

Abstract
Bromodomain-containing proteins are considered atypical kinases, but their potential to interact with kinase inhibitors is unknown. Dinaciclib is a potent inhibitor of cyclin-dependent kinases (CDKs) which recently advanced to Phase III clinical trials for the treatment of leukemia. We determined the crystal structure of dinaciclib in complex with CDK2 at 1.7 Å resolution, revealing an elaborate network of binding interactions in the ATP site which explains the extraordinary potency and selectivity of this inhibitor. Remarkably, dinaciclib also interacted with the acetyl-lysine recognition site of the bromodomain testis-specific protein BRDT, a member of the BET family of bromodomains. The binding mode of dinaciclib to BRDT at 2.0 Å resolution suggests that general kinase inhibitors (“hinge binders”) possess a previously unrecognized potential to act as protein-protein inhibitors of bromodomains. The findings may provide a new structural framework for the design of next-generation bromodomain inhibitors using the vast chemical space of kinase inhibitors.
Bromodomain (BRD)-containing proteins are essential for the recognition of acetylated lysine (KAc) residues of histones during transcriptional activation(1). Sixty-one different BRDs have been identified from 46 different proteins to date, grouped into eight families(2, 3). Members of the bromodomain and extra terminal (BET) protein family (BRD2, BRD3, BRD4, and BRDT) have been implicated in a number of disease pathways, and have therefore emerged as potential drug targets(4). The feasibility of targeting bromodomains with small molecules has been demonstrated for a series of benzodiazepine inhibitors against BRD2, BRD3, and BRD4(5), some of which have since progressed to clinical trials(6). The thienodiazepine (+)-JQ1, which specifically targets BET family proteins with IC50 values ranging from 50–90 nM(7), has recently been utilized to validate the bromodomain testis-specific protein (BRDT) as a promising male contraceptive target(3). Other BRD inhibitors have since been developed, including phenylisoxazole sulfonamides, quinoline isoxazole, and 2-thiazolidinones scaffolds(8–10). Notably, BRDs are considered atypical kinases(11, 12), and cell-based studies provided evidence that RNA polymerase II (Pol II) is subject to phosphorylation by full-length and truncated versions of BRD4(12). However, the potential of BRDs to interact with ATP or ATP site-directed small molecule kinase inhibitors has not been validated by biochemical or biophysical methods.
Dinaciclib (Merck, SCH727965) is a new-generation inhibitor of cyclin-dependent kinases (CDKs) which recently advanced to Phase III clinical trials for refractory chronic lymphocytic leukemia(13–15). CDKs are serine/threonine kinases involved in cell cycle progression and transcription, and deregulation of CDKs has been associated with a number of medical conditions(16). Cell-cycle progression depends on the activity of CDK1, CDK2, CDK4, and CDK6. S-phase entry is promoted by CDK4 and CDK6 in complex with cyclin D1, D2, or D3, together with CDK2 in complex with cyclin E, leading to phosphorylation and inactivation of the retinoblastoma (Rb) protein(17). CDK1-cyclin A and CDK2-cyclin A propel cells through the S-phase, while CDK1-cyclin B is responsible for mitosis(18, 19). Therefore, CDK-specific inhibitors induce apoptosis by repressing transcription, perturbing the cell cycle, or both(15). First-generation CDK inhibitors such as flavopiridol, (R)-roscovitine, SNS-032(20), and PHA-793887(21) were discontinued in clinical trials, due in part to their lack of potency and target specificity. In contrast, dinaciclib is a highly potent and selective inhibitor of CDK1, CDK2, CDK5, and CDK9 with low nanomolar anti-proliferative activity against most cancer cells(13, 14).
During the course of a project aimed at the structure-guided development of CDK2 inhibitors (22), we realized that the structural basis for the inhibition of CDKs by dinaciclib was unknown. We therefore determined the crystal structure of the CDK2-dinaciclib complex at 1.7 Å resolution (Figure 1, Supplementary Table S1). Dinaciclib binds to the ATP site through an intricate network of binding interactions, explaining its high potency and selectivity towards CDK2. The pyrazolo-pyrimidine moiety forms hydrogen bonds with residues 81–83 of the hinge region in the ATP site. The piperidine ring adopts a chair conformation, and the 2-hydroxyethyl group interacts with the ε-amino group of the strictly conserved Lys33 residue, which is positioned midway (2.7 Å) between the inhibitor and residue Asp145 of the so-called DFG motif of kinases (Asp-Phe-Gly) (Figure 1a). The 3-ethyl group of the pyrazolo-pyrimidine establishes hydrophobic, van der Waals (VDW) interactions with the gatekeeper residue Phe80. Several additional potential VDW interactions exist between the inhibitor molecule and residues Ile10, Gly11, Val18, Ala31, Val64, Phe82 and Leu134. The pyridine oxide ring is positioned in the front specificity pocket and is partly exposed to solvent; the nitroxy group appears to interact with the ε-amino group of Lys89. Notably, regions such as the activation loop which normally exhibit high conformational flexibility are well-ordered in the CDK2-dinaciclib complex. It appears that the elaborate network of hydrogen bonding and VDW interactions in the active site rigidifies the enzyme-inhibitor complex, providing the structural basis for the high potency and selectivity of dinaciclib against CDK2 and structurally similar CDKs.
Figure 1. Crystal structures of dinaciclib bound to CDK2 and BRDT.

(a) Crystal structure of the CDK2-dinaciclib complex determined at 1.7 Å resolution. The exploded view details the hydrogen bonding interactions of dinaciclib (magenta) within the ATP site. The hinge region, gatekeeper residue, and DFG motif are colored in orange, red, and cyan, respectively. The 2Fo-Fc electron density, contoured at 1σ around the inhibitor and residues Lys33, Asp145, and Lys89, is displayed as blue mesh. (b) Crystal structure of the BRDT-dinaciclib complex determined at 2.0 Å resolution. The exploded view details the hydrogen bonding interactions of dinaciclib (yellow) within the KAc site. The critical residue Asn109, ZA channel residues, and the WFP shelf are indicated in slate blue, green, and magenta, respectively. The 2Fo-Fc electron density, contoured at 1σ around the inhibitor and residues Asn109, Tyr66, and the water molecules of the ZA channel, is displayed as blue mesh. The panel to the right shows schematic representations of the binding sites, with VDW interactions indicated in green. The Fo-Fc electron density maps omitting the inhibitor during refinement are shown in Supplementary Figures 1 and 2.
Intrigued by a recent report that BRD4 exerts kinase activity against Pol II(12), we decided to study the potential of dinaciclib as a representative kinase inhibitor to interact with bromodomains by crystallography. The first bromodomain of BRDT, BRDT(1), was chosen because conditions suitable for co-crystallization studies with this protein were recently established in our laboratory. The resulting 2.0 Å resolution crystal structure revealed dinaciclib bound to the KAc recognition site of BRDT, which is the target site of known BET bromodomain inhibitors such as JQ1(23) and IBET-151(24) (Figure 1b, Supplementary Table 1). Notably, dinaciclib was bound with full occupancy to both KAc sites of the two BRDT(1) molecules comprising the asymmetric unit. The pyridine oxide ring appears to act as a KAc mimic through interaction with the critical residue Asn109, the target residue of the triazole ring of JQ1 and the isoxazole ring of IBET-151. However, the distance of 3.5 Å between the nitroxide and Asn109, as well as the relatively weak electron density of the nitroxide oxygen atom indicate that this interaction is suboptimal. By comparison, the distances between Asn109 and the triazole of JQ1 or the isoxazole of IBET-151 are 3.0 Å and 3.2 Å, respectively. The pyrazolo-pyrimidine moiety lies parallel to the WFP shelf, stabilized by VDW interactions with Pro51 and Phe52. The overall interaction pattern between dinaciclib and JQ1 is remarkably different (Figure 2). In particular, dinaciclib establishes hydrogen bonding interactions with the backbone carbonyl oxygens of Pro55 and Val56 through two highly coordinated bridging water molecules of the so-called ZA channel of bromodomains(6). In BRDT, the ZA channel consists of an intricate network of structurally conserved water molecules within the KAc binding pocket that connects the conserved Asn109 and Tyr66 residues with the WPF shelf (Figure 2). While JQ1 does not interact with water molecules of the ZA channel, the quinoline nitrogen of IBET-151 interacts with one water molecule in the equivalent region in BRD2(9) and BRD4(24) (Figure 2c). The involvement of water molecules to satisfy the hydrogen bonding potential of the dinaciclib pharmacophore, suggests that common kinase inhibitor scaffolds (“hinge binders”) have a similar potential to interact with the ZA channel of bromodomains (Figure 3). Although IBET-151 is not known to inhibit protein kinases, compounds with a quinoline hinge binding scaffold have been reported as inhibitors of VEGFR and c-Met(25). In both these kinases the nitrogen of the quinoline forms a single hydrogen bond with the hinge region.
Figure 2. Binding modes of dinaciclib, JQ1, and IBET-151 in bromodomains.
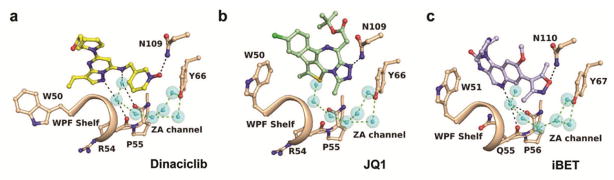
The hinge binding scaffold of dinaciclib interacts with two water molecules of the ZA channel (a). While JQ1 is not capable of forming hydrogen bonds with the ZA channel in BRDT (b, PDB entry 4FLP), the quinoline ring of IBET-151 interacts with one water molecule in BRD2 (c, PDB entry 4ALG). The seven water sites of the ZA channel are conserved throughout the known BET bromodomain structures, connecting the conserved Tyr66 residue with the WPF shelf through an intricate hydrogen bonding network.
Figure 3. Binding modes of dinaciclib in CDK2 and BRDT.

Similarity of hydrogen bonding interactions of dinaciclib with the hinge region of CDK2 (a) and residues of the ZA channel of BRDT through bridging water molecules (b). The schematic drawings in the right panel summarize the interactions of dinaciclib with subsites or solvent in CDK2 and BRDT.
Comparison between the structures of dinaciclib bound to CDK2 and BRDT reveals substantial differences in the interaction potential of the ethyl, piperidine and pyridine oxide moieties. While the ethyl and piperidine moieties establish binding interactions within the active site of CDK2, they are largely solvent exposed in the KAc site of BRDT. Importantly, the piperidine ring induces a conformational change of Trp50 due to steric hindrance (Figure 2). In most of the known liganded bromodomain structures, Trp50 partially shields the inhibitor from solvent. In the BRDT-dinaciclib complex, the side chain of Trp50 swings away from the binding pocket, leaving the inhibitor exposed to solvent. This imperfect shape complementarity renders dinaciclib a relatively weak inhibitor of BRDT, and dose-response analysis using a qPCR-based assay by DiscoveRx Corp. revealed a dissociation constant of Kd = 37 μM (Supplementary Figure 3), as compared to JQ1 (Kd=0.05 μM)(23) and IBET (Kd=0.6 μM)(9). Modification of the 2-piperidine moiety to avoid steric hindrance with Trp50 is likely to increase the binding potential of dinaciclib towards BRDT.
To our knowledge, the data presented herein provide the first evidence for the potential of kinase inhibitors to interact with bromodomains. The binding pattern of dinaciclib with BRDT suggests an intrinsic property of bromodomains to interact with ATP site-directed kinase inhibitors (hinge binders, Type I and II inhibitors) (Figure 3). A hinge-binding core such as the pyrazolo-pyrimidine in dinaciclib, paired with a moiety capable of interacting with the critical asparagine residue of the KAc site (e.g. triazole, isoaxole or pyridine oxide), may be sufficient to establish basic binding potential to most bromodomains. Therefore, the structure of the BRDT-dinaciclib complex provides a new framework for the rational design of next-generation bromodomain inhibitors using the vast chemical space of kinase inhibitors. Like kinase domains, bromodomains show a high degree of structural similarity suggesting that dinaciclib also interacts with other bromodomains. As expected, profiling against a panel of 24 bromodomains established that dinaciclib predominantly binds to members of the BET-family (40 – 80 % inhibition at 100 μM) with a preference for BRD3 (Figure 4). Notably, the only other bromodomains with binding potential for dinaciclib were TAF1 and TAF1L. It is unlikely that dinaciclib affects bromodomains at clinically relevant doses, as it binds to CDK2 with 30,000-fold higher affinity. However, it is conceivable that less potent kinase inhibitors administered at higher doses may cross-react with bromodomain-containing proteins.
Figure 4. Profiling of dinaciclib against a panel of 24 bromodomains.
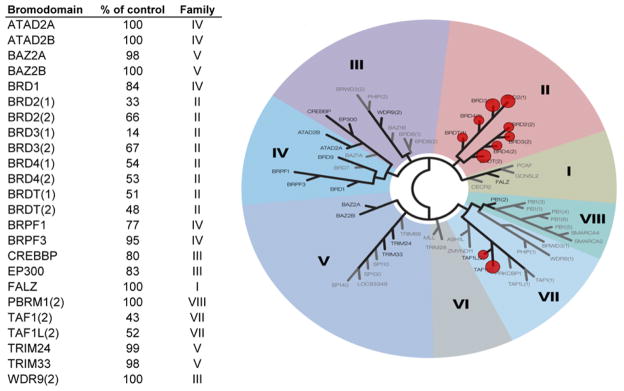
The binding potential of dinaciclib to other bromodomains was determined in duplicate at a single compound concentration of 100 μM. Binding activity is expressed as percentage of the positive control, with lower numbers indicating higher binding affinity. The right panel is a representation of the human bromodomain phylogenetic tree. Bromodomains tested are highlighted in black, and those interacting with dinaciclib are marked with red circles (cutoff ≤ 60% of the control), where larger circles indicate higher binding affinity. The experiment was performed by DiscoveRx Corp. using the binding assay described under Methods
METHODS
Reagents and compounds for biochemical and crystallographic experiments were purchased from Sigma–Aldrich and Hampton Research unless otherwise indicated. Dinaciclib was purchased from Selleck Chemicals. The gene encoding the first bromodomain domain of human BRDT (residues 21–137) was received in a pNIC28-Bsa4 vector from Addgene (plasmid 38898)(2), and was expressed in E. coli BL21 (DE3) after 24 h induction with 0.1mM IPTG at 16 °C. Full length human recombinant CDK2 was expressed, purified, and crystallized as described previously(26).
Protein purification
All purification steps were performed by FPLC at 4 °C. For BRDT, harvested bacterial cells were resuspended in 100 mM Na/K phosphate buffer (pH 7.4) containing 300 mM NaCl, 10 mM imidazole, 0.5 mg mL−1 lysosyme, and 0.01 % Triton X-100 at 4 °C for 1 h. After sonication and centrifugation (1 h at 29000 × g), the supernatant was purified by immobilized Ni2+-ion affinity chromatography (GE LifeSciences). Following incubation of peak fractions with PreScission (20:1) at 4°C, the cleaved His-tag was separated by size exclusion chromatography using a Superdex 75 (26/60) column, and eluted with 50 mM HEPES buffer (pH 7.4) containing 200 mM NaCl and 1 mM DTT(26). Eluted protein was concentrated to 35 mg/ml for crystallization studies.
Protein crystallography
Crystallization was performed at 18 °C using the sitting drop vapor diffusion method. Crystals of BRDT were grown in the presence of 3 mM dinacilib from 0.2 M KSCN, 20% PEG 3350 and 10 % ethylene glycol. Crystals were harvested in cryoprotectant (0.2 M KSCN, 20% PEG 3350 and 25% (v/v) ethylene glycol, 0.5 mM inhibitor) and flash frozen in a stream of nitrogen gas. Crystals of the CDK2-dinaciclib complex were obtained by in-diffusion of 3 mM dinaciclib into unliganded CDK2 crystals for 24 h. X-ray diffraction data were recorded at −180°C at the beamline 22-ID, SER-CAT, Advanced Photon Source, Argonne National Laboratories. Data were reduced with XDS(27); PHENIX(28) was employed for phasing and refinement, and model building was performed using Coot(29). The BRDT-dinaciclib structure was solved by molecular replacement (MolRep of the CCP4 suite(30, 31) using the monomer of PDB entry 4FLP(23) as the search model. The CDK2-dinaciclib structure was solved by molecular replacement using PDP entry 3QXP as the search model. Figures were prepared using PyMOL (Schrödinger, LLC).
Bromodomain binding assay
The dissociation constant of dinaciclib binding to BRDT and the profiling of dinaciclib against a panel of 24 bromodomain proteins was determined by DiscoveRx Corp. The amount of bromodomain captured on an immobilized ligand in the presence or absence of dinaciclib was measured using a quantitative real-time polymerase chain reaction (qPCR) method that detects the associated DNA label tagged to the bromodomain. Bromodomain-tagged T7 phage strains were grown in parallel in 24-well blocks in an E. coli host derived from the BL21 strain. E. coli were grown to log-phase and infected with T7 phage from a frozen stock (multiplicity of infection = 0.4) and incubated with shaking at 32 °C until lysis (90–150 minutes). The lysates were centrifuged (5,000 × g) and filtered (0.2μm) to remove cell debris. Streptavidin-coated magnetic beads were treated with biotinylated affinity ligands for 30 minutes at room temperature to generate affinity resins. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1 % BSA, 0.05 % Tween 20, 1 mM DTT) to remove unbound ligand and to reduce non-specific phage binding. Binding reactions were assembled by combining bromodomains, liganded affinity beads, and dinaciclib in 1× binding buffer (16 % SeaBlock, 0.32× PBS, 0.02 % BSA, 0.04 % Tween 20, 0.004 % Sodium azide, 7.9 mM DTT). Dinaciclib was prepared as 400× stock in 100 % DMSO and subsequently diluted 1:10 into ethylene glycol to 40× screening concentration and then diluted into the assay. All reactions were performed in polypropylene 384- well plates in a final volume of 0.02 ml. The assay plates were incubated at room temperature with shaking for 1 hour and the affinity beads were washed with wash buffer (1× PBS, 0.05 % Tween 20). The beads were then re-suspended in elution buffer (1× PBS, 0.05 % Tween 20, 2 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The bromodomain concentration in the eluates was measured by qPCR. For profiling, dinaciclib was screened at a single concentration of 100 μM and results are reported as:
Supplementary Material
Acknowledgments
We thank the Moffitt Chemical Biology Core for use of the protein crystallography facility, the Southeast Regional Collaborative Access Team (SER-CAT, University of Georgia) for assistance with Synchrotron data collection at Argonne National Laboratory, and N. Burgess-Brown (SGC, Oxford, UK) for providing the expression plasmid of BRDT. We thank D.J. Ingles (Moffitt) for manuscript editing. This work was supported in part by the Reproductive Branch of the National Institute of Child Health and Human Development Grant 3U01HD076542-01S1.
Footnotes
The authors declare no competing financial interests.
Author Contributions
M.P.M., G.I.G. and E.S. designed the experiments; M.P.M. and S.O. performed the protein production and purification; M.P.M. and E.S. performed the crystallographic studies; M.P.M and E.S. wrote the manuscript. All authors read and approved the manuscript.
Accession codes
The coordinates and structure factors of the CDK2-dinaciclib and BRDT-dinaciclib structures have been deposited in the PDB under accession numbers 4KD1 and 4KCX, respectively.
Summary of crystallographic data collection and structure refinement; electron density maps; Kd determination. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Owen DJ, Ornaghi P, Yang JC, Lowe N, Evans PR, Ballario P, Neuhaus D, Filetici P, Travers AA. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000;19:6141–6149. doi: 10.1093/emboj/19.22.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586:2692–2704. doi: 10.1016/j.febslet.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 4.Denis GV. Bromodomain coactivators in cancer, obesity, type 2 diabetes, and inflammation. Discov Med. 2010;10:489–499. [PMC free article] [PubMed] [Google Scholar]
- 5.Chung CW, Coste H, White JH, Mirguet O, Wilde J, Gosmini RL, Delves C, Magny SM, Woodward R, Hughes SA, Boursier EV, Flynn H, Bouillot AM, Bamborough P, Brusq JM, Gellibert FJ, Jones EJ, Riou AM, Homes P, Martin SL, Uings IJ, Toum J, Clement CA, Boullay AB, Grimley RL, Blandel FM, Prinjha RK, Lee K, Kirilovsky J, Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J Med Chem. 2011;54:3827–3838. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- 6.Hewings DS, Rooney TP, Jennings LE, Hay DA, Schofield CJ, Brennan PE, Knapp S, Conway SJ. Progress in the development and application of small molecule inhibitors of bromodomain-acetyl-lysine interactions. J Med Chem. 2012;55:9393–9413. doi: 10.1021/jm300915b. [DOI] [PubMed] [Google Scholar]
- 7.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bamborough P, Diallo H, Goodacre JD, Gordon L, Lewis A, Seal JT, Wilson DM, Woodrow MD, Chung CW. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J Med Chem. 2012;55:587–596. doi: 10.1021/jm201283q. [DOI] [PubMed] [Google Scholar]
- 9.Seal J, Lamotte Y, Donche F, Bouillot A, Mirguet O, Gellibert F, Nicodeme E, Krysa G, Kirilovsky J, Beinke S, McCleary S, Rioja I, Bamborough P, Chung CW, Gordon L, Lewis T, Walker AL, Cutler L, Lugo D, Wilson DM, Witherington J, Lee K, Prinjha RK. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A) Bioorg Med Chem Lett. 2012;22:2968–2972. doi: 10.1016/j.bmcl.2012.02.041. [DOI] [PubMed] [Google Scholar]
- 10.Zhao L, Cao D, Chen T, Wang Y, Miao ZH, Xu Y, Chen W, Wang X, Li Y, Du Z, Xiong B, Li J, Xu C, Zhang N, He J, Shen J. Fragment-based Drug Discovery of 2-thiazolidinones as Inhibitors of the Histone Reader BRD4 Bromodomain. J Med Chem. 2013 doi: 10.1021/jm301793a. [DOI] [PubMed] [Google Scholar]
- 11.LaRonde-LeBlanc N, Wlodawer A. The RIO kinases: an atypical protein kinase family required for ribosome biogenesis and cell cycle progression. Biochim Biophys Acta. 2005;1754:14–24. doi: 10.1016/j.bbapap.2005.07.037. [DOI] [PubMed] [Google Scholar]
- 12.Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, Ozato K, Sims RJ, 3rd, Singer DS. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci USA. 2012;109:6927–6932. doi: 10.1073/pnas.1120422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R, Nomeir A, Windsor W, Fischmann T, Wang Y, Oft M, Chen T, Kirschmeier P, Lees EM. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9:2344–2353. doi: 10.1158/1535-7163.MCT-10-0324. [DOI] [PubMed] [Google Scholar]
- 14.Paruch K, Dwyer MP, Alvarez C, Brown C, Chan TY, Doll RJ, Keertikar K, Knutson C, McKittrick B, Rivera J, Rossman R, Tucker G, Fischmann T, Hruza A, Madison V, Nomeir AA, Wang Y, Kirschmeier P, Lees E, Parry D, Sgambellone N, Seghezzi W, Schultz L, Shanahan F, Wiswell D, Xu X, Zhou Q, James RA, Paradkar VM, Park H, Rokosz LR, Stauffer TM, Guzi TJ. Discovery of Dinaciclib (SCH 727965): A Potent and Selective Inhibitor of Cyclin-Dependent Kinases. ACS Med Chem Lett. 2010;1:204–208. doi: 10.1021/ml100051d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guha M. Cyclin-dependent kinase inhibitors move into Phase III. Nat Rev Drug Discov. 2012;11:892–894. doi: 10.1038/nrd3908. [DOI] [PubMed] [Google Scholar]
- 16.Johnson LN. Protein kinase inhibitors: contributions from structure to clinical compounds. Q Rev Biophys. 2009;42:1–40. doi: 10.1017/S0033583508004745. [DOI] [PubMed] [Google Scholar]
- 17.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1777. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 18.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 19.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 20.Cicenas J, Valius M. The CDK inhibitors in cancer research and therapy. J Cancer Res Clin Oncol. 2011;137:1409–1418. doi: 10.1007/s00432-011-1039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massard C, Soria JC, Anthoney DA, Proctor A, Scaburri A, Pacciarini MA, Laffranchi B, Pellizzoni C, Kroemer G, Armand JP, Balheda R, Twelves CJ. A first in man, phase I dose-escalation study of PHA-793887, an inhibitor of multiple cyclin-dependent kinases (CDK2, 1 and 4) reveals unexpected hepatotoxicity in patients with solid tumors. Cell Cycle. 2011;10:963–970. doi: 10.4161/cc.10.6.15075. [DOI] [PubMed] [Google Scholar]
- 22.Schonbrunn E, Betzi S, Alam R, Martin MP, Becker A, Han H, Francis R, Chakrasali R, Jakkaraj SR, Kazi A, Sebti SM, Cubitt CL, Gebhard AW, Hazlehurst L, Tash JS, Georg GI. Development of highly potent and selective diaminothiazole inhibitors of cyclin-dependent kinases. J Med Chem. 2013 doi: 10.1021/jm301234k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, Knapp S, Bradner JE. Small-Molecule Inhibition of BRDT for Male Contraception. Cell. 2012;150:673–684. doi: 10.1016/j.cell.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJ, Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Norman MH, Liu L, Lee M, Xi N, Fellows I, D’Angelo ND, Dominguez C, Rex K, Bellon SF, Kim TS, Dussault I. Structure-based design of novel class II c-Met inhibitors: 1. Identification of pyrazolone-based derivatives. J Med Chem. 2012;55:1858–1867. doi: 10.1021/jm201330u. [DOI] [PubMed] [Google Scholar]
- 26.Betzi S, Alam R, Martin M, Lubbers DJ, Han H, Jakkaraj SR, Georg GI, Schonbrunn E. Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem Biol. 2011;6:492–501. doi: 10.1021/cb100410m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 28.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The CCP4 suite: programs for protein crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.