

Significance
Our studies combining genetic and pharmacological manipulations provide convincing evidence that exchange protein directly activated by cAMP (Epac) 1 plays a critical role in fatal spotted fever group rickettsioses. Inhibition of Epac1 suppresses bacterial adhesion and/or invasion. Most importantly, we show that a small-molecule Epac inhibitor can prevent and suppress rickettsial infection. Our results demonstrate that Epac1-mediated signaling represents a mechanism for host–pathogen interactions and that Epac1 is a potential target for the prevention and treatment of fatal rickettsioses. This is significant from the biodefense viewpoint because it suggests that Epac1 inhibitor can be potentially used as a prophylaxis to thwart initial bacterial infection in the event of a bioterrorism threat.
Keywords: rickettsial infection, cyclic AMP, Epac inhibitor, therapeutics, prophylaxis
Abstract
Rickettsiae are responsible for some of the most devastating human infections. A high infectivity and severe illness after inhalation make some rickettsiae bioterrorism threats. We report that deletion of the exchange protein directly activated by cAMP (Epac) gene, Epac1, in mice protects them from an ordinarily lethal dose of rickettsiae. Inhibition of Epac1 suppresses bacterial adhesion and invasion. Most importantly, pharmacological inhibition of Epac1 in vivo using an Epac-specific small-molecule inhibitor, ESI-09, completely recapitulates the Epac1 knockout phenotype. ESI-09 treatment dramatically decreases the morbidity and mortality associated with fatal spotted fever rickettsiosis. Our results demonstrate that Epac1-mediated signaling represents a mechanism for host–pathogen interactions and that Epac1 is a potential target for the prevention and treatment of fatal rickettsioses.
Rickettsiae are responsible for some of the most devastating human infections (1–4). It has been forecasted that temperature increases attributable to global climate change will lead to more widespread distribution of rickettsioses (5). These tick-borne diseases are caused by obligately intracellular bacteria of the genus Rickettsia, including Rickettsia rickettsii, the causative agent of Rocky Mountain spotted fever (RMSF) in the United States and Latin America (2, 3), and Rickettsia conorii, the causative agent of Mediterranean spotted fever endemic to southern Europe, North Africa, and India (6). A high infectivity and severe illness after inhalation make some rickettsiae (including Rickettsia prowazekii, R. rickettsii, Rickettsia typhi, and R. conorii) bioterrorism threats (7). Although the majority of rickettsial infections can be controlled by appropriate broad-spectrum antibiotic therapy if diagnosed early, up to 20% of misdiagnosed or untreated (1, 3) and 5% of treated RMSF cases (8) result in a fatal outcome caused by acute disseminated vascular endothelial infection and damage (9). Fatality rates as high as 32% have been reported in hospitalized patients diagnosed with Mediterranean spotted fever (10). In addition, strains of R. prowazekii resistant to tetracycline and chloramphenicol have been developed in laboratories (11). Disseminated endothelial infection and endothelial barrier disruption with increased microvascular permeability are the central features of SFG rickettsioses (1, 2, 9). The molecular mechanisms involved in rickettsial infection remain incompletely elucidated (9, 12). A comprehensive understanding of rickettsial pathogenesis and the development of novel mechanism-based treatment are urgently needed.
Living organisms use intricate signaling networks for sensing and responding to changes in the external environment. cAMP, a ubiquitous second messenger, is an important molecular switch that translates environmental signals into regulatory effects in cells (13). As such, a number of microbial pathogens have evolved a set of diverse virulence-enhancing strategies that exploit the cAMP-signaling pathways of their hosts (14). The intracellular functions of cAMP are predominantly mediated by the classic cAMP receptor, protein kinase A (PKA), and the more recently discovered exchange protein directly activated by cAMP (Epac) (15). Thus, far, two isoforms, Epac1 and Epac2, have been identified in humans (16, 17). Epac proteins function by responding to increased intracellular cAMP levels and activating the Ras superfamily small GTPases Ras-proximate 1 and 2 (Rap1 and Rap2). Accumulating evidence demonstrates that the cAMP/Epac1 signaling axis plays key regulatory roles in controlling various cellular functions in endothelial cells in vitro, including cell adhesion (18–21), exocytosis (22), tissue plasminogen activator expression (23), suppressor of cytokine signaling 3 (SOCS-3) induction (24–27), microtubule dynamics (28, 29), cell–cell junctions, and permeability and barrier functions (30–37). Considering the critical importance of endothelial cells in rickettsioses, we examined the functional roles of Epac1 in rickettsial pathogenesis in vivo, taking advantage of the recently generated Epac1 knockout mouse (38) and Epac-specific inhibitors (39, 40) generated from our laboratory. Our studies demonstrate that Epac1 plays a key role in rickettsial infection and represents a therapeutic target for fatal rickettsioses.
Results
Deletion of the Epac1 Gene in Mice Protects Them from Fatal Rickettsiosis.
To investigate the functional role of Epac1 in rickettsiosis, we generated an Epac1 knockout mouse (38) and challenged both Epac1−/− and C57BL/6 wild-type (WT) mice with Rickettsia australis. The C57BL/6 mouse–R. australis model is an established animal model of human spotted fever group (SFG) rickettsiosis because the pathology involves disseminated endothelial infection and pathological lesions, including vasculitis in multiple organs, similar to what is observed in human RMSF (41, 42). All of the WT mice became progressively ill starting on day 3 after inoculation, with typical signs of markedly ruffled fur, a hunched posture, and partially closed eyelids (Fig. 1A). Eight of 12 WT mice died by the end of the 8-d experiment (67% mortality) (Fig. 1B). To our surprise, most of the Epac1−/− mice suffered only a very mild illness and only 2 of 17 Epac1−/− mice died (12% mortality). Consistent with the morbidity and mortality observations, histological examination of WT mice tissues revealed severe vasculitis and perivasculitis in the testes and liver, interstitial pneumonia, and multifocal hepatocellular necrosis (Fig. 1C). Immunofluorescent (IF) staining for rickettsial antigens and von Willebrand factor in tissue confirmed SFG rickettsiae infection in endothelial cells (Fig. S1). Conversely, these typical pathological lesions associated with rickettsioses (41, 42) were largely absent from the Epac1−/− mice given a dose of R. australis that normally kills at least half of the mice (Fig. 1C).
Fig. 1.

Mice lacking Epac1 are protected from rickettsial infection. (A and B) Disease progression (A) and survival (B) of Epac1+/+ (WT) (n = 12) and Epac1−/− (KO) (n = 17) mice were monitored daily for 8 d postinfection following infection with R. australis (R. a) or mock infection. (C) Representative IHC staining of SFG rickettsiae (red) in foci of necrotic infected liver, lung, and testis from WT C57BL/6 (Epac1+/+) and Epac1−/− mice. Rickettsiae (red) were stained using alkaline phosphatase–fast red, whereas nuclei of mouse cells were counterstained with hematoxylin (blue). The light pink color in the periportal hepatocytes and connective tissue of the testis interstitium is nonspecific staining by the alkaline phosphatase–fast red. (Scale bars: 20 µm.)
Rickettsial Infection Induces Increased Expression of Epac1 in Rickettsial Lesions.
Expression profiling of endogenous Epac1 provided evidence that in WT C57BL/6 mice infection with R. australis induced increased expression of Epac1 in cells of the interstitium of mouse lung and testis where there are rickettsial antigen signals (Fig. 2A) 8 d after inoculation. Interestingly, dual-target IF studies on tissues from two cases of archival pediatric fatal RMSF also revealed increased Epac1 immunostaining signals within multiple rickettsial lesions located in the blood–brain barrier areas (Fig. 2B). These in vivo observations of enhanced expression of Epac1 within rickettsial lesions, coupled with the fact that deficiency of Epac1 protects mice against rickettsial infection, suggest that Epac1 plays a critical role in fatal SFG rickettsiosis.
Fig. 2.
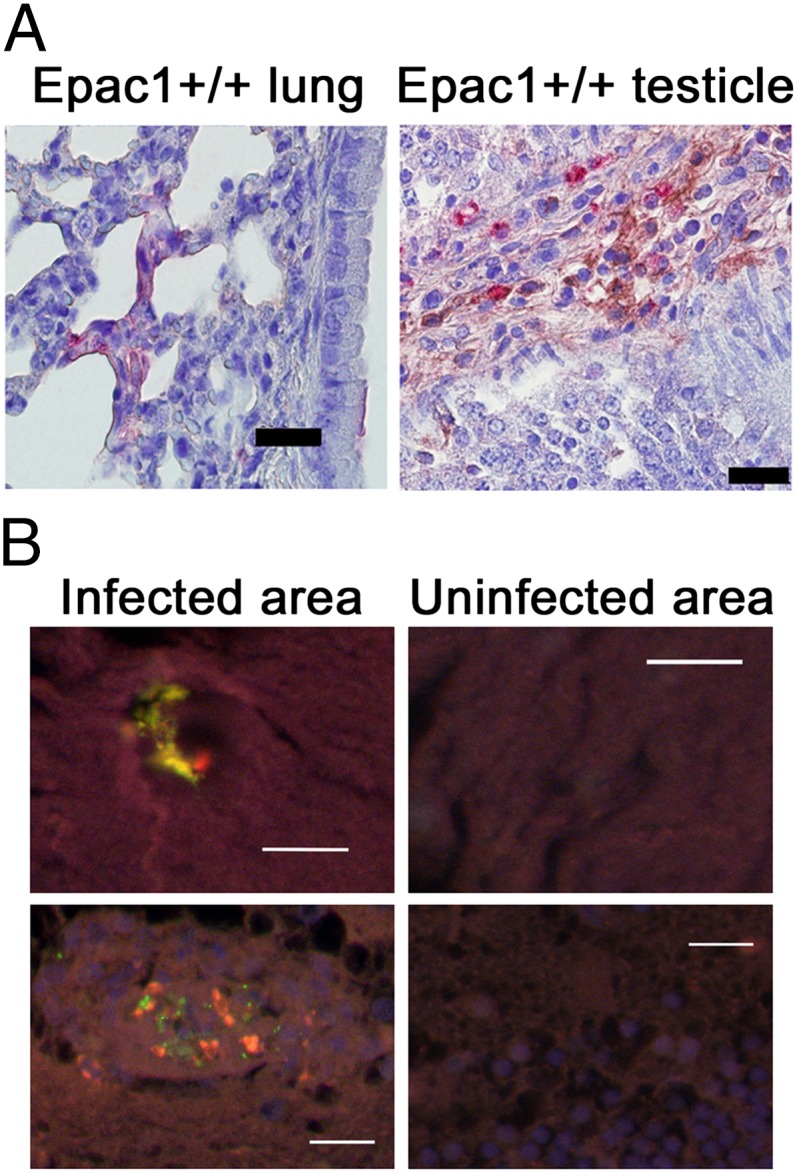
Rickettsial infection induces increased expression of Epac1 in rickettsial lesions. (A) Representative dual-target IHC staining of rickettsiae (red) and Epac1 (brown) in lung and testis from Epac1+/+ mice (n = 12). Rickettsiae and Epac1 were stained using alkaline phosphatase–fast red and peroxidase–DAB, respectively. Nuclei of mouse cells were counterstained with hematoxylin (blue). (B) Representative dual-target IF staining of rickettsiae (green) and Epac1 (red) in brain from an archived pediatric case of fatal RMSF. Nuclei of human cells are counterstained with DAPI (blue). (Scale bars: 20 µm.)
Deletion of Epac1 Impedes Rickettsial Attachment and/or Invasion into the Endothelial Cells ex Vivo.
We next sought to determine the molecular and cellular mechanisms by which Epac1 might be involved in rickettsial pathogenesis. To determine whether lack of Epac1 directly impedes bacterial infection, we established an ex vivo vascular endothelial culture rickettsial infection model using aortic rings prepared from WT C57BL/6 and Epac1-null mice (Materials and Methods). Deletion of Epac1 nearly completely blocked rickettsial attachment and/or invasion into the endothelial layer of the aortic ring at 30 min postexposure to R. australis at a dose of 1 × 105 pfu per aortic ring while the endothelium of the aortic ring from WT mice was highly decorated by rickettsia (Fig. 3A).
Fig. 3.

Epac1 plays critical role in rickettsial attachment and invasion into nonphagocytic host endothelial cells. (A) Representative dual-target IF staining of rickettsiae (red) and Epac1 (green) in frozen section of ex vivo aortic ring prepared from Epac1+/+ and Epac1−/− mice 30 min postinfection with R. australis. Nuclei of mouse cells were counterstained with DAPI (blue). (B) Total bacteria in EIS-09– and vehicle-exposed HUVECs 30 min postinfection with R. australis were enumerated by IF microscopy. The data presented are representative of three independent experiments. The error bar is SD. *P < 0.01. (C) Representative dual-target IF staining of rickettsiae (red) and Epac1 (green) in ESI-09– and vehicle-exposed HUVECs 30 min postinfection with R. australis. Nuclei of HUVECs were counterstained with DAPI (blue). (Scale bars: A, 50 µm; C, 10 µm.)
Inhibition of Epac1 Blocks Rickettsial Attachment and Invasion into Nonphagocytic Host Cells.
Taking advantage of a recently discovered Epac-specific inhibitor, ESI-09 (39, 40), we monitored the rickettsial infection process in human umbilical vein endothelial cells (HUVECs) exposed to 5 µM ESI-09 or a vehicle sham. As shown in Fig. 3B and Fig. S2, exposure to ESI-09 significantly reduced intracellular and total bacterial counts in HUVECs at 30 min postinfection with 10 multiplicities of infection (MOI) of R. australis compared with similarly infected controls exposed to vehicle only. Moreover, IF staining revealed that Epac1 colocalized with rickettsiae (Fig. 3C) in the cytosol of HUVECs, whereas exposure to ESI-09 attenuated this interaction. An earlier investigation identified Ku70, a subunit of DNA-dependent protein kinase (DNA-PK), as a potential host cell receptor (43). Indeed, IF analysis showed that Ku70 colocalized with the bacteria inside the cytosol of the HUVECs (Fig. 4). Again, pharmacological inhibition of Epac1 using ESI-09 significantly reduced such interactions. Taken together, these data suggest that Epac1 plays a critical role in the early stage of rickettsial attachment and invasion into nonphagocytic host cells.
Fig. 4.

Epac1 inhibition blocks rickettsial invasion by impeding the expression of Ku70 outside of the nucleus in HUVECs. Representative dual-target IF staining of rickettsiae (red) and Ku70 (green) in ESI-09– and vehicle-exposed HUVECs 30 min postinfection with R. australis. Nuclei of HUVECs were counterstained with DAPI (blue). (Scale bars: 10 µm.)
Pharmacological Inhibition of Epac1 Protects WT Mice from Fatal SFG Rickettsiosis.
Encouraged by the host protection data using the Epac1 knockout mouse model, we further explored the potential of using Epac pharmacological intervention as a therapeutic strategy for combating fatal SFG rickettsiosis. WT C57BL/6 mice, randomly divided into two groups, were treated with ESI-09 (10 mg⋅kg−1⋅d−1) or vehicle via i.p. injection for 5 d, followed by i.v. inoculation of R. australis. ESI-09 treatment was continued for another 7 d. As shown in Fig. 5, treatment with ESI-09 dramatically protected WT mice against R. australis infection with much milder disease manifestations (Fig. 5A) and significantly improved survival (Fig. 5B). Only 1 of 11 ESI-09–treated mice died (9% mortality), compared with those of the vehicle-only group, in which 6 of 10 WT mice died (60% mortality) at the end of experiment. Histological evaluation confirmed that pharmacological inhibition of Epac significantly attenuated the pathological responses, resulting in milder vasculitis in the testis, occasional microvesicular hepatocellular fatty change, less interstitial inflammation in the lung, and immunohistochemical evidence of significantly less rickettsial antigen in tissues compared with the control group (Fig. 5C). Such observations from in vivo pharmacological inhibition of Epac recapitulated data based on genetic Epac1 knockout mice. Taken together, these results demonstrated that Epac1 inhibition is an effective therapeutic strategy for fatal SFG rickettsiosis.
Fig. 5.

Pharmacological inhibition of Epac1 protects WT mice against rickettsial infection. Disease progression (A) and survival (B) of ESI-09–treated (n = 11) and vehicle-treated (n = 10) Epac1+/+ (WT) mice were monitored daily for 8 d postinfection with R. australis or mock infection. (C) Representative IHC staining of SFG rickettsiae (red) in liver, lung, and testis from ESI-09– and vehicle-treated Epac1+/+ (WT) mice. Rickettsiae were stained using alkaline phosphatase–fast red, whereas nuclei of mouse cells were counterstained with hematoxylin (blue). (Scale bars: 20 µm.)
Discussion
In this study, we probed the role of Epac1, a newly discovered family member of eukaryotic cAMP receptors, in the pathogenesis of rickettsiosis using both genetic and pharmacological approaches in vivo. The rationale behind the study is twofold. First, cAMP signaling has been extensively manipulated by microbial pathogens to facilitate their virulence both from functions within the pathogens themselves and their mammalian host cells (14). Second, Epac1 has been implicated as a key player in regulating various functions in endothelial cells, a major target of rickettsial infection. The findings that the genetic deletion of Epac1 and its pharmacological inhibition protected mice from fatal rickettsiosis are noteworthy. Based on Epac1’s published functions in cell–cell junctions and barrier functions (30–37), our initial prediction was that deletion or inhibition of Epac1 in mice would compromise the endothelial barrier and lead to a more severe response to rickettsial infection. To the contrary, both genetic deletion and pharmacological inhibition of Epac1 protect mice from an ordinarily lethal dose of rickettsiae.
Rickettsiae–endothelial cell interactions are associated with several major steps, namely bacterial adhesion, invasion through induced phagocytosis, phagosomal escape for intracellular survival, bacterial replication, and motility enhancement for cell-to-cell spreading (6, 9, 43, 44). Because Epac1 is known to regulate functions associated with cell adhesion and formation of intercellular junctions, particularly barrier functions in endothelial cells (45), we developed an ex vivo rickettsial infection model to further identify at which step Epac1 plays a role in the bacteria–host cell interaction during rickettsioses. Studies using aortic rings prepared from WT C57BL/6 and Epac1−/− (null) mice allow us to demonstrate that deletion of Epac1 prevents adhesion and/or invasion of rickettsiae into endothelial cells. This observation is further validated using ESI-09 and an in vitro rickettsial infection model that uses HUVECs (12). Considering the lack of effective animal models for many microbial pathogens, the establishment of aortic rings ex vivo as an endothelial infection model will have a major impact not just on rickettsiosis but also on other studies of endothelial-related microbial pathogenesis. It represents a valuable tool for examining cellular and molecular mechanisms of pathogen infection, as well as for testing potential treatments for endothelial-related microbial pathogenesis that would not be possible otherwise.
Rickettsiae induce their internalization into host cells by a receptor-mediated invasion mechanism using Ku70 as a potential host cell receptor for rickettsial autotransporter protein outer membrane protein B (OmpB) encoded by the surface cell antigen 5 (sca5) gene (43, 46). Considering that Epac1 is capable of promoting the nuclear exit of DNA-PK in various cell types (47), it is conceivable that Epac1 may mediate rickettsial adhesion and/or invasion by modulating the cellular translocation of Ku70. However, in addition to Ku70, previous studies show that that entry of R. conorii into nonphagocytic mammalian cells also depends on actin and microtubule dynamics, the host endocytic machinery, and the activation of protein tyrosine kinases and lipid kinases (44). Martinez and coworkers have reported that bacterial internalization correlates with cCbl-induced rapid ubiquitination of Ku70 and depletion of host clathrin and caveolin-2 inhibits OmpB-mediated rickettsial invasion of mammalian cells (43, 46). Recent data also suggest that another related rickettsial autotransporter protein, OmpA (encoded by sca0), is sufficient to mediate integrin-dependent invasion of mammalian cells (48). Epac1 is known to regulate all of the aforementioned cellular functions (45), but the precise molecular mechanism(s) by which Epac1 controls rickettsial adhesion and/or invasion is not clear at this time. Further research into understanding the signal cross-talk between Epac1 and endocytic pathways hijacked by rickettsiae is currently ongoing.
In conclusion, our studies combining genetic knockout and pharmacological manipulations provide convincing evidence that Epac1 plays a critical role in fatal SFG rickettsioses. Studies based on genetic manipulations, such as knockout mouse models, are invaluable for unveiling physiological functions of a target gene through phenotypic characterization but have several potential limitations, such as secondary effects attributable to disruption of protein complexes, compensation, and/or adaptation. In addition, there are now many examples where genetics and pharmacology produce different phenotypes for the same target protein. Considering that many therapeutics are small molecules (49), the combination of animal models and small molecular inhibitors enables us to reveal in vivo functions of Epac1 and to conclude that Epac1 plays a critical role in rickettsial infection. The fact that Epac1 knockout mice are viable and overall healthy suggests that the on-target toxicity of Epac1 inhibition should not be a major issue for the next phase of drug development. Taken together, these findings reveal an effective therapeutic target that can be used in fatal rickettsiosis. Most importantly, we show that a small-molecule Epac inhibitor can prevent and suppress rickettsial infection. This is significant from the biodefense viewpoint because it suggests that Epac1 inhibitors are capable of providing protection against bacterial infection and potentially be used as a small-molecule prophylaxis to thwart initial bacterial infection in the event of a bioterrorism threat.
Materials and Methods
Rickettsiae.
R. australis (Cutlack strain) was provided by C. Pretzman (Department of Health Laboratory, Columbus, OH) and was passaged three times in Vero cells (ATCC CCL81; American Type Culture Collection) and four times in embryonated chicken yolk sacs in our laboratory. For ex vivo and in vitro experiments, a 10% (vol/vol) yolk sac suspension of R. australis was propagated through two passages in Vero cells. R. australis were isolated using a bead-based protocol, as described previously (12). Purified rickettsiae were frozen in sucrose–phosphate–glutamate buffer at −80 °C (12, 42). Rickettsial counts and infectivity of the frozen stocks were determined by plaque assay and tissue culture ID50 assays on Vero cells or HUVECs using established protocols (12, 42, 50) and yielded ∼1 × 109 infectious bacterial cells per milliliter. Uninfected Vero cells were processed by the same procedure as control material. The rickettsial stock was mycoplasma-free. All biosafety level (BSL) 3 or animal BSL3 experiments were performed in Centers for Disease Control and Prevention-certified facilities in the Galveston National Laboratory at the University of Texas Medical Branch (UTMB) using standard operating procedures and precautions.
Mice.
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of UTMB. C57BL/6 Epac1 null mice were derived as described previously (38). All mice used in this study were 8- to 12-wk-old males. C57BL/6 mice are highly susceptible to R. australis; therefore, this organism was chosen as the SFG rickettsial agent (41, 42).
Rickettsial Challenge Study.
Experimental animals (12 WT and 17 Epac1−/− mice) were inoculated i.v. with 2 × 106 pfu per mouse of R. australis and observed daily for illness and survival. Five WT and eight Epac1−/− mice were mock-infected and used as controls. Mice with markedly ruffled fur, a hunched posture, and partially closed eyelids were defined as severely ill and counted in the “ill” category (41, 42).
Antibodies and Other Reagents.
Anti-Epac1 mouse monoclonal antibody (mAb) (5D3) was purchased from Cell Signaling. Anti-Ku70 mAb (Clone 162) was purchased from Thermo Scientific. A rabbit polyclonal antibody against SFG rickettsiae was described previously (12, 42). The unconjugated AffiniPure Fab fragment goat anti-mouse IgG (H+L) for immunohistochemistry (IHC) or IF using mouse tissues was purchased from Jackson ImmunoResearch Labs. Biotinylated goat anti-mouse and rabbit IgG, a fast red alkaline phosphatase substrate kit, and a 3,3′-diaminobenzidine (DAB) peroxidase substrate kit were purchased from Vector Laboratories. AlexaFluor 488- and AlexaFluor 594-conjugated goat anti-mouse and anti-rabbit IgG and ProLong Gold Antifade Reagent with DAPI were purchased from Invitrogen.
Histopathology and Immunohistology.
Complete necropsies were performed on all experimentally infected and control mice. Samples of liver, lung, and testis were fixed in a 4% (vol/vol) neutral buffered solution of formaldehyde, embedded in paraffin, sectioned at 5-μm thickness, and processed by staining with hematoxylin and eosin for evaluation of histopathology and by IHC or IF microscopy for detection of SFG rickettsiae and Epac1 as described previously (12). For IHC or IF studies on mouse tissue samples using mAbs, deparaffinized and rehydrated sections were blocked with unconjugated AffiniPure Fab fragment goat anti-mouse IgG (H+L) for 1 h at room temperature before incubation with mAb against Epac1 (dilution, 1:500) and rabbit polyclonal antibody against SFG rickettsiae (1:1,000) overnight at 4 °C. Epac1 and rickettsiae were detected with biotinylated goat anti-mouse and anti-rabbit antibodies, respectively, followed by alkaline phosphatase–fast red and DAB reactions, respectively. For IF assays using archival human brain tissues, AlexaFluor 594 goat anti-mouse and AlexaFluor 488 goat anti-rabbit antibodies were used. Nuclei were counterstained with DAPI. Fluorescent images were analyzed using an Olympus BX51 epifluorescence or Olympus IX81 confocal microscope.
Ex Vivo Mouse Vascular Endothelial Assay for Rickettsial Infection.
We established an ex vivo vascular endothelial culture model for rickettsial infection using aortic rings isolated from three WT and three Epac1−/− mice. Briefly, aortae were first dissected from mice and then cleaned of adipose tissue and cut into five rings per mouse aorta. A total of 30 aortic rings were cultured for 48 h in Prigrow I medium (Applied Biological Materials) supplemented with 10% (vol/vol) FBS. Three rings per mouse were exposed to R. australis at 1 × 105 pfu per ring in medium, and the other two rings were incubated in uninfected medium until microscopic evidence of neocellular growth was present after 1 wk to confirm the viability of the cultures. At the end of the experiment, all aortic rings were fixed in cold methanol for 24 h at −20 °C, cryosectioned at 5-μm thickness, and processed using IF reagents to detect SFG rickettsiae and Epac1 as described above.
In Vitro Rickettsial Infection Assay.
For in vitro studies, HUVECs (Cell Application) were cultivated in Prigrow I medium supplemented with 10% (vol/vol) heat-inactivated FBS in 5% (vol/vol) CO2 at 37 °C. All experiments were performed between passages 5 and 7, and cells were maintained in Prigrow I medium with 3% (vol/vol) FBS. Before infection with 10 MOI of rickettsiae, HUVECs were exposed to 5 µM EIS-09 in Prigrow I media for 1 h and were kept exposed at this concentration throughout the infection. For IF staining to detect rickettsiae, Epac1, and Ku70, the cells on the coverslips were washed extensively at least three times in PBS before the cells were fixed with cold methanol for 24 h and processed according to IF protocols detailed previously for ex vivo and in vivo models. Cells were examined and IF images were captured with an Olympus BX51 image system using a final 100× optical zoom. The number of HUVEC nuclei and total bacteria in each microscopic field were manually enumerated (51, 52). The results were expressed as the ratio of R. australis organisms to HUVECs (nuclei) (51, 52). Ten microscopic fields were examined for each experiment. Data are representative of at least three experiments. P values were determined using a standard Student t test.
Extracellular and total rickettsiae in host cell preparation were determined as described previously (44, 51). Briefly, at the end of the experiment, HUVECs were washed extensively and fixed in 4% (vol/vol) paraformaldehyde at room temperature before being subjected to the above described IF staining procedure. For detection of extracellular bacterial signaling, fixed HUVECs were incubated with rabbit polyclonal antibody against SFG rickettsiae (1:1,000) for 2 h at room temperature and then incubated with AlexaFluor 594 goat anti-rabbit antibody. To detect total bacterial signaling, stained HUVECs were permeabilized in 0.1% Triton X-100 in PBS before reincubation with rabbit polyclonal antibody against SFG rickettsiae antibody and AlexaFluor 488 goat anti-rabbit antibody. Cells were examined and IF images were captured with an Olympus BX51 image system using a final 100× optical zoom. The number of HUVEC nuclei, extracellular adherent rickettsiae (detected by red fluorescence emission), and total rickettsiae (detected by green fluorescence emission) in each microscopic field were manually enumerated (51, 53). Intracellular invaded rickettsiae were calculated subsequently (44, 51). Ten microscopic fields were examined for each experiment. The results were expressed as the ratio of R. australis organisms to HUVECs (nuclei) per examined field (51, 53, 54). Data are representative of at least three experiments. P values were determined using a standard Student t test.
In Vivo ESI-09 Treatment of R. australis-Infected Epac1+/+ Mice.
Thirty-three WT C57BL/6 mice were divided into four groups [11 mice (group A), 10 mice (group B), 6 mice each (groups C and D)]. Groups A and C were treated with the Epac-specific inhibitor ESI-09 [10 mg⋅kg−1⋅d−1 dissolved in buffered saline containing 10% (vol/vol) ethanol and 10% (vol/vol) Tween-80] via i.p. injection for 5 d before infection, whereas groups B and D were treated with vehicle, followed by i.v. inoculation of R. australis (2 × 106 pfu per mouse) for groups A and B or mock inoculation for groups C and D. ESI-09 or vehicle treatment was continued for another 7 d until mice were killed on day 8. During the course of the experiments, animals were monitored daily for signs of illness and mortality.
Supplementary Material
Acknowledgments
We thank Dr. Kimberly Schuenke for critical review and editing of the manuscript. We thank Nicole Mendell, Qinyu Gong, Luciano Victor, Tais Saito, and Yan Liu for technical assistance. This work was supported by the Center for Biodefense and Emerging Infectious Diseases (B.G.), the Carmage and Martha Walls Distinguished University Chair in Tropical Diseases (to B.G.), and National Institutes of Health Grant GM066170 (to X.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1314400110/-/DCSupplemental.
References
- 1.Dumler JS, Walker DH. Rocky Mountain spotted fever—changing ecology and persisting virulence. N Engl J Med. 2005;353(6):551–553. doi: 10.1056/NEJMp058138. [DOI] [PubMed] [Google Scholar]
- 2.Chapman AS, et al. Rocky Mountain spotted fever in the United States, 1997-2002. Ann N Y Acad Sci. 2006;1078:154–155. doi: 10.1196/annals.1374.026. [DOI] [PubMed] [Google Scholar]
- 3.Walker DH, Paddock CD, Dumler JS. Emerging and re-emerging tick-transmitted rickettsial and ehrlichial infections. Med Clin North Am. 2008;92(6):1345–1361. doi: 10.1016/j.mcna.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Mansueto P, et al. New insight into immunity and immunopathology of Rickettsial diseases. Clin Dev Immunol. 2012;2012:967852. doi: 10.1155/2012/967852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parola P, et al. Warmer weather linked to tick attack and emergence of severe rickettsioses. PLoS Negl Trop Dis. 2008;2(11):e338. doi: 10.1371/journal.pntd.0000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan YG, Riley SP, Martinez JJ. Adherence to and invasion of host cells by spotted fever group rickettsia species. Front Microbiol. 2010;1:139. doi: 10.3389/fmicb.2010.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker DH. Principles of the malicious use of infectious agents to create terror: Reasons for concern for organisms of the genus Rickettsia. Ann N Y Acad Sci. 2003;990:739–742. doi: 10.1111/j.1749-6632.2003.tb07453.x. [DOI] [PubMed] [Google Scholar]
- 8.Openshaw JJ, et al. Rocky Mountain spotted fever in the United States, 2000-2007: Interpreting contemporary increases in incidence. Am J Trop Med Hyg. 2010;83(1):174–182. doi: 10.4269/ajtmh.2010.09-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker DH, Ismail N. Emerging and re-emerging rickettsioses: Endothelial cell infection and early disease events. Nat Rev Microbiol. 2008;6(5):375–386. doi: 10.1038/nrmicro1866. [DOI] [PubMed] [Google Scholar]
- 10.de Sousa R, Nóbrega SD, Bacellar F, Torgal J. Mediterranean spotted fever in Portugal: Risk factors for fatal outcome in 105 hospitalized patients. Ann N Y Acad Sci. 2003;990:285–294. doi: 10.1111/j.1749-6632.2003.tb07378.x. [DOI] [PubMed] [Google Scholar]
- 11.Walker DH. The realities of biodefense vaccines against Rickettsia. Vaccine. 2009;27(Suppl 4):D52–D55. doi: 10.1016/j.vaccine.2009.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong B, et al. Rickettsiae induce microvascular hyperpermeability via phosphorylation of VE-cadherins: Evidence from atomic force microscopy and biochemical studies. PLoS Negl Trop Dis. 2012;6(6):e1699. doi: 10.1371/journal.pntd.0001699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beavo JA, Brunton LL. Cyclic nucleotide research — still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3(9):710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 14.McDonough KA, Rodriguez A. The myriad roles of cyclic AMP in microbial pathogens: From signal to sword. Nat Rev Microbiol. 2012;10(1):27–38. doi: 10.1038/nrmicro2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 2008;40(7):651–662. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Rooij J, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396(6710):474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki H, et al. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282(5397):2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 18.Netherton SJ, Sutton JA, Wilson LS, Carter RL, Maurice DH. Both protein kinase A and exchange protein activated by cAMP coordinate adhesion of human vascular endothelial cells. Circ Res. 2007;101(8):768–776. doi: 10.1161/CIRCRESAHA.106.146159. [DOI] [PubMed] [Google Scholar]
- 19.Noda K, et al. Vascular endothelial-cadherin stabilizes at cell-cell junctions by anchoring to circumferential actin bundles through alpha- and beta-catenins in cyclic AMP-Epac-Rap1 signal-activated endothelial cells. Mol Biol Cell. 2010;21(4):584–596. doi: 10.1091/mbc.E09-07-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen A, et al. Activation of GPR4 by acidosis increases endothelial cell adhesion through the cAMP/Epac pathway. PLoS ONE. 2011;6(11):e27586. doi: 10.1371/journal.pone.0027586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park TY, Baik EJ, Lee SH. Prostaglandin E2-induced intercellular adhesion molecule-1 expression is mediated by cAMP/Epac signalling modules in bEnd.3 brain endothelial cells. Br J Pharmacol. 2013;169(3):604–618. doi: 10.1111/bph.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Hooren KW, et al. The Epac-Rap1 signaling pathway controls cAMP-mediated exocytosis of Weibel-Palade bodies in endothelial cells. J Biol Chem. 2012;287(29):24713–24720. doi: 10.1074/jbc.M111.321976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang F, et al. PDE4 regulates tissue plasminogen activator expression of human brain microvascular endothelial cells. Thromb Res. 2012;129(6):750–753. doi: 10.1016/j.thromres.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sands WA, Woolson HD, Milne GR, Rutherford C, Palmer TM. Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells. Mol Cell Biol. 2006;26(17):6333–6346. doi: 10.1128/MCB.00207-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woolson HD, Thomson VS, Rutherford C, Yarwood SJ, Palmer TM. Selective inhibition of cytokine-activated extracellular signal-regulated kinase by cyclic AMP via Epac1-dependent induction of suppressor of cytokine signalling-3. Cell Signal. 2009;21(11):1706–1715. doi: 10.1016/j.cellsig.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 26.Sands WA, Woolson HD, Yarwood SJ, Palmer TM. Exchange protein directly activated by cyclic AMP-1-regulated recruitment of CCAAT/enhancer-binding proteins to the suppressor of cytokine signaling-3 promoter. Methods Mol Biol. 2012;809:201–214. doi: 10.1007/978-1-61779-376-9_14. [DOI] [PubMed] [Google Scholar]
- 27.Yarwood SJ, Borland G, Sands WA, Palmer TM. Identification of CCAAT/enhancer-binding proteins as exchange protein activated by cAMP-activated transcription factors that mediate the induction of the SOCS-3 gene. J Biol Chem. 2008;283(11):6843–6853. doi: 10.1074/jbc.M710342200. [DOI] [PubMed] [Google Scholar]
- 28.Sehrawat S, Cullere X, Patel S, Italiano J, Jr, Mayadas TN. Role of Epac1, an exchange factor for Rap GTPases, in endothelial microtubule dynamics and barrier function. Mol Biol Cell. 2008;19(3):1261–1270. doi: 10.1091/mbc.E06-10-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mei FC, Cheng X. Interplay between exchange protein directly activated by cAMP (Epac) and microtubule cytoskeleton. Mol Biosyst. 2005;1(4):325–331. doi: 10.1039/b511267b. [DOI] [PubMed] [Google Scholar]
- 30.Pannekoek WJ, et al. Epac1 and PDZ-GEF cooperate in Rap1 mediated endothelial junction control. Cell Signal. 2011;23(12):2056–2064. doi: 10.1016/j.cellsig.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 31.Fukuhara S, et al. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol. 2005;25(1):136–146. doi: 10.1128/MCB.25.1.136-146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kooistra MR, Corada M, Dejana E, Bos JL. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579(22):4966–4972. doi: 10.1016/j.febslet.2005.07.080. [DOI] [PubMed] [Google Scholar]
- 33.Schnoor M, et al. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med. 2011;208(8):1721–1735. doi: 10.1084/jem.20101920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cullere X, et al. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105(5):1950–1955. doi: 10.1182/blood-2004-05-1987. [DOI] [PubMed] [Google Scholar]
- 35.Spindler V, et al. The extent of desmoglein 3 depletion in pemphigus vulgaris is dependent on Ca(2+)-induced differentiation: A role in suprabasal epidermal skin splitting? Am J Pathol. 2011;179(4):1905–1916. doi: 10.1016/j.ajpath.2011.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rampersad SN, et al. Cyclic AMP phosphodiesterase 4D (PDE4D) Tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeability. J Biol Chem. 2010;285(44):33614–33622. doi: 10.1074/jbc.M110.140004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheung TM, Ganatra MP, Peters EB, Truskey GA. Effect of cellular senescence on the albumin permeability of blood-derived endothelial cells. Am J Physiol Heart Circ Physiol. 2012;303(11):H1374–H1383. doi: 10.1152/ajpheart.00182.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan J, et al. Enhanced leptin sensitivity, reduced adiposity, and improved glucose homeostasis in mice lacking exchange protein directly activated by cyclic AMP isoform 1. Mol Cell Biol. 2013;33(5):918–926. doi: 10.1128/MCB.01227-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsalkova T, et al. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci USA. 2012;109(45):18613–18618. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Almahariq M, et al. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol Pharmacol. 2013;83(1):122–128. doi: 10.1124/mol.112.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng HM, Wen J, Walker DH. Rickettsia australis infection: A murine model of a highly invasive vasculopathic rickettsiosis. Am J Pathol. 1993;142(5):1471–1482. [PMC free article] [PubMed] [Google Scholar]
- 42.Xin L, et al. Systemic treatment with CpG-B after sublethal rickettsial infection induces mouse death through indoleamine 2,3-dioxygenase (IDO) PLoS ONE. 2012;7(3):e34062. doi: 10.1371/journal.pone.0034062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinez JJ, Seveau S, Veiga E, Matsuyama S, Cossart P. Ku70, a component of DNA-dependent protein kinase, is a mammalian receptor for Rickettsia conorii. Cell. 2005;123(6):1013–1023. doi: 10.1016/j.cell.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 44.Martinez JJ, Cossart P. Early signaling events involved in the entry of Rickettsia conorii into mammalian cells. J Cell Sci. 2004;117(Pt 21):5097–5106. doi: 10.1242/jcs.01382. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): A multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol Rev. 2013;65(2):670–709. doi: 10.1124/pr.110.003707. [DOI] [PubMed] [Google Scholar]
- 46.Chan YG, Cardwell MM, Hermanas TM, Uchiyama T, Martinez JJ. Rickettsial outer-membrane protein B (rOmpB) mediates bacterial invasion through Ku70 in an actin, c-Cbl, clathrin and caveolin 2-dependent manner. Cell Microbiol. 2009;11(4):629–644. doi: 10.1111/j.1462-5822.2008.01279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huston E, et al. EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc Natl Acad Sci USA. 2008;105(35):12791–12796. doi: 10.1073/pnas.0805167105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hillman RD, Jr, Baktash YM, Martinez JJ. OmpA-mediated rickettsial adherence to and invasion of human endothelial cells is dependent upon interaction with α2β1 integrin. Cell Microbiol. 2013;15(5):727–741. doi: 10.1111/cmi.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knight ZA, Shokat KM. Chemical genetics: Where genetics and pharmacology meet. Cell. 2007;128(3):425–430. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 50.Hanson B. Improved plaque assay for Rickettsia tsutsugamushi. Am J Trop Med Hyg. 1987;36(3):631–638. doi: 10.4269/ajtmh.1987.36.631. [DOI] [PubMed] [Google Scholar]
- 51.Cardwell MM, Martinez JJ. Identification and characterization of the mammalian association and actin-nucleating domains in the Rickettsia conorii autotransporter protein, Sca2. Cell Microbiol. 2012;14(9):1485–1495. doi: 10.1111/j.1462-5822.2012.01815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riley SP, et al. The Rickettsia conorii autotransporter protein Sca1 promotes adherence to nonphagocytic mammalian cells. Infect Immun. 2010;78(5):1895–1904. doi: 10.1128/IAI.01165-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riley SP, Patterson JL, Martinez JJ. The rickettsial OmpB β-peptide of Rickettsia conorii is sufficient to facilitate factor H-mediated serum resistance. Infect Immun. 2012;80(8):2735–2743. doi: 10.1128/IAI.00349-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Read SC, Serio AW, Welch MD. Rickettsia parkeri invasion of diverse host cells involves an Arp2/3 complex, WAVE complex and Rho-family GTPase-dependent pathway. Cell Microbiol. 2012;14(4):529–545. doi: 10.1111/j.1462-5822.2011.01739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.