

Abstract
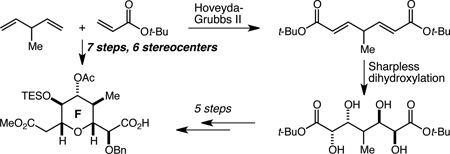
A concise and efficient synthesis of the F-ring fragment of the potent anti-mitotic marine macrolide spongistatin 1 has been developed. The key sequence involves double cross-metathesis/Sharpless asymmetric dihydroxylation reactions to establish four stereocenters in a pseudo C2-symmetric array, followed by a selective protection reaction that breaks the pseudo-symmetry, establishes a fifth stereocenter, and effectively differentiates the ester termini. Overall, the six contiguous stereocenters in the C(37)–C(45) F-ring fragment are established in just seven steps.
The spongistatins – structurally complex marine macrolides possessed of extraordinarily potent anti-mitotic activity – have elicited a great deal of attention from synthetic chemists since their isolation in 19931,2,3 resulting in seven groups reporting syntheses of spongistatin 1 and/or 2.4,5,6,7,8,9,10 We are pursuing the design, synthesis, and evaluation of a series of analogs of spongistatin 1, with the CD spiroketal region the main focus of those efforts.11 In order to synthesize the completed CD spiroketal-modified analogs of spongistatin 1, we will of course require a supply of the C(29)–C(51) EF fragment12 (Fig. 1A), and our initial explorations into the development of an efficient, step-economical, and scalable synthesis of that fragment are the subject of this report.
Figure 1.

A The C(29)–C(51) EF fragment of spongistatin 1. B Retrosynthetic analysis of the C(37)–C(45) F-ring fragment reveals pseudo C2-symmetry.
We set as our initial goal the development of an efficient synthesis of penta-substituted tetrahydropyran F-ring fragment 1 with an ester at C(45) from which to construct the chlorodiene side-chain and a methyl ketone at C(37) for introduction of the E-ring by way of an aldol reaction according to the Paterson precedent7c (Figure 1B). Within 1 there may be found a latent element of pseudo C2-symmetry, and we became intrigued by the possibility of employing a two-directional chain synthesis strategy.13 To this end, we envisioned that the C(43) stereocenter might arise from reduction of a lactol and, in the first iteration of our retrosynthesis, that the C(37) methyl ketone might arise from decarboxylation of the corresponding β-ketoester. These operations lead retrosynthetically to lactol 2, and in turn to tetraol 3, revealing the pseudo C2-symmetry. We were optimistic that spontaneous lactol formation from a tetraol such as 3 would prove an effective and straightforward method for termini differentiation, as lactol 2 should be favored over the alternative, lactol 4, in which the C(40) methyl group would be axially disposed. Double Sharpless asymmetric dihydroxylation (AD)14,15 leads retrosynthetically to 5, which we hoped might be accessible by double cross-metathesis (CM)16 with vinyl ketone 6 and 3-methyl-1,4-pentadiene 7. The possibility that we might access 2 in just two steps from 6 and 7 provided all the impetus we needed to initiate this investigation.
Initial attempts to accomplish the double CM reaction of diene 7 with 6a (R = Me) resulted only in dimerization of 6a (Scheme 1). Various protected/masked versions of 6a (among them alcohol 8 and dioxolane 9) also failed to provide any of the desired CM products. We turned next to the use of simple acrylate esters (methyl and t-butyl) and were delighted to find not only that the double cross metathesis reaction worked well with 10 mol % of the second generation Hoveyda-Grubbs catalyst (HG-II),17 but also that the products, 10a and 10b, were stable to standard work-up and purification procedures. Initial attempts to establish the feasibility of the double Sharpless AD reaction using AD-Mix-β revealed that while no tetraol product could be isolated from the reaction of 10a, tetraol 11 could indeed be isolated, albeit in low yield, from the reaction of 10b. The t-butyl ester was therefore selected for optimization, and in the case of the CM reaction, the primary goal was a significantly reduced HG-II catalyst loading. By adding the catalyst portion-wise over seven days it proved possible to achieve an 80% yield of 10b with a catalyst loading of 2.25 mol %. While this is a quite low catalyst loading, especially for a double CM reaction, and while this procedure proved reliable on multi-gram scale (22 g of 10b were produced in a single run), the inconveniently long reaction time motivated us to optimize further. Upsing t-butyl acrylate as the solvent, we found that a total loading of 0.69 mol % of the HG-II catalyst added portion wise (3×0.23 mol %) over a total reaction time of just 5 h allowed the isolation of 10b in 58% yield, also on multi-gram scale. Despite the lower yield, this protocol is a significant improvement in terms of the amount of product produced per unit of HG-II catalyst. For the double AD reaction, the low yield was the primary problem, and this was addressed by increasing the loading of both the osmium source (K2OsO4•2 H2O) and the chiral ligand ((DHQD)2PHAL). Using 4 mol % and 5 mol %, respectively, tetraol 11 could be isolated as the major product of a 4.5:1 mixture of diastereomers18 in 62% yield. Importantly, this reaction proved reliable on multi-gram scale as well, and was used to prepare 16.4 g of 11 in a single run.
Scheme 1.

Development of a scalable two-step synthesis of pseudo C2-symmetric tetraol 11.
With efficient access to large quantities of 11 in just two steps secured, we turned our attention to its elaboration into a compound such as 3 so as to be able to investigate the effectiveness of the pseudo symmetry-breaking cyclization reaction. To that end, tetraol 11 was protected in the form of bis-acetonide 12 in 75% yield (Scheme 2). Saponification and alkylation provided bis-methyl ester 13 in 84% overall yield. Double Claisen condensation of 13 with methyl cetate then delivered bis-β-ketoester 14 in 57% yield. Hydrolysis/methanolysis of the acetonides with 1 N HCl in methanol led to the relatively clean production of a compound that we have assigned as 15. There was some irreproducibility in this transformation, and the product was difficult to work with, but we were able to characterize 15 by1H NMR spectroscopy including a NOESY experiment which supported the stereochemical assignment at the methyl-bearing C(40) stereocenter. Though this route was abandoned due to the difficulties in doing anything productive with 15, we were delighted to secure this evidence that the C(40) stereocenter could indeed be controlled in this fashion.
Scheme 2.

Demonstration of termini differentiation by way of a selective lactolization reaction.
Further experimentation revealed other transformations of tetraol 11 were possible, such as its completely selective conversion to orthoester 16 in 72% yield19 (Scheme 3). Although the fact that the C(40) methyl group is equatorial in 16 and axial in 17 provides a simple rationalization for this selectivity, the A value of a methyl group in the 5-position of a 2-methyl-1,3-dioxane is only 0.97 kcal/mol,20 and it thus appears that other less obvious interactions are relevant as well. Benzylation of the remaining alcohol provided fully protected tetraol 18 in 78% yield. Prior to replacing the t-butyl esters with methyl esters in anticipation of attempting a double Claisen condensation (cf. 13 to 14), we decided to see if 18 itself was a viable substrate. Interestingly, while 18 did undergo a Claisen condensation, only the C(43) ester was reactive under these conditions. Upon optimization, this reaction provided 19 in 80% yield. Treatment of 19 with pyridinium p-toluene sulfonate (PPTS) in MeOH/H2O resulted in smooth and selective partial hydrolysis of the orthoester and cyclization to give lactol 20 in 98% yield. With access to 20 secured, we were finally in a position to examine the lactol reduction21 to set the final stereocenter of the F-ring. Initial attempts using Et3SiH and BF3•OEt2 proved messy and inefficient, while the use of triethylsilyl triflate (TESOTf) as the Lewis acid led to partial t-Bu ester cleavage in addition to the desired reduction. Rather than attempting to avoid this cleavage, we decided to drive it to completion by using excess TESOTf and letting the reaction mixture warm to 0 °C. A triethylamine quench further resulted in alcohol silylation, and when this process was optimized, it led to the isolation of 21 in 72% yield.
Scheme 3.

Termini differentiation by selective protection and F-ring completion.
We turned our attention next to the transformation of 21 into a compound that was appropriately functionalized for attachment of the E-ring and the chlorodiene sidechain. As discussed above, this meant transformation of the carboxylic acid into a methyl ketone, while we envisioned that an allylsilane might prove a versatile functionality with which to install the chlorodiene sidechain. Thus, acid 21 was transformed into Weinreb amide22 2223 in 82% yield (Scheme 4). Attempts to transform the methyl ester into an allylsilane using Bunnelle’s method24 at this stage were completely unsuccessful, a failure we attributed to severe steric hindrance around the ester, mainly due to the TES ether. Cleavage of the TES ether and the acetate could be carried out with n-Bu4NF to give in 88% yield diol 23, which was transformed into acetonide 24 in 69% yield. We recovered 21% of 23 from this latter reaction, whose reluctance to go to completion is attributable to ring strain in the trans-fused acetonide. Gratifyingly, conversion of less sterically encumbered ester 24 to allylsilane 25 using Bunnelle’s procedure could be carried out at this stage, albeit with moderate efficiency (59% yield). Finally, addition of MeMgBr to the Weinreb amide delivered 26, ready for coupling at both ends to install the E-ring and the chlorodiene sidechain.
Scheme 4.

Elaboration of the F-ring for installment of the E-ring and the chlorodiene sidechain.
We have described an efficient synthesis of the F-ring of the spongistatins. A previously unrecognized latent element of pseudo C2-symmetry was identified and exploited by way of a double CM and double Sharpless AD sequence that quickly established 4 carbinol stereocenters in just two steps. Diastereoselective protection of the tetraol broke the pseudo C2-symmetry and established the C(40) methyl-bearing stereocenter, and effectively differentiated the ester termini. Just four additional steps delivered completed F-ring fragment 21, which contains six of the 11 stereocenters found in the EF fragment of the spongistatins, in seven steps and 16% overall yield from commercially available starting materials. Five additional steps produced a fully elaborated F-ring fragment appropriately functionalized and protected for installation of the E-ring and the chlorodiene sidechain. Efforts to refine and streamline this sequence – especially with respect to minimizing functional group and protecting group manipulations – for use in an efficient, step-economical, and scalable synthesis of the EF fragment of the spongistatins are ongoing.
Supplementary Material
Acknowledgment
The National Institute of General Medical Sciences is gratefully acknowledged for their support of this work (GM058133), and for a postdoctoral fellowship award (F32GM090487) to P.S.T. We thank our colleagues Prof. Gerard Parkin and Dr. Aaron Sattler (Columbia University) for the X-ray structure analysis of 22, and the National Science Foundation (CHE-0619638) is thanked for acquisition of an X-ray diffractometer.
Footnotes
Supporting Information Available: Experimental procedures, characterization data, and stereochemical proofs as well as X-ray crystallographic data for compound 22. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pettit GR, Cichacz ZA, Gao F, Herald CL, Boyd MR, Schmidt JM, Hooper JNA. J. Org. Chem. 1993;58:1302–1304. [Google Scholar]
- 2.Fusetani N, Shinoda K, Matsunaga S. J. Am. Chem. Soc. 1993;115:3977–3981. [Google Scholar]
- 3.a Kobayashi M, Aoki S, Sakai H, Kawazoe K, Kihara N, Sasaki T, Kitagawa I. Tetrahedron Lett. 1993;34:2795–2798. [Google Scholar]; b Kobayashi M, Aoki S, Sakai H, Kihara N, Sasaki T, Kitagawa I. Chem. Pharm. Bull. 1993;41:989–991. doi: 10.1248/cpb.41.989. [DOI] [PubMed] [Google Scholar]; c Kobayashi M, Aoki S, Kitagawa I. Tetrahedron Lett. 1994;35:1243–1246. [Google Scholar]
- 4.a Evans DA, Coleman PJ, Dias LC. Angew. Chem. Int. Ed. 1997;36:2738–2741. [Google Scholar]; b Evans DA, Trotter BW, Côté B, Coleman PJ. Angew. Chem. Int. Ed. 1997;36:2741–2744. [Google Scholar]; c Evans DA, Trotter BW, Côté B, Coleman PJ, Dias LC, Tyler AN. Angew. Chem. Int. Ed. 1997;36:2744–2747. [Google Scholar]; d Evans DA, Trotter BW, Coleman PJ, Côté B, Dias LC, Rajapakse HA, Tyler AN. Tetrahedron. 1999;55:8671–8726. [Google Scholar]
- 5.a Guo J, Duffy KJ, Stevens KL, Dalko PI, Roth RM, Hayward MM, Kishi Y. Angew. Chem. Int. Ed. 1998;37:187–192. [Google Scholar]; b Hayward MM, Roth RM, Duffy KJ, Dalko PI, Stevens KL, Guo J, Kishi Y. Angew. Chem. Int. Ed. 1998;37:192–196. [Google Scholar]
- 6.a Smith AB, III, Doughty VA, Lin Q, Zhuang L, McBriar MD, Boldi AM, Moser WH, Murase N, Nakayama K, Sobukawa M. Angew. Chem. Int. Ed. 2001;40:191–195. doi: 10.1002/1521-3773(20010105)40:1<191::AID-ANIE191>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; b Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Brook CS, Murase N, Nakayama K. Angew. Chem. Int. Ed. 2001;40:196–199. [PubMed] [Google Scholar]; c Smith AB, III, Tomioka T, Risatti CA, Sperry JB, Sfouggatakis C. Org. Lett. 2008;10:4359–4362. doi: 10.1021/ol801792k. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Boldi AM, Murase N, Moser WH, Brook CS, Bennett CS, Nakayama K, Sobukawa M, Trout REL. Tetrahedron. 2009;65:6470–6488. doi: 10.1016/j.tet.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Smith AB, III, Sfouggatakis C, Risatti CA, Sperry JB, Zhu W, Doughty VA, Tomioka T, Gotchev DB, Bennett CS, Sakamoto S, Atasoylu O, Shirakami S, Bauer D, Takeuchi M, Koyanagi J, Sakamoto Y. Tetrahedron. 2009;65:6489–6509. doi: 10.1016/j.tet.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Paterson I, Chen DY-K, Coster MJ, Aceña JL, Bach J, Gibson KR, Keown LE, Oballa RM, Trieselmann T, Wallace DJ, Hodgson AP, Norcross RD. Angew. Chem. Int. Ed. 2001;40:4055–4060. [PubMed] [Google Scholar]; b Paterson I, Coster MJ, Chen DY-K, Oballa RM, Wallace DJ, Norcross RD. Org. Biomol. Chem. 2005;3:2399–2409. doi: 10.1039/b504146e. [DOI] [PubMed] [Google Scholar]; c Paterson I, Coster MJ, Chen DY-K, Gibson KR, Wallace DJ. Org. Biomol. Chem. 2005;3:2410–2419. doi: 10.1039/b504148a. [DOI] [PubMed] [Google Scholar]; d Paterson I, Coster MJ, Chen DY-K, Aceña JL, Bach J, Keown LE, Trieselmann T. Org. Biomol. Chem. 2005;3:2420–2430. doi: 10.1039/b504149j. [DOI] [PubMed] [Google Scholar]; e Paterson I, Chen DY-K, Coster MJ, Aceña JL, Bach J, Wallace DJ. Org. Biomol. Chem. 2005;3:2431–2440. doi: 10.1039/b504151a. [DOI] [PubMed] [Google Scholar]
- 8.Crimmins MT, Katz JD, Washburn DG, Allwein SP, McAtee LF. J. Am. Chem. Soc. 2002;124:5661–5663. doi: 10.1021/ja0262683. [DOI] [PubMed] [Google Scholar]
- 9.Heathcock CH, McLaughlin M, Medina J, Hubbs JL, Wallace GA, Scott R, Claffey MM, Hayes CJ, Ott GR. J. Am. Chem. Soc. 2003;125:12844–12849. doi: 10.1021/ja030317+. [DOI] [PubMed] [Google Scholar]
- 10.a Ball M, Gaunt MJ, Hook DF, Jessiman AS, Kawahara S, Orsini P, Scolaro A, Talbot AC, Tanner HR, Yamanoi S, Ley SV. Angew. Chem. Int. Ed. 2005;44:5433–5438. doi: 10.1002/anie.200502008. [DOI] [PubMed] [Google Scholar]; b Kraus H, Français A, O’Brien M, Frost J, Diéguez-Vázquez A, Polara A, Baricordi N, Horan R, Hsu D-S, Tsunoda T, Ley SV. Chem. Sci. 2013;4:1989–1994. [Google Scholar]
- 11.a Reznik SK, Marcus BS, Leighton JL. Chem. Sci. 2012;3:3326–3330. doi: 10.1039/C2SC21325G. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Reznik SK, Leighton JL. Chem. Sci. 2013;4:1497–1501. doi: 10.1039/C3SC22186E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For other approaches to the EF fragment of the spongistatins see: Kary PD, Roberts SM. Tetrahedron Lett. 1999;40:217–219. Micalizio GC, Roush WR. Tetrahedron Lett. 1999;40:3351–3354. Anderson JC, McDermott BP. Tetrahedron Lett. 1999;40:7135–7138. Kim H, Hoffmann HMR. Eur. J. Org. Chem. 2000:2195–2201. Samadi M, Munoz-Letelier C, Poigny S, Guyot M. Tetrahedron Lett. 2000;41:3349–3353. Lemaire-Audoire S, Vogel P. J. Org. Chem. 2000;65:3346–3356. doi: 10.1021/jo991642b. Micalizio GC, Pinchuk AN, Roush WR. J. Org. Chem. 2000;65:8730–8736. doi: 10.1021/jo001236o. Terauchi T, Morita M, Kimijima K, Nakamura Y, Hayashi G, Tanaka T, Kanoh N, Nakata M. Tetrahedron Lett. 2001;42:5505–5508. Terauchi T, Tanaka T, Terauchi T, Morita M, Kimijima K, Sato I, Shoji W, Nakamura Y, Tsukada T, Tsunoda T, Hayashi G, Kanoh N, Nakata M. Tetrahedron Lett. 2003;44:7747–7751. Ciblat S, Kim J, Stewart CA, Wang J, Forgione P, Clyne D, Paquette LA. Org. Lett. 2007;9:719–722. doi: 10.1021/ol063083i.
- 13.a Still WC, Barrish JC. J. Am. Chem. Soc. 1983;105:2487–2489. [Google Scholar]; b Poss CS, Schreiber SL. Acc. Chem. Res. 1994;27:9–17. [Google Scholar]
- 14.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483–2547. [Google Scholar]
- 15.It should be noted that Vogel employed sequential dihydroxylation reactions to access a related tetraol. See ref. 12f.
- 16.Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH. J. Am. Chem. Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 17.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168–8179. [Google Scholar]
- 18.We have been unable to isolate a pure sample of the minor diastereomer, but the1H NMR spectrum of the mixture is consistent with it being, as expected, one of the two possible meso tetraol diastereomers.
- 19.The starting material (11) used in this experiment was the 4.5:1 mixture of diastereomers produced in the double AD reaction (Scheme 1). No evidence for the formation of a second orthoester was observed, suggesting that the minor tetraol diastereomer does not successfully cyclize to an orthoester.
- 20.Eliel EL, Knoeber MC. J. Am. Chem. Soc. 1968;90:3444–3458. [Google Scholar]
- 21.Lewis MD, Cha JK, Kishi Y. J. Am. Chem. Soc. 1982;104:4976–4978. [Google Scholar]
- 22.Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]
- 23.Compound 22 proved crystalline, and an X-ray structural analysis was carried out which confirmed its assigned stereostructure. See the Supporting Information for details.
- 24.a Narayanan BA, Bunnelle WH. Tetrahedron Lett. 1987;28:6261–6264. [Google Scholar]; b Bunnelle WH, Narayanan BA. Organic Syntheses. Vol. 8. New York: Wiley & Sons; 1993. pp. 602–605. Collect. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.