

Abstract
The voltage-sensing domain (VSD) is the common scaffold responsible for the functional behavior of voltage gated ion channels, voltage sensitive enzymes and proton channels. Because of the position of the voltage dependence of the available VSD structures, at present, they all represent the activated state of the sensor. Yet, in the absence of a consensus resting state structure, the mechanistic details of voltage sensing remain controversial. The voltage dependence of the VSD from Ci-VSP (Ci-VSD) is dramatically right shifted, so that at 0 mV It presumably populates the putative resting state. Appropriate biochemical methods are an essential prerequisite to generate sufficient amounts of Ci-VSD protein for high-resolution structural studies. Here, we present a simple and robust protocol for the Escherichia coli expression of eukaryotic Ci-VSD at milligram levels. The protein is pure, homogeneous, mono-disperse and well folded after solubilization in Anzergent 3-14 at the analyzed concentration (~ 0.3 mg/mL). Ci-VSD can be reconstituted into liposomes of various compositions and initial site-directed spin labeling and EPR spectroscopic measurements indicate its first transmembrane segment folds into an α-helix, in agreement to the homologous region of other VSDs. Based on current results and enhanced relaxation EPR spectroscopy measurement, Ci-VSD reconstitutes essentially randomly in proteo-liposomes, precluding straightforward application of transmembrane voltages in combination with spectroscopic methods. Nevertheless, the present results represent an initial step that makes the resting state of a VSD accessible to a variety of biophysical and structural approaches, including X-ray crystallography, spectroscopic methods and electrophysiology in lipid bilayers.
In most members of the voltage gated cationic channel super family, the voltage-sensing domain (VSD) is responsible for the electromechanical transduction that couples transmembrane protein motion into an increase in membrane permeability (1, 2). The identification of the Ciona intestinalis voltage-dependent phosphatase (Ci-VSP) (3), where the activity of a membrane-bound enzyme is directly modulated by transmembrane voltage via coupling with a bona-fide VSD (CiVSD), suggests that VSDs can operate as independent functional domains. Furthermore, VSDs have been also proven to be the key structural scaffold for proton channels (4, 5). In all known VSDs, both the architecture and voltage-sensing mechanism are conserved (6).
The conserved VSD scaffold is formed by four transmembrane segments (S1–S4), arranged in an antiparallel four-helical bundle. Positively charged residues (R/K) on the fourth transmembrane segment S4 (7) reorient in response to voltage changes across the membrane, leading to subsequent physiological responses. This conformational change produce a transient charge movement that generates a nonlinear capacitive current described in the early seventies as the gating current (8), and represents the electrical expression of the structural rearrangements in all VSDs (9). Our knowledge of the functional properties of VSD comes predominantly from the gating current measurements in Shaker (10, 11), a eukaryotic potassium channel from Drosophila (12). Long term efforts to structurally characterize Shaker have been limited by the lack of a robust biochemical prep, and at the same time, VSDs with available high-resolution structures have had limited or no gating current data. That Ci-VSD gating current measurements can be readily obtained (3, 13) makes this system an attractive candidate to correlate functional and structural information in the same VSD system. Clearly, this would help sort out the potential differences between VSD homologs and create a reference point to re-interpret the available information in the field.
Ultimately, determining the mechanism of voltage sensing will require high-resolution structures of VSDs in at least two functionally relevant states. First, the activated (“Up”) conformation, found when the transmembrane field intensity is eliminated or reverses it polarity. This conformation is best represented by the available VSD structures KvAP (14, 15), Kv1.2 chimera (16), NavAb (17) and NavRh (18), all obtained at 0 mV. Second, the elusive resting (“Down”) conformation, where charges are directly under the influence of the transmembrane electric field and is, still, experimentally unavailable. At a minimum, determining the structure of an alternative state (other than the Up state) would go a long way to clarify a great deal of the existing controversies. The voltage dependence of Ci-VSD is significantly right shifted (Figure 1A) a fact that would make the Down state VSD accessible to experimental manipulations in the absence of an asymmetric voltage.
Figure 1.

Sequence alignment and function comparison of Ci-VSD with existing VSDs. (A) Gene of Ci-VSP consists of N-terminal, voltage sensing domain, a linker and phosphatase domain. The first 260 residues, indicated by the arrow on the top, is our current interest for biochemical preparation. Sequence of the fourth transmembrane segment (S4) of Ci-VSP was aligned with homologous sequences from voltage-gated ion channels. The positive charged residues (R/K) were distributed at every third position, and primarily responsible for voltage sensing and highly conserved. (B) Relationship between the charge movement and the test pulse amplitude (Q-V curve) of VSDs from a typical potassium channel Shaker (black) and Ci-VSP (red). The curves was simulated with V1/2 = −48 mV and z = 4.7 for Shaker (38), and V1/2 = 71.8 mV and z = 1.1 for Ci-VSP (3). The dotted line at 0 mV indicates the potential states of VSDs under biochemical conditions in absence of asymmetric voltage across the proteins: Shaker’s VSD at activated state (or Up state); Ci-VSD at resting state (or Down state).
Ci-VSD is an interesting model not only in regards to the voltage sensing mechanism, but also as a structural stand-in for the proton channel Hv1. A point mutation in the VSD of Shaker is able to generate a hyperpolarization-driven ion current (the omega current (19)) that allows proton permeation under some conditions (20). Based on sequence and functional correlations, it has been suggested that Ci-VSD might represent an evolutionary intermediate between the voltage sensor in voltage-gated ion channels and proton channels (21). Though Ci-VSD functions as a voltage sensor, its sequence is closer to that of proton channels than to all four of the VSDs with available high-resolution structures. Thus, structural studies on the resting state VSD of Ci-VSD could not only yield valuable information about the basic principles of a voltage sensing mechanism, but it could also provide a better homology model for the proton channel, for which there are no high resolution structures so far.
Encouraged by the potential of Ci-VSD as a pathway to solve key questions regarding voltage sensing in cationic and proton channels, we have developed a biochemical approach to generate milligram-scale preparations of homogeneous CiVSD for potential biophysical studies. Initial characterization of solubilized preparations suggests that Ci-VSD is stable in five detergents at high concentration (20 mg/mL) for at least a week. Ci-VSD can be reconstituted into liposomes of various compositions and is mono-disperse in POPC:POPG in the long term, a fact that provides suitable conditions for EPR, solid state NMR and electrophysiologcal approaches. The present methodology shall play a key role in our attempts to understand the molecular basis of voltage-dependent gating in a variety of biological systems.
Experimental Procedures
Materials
cDNA of Ci-VSP-1-576 in Sp6 vector was kindly provided by Francisco Benzanilla from the University of Chicago (Chicago, IL). E. coli strains M15, SG1300, and the plasmid vectors pQE32 and pQE70 were bought from Qiagen (Valencia, CA). Penta-His and RGS-His antibodies were bought from Qiagen (Valencia, CA). GST antibody was bought from GE Health Care. The IPTG and the detergents were obtained from Anatrace (Maumee, OH). Talon cobalt resin was bought from Clontech (Mountain View, CA). The reducing agent TCEP was purchased from the Pierce (Rockford, IL). The reducing reagent TCYP, along with the fluorophores fluorescein 5-maleimide and tetramethylrhodamine 5-maleimide, were bought from Molecular Probes (Carlsbad, CA). The spin label (1-oxyl-2,2,5,5-tetramethylpyrrolidin-3-yl) methyl methanethiosulfonate was purchased from Toronto Research Chemicals (North York, ON, Canada). The lipids POPC, POPE, POPG, E. coli polar lipid extract, chicken egg extract, and soy bean extract Asolectin were bought from Avanti Polar Lipids (Alabaster, AL). All other reagents were purchased from Sigma or Fisher.
Molecular Biology and Expression Test
The DNA fragment containing the first 260 amino acids of the voltage sensing domain of Ci-VSP (Ci-VSD-1-260, Figure 1A) was amplified by PCR and cloned into pET28b, pQE32, pQE70 and pGEX-6p-1 vectors. pET28b vector was pre-modified into two individual vectors which contain one His-tag at either N- or C- terminal. The Ci-VSD containing plasmids were transformed into fresh E. coli competent cells, and grown overnight at 37 °C for 12~16 hours. The saturated overnight cultures were diluted at 1:100 ratio into LB medium with 1 % glycerol and grown at 37 °C for 2~4 hours, and the expression was induced at OD600~1 with 1 mM IPTG for 3 hours at 37 °C. One mL cell samples were collected before induction and at the end of the expression run. Cells were spun down and the pellets re-suspended in SDS loading dye. Viscous samples were broken down by shear force with a syringe fitted with a 30 Gauge needle. SDS-dissolved cell samples were loaded for SDS PAGE and developed for Western blot. The penta-His antibody was used as primary antibody for pET28b and pQE32, the RGS-His antibody for pQE70 and a GST antibody for pGEX-6P-1. In all cases, the used secondary antibody was anti-mouse Alexa fluor 647, followed by detection on a ChemiDoc (BioRad). Expression levels were compared roughly by intensity from Western blots. Initial expression conditions were further optimized for E. coli strain, temperature, IPTG amount and expression time, following the above procedures.
The only native cysteine residue at position 159 was mutated to a serine by site-directed mutagenesis methods following standard procedures to give a cysteine-less Ci-VSD, which was used as template to generate single cysteine mutant for probing methods with fluorescence or EPR spectroscopy. A total of 44 plasmids, each with a single cysteine residue scanning positions 110–142 and 214, were prepared on the optimized expression construct as shown in below.
Expression and Purification of Ci-VSD
Fresh XL10-Gold competent cells were transformed with Ci-VSD-1-260 in pQE32. The transformation mixture was grown overnight in the presence of the 100 mg/L ampicillin. Overnight cultures were diluted 1:100 in regular LB medium in presence of 0.2% glucose. Cells were grown at 37 °C for 3~4 hours to an OD600 of 0.5–0.7, and protein expression was induced with 0.65 mM IPTG for 3 hours at 30 °C. The cells were harvested by centrifugation and the pellet was re-suspended in Tris buffer (20 mM Tris pH 8.0, 150 mM NaCl) in presence of 1 mM phenylmethanesulfonyl fluoride (PMSF), 0.1 mg/mL DNAse, 5 mM MgCl2, 0.1 ug/mL pepstatin, 1 ug/mL aprotinin and 1 ug/mL leupeptin. The mixture was homogenized and spun down at 100,000g for 35 min and the the membrane fraction was re-suspended in Tris buffer and solubilized with 10 mM Anzergent 3-14 in the presence of 1 mM PMSF, 5 mM imidazole and 1 mM β-mercaptoethanol at room temperature for 1 hour. The homogenate was then spun down at 100,000g for 35 min, and the supernatant loaded onto a cobalt metal-affinity resin (2 mL resin per liter of cell culture). The resin was washed with 20 times resin volume of Tris buffer in presences of 1 mM Anzergent 3-14 and 5 mM imidazole. Ci-VSD was eluted with 300 mM imidazole in presence of 1 mM Anzergent 3-14 and Tris buffer pH 8.0. For the purification of single cysteine mutants, all washing buffers contained additional 0.5 mM Tris(2-carboxyethyl)phosphine hydrochloride and 0.5 mM Tris(2-cyanoethyl)phosphine. The eluate from the cobalt column was further purified by size exclusion chromatography (SEC) using a Superdex 200 HR 10/30 column (GE Health Care) in an AKTA FPLC system from the same company.
Detergent Screen
All detergents were from Anatrace and of the highest purity. Following the methods above, the E. coli pellet containing the membrane fraction, from a large-scale Ci-VSD expression, was re-suspended in Tris buffer as crude membranes. 14 detergents were individually added to the membrane suspension at 10 times their critical micelle concentration. After a 3-hour solubilization at room temperature, the mixture was centrifuged at 100,000g for 1 hour. The pellet was completely dissolved in SDS loading dye to the same initial volume and both the supernatant and the dissolved pellet were run on SDS PAGE and detected by Western blot. The extraction efficiency of Ci-VSD from membrane was estimated by the relative amount of protein in the supernatant fraction over the pellet fraction.
After extracting Ci-VSD out of E. coli membranes, the solubilization detergent was exchanged into 18 detergents to evaluate Ci-VSD’s homogeneity. The Superdex 200 HR 10/30 column was individual pre-equilibrated with various detergents, and ~0.2 mg of Ci-VSD from the cobalt column eluate were loaded for SEC. Peak homogeneity was calculated with the UV elution profile by the ratio of weighted 1 mL main peak over the weighted entire protein elution region using the following equation:
where A is the UV absorbance at 280 nm, Vp is the elution volume of the main peak which is between 13.5–14.2 mL for all tested detergents, Vv = 7 mL is the void volume of column. Ve = 17 mL is the end volume for protein related absorbance in the elution profile. Peak homogeneity from Vp-0.5 mL to Vp+0.5 mL was calculated for each elution profile for 18 detergents. Detergents with top homogeneity probabilities were selected to test for stability at high concentrations.
For stability determination, Ci-VSD was purified with the chosen detergents and concentrated to 20 mg/mL (~600 μM). Protein samples were kept at room temperature (~22 °C) for a week and their concentrations monitored by UV/Visible spectroscopy (Nanodrop, Thermo Scientific) using 32,430 M−1 cm−1 as extinction coefficient, following sample filtering through a centrifuge filter of 0.22 μm pore size, to remove potential aggregates. After a week, the remaining protein samples were loaded back onto SEC to check its homogeneity. Detergents that kept Ci-VSD homogenous at 20 mg/mL for a week were considered compatible with subsequent high-resolution structural studies.
Protein Characterization
The purity of Ci-VSD was established by SDS PAGE, while its secondary structure in detergent was determined by circular dichroism (CD) spectroscopy. Protein concentration was adjusted to ~10 μM and the spectra were measured on an AVIV circular dichroism spectrophotometer model 202 (AVIV Instrument Inc.) between 190 to 260 nm at a 1 nm interval in a 1 mm path length strain-free cylindrical cuvette (Hellma, Jamica, NY). Differential absorbance was converted into molar ellipticity [θ]. The percent helicity of Ci-VSD was calculated from θ222 following standard methods (22).
The molecular mass and homogeneity of Ci-VSD-detergent complexes were determined by multiple angle light scattering (MALS) methods. An AKTA FPLC system equipped with a Superdex 200 HR 10/30 size exclusion column was coupled to a MALS system with a flow cell. Light scattering was detected with a HELEOS system (Wyatt Technology Corp.) equipped with 60 mW GaAs laser at 658 nm and eighteen detectors at angles from 22.5° to 147.0°. Refractive index was determined from an Opilab rEX unit (Wyatt Technology Corp.). The refractive index increment (dn/dc) of Anzergent 3-14 was determined by linear fit of its refractive indexes at incremental concentrations, to be 0.1534 mL/g. UV absorbance was measured at 280 nm by the detector from AKTA system (GE Health Care) and converted to analog between 0–1 Volts into HELEOS system. Data was acquired and analyzed using the ASTRA software package (Wyatt Technology Corp.). Even though self-correlation between the intensity of light scattering at discrete angles is sufficient to determine the molecular mass without a standard, a BSA sample was used to ensure correct settings and data analysis. Both, the molecular weight of the protein-detergent complex and the protein content of the complex were analyzed for the peak of interest by the system template Protein Conjugate of ASTRA.
Reconstitution and Fluorescence Spectroscopy
Ci-VSD was reconstituted into liposomes with five different compositions: Asolectin, E. coli extract, chicken egg extract, POPE:POPG = 3:1 and POPC:POPG = 3:1. The degree of aggregation of Ci-VSD in liposomes was determined by an established fluorescence energy transfer method (23–25). Liposomes were prepared from stock lipids in chloroform following standard protocols (26) into 10 mg/mL stock solutions. Concentrated Ci-VSD (~ 5 mg/mL) samples were incubated with stock liposome for 30 min. The mixture was diluted 20 times and detergents were removed using bio-beads (Bio-Rad) overnight at room temperature.
Ci-VSD G214C was purified and individually labeled with either fluorescein 5-maleimide (excitationmax= 494 nm; emissionmax= 518 nm) or tetramethylrhodamine 5-maleimide (excitationmax= 544 nm; emissionmax= 572 nm) fluorescence dye. The individually labeled Ci-VSD 214C was mixed at donor: acceptor = 1:1 ratio, and reconstituted into liposomes. The fluorescence emission was recorded from 500 to 650 nm while excited at 494 nm in a PTI fluorimeter (PTI technology). The relative FRET intensity in the 570–580 nm range was used as an indicator of the closeness between donor and acceptor, and thus, the relative aggregation level of Ci-VSD. Lipid compositions, protein to lipid ratio and the stability upon time were tested and optimized for minimal FRET values.
EPR spectroscopy
Ci-VSD single cysteine mutants were purified and spin-labeled using a 1:20 molar excess of spin label at room temperature for 20 minutes. Excess spin label was removed with a PD10 column (GE Health Care). After liposome reconstitution, continuous-wave (CW) EPR measurements were carried following standard protocol (23, 25, 27). The mobility (ΔHo−1), Ni-EDDA accessibility (ΠNi) and oxygen accessibility (ΠO2) were obtained to deduce the dynamic and structural information of Ci-VSD inside the liposome. Periodic analysis was done following standard methods (27). Briefly, the oscillation of ΠO2 along the residue number was Fourier transformed into an angular power spectra P(ϖ) in the 1–180° range. The ideal alpha helix (3.6 residues per turn) is expected to have a maximum peak around 360°/3.6 = 100° with weighted probability αPI above 2.
To determine the directionality of Ci-VSD reconstitution in liposomes, we followed the percentage of quenching on the spin label signals at two positions, 110 and 141, on the opposite sites of the membrane. Ci-VSD 110C and 141C were labeled with spin label and reconstituted into multiple-layered liposomes as the standard procedure for EPR. Reconstituted unilamellar proteo-liposomes were generated by extrusion (30 times) through a 100 nm polycarbonate double membrane (Avanti Polar Lipids). 20 μL samples were loaded to sealed glass capillaries and CW-EPR spectra were measured with a high-Q cavity (Bruker Biospin). The impermeable spin label quencher Potassium Tris(Oxalato) Chromate (III) (CrOx) was used to evaluate VSD orientation. Due to its high charge (3+), Chromium (III) is essentially impermeable through the lipid bilayer. The percent reduction of the peak amplitude of Ci-VSD in the presence and absence of 30 mM CrOx directly indicates the amount of spin label on the outside surface of liposome. For the method to be internally consistent, two opposite sites on each Ci-VSD surface should have complementary quenching percentages.
Results
Protein Expression
During initial expression tests, five out of seven Ci-VSD-1-260 constructs were successfully expressed in E. coli strains (Table 1). The two best expressors (highlighted in Table 1) were further optimized by exploring E. coli strains, temperature, IPTG concentration and expression time. Expression conditions were tested for the N-His-Ci-VSD-260 in pET28b construct in four different BL21 DE3 strains: BL21 DE3, BL21 Gold DE3, BL21 codon + RPIL, and BL21 DE3 pLysS; the construct Ci-VSD-1-260-C-His in pQE32 was tested in five different E. coli strains: XL10-Gold, XL1-Blue, DH5a, M15, SG1300. The two best conditions were carried out for preparative scale expression and optimized in regards to purity and yield. The two best outcomes from expression tests were: N-His-Ci-VSD-1-260 in pET28b in BL21 Gold, induced by 1 mM IPTG for 6 hours at 37 °C and Ci-VSD-1-260-C-His in pQE32 in XL10 Gold, induced with 0.65 mM IPTG for 3 hours at 30 °C.
Table 1.
E. coli strains and constructs for the initial expression test. + (positive) and −(negative) signs indicate the relative expression level from Western blot: − indicates no visible band of expression; + indicates visible band and the more signs indicate relative higher expression level. The two highlighted conditions from initial test were chosen for further optimization for expression.
| Sequence | Affinity Tag | Vector | Promoter | E. coli strain | Expression Level |
|---|---|---|---|---|---|
| Ci-VSD-1-260 | N-terminal His-tag | pET28b | T7 | BL21 DE3 | + |
| BL21 DE3 pLysS | ++ | ||||
| C-terminal His-tag | pET28b | T7 | BL21 DE3 | − | |
| BL21 DE3 pLysS | − | ||||
| N-terminal His-tag | pQE70 | T5 | XL1-Blue | − | |
| XL10-Gold | − | ||||
| C-terminal His-tag | pQE32 | T5 | XL1-Blue | ++ | |
| XL10-Gold | +++ | ||||
| N-terminal GST-tag | pGEX-6P-1 | LAC | BL21 DE3 | + | |
| XL1-Blue | − |
Protein Purification
Preparative expression (1 liter culture) of Ci-VSD was carried out with the above two established constructs and conditions. The cells were harvested and homogenized and the supernatant was discarded after centrifugation at 100,000 g for 35 min. The pellet was re-suspended in Tris buffer as crude membranes. Solubilization with five out of 14 screened detergents (FC-10, FC-12, FC-14, Anzergent 3-12, Anzergent 3-14), allowed extraction of >90% of the Ci-VSD into the supernatant fraction from crude membrane (Figure 2A, top). No obvious protein degradation was observed during the solubilization stage judged by Western blots. FC-12 and Anzergent 3-14 were chosen for solubilization of larger scale expression and further optimization test against protein’s purity, homogeneity and stability. After the cobalt affinity column and SEC purification, SDS-PAGE was used to determine purity. N-His-Ci-VSD-1-260 in pET28b expressed from BL21 Gold show significant amounts of impurities seen as multiple bands on SDS-PAGE gel after purification (data not shown). On the other hand, Ci-VSD-1-260-C-His in pQE32 expressed in XL10 Gold gives suitable purity under the same conditions. Figure 3A shows an overloaded SDS PAGE gel of the Ci-VSD-1-260-C-His in pQE32 construct to amplify the impurities and demonstrate the purity resulting from the optimized procedures.
Figure 2.

Detergent screen. (A) Western blot of solubilization test: Top, supernatant fractions extracted by different detergents after ultra-centrifugation; Bottom: pellet fraction dissolved by SDS. FC-10, FC-12, FC-14, Anzergent 3-12 and Anzergent 3-14 were able to extract Ci-VSD efficiently out of crude cell membrane. FC-12 and Anzergent 3-14 were chosen for subsequent purification test. (B) Representative elution profile of crude eluate from cobalt affinity chromatography on to SEC with Anzergent 3-14. Its elution volume is 13.7 mL on the Superdex 200 HR 10/30 column. Homogeneity of main peak was calculated from the ratio of weighted main peak (grey area) over weighted whole region (black arrow on bottom). (C) Homogeneity probability of Ci-VSD eluted in eighteen detergents. Eight detergents (Anzergent 3-12, Anzergent 3-14, C8E4, FC-14, LDAO, Mega-10, NG and OG), whose homogeneity probability is higher than the reference line (dotted), were chosen for the further stability test.
Figure 3.

Ci-VSD characterization. (A) SDS-PAGE gel of Ci-VSD-1-260. The sample was overloaded to amplify the impurities. (B) Circular Dichroism spectra in molar ellipticity of Ci-VSD in Anzergent 3-14. The calculated helicity is 52%, which is consistent with high helical contents for the expected four transmembrane helices. It suggests that Ci-VSD maintained its secondary structure in the tested detergent and the N-terminal region 1–100 is most likely unstructured. (C) Ci-VSD’s molecular weight determined by multiple angle light scattering methods in Anzergent 3-14. The molecular weight (black line) is evenly distributed throughout the peak region (blue line) indicating a homogeneous protein-detergent complex.
Protein Characterization
Based on SDS PAGE (Figure 3A), the molecular weight of the Ci-VSD-1-260-C-His construct was 28 kDa determined by Rf value in reference with a maker (analysis not shown). This is slightly lower than its predicted molecular weight 31.0 kDa, and probably indicates that Ci-VSD retains at least partial secondary structure in SDS. This observation is not un-common among membrane proteins (28).
Ci-VSD’s homogeneity and stability were screened by SEC by loading the protein samples into columns pre-equilibrated with Tris buffer plus 18 individual detergents (Figure 2C). We report an elution volume of 13.7 mL in Anzergent 3-14 using a Superdex 200 HR 10/30 column (Figure 2B). The elution profiles for Ci-VSD in other detergents all contained predominant peaks around 13.4–14.2 mL, but displayed different amounts of large molecular weight components at 7–12 mL (void volume ~ 8.5 mL). Ranked by the homogeneity probabilities, the eluted proteins were collected from the 8 top detergents (above dotted line, Figure 2C) and concentrated to ~20 mg/mL. Protein concentration was monitored after removal of potential aggregation with a 0.22 μm filter. Ci-VSD continually precipitated in MEGA-10, NG and OG at high concentration, but became stable at concentrations of less than 1 mg/mL. The rest of the five concentrated samples were reloaded on to SEC after one week, with the aggregation portion increasing slightly and the predominant peak representing >90 % of homogenous protein. Criteria on homogeneity and stability determined a list of five detergents (Anzergent 3-12, Anzergent 3-14, C8E4, FC-14, LDAO) that are compatible with Ci-VSD at high concentration (20 mg/mL). Anzergent 3-14 was chosen for all subsequent sample preparations.
The homogeneity of the molecular mass was further evaluated by multiple angle light scattering after SEC separation of the main peak region. The determined molecular weight of the Ci-VSD/Anzergent 3-14 complex is 77.8 kDa of which 39% corresponds to the protein fraction. The molecular mass for Ci-VSD was determined at 30.3 kD evenly distributed throughout the main peak (Figure 3C). This indicates that Ci-VSD is a stable monomer in complex with approximately 130 Anzergent 3-14 molecules at the analyzed concentration ~0.5 mg/mL. Molecular weight and homogeneity of Ci-VSD analyzed with three other detergents (OG, LDAO and DM) were also similar to Anzergent 3-14.
We then evaluated the secondary structure of Ci-VSD in Anzergent 3-14 by CD spectra. The spectra show the typical features of alpha helical conformation (Figure 3B) with ellipticity minima near 208 and 222 nm. Based on standard basis set spectra, the calculated helical content of Ci-VSD is 52%. As expected, this points to a predominately helical conformation for Ci-VSD in detergent, and is consistent with the four transmembrane helices within residues 100–239 predicted by sequence alignment. The N-terminal region of first 100 residues seems to be unstructured under present conditions. These results suggest that the transmembrane region of Ci-VSD folds well in detergent, and appears to be stable and displays the characteristic secondary structure profile of all reported VSDs.
Reconstitution, Stability and Orientation
Membrane proteins have tendency to form small aggregates under non-physiological conditions. This crowding effect is usually more dramatic when reconstituted into bilayers and limited in two-dimensional space or as a consequence of hydrophobic mismatch (29). Most local dynamics and solvent accessibility measurements rely on the protein being mono-disperse on the plane of the lipid bilayer. In addition, the voltage dependent response of VSDs is known to be sensitive to lipid composition (30–32).
Based on single molecule fluorescence approaches, Ci-VSP has been shown to be mono-disperse in its functional state after heterologous expression in Xenopus Oocytes (33). We carried out a systematic search for reconstitution conditions that afforded Ci-VSD mono-dispersity in liposomes. To that end, Ci-VSD was reconstituted into pre-made liposomes of five different compositions: Asolectin, E. coli extract, chicken egg extract, POPE:POPG = 3:1 and POPC:POPG = 3:1. The extent of two-dimensional aggregation was estimated by a simple FRET based assay (Figure 4A) where a single cysteine mutant of the test protein was alternatively labeled with donor or acceptor fluorophores (23–25). Ci-VSD mutated at position 214 (G214C) was expressed and individually labeled with a fluorescence donor and an acceptor (fluorescein/tetramethylrhodamine pair), and mixed at 1:1 ratio and reconstituted into different liposomes with 1:2000 protein to lipids molar ratio. The FRET signal was monitored after 24 hours of reconstitution. FRET signals in the range of 570–580 nm were significantly lower in POPC:POPG and Asolectin (Figure 4B), which indicates that Ci-VSD is mono-disperse under those conditions. The protein-to-lipid ratio and Ci-VSD’s stability over time were also tested (data not shown). Ci-VSD didn’t show a significant increase in FRET signal in either POPC:POPG or Asolectin at a molar ratio up to 1:250 protein:lipid within 48 hours at room temperature.
Figure 4.

Ci-VSD reconstitution. (A) Cartoon representation of FRET assay to evaluate the aggregation behavior. Ci-VSD was individually labeled with fluorescence donor and acceptor, then mixed at 1:1 molar ratio and reconstituted into liposomes. FRET signal in the range of 560–580 nm indicates of closeness of fluorephores in liposome, thus the degree of Ci-VSD aggregation. (B) FRET signal of Ci-VSD in five different liposomes at 1:2000 protein to lipids ratio 24 hours after reconstitution. The signals from two liposomes POPC:POPG = 3:1 and Asolectin were significantly lower than other three compositions. (C) CW-EPR spectra of Ci-VSD in POPC:POPG (red) and Asolectin (black) at seven residues 123–128 in the first transmembrane segment (S1). Spectra features are distinctly different among residues, but essentially the same in both liposomes.
To compare the effect of lipid compositions on Ci-VSD conformation in lipids, we carried out limited site-directed spin labeling on six residues (123–128) in the first transmembrane segment (S1) of Ci-VSD and under two different conditions: POPC:POPG and Asolectin. The spin labeled cysteine mutants were subject to CW-EPR spectroscopic measurements on liposomes in the absence of a nominal transmembrane voltage. These conditions stabilize Ci-VSD in its resting conformation. In Figure 4C, the EPR spectra show the expected difference among residues scanned along a single helix of the four-helix bundle. However, this sequential pattern is independent of whether the measurement is carried out in Asolectin or POPC:POPG (Figure 4C), as expected from a well-behaved non-aggregated reconstituted sample. Given that earlier EPR studies from this laboratory have been carried out using POPC:POPG mixtures for KvAP (23, 25) and NaChBac (34) voltage sensors, all subsequent spectroscopic studies were also carried out in this lipid mixture.
Transduction of the electric field energy by VSDs is, by necessity, vectorial in nature. Therefore, in planning and interpreting spectroscopic data from reconstituted protein, it is of paramount importance to determine whether an external electric field is applied to a molecule population that has been vectorially reconstituted or to one that displays a random orientation. In the first case, the ensemble will respond synchronously and any resulting signal corresponds to one (or very few) conformational state. The second case generates a mixed and complicated signal mix composed of at least a fraction of the population that responds to the external electric field and a presumably unresponsive fraction. The Ci-VSD reconstitution orientation was determined using the ability of certain paramagnetic metals to quench (broaden) nitroxide signals in a collision-dependent way. Chromium (III) Oxalate (CrOx) is one of the most effective paramagnetic relaxing reagents, and due to its net charge (3+), essentially impermeable through a lipid bilayer (35). We determined the fractional quenching by the amplitude reduction of CW-EPR signals in Ci-VSD spin labeled at two positions, 110 and 141, on the opposite sides of the membrane. Spin labeled sensors were individually reconstituted into POPC:POPG liposome and quenched by 30 mM CrOx. We find that the reduction of peak amplitude was, on average, 36% for position 110C and 44% for 141C (Figure 5B). Since 110C and 141C are on the opposite sites of bilayer (Figure 6A, top), the percent of quenching should be complementary. Under these conditions, if sensors are randomly reconstituted, the theoretical values for quenching would be close to 50%, but close to either 0% or 100% for a fully vectorial reconstitution. The values for 110C and 141C were clearly closer to 50% in both our samples, unambiguously suggesting that Ci-VSD reconstitution shows little vectoriality. The ~10% discrepancy was likely from the imperfect sample preparation and partial shielding effect from the liposome, which could somewhat reduce the collision efficiency between spin label and CrOx.
Figure 5.

Direction of Ci-VSD in liposome. (A) Cartoon representation of Chromium (III) quenching methods to access the direction of Ci-VSD in liposome. Addition of 30 mM CrOx in the exterior solution will quench spin label signal at the outside surface of liposome. Percent of quenching illustrates the direction of the labeled position in reference to liposome: inside, outside, or randomly distributed. (B) Spectra of spin labeled Ci-VSD 141C in POPC:POPG liposome (black) and in the presence of 30 mM CrOx (red). Percent of amplitude reduction is 44%.
Figure 6.
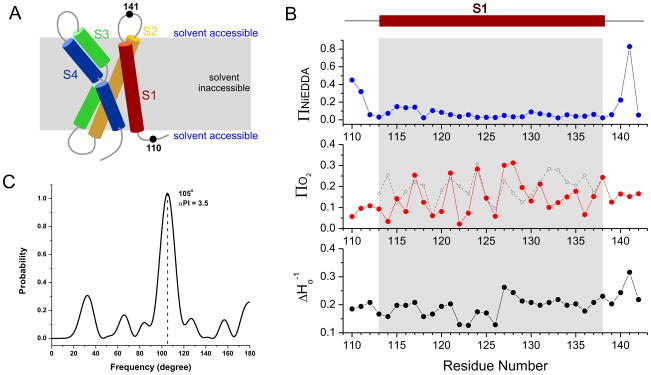
Structure of S1 of Ci-VSD in liposome studied by EPR spectroscopy. (A) Cartoon representation of VSD’s scaffold of four helices. Relative position of membrane was shown in grey, where it is solvent inaccessibility. The positions 110 and 141 are outside of lipid bilayer and on the opposite sides. (B) Ni-EDDA accessibility (ΠNi, blue), Oxygen accessibility (ΠO2, red) and Mobility (ΔHo−1, black) for the S1 of Ci-VSD. The grey region represents the transmembrane region identified by ΠNi. The ΠO2 of KvAP in the homologous region were plotted in grey as a reference. Two sets of ΠO2 values oscillate in the same range with almost the same pattern, with maximum values at every three or four residues. They indicate the S1 of Ci-VSD folds into α-helix in liposome as the same way as KvAP. Clearly, Ci-VSD folds into the same scaffold as existing VSDs. (C) Periodic probability analysis for ΠO2 of the transmembrane region 113–138 shows Pmax = 105° and αPI = 3.5. It suggests α-helical conformation as 3.4 residues per turn (360°/105°) with high probability (αPI > 2).
Local Structure and Dynamics
Once conditions were established to reliably generate a stable, well-behaved, reconstituted Ci-VSD preparation, we initiated an exploration of the overall structure and dynamics of the sensor in liposomes. As an initial approximation, we carried out a series of sequential cysteine mutations in the first transmembrane segment, S1 and adjacent extracellular loop (residues 110–142, Figure 6A). Each one of these cysteines was subsequently spin labeled and their CW-EPR spectra measured to characterize their local environmental properties: local dynamics from mobility measurements (ΔHo−1) and accessibility to contrast agents Ni-EDDA (ΠNi), and Oxygen (ΠO2). ΔHo−1 is an empirical measure of local dynamics of the labeled site, by the spectral anisotropy affected through tertiary contacts. ΔHo−1 is expected to be higher at loop regions and oscillate in transmembrane regions involved in periodic tertiary interactions between lipids and other parts of protein. Ni-EDDA is a water-soluble chelated paramagnetic ion and reflects accessibility to extra-membrane aqueous environments. ΠNi is expected to be high for exposed loop regions, but extremely low or absent for transmembrane region. In contrast, O2 shows higher solubility in lipids, so that high ΠO2 reflects lipid-exposed regions.
Residues 110, 111 and 140–141 clearly displayed high Ni-EDDA accessibilities as is characteristic of water exposed regions, while positions 113–139 show very limited water accessibility (Figure 6B, top, grey region). This data unambiguously defines the boundary of the membrane-embedded boundaries of the first transmembrane region, fully consistent with the expected range for the S1 segment from sequence alignments. The ΠO2 and ΔHo−1 values, on the other hand, show clearly periodic behavior with maximal values at every three or four residues, in confirmation of its α-helical secondary structure. As a comparison, previously determined ΠO2 of the KvAP S1 segment was plotted and aligned with the present Ci-VSD data. The values of ΠO2 show general agreement in regards to range and oscillation pattern. The periodicity of the ΠO2 oscillation within residues 113–138 can be quantitatively analyzed by evaluating its angular power spectrum after a Fourier transform (27). Figure 6C shows the power spectrum from the ΠO2 oscillation data in Figure 6B. The angular periodicity probability is characterized by a single dominating peak around 105°, close to the ideal alpha helix value of 100°. The alpha helical probability (αPI = 3.5 > 2) also points to the strong heliciity of the S1 segment. Overall, present data set strongly supports the idea that the S1 region of Ci-VSD folds into the same way and in a similar conformation as that of the structurally characterized KvAP sensor.
Discussion
The mechanism of voltage sensing remains one of the most challenging subjects in modern biophysics. Several models have been proposed to explain the mechanism of voltage sensing (36, 37). With recent advances in membrane protein crystallography, structures of voltage gated ion channels are being obtained at unprecedented rate (14, 16–18). Yet, because these are biochemically processed membrane protein samples, all available structures have been obtained in the absence of a nominal transmembrane electric field. Perhaps, not surprisingly, all of the available VSD structures appear to converge on one stable conformation, that of the activated or “up” state. The discovery of VSD-linked phosphatases like Ci-VSP (3) has not only demonstrated the universality of the VSD scaffold as a mean to electromechanical coupling, but it might offer an alternative to pursue multiple VSD conformations using structural approaches.
In reference to VSDs from canonical voltage-gated ion channels, the voltage dependent behavior of Ci-VSP is unique in two clear ways. First, its voltage dependence is significantly right shifted. Second, it shows a much shallower activation slope (z = 1~2) suggesting that less charge might be transferred during the gating process. These properties indicate that under biochemical conditions (close to 0 mV) most of the Ci-VSD molecules would populate the resting or “down” state. Consequently, a viable Ci-VSP biochemical preparation would provide access to alternative VSD conformations using high-resolution structural approaches.
As an initial approximation, we have focused efforts on developing a bacterial expression system for the eukaryotic Ci-VSD. Expression tests included constructs containing a combination of bacterial promoters (T7, T5 and LAC) and affinity tags (N-terminal His tag, C-terminal His tag and GST tag) in various E. coli strains (BL21 DE3, BL21 pLysS, XL-Blue and XL-Gold). Early tests successfully showed that Ci-VSD could be over-expressed in E. coli. 14 detergents were screened by their ability to extract the most protein out of the crude membrane, while 18 detergents were screened for Ci-VSD’s homogeneity and stability. Here, we show that Ci-VSD can be purified to homogeneity after a simple two-step purification through cobalt affinity chromatography and size exclusion chromatography. The yield of our final optimized protocol is about 0.5 mg/L culture with the detergent Anzergent 3-14.
The native environment for membrane proteins is defined by a complex interplay between lipid composition and the local physical properties of the membrane. Topology and three-dimensional architecture determinations of membrane proteins by spectroscopic approaches rely on the fact that proteins have to be mono-disperse on the plane of the bilayer; otherwise, the acquired information will be blended by the effect of non-specific protein-protein contacts. Using a simple FRET assay, we showed Ci-VSD can be reconstituted as a monomer using various phospholipids compositions and following standard procedures. However, the relative FRET signals were shown to be quite different for different tested liposomes, suggesting that the aggregation behavior of Ci-VSD is very sensitive to the lipid compositions. Ci-VSD is remarkably stable in liposomes made of POPC:POPG and Asolectin. Puzzlingly, while Ci-VSD can be over-expressed in E. coli, yet it severely aggregates in E. coli lipid extract. It should be noted that membrane protein handling might not be well defined under overexpression conditions, thus emphasizing the necessity for lipid composition screens as a prerequisite to structural or functional assays.
Under physiological conditions, membrane proteins are anchored to lipid bilayers in a preferred direction and respond to specific stimulus from both sides of membrane. This is particularly relevant when the external stimulus is vectorial in nature, as is the case for the transmembrane electric fields. Establishing the overall orientation of Ci-VSD after reconstitution is of utmost importance, for it defines the types of experimental approaches that can be pursued to determine voltage-driven conformational changes. Our current results from ΠNi determinations and enhanced paramagnetic relaxation methods indicated that, as is the case for many voltage-gated channels or isolated VSDs, Ci-VSD reconstitutes essentially in a random orientation. For spectroscopy-based approaches, this result limits experiments to measurements in the absence of an external transmembrane voltage. However, other liposome preparation and reconstitution protocols will need to be further developed and optimized if external transmembrane voltages are to be applied on the proteo-liposomes population.
That the purified and reconstituted Ci-VSD preparation represents a properly folded and structurally stable system was demonstrated by limited site-directed spin labeling analysis of the first transmembrane segment (S1). We focused on this region, as it tends to be structurally conserved among all available VSD structures. Local environment parameters reflecting overall dynamics (ΔHo−1), accessibility to the lipid environment (ΠO2) or accessibility to the aqueous milieu (ΠNi) provide a powerful evaluation of the overall structure of lipid-embedded Ci-VSD. Data shown in Figure 6B–C unambiguously demonstrates not only that S1 has a transmembrane topology, but it defines its membrane boundaries and reveals an α-helical secondary structure. Comparison of the Ci-VSD environmental data with the relevant region of KvAP demonstrates the structural equivalence of the two molecules. The data were in the same range and its oscillation pattern almost overlapped, strongly suggesting that in POPC:POPG liposomes Ci-VSD conforms to a scaffold common to that of known VSDs.
In summary, we have established a reliable prokaryotic expression system and biochemical preparation methods for the voltage-sensing domain of eukaryotic Ci-VSP. The protein is well-folded into the same scaffold as existing VSDs in a variety of detergents and lipid compositions. The interplay between functional and structural information from Ci-VSD should allow us to gain insights into the basic underpinnings of voltage sensor function. Furthermore, Ci-VSD’s sequence is closer to that of the proton channel than all four available VSDs with successful biochemical preparations and its structural information could provide a viable homology model to proton channel (4,5).
Acknowledgments
This work was supported in part by National Institutes of Health Grants to E.P. R01-GM057846 and U54-GM087519.
We thank Francisco Benzanilla for kindly providing the cDNA of Ci-VSP in Sp6 vector, and allowing unfettered access to the PTI fluorimeter. We thank Sudha Chakrapani and Jose Santos for providing comments and experimental advice in the early phases of this work. This work was supported in part by NIH grants R01-GM57846, and U54 GM74946.
Abbreviations and Textual Footnotes
- C8E4
n-octyltetraoxyethylene
- Ci-VSD
voltage sensor domain of Ci-VSP
- Ci-VSP
Ciona intestinalis voltage-sensor-containing phosphatase
- CW-EPR
continuous wave electron paramagnetic resonance
- ΔHo−1
mobility as the inverse of central line width of CW-EPR spectra
- DDM
dodecyl D-maltoside
- DM
decyl D-maltoside
- FC-10
n-decylphosphocholine
- GST
glutathione S-transferase
- LDAO
lauryldimethylamine oxide
- IPTG
isopropyl β-D-thiogalactopyranoside
- MEGA-9
nonanoyl-N-methylglucamide
- Ni-EDDA
nickel (II) ethylenediaminedi(o-hydroxyphenylacetic acid)
- OG
octyl D-glucoside
- ΠNi
Ni-EDDA accessibility
- ΠO2
Oxygen accssibility
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolaime
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-[phosphor-rac-(1-glycerol)]
- SEC
size exclusion chromatography
- VSD
voltage sensing domain
References
- 1.Bezanilla F. How membrane proteins sense voltage. Nat Rev Mol Cell Biol. 2008;9:323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- 2.Swartz KJ. Sensing voltage across lipid membranes. Nature. 2008;456:891–897. doi: 10.1038/nature07620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435:1239–1243. doi: 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki M, Takagi M, Okamura Y. A voltage sensor-domain protein is a voltage-gated proton channel. Science. 2006;312:589–592. doi: 10.1126/science.1122352. [DOI] [PubMed] [Google Scholar]
- 5.Ramsey IS, Moran MM, Chong JA, Clapham DE. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev. 2000;80:555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- 7.Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, et al. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- 8.Armstrong CM, Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature. 1973;242:459–461. doi: 10.1038/242459a0. [DOI] [PubMed] [Google Scholar]
- 9.Mannuzzu LM, Moronne MM, Isacoff EY. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- 10.Tempel BL, Papazian DM, Schwarz TL, Jan YN, Jan LY. Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science. 1987;237:770–775. doi: 10.1126/science.2441471. [DOI] [PubMed] [Google Scholar]
- 11.Bezanilla F, Stefani E. Gating currents. Methods Enzymol. 1998;293:331–352. doi: 10.1016/s0076-6879(98)93022-1. [DOI] [PubMed] [Google Scholar]
- 12.Schwarz TL, Tempel BL, Papazian DM, Jan YN, Jan LY. Multiple potassium-channel components are produced by alternative splicing at the Shaker locus in Drosophila. Nature. 1988;331:137–142. doi: 10.1038/331137a0. [DOI] [PubMed] [Google Scholar]
- 13.Villalba-Galea CA, Sandtner W, Starace DM, Bezanilla F. S4-based voltage sensors have three major conformations. Proc Natl Acad Sci U S A. 2008;105:17600–17607. doi: 10.1073/pnas.0807387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 15.Butterwick JA, MacKinnon R. Solution structure and phospholipid interactions of the isolated voltage-sensor domain from KvAP. J Mol Biol. 2010;403:591–606. doi: 10.1016/j.jmb.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 17.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Ren W, DeCaen P, Yan C, Tao X, Tang L, Wang J, Hasegawa K, Kumasaka T, He J, Clapham DE, Yan N. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature. 2012;486:130–134. doi: 10.1038/nature11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tombola F, Pathak MM, Gorostiza P, Isacoff EY. The twisted ion-permeation pathway of a resting voltage-sensing domain. Nature. 2007;445:546–549. doi: 10.1038/nature05396. [DOI] [PubMed] [Google Scholar]
- 20.Starace DM, Bezanilla F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. 2004;427:548–553. doi: 10.1038/nature02270. [DOI] [PubMed] [Google Scholar]
- 21.Sutton KA, Jungnickel MK, Jovine L, Florman HM. Evolution of the Voltage Sensor Domain of the Voltage-Sensitive Phosphoinositide Phosphatase, VSP/TPTE, Suggests a Role as a Proton Channel in Eutherian Mammals. Mol Biol Evol. 2012 doi: 10.1093/molbev/mss083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Fung LW. Structural and dynamic study of the tetramerization region of non-erythroid alpha-spectrin: a frayed helix revealed by site-directed spin labeling electron paramagnetic resonance. Biochemistry. 2009;48:206–215. doi: 10.1021/bi8013032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chakrapani S, Cuello LG, Cortes DM, Perozo E. Structural dynamics of an isolated voltage-sensor domain in a lipid bilayer. Structure. 2008;16:398–409. doi: 10.1016/j.str.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vasquez V, Cortes DM, Furukawa H, Perozo E. An optimized purification and reconstitution method for the MscS channel: strategies for spectroscopical analysis. Biochemistry. 2007;46:6766–6773. doi: 10.1021/bi700322k. [DOI] [PubMed] [Google Scholar]
- 25.Cuello LG, Cortes DM, Perozo E. Molecular architecture of the KvAP voltage-dependent K+ channel in a lipid bilayer. Science. 2004;306:491–495. doi: 10.1126/science.1101373. [DOI] [PubMed] [Google Scholar]
- 26.Cuello LG, Romero JG, Cortes DM, Perozo E. pH-dependent gating in the Streptomyces lividans K+ channel. Biochemistry. 1998;37:3229–3236. doi: 10.1021/bi972997x. [DOI] [PubMed] [Google Scholar]
- 27.Perozo E, Cortes DM, Cuello LG. Three-dimensional architecture and gating mechanism of a K+ channel studied by EPR spectroscopy. Nat Struct Biol. 1998;5:459–469. doi: 10.1038/nsb0698-459. [DOI] [PubMed] [Google Scholar]
- 28.Cortes DM, Perozo E. Structural dynamics of the Streptomyces lividans K+ channel (SKC1): oligomeric stoichiometry and stability. Biochemistry. 1997;36:10343–10352. doi: 10.1021/bi971018y. [DOI] [PubMed] [Google Scholar]
- 29.Killian JA. Hydrophobic mismatch between proteins and lipids in membranes. Biochim Biophys Acta. 1998;1376:401–415. doi: 10.1016/s0304-4157(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 30.Lee SY, Lee A, Chen J, MacKinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc Natl Acad Sci U S A. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milescu M, Bosmans F, Lee S, Alabi AA, Kim JI, Swartz KJ. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat Struct Mol Biol. 2009;16:1080–1085. doi: 10.1038/nsmb.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt D, Jiang QX, MacKinnon R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 2006;444:775–779. doi: 10.1038/nature05416. [DOI] [PubMed] [Google Scholar]
- 33.Kohout SC, Ulbrich MH, Bell SC, Isacoff EY. Subunit organization and functional transitions in Ci-VSP. Nat Struct Mol Biol. 2008;15:106–108. doi: 10.1038/nsmb1320. [DOI] [PubMed] [Google Scholar]
- 34.Chakrapani S, Sompornpisut P, Intharathep P, Roux B, Perozo E. The activated state of a sodium channel voltage sensor in a membrane environment. Proc Natl Acad Sci U S A. 2010;107:5435–5440. doi: 10.1073/pnas.0914109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vistnes AI, Puskin JS. A spin label method for measuring internal volumes in liposomes or cells, applied to Ca-dependent fusion of negatively charged vesicles. Biochim Biophys Acta. 1981;644:244–250. doi: 10.1016/0005-2736(81)90381-3. [DOI] [PubMed] [Google Scholar]
- 36.Chanda B, Asamoah OK, Blunck R, Roux B, Bezanilla F. Gating charge displacement in voltage-gated ion channels involves limited transmembrane movement. Nature. 2005;436:852–856. doi: 10.1038/nature03888. [DOI] [PubMed] [Google Scholar]
- 37.Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 38.Xu Y, Ramu Y, Lu Z. A shaker K+ channel with a miniature engineered voltage sensor. Cell. 2010;142:580–589. doi: 10.1016/j.cell.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]