

Abstract
We studied the microsecond folding dynamics of three β hairpins (Trp zippers 1–3, TZ1–TZ3) by using temperature-jump fluorescence and atomistic molecular dynamics in implicit solvent. In addition, we studied TZ2 by using time-resolved IR spectroscopy. By using distributed computing, we obtained an aggregate simulation time of 22 ms. The simulations included 150, 212, and 48 folding events at room temperature for TZ1, TZ2, and TZ3, respectively. The all-atom optimized potentials for liquid simulations (OPLSaa) potential set predicted TZ1 and TZ2 properties well; the estimated folding rates agreed with the experimentally determined folding rates and native conformations were the global potential-energy minimum. The simulations also predicted reasonable unfolding activation enthalpies. This work, directly comparing large simulated folding ensembles with multiple spectroscopic probes, revealed both the surprising predictive ability of current models as well as their shortcomings. Specifically, for TZ1–TZ3, OPLS for united atom models had a nonnative free-energy minimum, and the folding rate for OPLSaa TZ3 was sensitive to the initial conformation. Finally, we characterized the transition state; all TZs fold by means of similar, native-like transition-state conformations.
Protein- and peptide-folding events on time scales of 1–10 μs are accessible to both the fastest time-resolved experiments, such as laser temperature-jump (T-jump) spectroscopy, and to advanced simulation techniques, such as distributed computing (1–6). Combining simulation and experimental techniques in studying such systems can lead to a detailed description of folding at the molecular level, along with experimental confirmation of the predicted kinetics and thermodynamics. The β hairpin, a common element in protein structures, is an important test system and a potential source of insight into the folding kinetics of larger proteins. Consequently, we have seen many inquiries into the structure and folding dynamics of β hairpins in recent years (7–17). Here, we have studied Trp zippers 1–3 (TZ1–TZ3), a series of unusually stable 12-residue hairpins designed by Cochran et al. (ref. 18 and Table 1).
Table 1. TZ thermodynamics.
| TZ | ΔH, kJ/mol | ΔS, J/mol·K | ΔG, kJ/mol |
|---|---|---|---|
| TZ1 | 19.2 | 55.9 | 2.65 |
| TZ2 | 12.7 | 23.8 | 5.67 |
| TZ3 | 9.07 | 14.3 | 4.83 |
Thermodynamic quantities at 23°C are reproduced from Cochran et al. (18).
These TZs (“TrpZips”) differ only at the turn (types II′, I′, and d-Pro-enhanced II′) and form a unique hairpin conformation in which the indole side chains from opposing pairs of Trp residues interlace to form a non-hydrogen-bonded stack or zipper along the hairpin. Our objective in this work was to explore the folding process for these peptides, as observed in hundreds of folding events simulated in atomistic molecular dynamics. To test the predicted dynamics, we compared the folding rates obtained from our simulations with experimental results from laser T-jump spectroscopy by using both Trp-fluorescence and IR-absorbance probes.
Materials and Methods
Simulation Methodology. Our molecular dynamics simulations used software adapted from the tinker 3.8 (J.W. Ponder, available at http://dasher.wustl.edu/tinker) molecular-modeling package (19). We used the united-atom optimized potentials for liquid simulations (OPLSua) and all-atom OPLS (OPLSaa) parameter sets (20, 21) without 1–4 scaling. We modeled solvation with the generalized Born/surface area implicit-solvent model (22), which incorporates solvent entropy. Accordingly, we defined the internal free energy of a given conformation as the sum of the internal potential energy of the protein and its interactions with solvent (including solvent entropy). Constant temperature stochastic dynamics modeled the viscous drag of water (frictional coefficient, 91 ps–1). Only the initial OPLSua simulations (TZ1 and TZ2) used electrostatic cutoffs (16 Å with 12-Å tapers). The bond lengths were constrained with the rattle algorithm, allowing time steps of 2 fs (23). Trajectory conformations were recorded at 250-ps intervals.
Models for TZ1 and TZ2 were taken from Protein Data Bank coordinates 1LE0 and 1LE1 (18). We used the first structure from each NMR ensemble. A model for TZ3 was prepared from the TZ1 model by replacing Gly-6 with a d-proline. The TZ3 model was briefly energy-minimized to relax the new bond lengths and angles, including the Born/surface area energy at each step. To obtain initial unfolded conformations, fully extended conformers were generated by using tinker 3.8 (ϕ, ψ) = (–135, 135). Before distributed simulation, each model was equilibrated with 5–100 ps of molecular dynamics.
Simulation Analysis. To quantify the degree of tertiary structure, we aligned each conformation to the Cα positions of the relevant NMR structure and calculated the root-mean-square α-carbon deviation (RMSDcα) from the NMR structure by using the McLachlan algorithm (24), as implemented in the program profit (A. C. R. Martin, available at www.bioinf.org.uk/software/profit). An order parameter, L, was defined to be the sum of the four inner native hydrogen-bond distances from nitrogen to oxygen and the distances between the CD2 atoms of the three neighboring Trp pairs. The sum of distances L measured how tightly the hairpin was interlaced. Finally, a conformation contained symmetric β structure if it had either a β-bridge or β-strand assignment at a pair of the native hairpin locations (residues 2–11, 3–10, 4–9, or 5–8), according to ref. 25, with the default hydrogen-bond cutoff.
The conformational space of even small polypeptides like the TZs has many degrees of freedom. To study the space, it is helpful to choose order parameters and project the ensemble into two dimensions. We constructed potential of mean force (PMF) surfaces as the negative natural-log probability of bin occupancy, with the axis of RMSDcα and L. The surfaces had contours of 0.75 RT (molar gas constant × temperature) and were shifted such that the most populated bin was at 0. It is important to note that the PMF surfaces are suggestive of the unfolded portion of the free-energy surface but will not represent a free-energy landscape until the underlying ensemble is at equilibrium.
We estimated folding rates from the linear growth of the folded population with time (see Figs. 5–15, which are published as supporting information on the PNAS web site). To improve our folding rate constant estimates we considered folding to be irreversible. The rate estimates (Table 2) depend more strongly on cutoffs than on fitting error; therefore, we reported rate estimates by using a range of folding and unfolding cutoffs. To prevent experimental data from biasing the folding criteria, the cutoffs were chosen before the authors exchanged estimates of the folding rates. We selected folded conformations by requiring a symmetric β structure and RMSDcα < 1.4–1.8 Å. The folding-RMSDcα cutoffs were chosen to be approximately one SD above the mean RMSDcα of the native-state simulations. The unfolding cutoff range (RMSDcα + 0.125 L < 9.5 ± 0.5) was selected after inspection of the L-RMSDcα PMF surfaces for unfolding.
Table 2. TZ kinetics.
| Characteristic times, μs
|
|||
|---|---|---|---|
| TZ | Fluorescence | IR | Simulation estimates |
 |
6.25 ± 0.34 | — | 5-7 |
 |
18.3 ± 3.14 | — | 9-11 |
 |
1.80 ± 0.01 | 2.47 ± 0.05 | 3-6 |
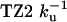 |
18.0 ± 1.40 | 24.7 ± 3.0 | 14-20 |
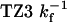 |
0.83 ± 0.13 | — | 100-300, 2-6 |
 |
5.88 ± 0.84 | — | 9-12 |
Observed T-jump relaxation kinetics were fit to an Arrhenius model to calculate the characteristic folding and unfolding times at 23°C. The TZ3 fluorescence rates were, instead, based only on data at 21°C. The 23°C simulation estimates reflect varied folding and unfolding cutoffs. The second simulation TZ3 folding rate estimate was based on refolding from a relaxed initial unfolded ensemble (see Figs. 5-11).
Finally, we selected folding transition state structures by inspecting the folding PMF surfaces and defining a strict cutoff (RMSDcα < 1.5 and L < 38). Then, for each folding trajectory, we considered the last conformation that did not meet the cutoff. To study the transition state ensemble we considered the 40 structures that are closest to the PMF saddlepoint (RMSDcα ≈ 2.0 Å, L ≈ 38 Å).
Laser T-Jump Spectroscopy. Laser T-jump spectroscopy provides the time resolution needed for experimental studies of TrpZip folding kinetics. Here, we used two different T-jump instruments to probe both Trp-fluorescence and IR absorbance (2, 26). Both T-jump instruments used an IR-laser pulse (1,900 nm) to trigger rapid T-jumps (δT ≈10–15°C) in an aqueous sample of protein. The fluorescence instrument probed the subsequent relaxation by exciting the Trp fluorescence with a 266-nm laser and detecting fluorescence with a photomultiplier. The IR instrument monitored the relaxation kinetics by means of the amide I′ absorbance of the polypeptide backbone at 1,624 cm–1 (40.5–80°C). The transient absorbance change of a tunable IR diode laser induced by the T-jump pulse was then detected by a 50-MHz mercury–cadmium–telluride detector. The T-jump-induced relaxation was obtained by subtracting the D2O-absorbance change, measured under identical conditions.
We used far-UV circular dichroism to measure the equilibrium folding and unfolding properties of TZ1–TZ3 under thermal denaturation. We fit the denaturation curves to a two-state model and obtained thermodynamic parameters in close agreement with those reported by Cochran et al. (18). Equilibration of the peptide after a jump to temperature T generated a relaxation with a characteristic rate k, where k(T) is the sum of the folding and unfolding rates, k = kf + ku, in a two-state kinetic model. With the known equilibrium constant Keq = kf/ku, we could then derive the folding rate kf = k/(1 + Keq–1) from the observed relaxation, even though the T-jump primarily triggers unfolding of the protein.
Laser T-jump fluorescence studies provided the folding rate of TZ1 in water and over a wide range of final temperatures (19.6–60°C). However, because of a weak temperature dependence of the free energy of folding, TZ2 and TZ3 produced only a small relaxation signal in water at room temperature; at ≈20°C, the folded population scarcely changed in response to a small T-jump. Accordingly, for TZ2–TZ3, we measured the relaxation rate k in the presence of small concentrations of denaturant (1–3 M guanidine·HCl), which destabilized the folded state enough to generate a measurable signal. For both TZ2 and TZ3, the denaturant enhanced the signal amplitude without greatly affecting the observed relaxation rate (d ln k/d [GdnHCl] ≈ –0.08 ± 0.11 M–1 for TZ2 and approximately –0.22 ± 0.07 M–1 for TZ3), such that we could obtain the zero-denaturant relaxation rate from a short extrapolation.
Peptide Synthesis. C-terminal amide peptides were synthesized by fluorenylmethoxycarbonyl chemistry, purified by reverse-phase HPLC, and characterized by mass spectroscopy. For fluorescence studies, lyophilized TZ1–TZ3 were dissolved in 20 mM phosphate buffer (pH 7) at ≈50 μM concentrations, at which self-association does not occur (18). We found that at the concentration used in IR studies (1–4 mM), both TZ1 and TZ3 aggregate. Therefore, only TZ2 was studied by IR. After the residual trifluoroacetic acid from peptide synthesis was removed by lyophilization against 0.1 M DCl solution, samples for IR experiments were dissolved into 20 mM phosphate D2O buffer solution (pH 7) to give a final concentration of 2–3 mM.
Results and Discussion
Simulation Results. The small size of TZ1–TZ3 allows detailed simulations to reach exceptional lengths. The aggregate simulation time reported here exceeds 22 ms (more than all comparable previous folding simulations combined). The individual trajectories ranged in length from 10 ns to >1.5 μs. The aggregate simulation time included simulations of each related TrpZip system, for several models and potential sets, starting from both extended and native conformations, at water-like and lower viscosities, and at various temperatures (see Figs. 5–15 and Table 4, which is published as supporting information on the PNAS web site). For each initial condition, tens of thousands of molecular-dynamics trajectories of varying lengths were calculated by using the folding@home (version 2.0; available at http://folding.stanford.edu) distributing computing project (27). In total, we sampled the formation of the expected β hairpin in several hundred room-temperature trajectories.
Native-state simulations of TZ1–TZ3 were exceedingly stable at 23°C, with RMSDcα distributions centered around average values of ≈1.0 Å after 50 ns. Each ensemble of native simulations contained rare unfolding events (except native ensembles at 0°C), leading to estimated unfolding rates on the multiple-microsecond time scale.
To create unique unfolded conformations for each trajectory with no native-state bias, simulations were started from completely extended conformations. Within nanoseconds, the ensemble of trajectories reached a diverse, relaxed ensemble of compact conformations. The distribution of average α-carbon distances in the (TZ1) unfolded ensemble matched that of a random-walk polymer with a persistence length of 1 aa (28).
Rare transition events in simulations of tens or hundreds of nanoseconds can accurately reflect slow (in μs) two-state transitions. The requisite conditions have been discussed (29–31). In particular, each trajectory must not be too short. Each simulation must exceed the time needed to relax into a diverse unfolded state (trelaxU) and also to allow rearrangement of the protein into a folded conformation, known as the “barrier-crossing” time (tcross). Normally, these time scales will be much shorter than the characteristic folding time of the system (tf = 1/kf).
Here, to predict folding rates and mechanism, we took advantage of the fact that the simulation times (tsim) exceeded the relaxation time scales, such that trelaxU + tcross < tsim < tf. The relaxation times will likely depend on the type of model, viscosity, initial conformation, and particularly protein length. The TZ collapse and minimum folding times suggested that trelaxU and tcross were on the multiple-nanosecond time scale (Fig. 1), similar to previous simulations made by V.S.P. and coworkers (1, 3, 5) and Caflisch and coworkers (31). These multiple-nanosecond relaxation times match the order of magnitude of experimental estimates of the minimum diffusion-limited loop-formation time for short peptides (32). Whereas Paci et al. (31) report the duration of the early biased regime as a fraction of the folding time, we stress that the duration will likely be related directly to the relaxation times trelaxU and tcross and related only indirectly to the folding time (tf). For example, tf values for various CI2 mutants span three and a half orders of magnitude (33), but relaxation within the unfolded state (almost a random coil) would not be likely to vary significantly.
Fig. 1.

The folding ensembles were tens of thousands of molecular dynamics trajectories of varying lengths (>8,000 to 50 ns and >1,000 to 200 ns). The ensemble average of the Rg (upper trace) and the internal free energy (lower trace) are shown in gray with light gray error bars representing the SD for the TZ1 (a), TZ2 (b), and TZ3 (c) unfolded ensembles. For each identified folding-transition conformation (see Materials and Methods), we show the Rg (○) and the internal free energy (•). The early folding events (<10 ns) resemble the later folding events (>100 ns), and both are similar to the unfolded ensemble in Rg and internal free energy.
In this study, we found that TZ1–TZ3 collapsed quickly to a diverse set of compact conformations. The average radius of gyration for TZ1–TZ3 came within 0.5 Å of the final value (100–200-ns average) within 5.5, 4.5, and 11.5 ns, respectively. As expected, collapse is more rapid than the ≈60-ns collapse recently observed experimentally for a 40-residue protein (34). The average internal free energy also came within 5 kJ/mol of the final value after 11.5, 7.25, and 16.5 ns, respectively (Fig. 1). TZ1 and TZ2 had a short lag time, with initial folding events occurring after ≈2 ns. Although the most rapid events (during the initial collapse trelaxU) were candidates to be unrepresentative, we found that the early folding events for these systems (<10 ns) were similar to later (>100 ns) folding events in all order parameters tested. The early transition conformations had similar Rg and internal free-energy values (Fig. 1) and similar distributions for L and RMSDcα (data not shown). Furthermore, early folding trajectories had similar heterogeneity in the hydrogen-bond and Trp–Trp distances in the last nanosecond before folding (see Figs. 5–15).
Comparison with Experiment. Table 2 shows strong agreement between the simulation prediction and the fluorescence T-jump experiment for the TZ1 folding rate (5–7 μs for simulation versus 6.3 ± 0.3 μs for experiment). For TZ2, the simulated and measured folding rates were in good agreement: 3–6 μs versus 1.8 μs (fluorescence) or 2.5 μs (IR). Both simulation and measurement found that TZ2 folds somewhat faster than TZ1.
Having both fluorescence and IR data for TZ2, we generated additional ensembles of TZ2-folding simulations at both 46°C and 69°C. With simulated folding and unfolding rate constants, we could compare an estimated fraction folded to the experimental Cochran et al. (18) results. Our simulations did not account for the temperature dependence of the hydrophobic effect, and as expected, the simulation stability of TZ2 was an underestimate of the experimental stability at each temperature. We also estimated Arrhenius activation enthalpies from the temperature dependence of the rate constants (Table 3 and Figs. 5–15). During the simulations, folding and unfolding were observed directly. In contrast, the experiments were constrained because the folding and unfolding rate constants were inferred from an observed relaxation rate by using Keq from Cochran et al. (18). The simulation folding rate increases slightly with temperature, leading to a small activation-energy estimate (4–7 kJ/mol), within an order of magnitude of the energies found in T-jump experiments [e.g., Ea = kB d ln kf/d (1/T) = 15.9 ± 0.3 kJ/mol for Trp fluorescence and 17.9 ± 1.1 kJ/mol for IR]. In contrast, the unfolding rate depends strongly on the temperature. Simulations give activation enthalpies of ≈49, ≈55, and ≈65 kJ/mol for TZ1, TZ2, and TZ3, respectively, quite close to experimental values (55 kJ/mol for TZ1 and 50 kJ/mol or 74 kJ/mol for TZ2) and roughly consistent with the internal free-energy gaps between the native and unfolded ensembles (Fig. 2).
Table 3. Activation enthalpy.
| ΔHu, kJ/mol
|
ΔHf, kJ/mol
|
|||||
|---|---|---|---|---|---|---|
| TZ | Fluorescence | IR | Simulation estimates | Fluorescence | IR | Simulation estimates |
| TZ1 | 55.2 ± 2.0 | — | 47-51 | 21.9 ± 1.3 | — | — |
| TZ2 | 50.2 ± 0.6 | 73.7 ± 1.4 | 53-57 | 15.9 ± 0.3 | 17.9 ± 1.1 | 4-7 |
| TZ3 | — | — | 65-66 | — | — | — |
Activation-enthalpy values from Arrhenius fits of the folding and unfolding kinetics.
Fig. 2.

(a) Each point is the internal free energy of a conformation (averaged over the previous 10 ns of molecular dynamics) versus RMSDcα for OPLSaa TZ1 (a), OPLSaa TZ2 (b), OPLSaa TZ3 (c), and OPLSua TZ1 (d). The red points indicate data taken from 23°C unfolded state ensembles after 200, 100, 100, and 100 ns. The gray points indicate data taken from 23°C native-state ensembles after 200, 50, 50, and 50 ns. The trace represents the running average of the internal free energy from 30 conformations.
For each OPLSaa TZ model, we found a favorable internal free-energy landscape for folding (Fig. 2). Specifically, the native state had a considerably more favorable internal free energy, and there did not appear to be large internal free energy traps hindering folding. Comparing TZ1 and TZ2, which differ only in the order of the turn residues, the native conformations for TZ2 were more tightly ordered and the internal free energy increases more rapidly with RMSDcα. Experimentally, TZ2 has a more cooperative transition.
Intuitively, one would expect TZ3 to fold rapidly because of its stability and the turn promoting d-Pro-6. Interestingly, the initial TZ3 simulations appeared to depart significantly from experiment and intuition: the estimated folding rate fell dramatically relative to TZ1 and TZ2, whereas experiment showed TZ3 to be the fastest folding system. Slow folding was especially surprising because there was no obvious internal free-energy barrier (Fig. 2c). Instead, the proline dramatically reduced the variation within the unfolded state and slow folding resulted from a kinetic trap that dominated the unfolded ensemble (see Figs. 5–15). In keeping with the kinetic-trap hypothesis, room-temperature unfolding trajectories did not populate the trap significantly, and a new folding ensemble, started from the highest RMSDcα conformations of 100 different room-temperature unfolding trajectories, avoided the trap and refolded with tf = 2–6 μs (see Figs. 5–15). This result indicates that replacing a glycine with a trans-d-proline slows the interconversion of unfolded states and that care must be taken with the preparation of the unfolded state because the folding rate is sensitive to the initial condition.
We found significant differences between the OPLSaa and OPLSua force fields. Specifically, the OPLSua global internal free-energy minimum corresponded to a high RMSDcα conformation in which Glu-5 associated, incorrectly, with the charged N terminus (Fig. 2d). Comparison of the OPLSua global minimum with similar OPLSaa unfolded structures suggested that OPLSua favored the decoys (relative to native-like states) by means of a combination of steric factors and more favorable electrostatic interactions (see Figs. 5–15). The OPLSua internal free-energy landscape resulted in very different dynamics: unfolding was rapid (≈70% of the trajectories have RMSDcα of >3 Å after 100 ns), and folding was too slow to observe. Despite massive sampling, there were no folding events (which one might expect if the folding events observed by means of massively parallel simulations were merely the result of thoroughly sampling random compact conformations).
Description of TrpZip Folding. To characterize the folding pathway of the TrpZips, we sought to identify the conformations that occupy the transitional region between the folded and unfolded basins of attraction. Projection of the folding ensembles into RMSDcα and L revealed a broad unfolded state, a distinct native state, and a saddlepoint between the minima (Fig. 3). To look at specific transition structures, we identified conformations that will fold within the next 250 ps for TZ1–TZ3 (see Materials and Methods). The TZ1–TZ3 ensembles were similar by eye (Fig. 4) and had comparable average interatom-distance matrices for equivalent heavy atoms (<0.75 Å for each pair of ensembles). As suggested for other proteins (35), the TrpZip transition state was quite similar to the native state; a small fluctuation in the native structure can be sufficient to activate the molecule for unfolding. This fluctuation can be characterized as a reorganization in which the outer Trp side chains, particularly Trp-2, reach suboptimal packing conformations. Fig. 4, for clarity, shows only the Trp Cβ atoms. It is apparent that the Trp-2 position varied more than the position of the inner Trp pair Trp-4 and Trp-9. From inspection of individual trajectories, we concluded that the timing of individual events, such as formation of hydrogen bonds or Trp contacts, was heterogeneous (see Figs. 5–15). On average, however, the folding pathway was a zipper: the inner Trp pair and inner hydrogen bonds generally formed first, and formation of the final hydrogen bonds usually occurred with correct Trp packing.
Fig. 3.

PMF surfaces for room-temperature folding ensembles, excluding conformations before 100 ns (150 ns for TZ3), for TZ1 (a), TZ2 (b), and TZ3 (c). Contours are drawn at intervals of the available thermal energy, 0.75 RT.
Fig. 4.

(a) Native conformations for TZ1, TZ2, and TZ3, showing the Trp side chains and the turn residues. The backbone of residues 2–4 and 9–11 is indicated in blue. b–d are stereodiagrams that highlight the β carbons of Trp-2 (yellow), Trp-4 (blue), Trp-9 (cyan), and Trp-11 (red). (b) The native state (TZ3) was similar to transition conformations (see Materials and Methods) from TZ1 (c), TZ2 (d), and TZ3 (e).
Conclusion
The computational biology community has worked for decades to develop reliable force fields for molecular dynamics, which, in favorable circumstances, do not immediately drive a native protein or nucleic acid structure away from the native state. Here, we used such models to reproduce entire folding pathways, in which comparison with experimental kinetics could reveal the simulation limitations. Most difficulties could be traced to the need for extensive sampling and the limitations of current potential energy functions. Like a chemical denaturant, a faulty potential energy surface will selectively stabilize certain regions of the phase space. Models do not need to be perfect to predict the features of interest. For example, simple models biased toward the native state (Gō models) can reproduce Φ values, an experimental description of the transition state (36). Reproducing the observed kinetics for slow barrier-crossing events such as folding and unfolding is also a demanding test; the results will be particularly sensitive to free-energy differences between models. For instance, the free-energy difference between the OPLSua and OPLSaa models resulted in a drastic change in folding dynamics. Therefore, to achieve folding with a reasonable time-scale is an important test for a given force field, and order-of-magnitude agreement between the predicted and measured folding rates is noteworthy.
To verify that the simulations probe the same pathway as experiment, it is valuable to compare them with many experimental probes. Notably, the IR and fluorescence probes produced slightly different derived rate constants in addition to different denaturation midpoints, as shown by Yang et al. (37). The implications for kinetics are an intriguing avenue for further study.
With the exception of the initial kinetically trapped TZ3 unfolded ensemble, the OPLSaa simulations produced reasonable folding and unfolding rates. In all cases, the global internal free-energy minimum corresponded to the native conformation. Furthermore, the temperature dependence of the rates was also reasonable. Because small free-energy discrepancies can lead to large kinetic discrepancies, we believe that the TZ1 and TZ2 models captured the major features of the free-energy landscape correctly. Furthermore, we used an enormous amount of sampling to ensure that our simulation predictions were quantitative rather than anecdotal.
The series of related TrpZips are well suited for an investigation of the role of the turn in hairpin folding. We know from experiment that reversing the turn (TZ1 to TZ2) increased the folding rate, which, in conjunction with the thermodynamic data, indicated a less favorable unfolded state for TZ2 rather than a shifted transition state. As discussed above, the simulation results indicated that the transition state was very close to the native state. The type of β turn (I′,II′, or d-Pro-enforced II′) did not change this primary feature of the folding pathway.
Alone, a coincidence of rate constants cannot validate the transition-state structural details. To verify these models further, additional experiments might engineer slightly perturbed TrpZip molecules to probe the transition state. Because rates are often the primary means of experimental characterization of folding, the ability to predict folding rates accurately is an important step in the development and validation of biomolecular-simulation methods and the final understanding of how proteins fold.
Supplementary Material
Acknowledgments
We thank the folding@home volunteers who made this work possible and members of the laboratory of V.S.P. for comments. C.D.S. was supported by a predoctoral fellowship from the Howard Hughes Medical Institute. The computation was supported by National Institutes of Health Grant R01GM62868, American Chemical Society Petroleum Research Fund Grant 36028-AC4, National Science Foundation Materials Research Science and Engineering Center on Polymer Interfaces and Macromolecular Assemblies Grant DMR-9808677, and gifts from Intel and Google. This work was also supported by National Science Foundation Molecular and Cellular Biosciences Grant 0077907 (to S.J.H. and L.Q.) and Chemistry Grant 0094077 (to F.G. and D.D.).
Abbreviations: TZ, Trp zipper; OPLSaa, all-atom optimized potentials for liquid simulations; OPLSua, united-atom OPLS; T-jump, temperature jump; RMSDcα, root-mean-square α-carbon deviation; PMF, potential of mean force.
References
- 1.Snow, C. D., Nguyen, H., Pande, V. S. & Gruebele, M. (2002) Nature 420, 102–106. [DOI] [PubMed] [Google Scholar]
- 2.Qiu, L., Pabit, S. A., Roitberg, A. E. & Hagen, S. J. (2002) J. Am. Chem. Soc. 124, 12952–12953. [DOI] [PubMed] [Google Scholar]
- 3.Snow, C. D., Zagrovic, B. & Pande, V. S. (2002) J. Am. Chem. Soc. 124, 14548–14549. [DOI] [PubMed] [Google Scholar]
- 4.Kubelka, J., Eaton, W. A. & Hofrichter, J. (2003) J. Mol. Biol. 329, 625–630. [DOI] [PubMed] [Google Scholar]
- 5.Wang, M., Tang, Y., Sato, S., Vugmeyster, L., McKnight, C. J. & Raleigh, D. P. (2003) J. Am. Chem. Soc. 125, 6032–6033. [DOI] [PubMed] [Google Scholar]
- 6.Zagrovic, B., Snow, C. D., Shirts, M. R & Pande, V. S. (2002) J. Mol. Biol. 323, 927–937. [DOI] [PubMed] [Google Scholar]
- 7.Muñoz, V., Thompson, P. A., Hofrichter, J. & Eaton, W. A. (1997) Nature 390, 196–199. [DOI] [PubMed] [Google Scholar]
- 8.Pande, V. S. & Rokhsar, D. S. (1999) Proc. Natl. Acad. Sci. USA 96, 9062–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrara, P., Apostolakis, J. & Caflisch, A. (2000) J. Phys. Chem. B 104, 5000–5010. [Google Scholar]
- 10.Zagrovic, B., Sorin, E. J. & Pande, V. S. (2001) J. Mol. Biol. 313, 151–169. [DOI] [PubMed] [Google Scholar]
- 11.Garcia, A. E. & Sanbonmatsu, K. Y. (2001) Proteins 42, 345–354. [DOI] [PubMed] [Google Scholar]
- 12.Daura, X., Gademann, K., Schafer, H., Jaun, B., Seebach, D. & Gunsteren, v. W. F. (2001) J. Am. Chem. Soc. 123, 2393–2404. [DOI] [PubMed] [Google Scholar]
- 13.Zhou, R. H. & Berne, B. J. (2002) Proc. Natl. Acad. Sci. USA 99, 12777–12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu, H. W., Wang, S. M. & Brooks, B. R. (2002) J. Am. Chem. Soc. 124, 5282–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, J. & Shin, S. (2002) J. Phys. Chem. B 106, 8796–8802. [Google Scholar]
- 16.Ramirez-Alvarado M., Blanco, F. J. & Serrano, L. (2001) Protein Sci. 10, 1381–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karanicolas, J. & Brooks, C. L., 3rd (2003) Proc. Natl. Acad. Sci. USA 100, 3954–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cochran, A. G., Skelton, N. J. & Starovasnik, M. A. (2001) Proc. Natl. Acad. Sci. USA 98, 5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ponder, J. W. & Richards, F. M. (1987) J. Comput. Chem. 8, 1016–1026. [Google Scholar]
- 20.Jorgensen, W. L. & Tirado-Rives, J. (1988) J. Am. Chem. Soc. 110, 1657–1666. [DOI] [PubMed] [Google Scholar]
- 21.Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. (1996) J. Am. Chem. Soc. 117, 11225–11236. [Google Scholar]
- 22.Qiu, Q., Shenkin, P. S., Hollinger, F. P. & Still, W. C. (1997) J. Phys. Chem. A 101, 3005–3014. [Google Scholar]
- 23.Andersen, H. C. (1983) J. Comput. Phys. 52, 24–34. [Google Scholar]
- 24.McLachlan, A. D. (1982) Acta Crystallogr. A 38, 871–873. [Google Scholar]
- 25.Kabsch, W. & Sander, C. (1983) Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 26.Huang, C., Getahun, Z., Zhu, Y., Klemke, J. W., DeGrado, W. F. & Gai, F. (2002) Proc. Natl. Acad. Sci. USA 99, 2788–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shirts, M. R. & Pande, V. S. (2000) Science 290, 1903–1904. [DOI] [PubMed] [Google Scholar]
- 28.Zagrovic, B. & Pande, V. S. (2003) Nat. Struct. Biol. 10, 955–961. [DOI] [PubMed] [Google Scholar]
- 29.Shirts, M. R. & Pande, V. S. (2001) Phys. Rev. Lett. 86, 4983–4987. [DOI] [PubMed] [Google Scholar]
- 30.Fersht, A. R. (2002) Proc. Natl. Acad. Sci. USA 99, 14122–14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paci, E., Cavalli, A., Vendruscolo, M. & Caflisch, A. (2003) Proc. Natl. Acad. Sci. USA 100, 8217–8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krieger, F., Fierz, B., Bieri, O., Drewello, M. & Kiefhaber, T. (2003) J. Mol. Biol. 332, 265–274. [DOI] [PubMed] [Google Scholar]
- 33.Daggett, V. & Fersht, A. R. (2003) Nat. Rev. Mol. Cell Biol. 4, 497–502. [DOI] [PubMed] [Google Scholar]
- 34.Sadqi, M., Lapidus, L. J. & Muñoz, V. (2003) Proc. Natl. Acad. Sci. USA 100, 12117–12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makarov, D. E. & Plaxco, K. W. (2003) Protein Sci. 12, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding, F., Dokholyan, N. V., Buldyrev, S. V., Stanley, H. E. & Shakhnovich, E. I. (2003) Biophys. J. 83, 3525–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang, W. Y., Pitera, J. W., Swope, W. C. & Gruebele, M. (2004) J. Mol. Biol. 336, 241–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}