebella of Ung−/− but not Ung+/+ mice (Fig. 3 C,D). Importantly, a similar pattern was observed in the heart (Ung+/+ ND, 10.2 ± 0.8; Ung−/− ND, 9.4 ± 1.0; Ung+/+ FD, 9.0 ± 1.0; Ung−/− FD, 4.1 ± 1.1) and kidneys (Ung+/+ ND, 40.1 ± 6.7; Ung−/− ND, 40.3 ± 6.7; Ung+/+ FD, 39.1 ± 4.0, Ung−/− FD, 29.2 ± 3.9).
Folate depletion increases mutant frequency in embryonic fibroblasts
Ung−/− and Ung+/+ mice were established on a homozygous Big Blue (Stratagene) background. Spontaneous mutation frequencies in the cll gene were similar in Ung−/− compared with Ung+/+ MEFs (Endres et al., 2004). Folate deficiency conferred increased mutant frequency in both Ung+/+ MEFs and more so Ung−/− MEFs compared with normal medium (Fig. 4 A).
Figure 4.

Folate deficiency confers increased mutant frequencies in MEFs but not in brains of Ung−/− mice. A, Mouse embryonic fibroblasts were cultured in folate-deficient medium for 6 d. Average mutant frequencies at the λ cII gene (left) and individual measurements (table) are shown. B, Ung−/−(BigBlue) and Ung+/+(BigBlue) mice were fed a normal or a folate-deficient diet. Average mutant frequencies in brain tissue were assessed after 3 months. Individual measurements are given in the table. pfu, Plaque-forming unit; MF, mutant frequency.
Folate depletion effect on mutant frequency in brains from Ung−/− and Ung+/+ mice
Ung−/−(BigBlue) and Ung+/+(BigBlue) mice were fed a normal or a folate-deficient diet for 3 months. Overall, spontaneous mutant frequencies in the cll gene were similar in brain tissue isolated from both Ung−/− and Ung+/+ mice. In contrast to the findings in MEFs, folate deficiency conferred no relevant increase in mutant frequencies in either the Ung+/+ or Ung−/− brains (Fig. 4 B).
Effects of folate depletion on neurotransmitter levels: folate deficiency decreases brain 5-HIAA/5-HT ratio independent of Ung genotype
Altered monoamine neurotransmitter metabolism has been identified in psychiatric patients with folate deficiency (Bottiglieri et al., 2000). In particular, low levels of serotonin metabolites were observed in the CSF of patients suffering from folate-responsive neuropsychiatric disease (Botez et al., 1982). The 5-HIAA/5-HT ratio is considered a sensitive index of utilization of 5-HT (Shannon et al., 1986). We observed an overall trend of lower 5-HIAA/5-HT ratios in folate-deficient animals, which was statistically significant in amygdala and striatum (supplemental Table 1, available at www.jneurosci.org as supplemental material). 5-HIAA levels were also significantly reduced in hippocampus, amygdala, and striatum of folate-deficient animals (Fig. 5 A). In addition, folate deficiency caused a significant increase in norepinephrine levels in amygdala and hippocampus (Fig. 5 B). Together, the effects of folate deficiency on neurotransmitter metabolism were largely independent of Ung genotype.
Figure 5.

Folate deficiency effects on neurotransmitter metabolism. A, Tissue content of 5-HIAA and of 5-HT. B, NA levels in amygdala and hippocampus. Tissue contents are reported per milligram of wet weight. *p < 0.05, ND versus FD; # p < 0.05, Ung+/+ versus Ung−/−. n = 8–10 animals per group.
Effect of chronic folate deficiency on neurogenesis: folate deficiency impairs adult hippocampal neurogenesis largely independent of Ung genotype
The effects of folate deficiency and impaired uracil excision repair were also investigated in proliferating neural precursor cells in vivo. Neurogenesis in the adult hippocampal dentate gyrus is a complex process regulated at a number of different levels (Kempermann et al., 2004). The number of BrdU-positive cells after a 6 d course of single daily intraperitoneal injections of BrdU, the number of DCX-immunoreactive cells, and the number of calretinin-immunoreactive cells were used to assess early steps of neurogenesis (Fig. 6 A). Animals had been placed on either a folate-deficient or control diet 3 months before they were killed. In this particular injection paradigm, BrdU numbers are a broad measure of cell proliferation. DCX immunoreactivity reflects neuronal lineage determination (Brown et al., 2003; Rao and Shetty, 2004), and calretinin is used as a marker of early postmitotic granule cells (Brandt et al., 2003). Consistently, folate deficiency led to a significant decrease of all these measures. An additional genotype effect on the level of DCX-immunoreactive cells emerged with lower numbers observed in Ung−/− animals (F (1,14) = 5.7; p = 0.03, post hoc test, Ung+/+ vs Ung−/−, p = 0.03).
Figure 6.
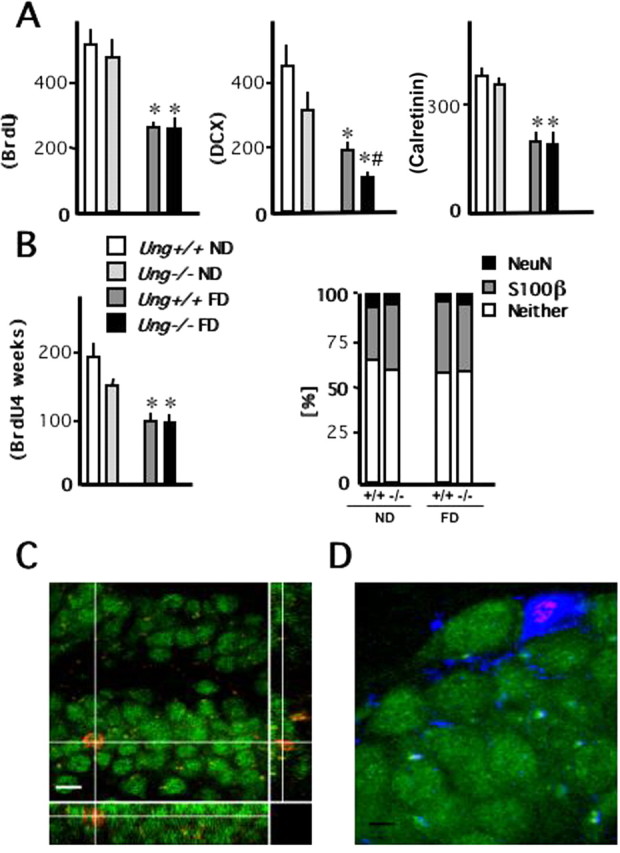
Hippocampal neurogenesis. A, Early steps of neuronal development in the adult dentate gyrus were assessed after 3 months of treatment (control or folate-deficient diet). Cell proliferation was quantified after a 6 d course of once daily intraperitoneal BrdU before the animals were killed. Doublecortin-expressing cells reflect neuronal lineage determination, whereas calretinin represents a marker of early postmitotic granule cells in the hippocampal dentate gyrus. n = 5 animals per group. B, To characterize the effects of folate deficiency on the fate of newly generated cells, animals received a 7 d course of once daily intraperitoneal BrdU 8 weeks after initiation of treatment (control or folate-deficient diet) and 4 weeks before the animals were killed. Neuronal marker NeuN (green in C) and astrocytic marker S100β (blue in D) were used for phenotypic analysis of BrdU-positive (red in C and D) cells. n = 8–10 animals per group. C, A three-dimensional reconstruction of a z-series through a NeuN+ cell along the y–z axis (right narrow panel) and x–z axis (bottom narrow panel), which confirms that BrdU and NeuN are present in the same cell. Scale bar, 15 μm. D, A newly generated S100β+ astrocyte. Scale bar, 5 μm. *p < 0.05, ND versus FD; # p < 0.05, Ung+/+ versus Ung−/−.
Net neurogenesis was analyzed 4 weeks after a 7 d course of single daily intraperitoneal injections of BrdU. Animals were placed on their respective diets 3 months before they were killed and 2 months before BrdU injections. In this injection paradigm, BrdU counts reflect a combination of initial proliferation and subsequent survival of newly generated cells. BrdU cell counts were significantly reduced as an effect of folate deficiency (Fig. 6 B) with no apparent effect of the genotype. BrdU-labeled cells were subjected to further phenotypic analysis. The majority of cells did not show colabeling with either astrocytic marker S100β or neuronal marker NeuN. Across groups, we did not observe a significant difference in the phenotypes of newly generated cells (Fig. 6 B–D).
Behavioral consequences of chronic folate deficiency
Folate deficiency impairs spatial learning
We analyzed the effects of folate deficiency in the Morris water maze, a hippocampus-dependent task modeling declarative learning and memory (Morris, 1984). In the place task (days 1–8), there was a significant effect of folate-deficient diet and number of trial on escape latency (F (1,37) = 5.46, p = 0.25; F (7,259) = 27.9, p < 0.0001, respectively; repeated-measures ANOVA) and distance (F (1,37) = 5.03, p = 0.31; F (7,259) = 26.0, p < 0.0001, respectively) (Fig. 7 A,B). Ung−/− FD mice showed inferior performance in the place task compared with the other groups, which, however, did not reach statistical significance. Similarly, in the probe trial there was a trend for fewer target crossings (Ung+/+ ND, 6 ± 1.1; Ung−/− ND, 5.5 ± 0.8; Ung+/+ FD, 5 ± 1.3; Ung−/− FD, 3.6 ± 0.7) and lower target preference (Fig. 7 C) in the folate-deficient Ung−/− group.
Figure 7.
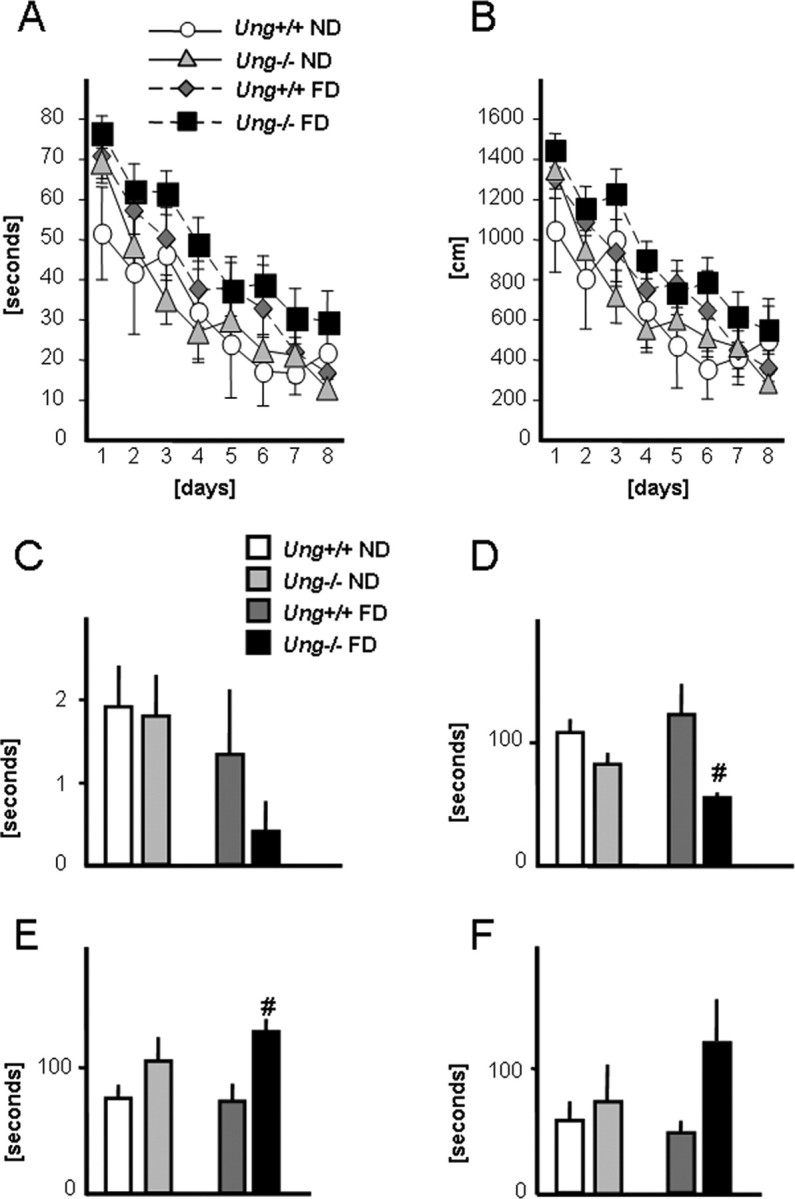
Behavioral analysis. Behavioral analyses were performed after 3 months on the experimental diet (control or folate-deficient diet). A, B, Latencies (A) and path lengths (B) of Ung+/+ and Ung−/− animals on folate-deficient or control diet during the acquisition phase of the Morris water maze. There were significant effects of diet and trial number on escape latency and path length (repeated-measures ANOVA; see Results). C, Target preference during probe trial. D, E, Latency to float (D) and time floating (E) in the behavioral despair test (Porsolt). F, Latency until first entry into the open arms of an elevated plus maze. # p < 0.05, Ung+/+ versus Ung−/−. n = 6–14 animals per group. For statistical analysis, see Results.
Folate deficiency induces an anxious phenotype and despair-like reactions in Ung−/− mice
In the Porsolt forced-swim test, latency to float (i.e., the time to “give up”) and the total time floating are measures of despair-related behavior (Cryan et al., 2002). There was a significant effect of genotype on latency to float and total time floating.
Post hoc testing revealed that this effect was significant in folate-deficient Ung−/− mice only (p < 0.005 for latency and total time floating) (Fig. 7 D,E).
The elevated plus maze is a behavioral assay based on an approach-avoidance conflict to test anxiety-related behavior. Two-way ANOVA revealed a significant effect of genotype on the number of open arm visits and time spent on the open arms [number of open arm entries: Ung+/+ ND, 9.8 ± 1.5; Ung−/− ND, 4.9 ± 1.2; Ung+/+ FD, 6.1 ± 0.9; Ung−/− FD, 4.4 ± 1.3; Tukey post hoc test for factor treatment within wild-type mice only, p < 0.05; factor genotype, p < 0.05; factor genotype within control diet, p < 0.05; time (in seconds) spent on the open arms: Ung+/+ ND, 9.7 ± 1.7; Ung−/− ND, 6.3 ± 1.3; Ung+/+ FD, 16.1 ± 4.7; Ung−/− FD, 6.6 ± 2.3; post hoc for factor genotype, p < 0.05; post hoc for factor genotype within folate-deficient animals, p < 0.05]. Similarly, folate-deficient Ung−/− mice displayed a longer latency to enter the open arms compared with controls (Fig. 7 F). Furthermore, two-way ANOVA revealed significant effects of both genotype and treatment on total distance moved (in centimeters) during the task (Ung+/+ ND, 1524 ± 126; Ung−/− ND, 1056 ± 86; Ung+/+ FD, 1096 ± 135; Ung−/− FD, 906 ± 105; post hoc for factor genotype, p < 0.01; post hoc for factor treatment, p < 0.05). The amount of time (in seconds) spent in the center of the plus maze apparatus did not differ significantly across groups (Ung+/+ ND, 52.2 ± 12.4; Ung−/− ND, 41.9 ± 9.7; Ung+/+ FD, 29.9 ± 4.4; Ung−/− FD, 45.9 ± 17.1). Although one-way ANOVA of the total number of entries (open and closed arms) demonstrated a significant difference between Ung+/+ ND and Ung−/− FD, two-way ANOVA for factors genotype and treatment did not yield significant effects (Ung+/+ ND, 40.4 ± 5.8; Ung−/− ND, 29.2 ± 3.8; Ung+/+ FD, 28.7 ± 3.3; Ung−/− FD, 24.1 ± 17.1).
Increased spontaneous locomotion has been reported in very different paradigms of brain damage, including transient ischemia, some forms of neonatal excitotoxic lesions, traumatic brain injury, or intrauterine exposure to toxins (Johns et al., 1982; Lewen et al., 1999; Flores et al., 2005; Winter et al., 2005). Animals were monitored for spontaneous activity in individual cages (total of 8 h overnight) in principle as described previously (Winter et al., 2005). Spontaneous activity (in minutes) was as follows: Ung+/+ ND, 42.8 ± 5.2; Ung−/− ND, 46.7 ± 6.5; Ung+/+ FD, 61.1 ± 12.2; Ung−/− FD, 57.9 ± 4.5 (two-way ANOVA for factor diet, p = 0.06). Similarly, the distance moved (in centimeters) was also increased in folate-deficient animals: Ung+/+ ND, 3936 ± 527; Ung−/− ND, 4703 ± 949; Ung+/+ FD, 6568 ± 1624; Ung−/− FD, 7276 ± 1476 (two-way ANOVA for factor diet, p = 0.06).
Discussion
We here show that impaired uracil excision repair contributes to neurodegeneration and brain dysfunction induced by folate depletion: exposing mice to a folate-deficient diet induced cognitive dysfunction and an anxious, despair-related phenotype, which was aggravated in animals lacking UNG. In vitro, folate antagonist methotrexate conferred cell death primarily in Ung−/− hippocampal neurons. In vivo, only the combination of Ung−/− genotype and folate depletion caused neurodegeneration in CA3, which was accompanied by decreased BDNF and GSH levels. These results identify uracil misincorporation as an important pathogenic mechanism of folate deficiency. However, not all effects of folate deficiency were found to be a function of Ung genotype. The reduction in hippocampal neurogenesis and metabolic sequelae like hyperhomocysteinemia and changes in monoamine levels were not prominently affected by Ung deficiency.
Neuronal populations even within a single brain region may show different susceptibility to different kinds of an insult. Strikingly, hippocampal neurodegeneration in folate-deficient Ung−/− mice was selective to CA3, as opposed to CA1 and additional brain areas investigated. This observation fits well with results from cell culture demonstrating greater susceptibility of primary hippocampal compared with cortical neurons to methotrexate or homocysteine. An earlier study of folate deprivation in amyloid precursor protein mutant transgenic mice likewise reported selective vulnerability of CA3 pyramidal neurons (Kruman et al., 2002). Converging evidence suggests that selective neuronal loss in CA3 might result from a specific vulnerability of these neurons to DNA damage and/or excitotoxicity, also in old age (Nadler et al., 1980; Mattson et al., 1992; Kesslak et al., 1995; Furukawa et al., 1997).
Folate deficiency increased mutant frequencies in Ung−/− MEFs but not in brains of either Ung+/+ or Ung−/− mice. However, data from cell lines cannot be correlated directly with in vivo data, because the impact of folate deficiency on an organism involves major metabolic changes throughout. Also, the metabolism of MEFs compared with postmitotic neurons will differ. Furthermore, an increase in mutations is likely observed in MEFs because they show greater resilience. In contrast, especially Ung−/− hippocampal neurons proved highly sensitive to folate deprivation. It is therefore likely that more severely damaged cells in Ung−/− brains degenerate before accumulation of mutations is observed.
Whereas the contribution of UNG deficiency was minor, folate deficiency strongly repressed neurogenesis, indicating similar cytotoxicity through uracil misincorporation (in Ung−/−) and base excision repair of misincorporated uracil (in Ung+/+) in hippocampal progenitors. Phenotypes of newborn cells did not differ across groups. A large fraction of BrdU+ cells lacked colabeling with neuronal or glial markers, in line with an earlier study in aged mice (Kempermann et al., 2002). Importantly, a significant decrease in neurogenesis has been reported long before an animal becomes aged (Heine et al., 2004; Leuner et al., 2007). Mechanisms underlying reduced cell proliferation under low-folate conditions likely involve hyperhomocysteinemia, inhibition of methyltransferase reactions, and decreased signaling through Ras pathways. Decreased Ras signaling through inhibition of carboxyl methylation mediates MTX's antiproliferative effect largely independent of thymidine biosynthesis (Winter-Vann et al., 2003). In line with our results, folate deficiency inhibits both proliferation of cultured fetal multipotent neuroepithelial cells and cell proliferation in the dentate gyrus in vivo (Kruman et al., 2005). In contrast, in postmitotic neurons folate deficiency (or methotrexate) alone did not have a strong effect on cell survival, whereas the combination of folate deficiency and Ung knock-out proved detrimental. Together, our results indicate that under low-folate conditions, defective uracil repair contributes to cell death in postmitotic differentiated cells, whereas the effects on proliferation are largely independent of base excision repair.
Here, 2′-dU levels were significantly elevated in Ung−/− compared with Ung+/+ mice and were further increased by folate deficiency. During folate deficiency, levels of dTTP are depleted, whereas levels of dUTP increase (Goulian et al., 1980a). Mechanisms of uracil misincorporation resulting from folate deficiency differ between proliferating and postmitotic cells. Large quantities of dUTP may be incorporated instead of dTTP during S-phase (or DNA repair). The resulting A:U mismatches, however, do not result in mutations, but may hamper recognition by DNA-binding proteins and thus alter physiological function (Courtemanche et al., 2004). Levels of uracil in the DNA of Ung−/− cells strongly depend on proliferation (Andersen et al., 2005). On the other hand, in mature postmitotic cells, low levels of genomic uracil may be generated during DNA repair or directly via deamination of cytosine. The resulting G:U mismatches are promutagenic and, if left unrepaired, lead to GC→AT transition mutations. Importantly, in the absence of S-adenosylmethionine (induced by folate deficiency), DNA (cytosine-5)-methyltransferase causes cytosine deamination in double-stranded DNA (Shen et al., 1992).
The Ung gene encodes mitochondrial (UNG1) and nuclear (UNG2) isoforms of UNG (Nilsen et al., 2000). When exposed to brain ischemia, Ung−/− animals develop enlarged lesions. The mechanism of cell death may relate to the lack of UNG1 in mitochondria [comparable with the recently described mitochondrial mutator phenotype (Chatterjee and Singh, 2001; Trifunovic et al., 2004; Kujoth et al., 2005)]. Indeed, the most significant increase of uracil-excising activity after ischemia was seen in mitochondrial/cytosolic extracts and was exclusively caused by UNG1 (Endres et al., 2004). Similarly, lack of DNA repair capacity was unmasked here as selective neurodegeneration after folate deprivation. We speculate that uracil misincorporation into mitochondrial DNA (mtDNA) and subsequent respiratory chain dysfunction is an important mechanism by which folate deficiency causes neuronal vulnerability, e.g., by provoking a bioenergy deficit or by lowering the signal threshold for cell death (Trifunovic et al., 2005). Notably, mutational damage of mtDNA, which has been implicated in neurodegenerative disease and aging (Tanhauser and Laipis, 1995), decreases with folate supplementation (Branda et al., 2002).
Clinical studies have suggested a role for folate and homocysteine in the pathogenesis of affective disorders (Reynolds et al., 1970; Reynolds, 2002; Papakostas et al., 2004a,b). There is also increasing evidence for a beneficial effect of folate in protecting against Alzheimer's disease and cognitive decline with age (Seshadri et al., 2002; Nurk et al., 2005; Durga et al., 2007; Luchsinger et al., 2007). Therefore, it is interesting to note within this context that in culture folate supplementation reversed increased susceptibility of the Ung−/− genotype to homocysteine.
The Porsolt forced-swim test is commonly used to screen for antidepressant agents and, more recently, also to assess despair-like behavior in rodents (Cryan et al., 2002; Bale and Vale, 2003; Govindarajan et al., 2006; Clapcote et al., 2007; Fukui et al., 2007). Whereas folate deficiency alone did not confer a clearly despair-like phenotype, folate-deficient Ung−/− mice demonstrated significantly increased immobile time and reduced latency to float. Similarly, in the elevated plus maze, folate-deficient Ung−/− animals spent significantly less time on the open arms compared with controls. Importantly, these results were attributable to an increase in specific anxious and despair-related behaviors and not accounted for by a decrease in spontaneous activity in Ung−/− FD.
Largely independent of genotype, folate deficiency reduced 5-HIAA levels and inhibited neurogenesis. In contrast, hippocampal BDNF was specifically reduced in Ung−/− FD, which also displayed increased anxiety and despair-related behavior. We therefore speculate that the distinct behavioral phenotype of Ung−/− folate-deficient animals is associated with pyramidal cell loss and reduced hippocampal BDNF, lending further support for a role of BDNF in regulating aspects of affective behavior (Schinder and Poo, 2000; Nestler et al., 2002; Lang et al., 2004; Ridder et al., 2005). Interestingly, Ung−/− mice on the control diet also showed a mild tendency toward anxiousness and despair-like reactions.
Spatial learning in the hippocampus-dependent Morris water maze was predominantly affected by folate deficiency. Several mechanisms, including neurodegeneration and disturbed neurotrophin and neurotransmitter signaling, may have contributed to this effect. Some previous studies have also described an association between acquisition in the place task and levels of neurogenesis, especially in old age (Kempermann and Gage, 2002; Drapeau et al., 2003; Wolf et al., 2006). Results reported here fit well with these earlier reports. However, other studies disrupting neurogenesis (Shors et al., 2002; Saxe et al., 2006) failed to demonstrate an effect on Morris water maze performance. The trend toward poorer performance of Ung−/− FD (place task, target preference in the probe trial) did not reach statistical significance, possibly because of sample size.
In summary, our study provides new insight into how folate deficiency impacts brain function. Folate deficiency leads to increased homocysteine, alters brain monoamine levels, and strongly impairs hippocampal neurogenesis. Whereas these effects of low folate are largely compensated for in wild-type animals, in animals with defective uracil excision repair, along with changes in BDNF, GSH, and selective neurodegeneration, anxious and despair-like behaviors develop.
Footnotes
This work was supported by the VolkswagenStiftung (Lichtenberg Program to M.E.), Deutsche Forschungsgemeinschaft (M.E.), Bundesministerium für Bildung und Forschung (Center for Stroke Research Berlin; M.E.), Schilling Foundation (M.E.), the Intramural Research Program of the National Institutes of Health (NIH), National Institute of Environmental Health Sciences (S.H.W.), a Research Scholar grant (RSG-05-246-01-GMC) from the American Cancer Society, and grants from the Susan G. Komen Breast Cancer Foundation (CTR0403276 to R.W.S.), NIH (1 R01 AG24364-01 and P20 CA103730 to R.W.S.; P20 RR015583-07 and P20RRP20RR017670-04 to F.C.P.), the University of Pittsburgh Medical Center Health System Competitive Medical Research Fund (R.W.S.), the University of Pittsburgh Cancer Institute (R.W.S.), and the National Institute on Aging (1R15AG023604-01 to F.C.P.).
References
- Andersen S, Heine T, Sneve R, König I, Krokan HE, Epe B, Nilsen H. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis. 2005;26:547–555. doi: 10.1093/carcin/bgh347. [DOI] [PubMed] [Google Scholar]
- Armstrong JS, Steinauer KK, Hornung B, Irish JM, Lecane P, Birrell GW, Peehl DM, Knox SJ. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002;9:252–263. doi: 10.1038/sj.cdd.4400959. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. Increased depression-like behaviors in corticotropin-releasing factor receptor-2-deficient mice: sexually dichotomous responses. J Neurosci. 2003;23:5295–5301. doi: 10.1523/JNEUROSCI.23-12-05295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- Beetstra S, Thomas P, Salisbury C, Turner J, Fenech M. Folic acid deficiency increases chromosomal instability, chromosome 21 aneuploidy and sensitivity to radiation-induced micronuclei. Mutat Res. 2005;578:317–326. doi: 10.1016/j.mrfmmm.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Bergman Y, Mostoslavsky R. DNA demethylation: turning genes on. Biol Chem. 1998;379:401–407. doi: 10.1515/bchm.1998.379.4-5.401. [DOI] [PubMed] [Google Scholar]
- Blom HJ, Shaw GM, den Heijer M, Finnell RH. Neural tube defects and folate: case far from closed. Nat Rev Neurosci. 2006;7:724–731. doi: 10.1038/nrn1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolin CM, Basha R, Cox D, Zawia NH, Maloney B, Lahiri DK, Cardozo-Pelaez F. Exposure to lead (Pb) and the developmental origin of oxidative DNA damage in the aging brain. FASEB J. 2006;20:788–790. doi: 10.1096/fj.05-5091fje. [DOI] [PubMed] [Google Scholar]
- Botez MI, Young SN, Bachevalier J, Gauthier S. Effect of folic acid and vitamin B12 deficiencies on 5-hydroxyindoleacetic acid in human cerebrospinal fluid. Ann Neurol. 1982;12:479–484. doi: 10.1002/ana.410120512. [DOI] [PubMed] [Google Scholar]
- Bottiglieri T, Laundy M, Crellin R, Toone BK, Carney MW, Reynolds EH. Homocysteine, folate, methylation, and monoamine metabolism in depression. J Neurol Neurosurg Psychiatry. 2000;69:228–232. doi: 10.1136/jnnp.69.2.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda RF, Brooks EM, Chen Z, Naud SJ, Nicklas JA. Dietary modulation of mitochondrial DNA deletions in copy number after chemotherapy in rats. Mut Res. 2002;501:29–36. doi: 10.1016/s0027-5107(02)00014-3. [DOI] [PubMed] [Google Scholar]
- Brandt MD, Jessberger S, Steiner B, Kronenberg G, Reuter K, Bick-Sander A, von der Behrens W, Kempermann G. Transient calretinin expression defines early postmitotic step of neuronal differentiation in adult hippocampal neurogenesis of mice. Mol Cell Neurosci. 2003;24:603–613. doi: 10.1016/s1044-7431(03)00207-0. [DOI] [PubMed] [Google Scholar]
- Brown JP, Couillard-Després S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003;467:1–10. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- Cardozo-Pelaez F, Song S, Parthasarathy A, Epstein CJ, Sanchez-Ramos J. Attenuation of age-dependent oxidative damage to DNA and protein in brainstem of Tg Cu/Zn SOD mice. Neurobiol Aging. 1998;19:311–316. doi: 10.1016/s0197-4580(98)00067-0. [DOI] [PubMed] [Google Scholar]
- Cardozo-Pelaez F, Brooks PJ, Stedeford T, Song S, Sanchez-Ramos J. DNA damage, repair, and antioxidant systems in brain regions: a correlative study. Free Radic Biol Med. 2000;28:779–785. doi: 10.1016/s0891-5849(00)00172-6. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Singh KK. Uracil-DNA glycosylase-deficient yeast exhibit a mitochondrial mutator phenotype. Nucleic Acids Res. 2001;29:4935–4940. doi: 10.1093/nar/29.24.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, Lerch JP, Trimble K, Uchiyama M, Sakuraba Y, Kaneda H, Shiroishi T, Houslay MD, Henkelman RM, Sled JG, Gondo Y, Porteous DJ, Roder JC. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Courtemanche C, Elson-Schwab I, Mashiyama ST, Kerry N, Ames BN. Folate deficiency inhibits the proliferation of primary human CD8+ T lymphocytes in vitro. J Immunol. 2004;173:3186–3192. doi: 10.4049/jimmunol.173.5.3186. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Markou A, Lucki I. Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci. 2002;23:238–245. doi: 10.1016/s0165-6147(02)02017-5. [DOI] [PubMed] [Google Scholar]
- D'Anci KE, Rosenberg IH. Folate and brain function in the elderly. Curr Opin Clin Nutr Metab Care. 2004;7:659–664. doi: 10.1097/00075197-200411000-00011. [DOI] [PubMed] [Google Scholar]
- Drapeau E, Mayo W, Aurousseau C, Le Moal M, Piazza PV, Abrous DN. Spatial memory performances of aged rats in the water maze predict levels of hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2003;100:14385–14390. doi: 10.1073/pnas.2334169100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durga J, van Boxtel MP, Schouten EG, Kok FJ, Jolles J, Katan MB, Verhoef P. Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomised, double blind, controlled trial. Lancet. 2007;369:208–216. doi: 10.1016/S0140-6736(07)60109-3. [DOI] [PubMed] [Google Scholar]
- Endres M, Biniszkiewicz D, Sobol RW, Harms C, Ahmadi M, Lipski A, Katchanov J, Mergenthaler P, Dirnagl U, Wilson SH, Meisel A, Jaenisch R. Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J Clin Invest. 2004;113:1711–1721. doi: 10.1172/JCI20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Ahmadi M, Kruman I, Biniszkiewicz D, Meisel A, Gertz K. Folate deficiency increases postischemic brain injury. Stroke. 2005;36:321–325. doi: 10.1161/01.STR.0000153008.60517.ab. [DOI] [PubMed] [Google Scholar]
- Felice LJ, Felice JD, Kissinger PT. Determination of catecholamines in rat brain parts by reverse-phase ion-pair liquid chromatography. J Neurochem. 1978;31:1461–1465. doi: 10.1111/j.1471-4159.1978.tb06573.x. [DOI] [PubMed] [Google Scholar]
- Flores G, Silva-Gómez AB, Ibáñez O, Quirion R, Srivastava LK. Comparative behavioral changes in postpubertal rats after neonatal excitotoxic lesions of the ventral hippocampus and the prefrontal cortex. Synapse. 2005;56:147–153. doi: 10.1002/syn.20140. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. San Diego: Academic; 1997. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. Ed 2. Washington, DC: ASM; 2006. DNA repair and mutagenesis. [Google Scholar]
- Fukui M, Rodriguiz RM, Zhou J, Jiang SX, Phillips LE, Caron MG, Wetsel WC. Vmat2 heterozygous mutant mice display a depressive-like phenotype. J Neurosci. 2007;27:10520–10529. doi: 10.1523/JNEUROSCI.4388-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Fu W, Li Y, Witke W, Kwiatkowski DJ, Mattson MP. The actin-severing protein gelsolin modulates calcium channel and NMDA receptor activities and vulnerability in hippocampal neurons. J Neurosci. 1997;17:8178–8186. doi: 10.1523/JNEUROSCI.17-21-08178.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulian M, Bleile B, Tseng BY. Methotrexate-induced misincorporation of uracil into DNA. Proc Natl Acad Sci U S A. 1980a;77:1956–1960. doi: 10.1073/pnas.77.4.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulian M, Bleile B, Tseng BY. The effect of methotrexate on levels of dUTP in animal cells. J Biol Chem. 1980b;255:10630–10637. [PubMed] [Google Scholar]
- Govindarajan A, Rao BS, Nair D, Trinh M, Mawjee N, Tonegawa S, Chattarji S. Transgenic brain-derived neurotrophic factor expression causes both anxiogenic and antidepressant effects. Proc Natl Acad Sci U S A. 2006;103:13208–13213. doi: 10.1073/pnas.0605180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailer MK, Slade PG, Martin BD, Sugden KD. Nei deficient Escherichia coli are sensitive to chromate and accumulate the oxidized guanine lesion spiroiminodihydantoin. Chem Res Toxicol. 2005;18:13. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harker KT, Whishaw IQ. Impaired spatial performance in rats with retrosplenial lesions: importance of the spatial problem and the rat strain in identifying lesion effects in a swimming pool. J Neurosci. 2002;22:1155–1164. doi: 10.1523/JNEUROSCI.22-03-01155.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms C, Lautenschlager M, Bergk A, Freyer D, Weih M, Dirnagl U, Weber JR, Hörtnagl H. Melatonin is protective in necrotic but not in caspase-dependent, free radical-independent apoptotic neuronal cell death in primary neuronal cultures. FASEB J. 2000;14:1814–1824. doi: 10.1096/fj.99-0899com. [DOI] [PubMed] [Google Scholar]
- He K, Merchant A, Rimm EB, Rosner BA, Stampfer MJ, Willett WC, Ascherio A. Folate, vitamin B6, and B12 intakes in relation to risk of stroke among men. Stroke. 2004;35:169–174. doi: 10.1161/01.STR.0000106762.55994.86. [DOI] [PubMed] [Google Scholar]
- Heine VM, Maslam S, Joëls M, Lucassen PJ. Prominent decline of newborn cell proliferation, differentiation, and apoptosis in the aging dentate gyrus, in absence of an age-related hypothalamus-pituitary-adrenal axis activation. Neurobiol Aging. 2004;25:361–375. doi: 10.1016/S0197-4580(03)00090-3. [DOI] [PubMed] [Google Scholar]
- Hellweg R, von Arnim CA, Büchner M, Huber R, Riepe MW. Neuroprotection and neuronal dysfunction upon repetitive inhibition of oxidative phosphorylation. Exp Neurol. 2003;183:346–354. doi: 10.1016/s0014-4886(03)00127-4. [DOI] [PubMed] [Google Scholar]
- Hellweg R, Lohmann P, Huber R, Kühl A, Riepe MW. Spatial navigation in complex and radial mazes in APP23 animals and neurotrophin signaling as a biological marker of early impairment. Learn Mem. 2006;13:63–71. doi: 10.1101/lm.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert V. Experimental nutritional folate deficiency in man. Trans Assoc Am Physicians. 1962;75:307–320. [PubMed] [Google Scholar]
- Ho PI, Ashline D, Dhitavat S, Ortiz D, Collins SC, Shea TB, Rogers E. Folate deprivation induces neurodegeneration: roles of oxidative stress and increased homocysteine. Neurobiol Dis. 2003;14:32–42. doi: 10.1016/s0969-9961(03)00070-6. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Gurol ME, Raju S, Diaz-Arrastia R, Locascio JJ, Tennis M, Hyman BT, Growdon JH, Greenberg SM, Bottiglieri T. Association of homocysteine with plasma amyloid beta protein in aging and neurodegenerative disease. Neurology. 2005;65:1402–1408. doi: 10.1212/01.wnl.0000183063.99107.5c. [DOI] [PubMed] [Google Scholar]
- Johns JM, Louis TM, Becker RF, Means LW. Behavioral effects of prenatal exposure to nicotine in guinea pigs. Neurobehav Toxicol Teratol. 1982;4:365–369. [PubMed] [Google Scholar]
- Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hortnagl H, Dirnagl U, Bhide PG, Endres M. Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci. 2001;21:5045–5053. doi: 10.1523/JNEUROSCI.21-14-05045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem. 2002;277:39926–39936. doi: 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Gage FH. Genetic determinants of adult hippocampal neurogenesis correlate with acquisition, but not probe trial performance, in the water maze task. Eur J Neurosci. 2002;16:129–136. doi: 10.1046/j.1460-9568.2002.02042.x. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Gage FH. Neuroplasticity in old age: sustained fivefold induction of hippocampal neurogenesis by long-term environmental enrichment. Ann Neurol. 2002;52:135–143. doi: 10.1002/ana.10262. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Jessberger S, Steiner B, Kronenberg G. Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 2004;27:447–452. doi: 10.1016/j.tins.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Kesslak JP, Yuan D, Neeper S, Cotman CW. Vulnerability of the hippocampus to kainate excitotoxicity in the aged, mature and young adult rat. Neurosci Lett. 1995;188:117–120. doi: 10.1016/0304-3940(95)11415-s. [DOI] [PubMed] [Google Scholar]
- Kronenberg G, Reuter K, Steiner B, Brandt MD, Jessberger S, Yamaguchi M, Kempermann G. Subpopulations of proliferating cells of the dentate gyrus respond differently to physiologic neurogenic stimuli. J Comp Neurol. 2003;467:455–463. doi: 10.1002/cne.10945. [DOI] [PubMed] [Google Scholar]
- Kronenberg G, Wang LP, Synowitz M, Gertz K, Katchanov J, Glass R, Harms C, Kempermann G, Kettenmann H, Endres M. Nestin-expressing cells divide and adopt a complex electrophysiologic phenotype after transient brain ischemia. J Cereb Blood Flow Metab. 2005;25:1613–1624. doi: 10.1038/sj.jcbfm.9600156. [DOI] [PubMed] [Google Scholar]
- Kruman II, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L, Mattson MP. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci. 2000;20:6920–6926. doi: 10.1523/JNEUROSCI.20-18-06920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, Mattson MP. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer's disease. J Neurosci. 2002;22:1752–1762. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Schwartz E, Kruman Y, Cutler RG, Zhu X, Greig NH, Mattson MP. Suppression of uracil-DNA glycosylase induces neuronal apoptosis. J Biol Chem. 2004;279:43952–43960. doi: 10.1074/jbc.M408025200. [DOI] [PubMed] [Google Scholar]
- Kruman II, Mouton PR, Emokpae R, Jr, Cutler RG, Mattson MP. Folate deficiency inhibits proliferation of adult hippocampal progenitors. Neuroreport. 2005;16:1055–1059. doi: 10.1097/00001756-200507130-00005. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lamberti P, Zoccolella S, Armenise E, Lamberti SV, Fraddosio A, de Mari M, Iliceto G, Livrea P. Hyperhomocysteinemia in L-dopa treated Parkinson's disease patients: effect of cobalamin and folate administration. Eur J Neurol. 2005;12:365–368. doi: 10.1111/j.1468-1331.2004.00973.x. [DOI] [PubMed] [Google Scholar]
- Lang UE, Jockers-Scherübl MC, Hellweg R. State of the art of the neurotrophin hypothesis in major psychiatric disorders: implications and limitations. J Neural Transm. 2004;111:387–411. doi: 10.1007/s00702-003-0100-0. [DOI] [PubMed] [Google Scholar]
- Laurence KM, James N, Miller MH, Tennant GB, Campbell H. Double-blind randomised controlled trial of folate treatment before conception to prevent recurrence of neural-tube defects. Br Med J (Clin Res Ed) 1981;282:1509–1511. doi: 10.1136/bmj.282.6275.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner B, Kozorovitskiy Y, Gross CG, Gould E. Diminished adult neurogenesis in the marmoset brain precedes old age. Proc Natl Acad Sci U S A. 2007;104:17169–17173. doi: 10.1073/pnas.0708228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewen A, Fredriksson A, Li GL, Olsson Y, Hillered L. Behavioural and morphological outcome of mild cortical contusion trauma of the rat brain: influence of NMDA receptor blockade. Acta Neurochir (Wien) 1999;141:193–202. doi: 10.1007/s007010050286. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Kim WK, Choi YB, Kumar S, D'Emilia DM, Rayudu PV, Arnelle DR, Stamler JS. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-d-aspartate receptor. Proc Natl Acad Sci U S A. 1997;94:5923–5928. doi: 10.1073/pnas.94.11.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger JA, Tang MX, Miller J, Green R, Mayeux R. Relation of higher folate intake to lower risk of Alzheimer disease in the elderly. Arch Neurol. 2007;64:86–92. doi: 10.1001/archneur.64.1.86. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millhouse OE, DeOlmos J. Neuronal configurations in lateral and basolateral amygdala. Neuroscience. 1983;10:1269–1300. doi: 10.1016/0306-4522(83)90112-4. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Gentry C, Cotman CW. Degeneration of hippocampal CA3 pyramidal cells induced by intraventricular kainic acid. J Comp Neurol. 1980;192:333–359. doi: 10.1002/cne.901920209. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell. 2000;5:1059–1065. doi: 10.1016/s1097-2765(00)80271-3. [DOI] [PubMed] [Google Scholar]
- Nurk E, Refsum H, Tell GS, Engedal K, Vollset SE, Ueland PM, Nygaard HA, Smith AD. Plasma total homocysteine and memory in the elderly: the Hordaland homocysteine study. Ann Neurol. 2005;58:847–857. doi: 10.1002/ana.20645. [DOI] [PubMed] [Google Scholar]
- Papakostas GI, Petersen T, Mischoulon D, Ryan JL, Nierenberg AA, Bottiglieri T, Rosenbaum JF, Alpert JE, Fava M. Serum folate, vitamin B12, and homocysteine in major depressive disorder, Part 1: predictors of clinical response in fluoxetine-resistant depression. J Clin Psychiatry. 2004a;65:1090–1095. doi: 10.4088/jcp.v65n0810. [DOI] [PubMed] [Google Scholar]
- Papakostas GI, Petersen T, Mischoulon D, Green CH, Nierenberg AA, Bottiglieri T, Rosenbaum JF, Alpert JE, Fava M. Serum folate, vitamin B12, and homocysteine in major depressive disorder, Part 2: predictors of relapse during the continuation phase of pharmacotherapy. J Clin Psychiatry. 2004b;65:1096–1098. doi: 10.4088/jcp.v65n0811. [DOI] [PubMed] [Google Scholar]
- Park EM, Choi JH, Park JS, Han MY, Park YM. Measurement of glutathione oxidation and 8-hydroxy-2′-deoxyguanosine accumulation in the gerbil hippocampus following global ischemia. Brain Res Brain Res Protoc. 2000;6:25–32. doi: 10.1016/s1385-299x(00)00033-7. [DOI] [PubMed] [Google Scholar]
- Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci. 2004;19:234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- Reynolds EH. Folic acid, ageing, depression, and dementia. BMJ. 2002;324:1512–1515. doi: 10.1136/bmj.324.7352.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds EH, Preece JM, Bailey J, Coppen A. Folate deficiency in depressive illness. Br J Psychiatry. 1970;117:287–292. [PubMed] [Google Scholar]
- Ridder S, Chourbaji S, Hellweg R, Urani A, Zacher C, Schmid W, Zink M, Hörtnagl H, Flor H, Henn FA, Schütz G, Gass P. Mice with genetically altered glucocorticoid receptor expression show altered sensitivity for stress-induced depressive reactions. J Neurosci. 2005;25:6243–6250. doi: 10.1523/JNEUROSCI.0736-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, Garcia AD, Sofroniew MV, Kandel ER, Santarelli L, Hen R, Drew MR. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci U S A. 2006;103:17501–17506. doi: 10.1073/pnas.0607207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- Selhub J, Jacques PF, Wilson PW, Rush D, Rosenberg IH. Vitamin status and intake as primary determinants of homocysteinemia in an elderly population. JAMA. 1993;270:2693–2698. doi: 10.1001/jama.1993.03510220049033. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D'Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- Shannon NJ, Gunnet JW, Moore KE. A comparison of biochemical indices of 5-hydroxytryptaminergic neuronal activity following electrical stimulation of the dorsal raphe nucleus. J Neurochem. 1986;47:958–965. doi: 10.1111/j.1471-4159.1986.tb00704.x. [DOI] [PubMed] [Google Scholar]
- Shea TB, Ortiz D, Rogers E. Differential susceptibility of transgenic mice lacking one or both apolipoprotein alleles to folate and vitamin E deprivation. J Alzheimers Dis. 2004;6:269–273. doi: 10.3233/jad-2004-6307. [DOI] [PubMed] [Google Scholar]
- Shen JC, Rideout WM, 3rd, Jones PA. High frequency mutagenesis by a DNA methyltransferase. Cell. 1992;71:1073–1080. doi: 10.1016/s0092-8674(05)80057-1. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E. Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus. 2002;12:578–584. doi: 10.1002/hipo.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithells RW, Sheppard S, Schorah CJ. Vitamin deficiencies and neural tube defects. Arch Child Dis. 1976;51:944–950. doi: 10.1136/adc.51.12.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperk G. Simultaneous determination of serotonin, 5-hydroxyindoleacetic acid, 3,4-dihydroxyphenylacetic acid and homovanillic acid by high performance liquid chromatography with electrochemical detection. J Neurochem. 1982;38:840–843. doi: 10.1111/j.1471-4159.1982.tb08708.x. [DOI] [PubMed] [Google Scholar]
- Sperk G, Berger M, Hörtnagl H, Hornykiewicz O. Kainic acid-induced changes of serotonin and dopamine metabolism in the striatum and substantia nigra of the rat. Eur J Pharmacol. 1981;74:279–286. doi: 10.1016/0014-2999(81)90046-7. [DOI] [PubMed] [Google Scholar]
- Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanhauser SM, Laipis PJ. Multiple deletions are detectable in mitochondrial DNA of aging mice. J Biol Chem. 1995;42:24769–24775. doi: 10.1074/jbc.270.42.24769. [DOI] [PubMed] [Google Scholar]
- Tchantchou F, Graves M, Rogers E, Ortiz D, Shea TB. N-acetyl cysteine alleviates oxidative damage to central nervous system of ApoE-deficient mice following folate and vitamin E-deficiency. J Alzheimers Dis. 2005;7:135–138. doi: 10.3233/jad-2005-7206. discussion 173–180. [DOI] [PubMed] [Google Scholar]
- Thoenen H. The changing scene of neurotrophic factors. Trends Neurosci. 1991;14:165–170. doi: 10.1016/0166-2236(91)90097-e. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bohlen und Halbach O, Unsicker K. Morphological alterations in the amygdala and hippocampus of mice during ageing. Eur J Neurosci. 2002;16:2434–2440. doi: 10.1046/j.1460-9568.2002.02405.x. [DOI] [PubMed] [Google Scholar]
- Winter B, Bert B, Fink H, Dirnagl U, Endres M. Dysexecutive syndrome after mild ischemia? Mice learn normally but have deficits in strategy switching. Stroke. 2004;35:191–195. doi: 10.1161/01.STR.0000107188.29688.2C. [DOI] [PubMed] [Google Scholar]
- Winter B, Juckel G, Viktorov I, Katchanov J, Gietz A, Sohr R, Balkaya M, Hörtnagl H, Endres M. Anxious and hyperactive phenotype following brief ischemic episodes in mice. Biol Psychiatry. 2005;57:1166–1175. doi: 10.1016/j.biopsych.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Winter-Vann AM, Kamen BA, Bergo MO, Young SG, Melnyk S, James SJ, Casey PJ. Targeting Ras signaling through inhibition of carboxyl methylation: an unexpected property of methotrexate. Proc Natl Acad Sci U S A. 2003;100:6529–6534. doi: 10.1073/pnas.1135239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SA, Kronenberg G, Lehmann K, Blankenship A, Overall R, Staufenbiel M, Kempermann G. Cognitive and physical activity differently modulate disease progression in the amyloid precursor protein (APP)-23 model of Alzheimer's disease. Biol Psychiatry. 2006;60:1314–1323. doi: 10.1016/j.biopsych.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Yanamoto H, Miyamoto S, Tohnai N, Nagata I, Xue JH, Nakano Y, Nakajo Y, Kikuchi H. Induced spreading depression activates persistent neurogenesis in the subventricular zone, generating cells with markers for divided and early committed neurons in the caudate putamen and cortex. Stroke. 2005;36:1544–1550. doi: 10.1161/01.STR.0000169903.09253.c7. [DOI] [PubMed] [Google Scholar]