

Abstract
This article introduces the computational procedure FamClash for analyzing incompatibilities in engineered protein hybrids by using protein family sequence data. All pairs of residue positions in the sequence alignment that conserve the property triplet of charge, volume, and hydrophobicity are first identified, and significant deviations are denoted as residue–residue clashes. This approach moves beyond earlier efforts aimed at solely classifying hybrids as functional or nonfunctional by correlating the rank ordering of these hybrids based on their activity levels. Experimental testing of this approach was performed in parallel to assess the predictive ability of FamClash. As a model system, single-crossover ITCHY (incremental truncation for the creation of hybrid enzymes) libraries were prepared from the Escherichia coli and Bacillus subtilis dihydrofolate reductases, and the activities of functional hybrids were determined. Comparisons of the predicted clash map as a function of crossover position revealed good agreement with activity data, reproducing the observed V shape and matching the location of a local peak in activity.
Keywords: protein engineering, dihydrofolate reductase, residue–residue clash, computational hybrid prescreening, incremental truncation
Recent advances in protein engineering (1–5) have allowed researchers to go beyond the limitations of homology-dependent directed evolution methods. The ability to freely explore protein sequence space has revealed a number of troublesome trends. First, the lower the sequence identity of the recombined parental sequences, the smaller the percentage of the combinatorial protein library that remains functional (2, 4). This has been reported in several studies (6–8) using differing protocols, thus implicating the global nature of this effect. More troublesome is the finding that the remaining functional hybrids tend to have only residual activities. Therefore, it appears that exploring protein sequence space freely comes at the expense of severely degrading the average stability and functionality of the combinatorial library. This has motivated the development of computational methods to prescreen hybrids for their potential of being stably folded (9) and functional. These analyses then serve to direct the sampling of protein sequences by the combinatorial library toward desirable regions in sequence space. Specifically, favorable positions for junctions between fragments from different parental sequences can be identified, and restrictions can be imposed on sets of parental sequences that contribute fragments to a particular junction.
Therefore, further improvements in the stability and functionality of hybrid proteins may be attained by developing quantitative methods that identify deleterious interactions arising from residue pairs within the gene fragment combinations. To this end, Monte Carlo simulations by Bogarad and Deem (10) suggested that swapping of low-energy structures is least disruptive to protein structure. The schema algorithm (11) postulates that contacting residue pairs in the hybrids that have different parental origins are unlikely to interact favorably and thus are preferentially avoided in functional hybrids. This hypothesis has been successfully applied to a number of experimental studies (5, 11, 12) to explain the distribution of functional crossover positions. Moore and Maranas (13) proposed the second-order mean-field approach to identify residue–residue clashes in hybrids that prohibit them from folding into the correct backbone structure. Interestingly, most of the clashes identified resulted from (i) electrostatic repulsion, (ii) steric hindrance or cavity formation, and (iii) disruption of hydrogen bonds. Subsequently, Saraf and Maranas (14) proposed a rapid method to identify directly such clashes between contacting residue pairs in the protein hybrids. Comparison with sequence data of functional clones derived from many studies (4, 11, 15–17) revealed that the method was capable of classifying hybrids (crossover combinations) as functional or nonfunctional accounting for mirror chimeras. However, neither this method nor any of those discussed earlier manage to a priori rank functional hybrids with respect to their level of activity. Given that the goal of directed evolution studies is not just to retain residual activity levels but rather to reach/improve on the parental levels of activity, the ability to move beyond active/nonactive classification and computationally rank-order hybrids defines the next key challenge.
Protein family sequence (18–21) and structure (22–24) data have often been used as a basis for predicting the presence or absence of functionality. Saraf et al. (18) have shown that residue pairs that are important for functionality frequently exhibit a correlated mutation pattern, implying that the physicochemical properties of these residue pairs are also coupled. Correlation in sequence alignment has also been inferred as structural constraints, translating to residue–residue contacts (25, 26). These correlation signals are stronger when obtaining measurements using ancestral sequences inferred from phylogenetic data (27, 28). In a similar effort, Govindarajan et al. (29) showed that for many pairs of positions in protein families certain residue combinations are highly preferred. It is reasonable to expect that the same correlation pattern may extend to the properties of specific residue pairs, e.g., size, hydrophobicity, and charge (30). For example, a lysine–lysine residue pair is often substituted for an arginine–arginine owing to the similarity in the charge, volume, and hydrophobicity between these residue pairs (31–35).
In this article, we introduce the FamClash procedure for inferring the rank ordering of the relative levels of activity of protein hybrids. FamClash is motivated by the method developed by Govindarajan et al. (29) that encompasses sequence information from not only the parental sequences but also from members composing the entire protein family to be engineered. In addition, because many studies have shown that the interactions of even distal residues can have a significant impact on the activity of the hybrids (36–38), we include such noncontacting pairs in our analysis. FamClash proceeds in three steps: (i) pairs of positions in the protein family sequence alignment are identified for which a particular property triplet of charge, volume, and hydrophobicity is preferentially retained; (ii) residue pairs at these positions in the hybrids are examined to check whether they retain the properties observed in the protein family; and (iii) ranking these hybrids with respect to their probable activity based on the extent of departure from the family sequences, measured in terms of number of clashes.
FamClash is experimentally tested by constructing single-crossover hybrids of Escherichia coli and Bacillus subtilis dihydrofolate reductases (DHFRs). Results demonstrate that the specific activities of the hybrids are qualitatively consistent with FamClash predictions. This combined experimental and computational study lays the groundwork for developing approaches to protein engineering using enzymes with low sequence identity. Furthermore, valuable information is derived as to which residue positions need to be redesigned.
Materials and Methods
Hybrid Construction and Functional Screening. Plasmid constructions. Plasmid pAZE was designed for combinatorial construction and genetic selection of DHFR hybrids. To build this plasmid, the lacIQ gene was PCR-amplified from pMAL-c2x (New England Biolabs) with NheI-tailed primers, digested with NheI, and ligated into the SpeI site of pZE12-luc (39). The ribosome-binding site and luc gene were removed with EcoRI (blunted) and XbaI, and this piece was replaced with a SacII (blunted), XbaI fragment from pDIM-N2 (40). Residues 1–120 of E. coli DHFR were PCR-amplified, digested with NdeI and BamHI, and ligated into pMAC (A.R.H. and S.J.B., unpublished results) cut with the same enzymes. Residues 31–168 of B. subtilis DHFR were PCR-amplified, digested with PstI and SpeI, and ligated downstream of the E. coli fragment on pMAC. The NdeI–SpeI piece was removed from pMAC and ligated into pAZE, and the resulting plasmid was named pAZE-EB and confirmed by DNA sequencing. A complementary plasmid for B. subtilis N-terminal DHFR hybrids, named pAZE-BE, was constructed from fragments 1–121 of B. subtilis and 31–159 of E. coli DHFRs. An additional plasmid with a fixed crossover at position 62 was constructed in vector pAZE by overlap extension (41). Primer sequences will be provided on request.
Construction of DHFR hybrid libraries. Plasmids pAZE-EB and pAZE-BE were linearized at a unique SalI site between the E. coli and B. subtilis fragments. The ITCHY (incremental truncation for the creation of hybrid enzymes) PCR technique was used to construct libraries of E. coli/B. subtilis (EB) DHFR hybrids in both orientations (42). Libraries were initially constructed and frozen in E. coli strain DH5α-E.
Selection of DHFR hybrids. E. coli strain MH829 has a deletion of the DHFR (folA) gene and was used for the in vivo selection of functional DHFR hybrids (43). Library plasmid was purified and electroporated into strain MH829. Transformed cells were washed twice in minimal media A (MMA) (41) and plated on 245 × 245-mm library plates of MMA supplemented with 0.5% glycerol, 0.6 mM arginine, 50 μg/ml thymidine, 25 μg/ml kanamycin, 100 μg/ml ampicillin, and 1 mM MgSO4. Selections were performed at room temperature, and isopropyl β-d-thiogalactoside was added to induce expression, usually at 250 μM final concentration. Isolates were restreaked onto the same media and grown at 30°C, and plasmids were sequenced to identify crossover positions. All DNA sequencing was performed at the Nucleic Acids Facility of Pennsylvania State University.
DHFR assays. DHFR ligands were prepared as described (44). The specific activities of WT and hybrid DHFRs were determined in cell-free lysates. The plasmid pAZE (described above) was used to express all DHFR proteins, and to increase expression, lacI was destroyed on all plasmids by EcoRV and SfoI digests. Plasmids were transformed into DHFR mutant strain MH829, and 50 ml of cultures was grown at 30°C in LB broth supplemented with 100 μg/ml ampicillin, 50 μg/ml thymidine, and 0.5 mM isopropyl β-d-thiogalactoside. Cultures were grown to an absorbance of 1.0 at 600 nm, centrifuged, and resuspended in 25 ml of 20 mM Tris·HCl, pH 7.7, with 2 mM DTT. Cells were centrifuged again, resuspended in 1 ml of buffer, and broken by sonication. Insoluble material was removed, and lysates were assayed on a Cary 100 Bio UV-Vis spectrophotometer (Varian), held at 25°C with a water-jacketed cuvette holder. Cell-free lysate was preincubated 3 min in MTEN buffer, pH 7.0, containing 1 mM DTT and 100 μM cofactor to avoid hysteresis (45), and the reaction was initiated by adding 100 μM substrate. To follow the reaction, the decrease in absorbance was monitored at 340 nm (Δε340 = 13.2 mM–1·cm–1).
FamClash Method. FamClash relies on identifying residue positions in the parental protein family sequences for which the sum of residue properties are conserved. Hybrids are then evaluated with respect to whether they conform to the identified conserved properties. Any deviations are denoted as residue–residue clashes. This is accomplished by first analyzing the family sequence alignment obtained from the pfam database (45) by using scoring matrices. These scoring matrices encode physicochemical properties of amino acids such as charge (46), volume (47), and hydrophobicity (48, 49). The additive charge (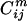 ), volume (
), volume (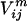 ), and hydrophobicity (
), and hydrophobicity (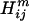 ) for a pair of residues k, l at positions i and j in sequence m is defined as the sum of the charge (c), volume (v), and normalized average hydrophobicity metric (h) of residues k and l:
) for a pair of residues k, l at positions i and j in sequence m is defined as the sum of the charge (c), volume (v), and normalized average hydrophobicity metric (h) of residues k and l:
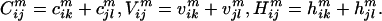 |
[1] |
All 20 × 20 pairwise residue combinations are partitioned into 3D property bins derived by subdividing the observed property ranges (see Fig. 1). A residue pair populates a particular bin φpqr if all of its properties lie within the rectangle defined by: [(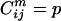 ), (
), ( ), (
), ( )] as shown in Fig. 1. Note that the total charge p of a residue pair can assume only one of five distinct values (i.e., –2, –1, 0, 1, and 2). In contrast, volume (q) and hydrophobicity (r) values may vary continuously within 10 equally sized bins ranging between 0 and 300 Å3 and –2.30 to 3.7 kcal/mol, resulting in ΔV and ΔH values of 30 Å3 and 0.6 kcal/mol, respectively.
)] as shown in Fig. 1. Note that the total charge p of a residue pair can assume only one of five distinct values (i.e., –2, –1, 0, 1, and 2). In contrast, volume (q) and hydrophobicity (r) values may vary continuously within 10 equally sized bins ranging between 0 and 300 Å3 and –2.30 to 3.7 kcal/mol, resulting in ΔV and ΔH values of 30 Å3 and 0.6 kcal/mol, respectively.
Fig. 1.

Residue pairs k, l whose properties are within a specified range in terms of charge (p), volume (q), and hydrophobicity (r) are said to belong to the same 3D property bin φpqr (i.e., Ckl = p, q ≤ Vkl < q + Δv, r ≤ Hkl < r + ΔH). Property values for the residue pair in the hybrid that are significantly different from those observed in the protein family denote a clash.
A pair of positions in the sequence alignment is deemed “conserved” if at least 20% of its residue pairs, including the parental residue pairs, populate the same 3D property bin φpqr. Conservation of additive property values signify that any significant deviations from the observed ranges may lead to residue–residue clashes (see Fig. 1). To safeguard against identifying conservation caused by chance, the mutual information index ( ) between all pairs of positions in the alignment for the corresponding property bin φpqr is calculated. Chance occurrences are revealed when the occupancy frequencies of residues k, l at two positions i and j (aik, ajl) are independent. In such a case, the joint probability P(aik, ajl) of observing an amino acid k at position i and at the same time amino acid l at position j is equal to the product P(aik)P(ajl) of the individual probabilities of occupancy for these two residues and position. The
) between all pairs of positions in the alignment for the corresponding property bin φpqr is calculated. Chance occurrences are revealed when the occupancy frequencies of residues k, l at two positions i and j (aik, ajl) are independent. In such a case, the joint probability P(aik, ajl) of observing an amino acid k at position i and at the same time amino acid l at position j is equal to the product P(aik)P(ajl) of the individual probabilities of occupancy for these two residues and position. The 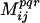 score quantifies the degree of dependence (or independence) between the distributions of residues at the two positions:
score quantifies the degree of dependence (or independence) between the distributions of residues at the two positions:
 |
[2] |
Note that completely independent residue positions will have a  score exactly equal to zero. The higher the value of
score exactly equal to zero. The higher the value of 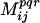 , the stronger the extent of covariance between positions i, j for property values within bin φpqr. A pair of positions i, j and bin φpqr is considered to be statistically significant if its
, the stronger the extent of covariance between positions i, j for property values within bin φpqr. A pair of positions i, j and bin φpqr is considered to be statistically significant if its  score is greater than a cutoff value (Mc).
score is greater than a cutoff value (Mc).
A bootstrap replicate analysis is used to determine the threshold value (Mc) for  scores. This establishes how likely is a
scores. This establishes how likely is a  score greater than Mc to occur by chance alone. First, two vectors are extracted from the sequence alignment (i.e., columns of residues at positions i, j from the alignment). Next, multiple copies (≈105) of each of these vectors (bootstrap replicates) are generated by randomly choosing residues by permuting the original vector. Finally,
score greater than Mc to occur by chance alone. First, two vectors are extracted from the sequence alignment (i.e., columns of residues at positions i, j from the alignment). Next, multiple copies (≈105) of each of these vectors (bootstrap replicates) are generated by randomly choosing residues by permuting the original vector. Finally, 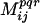 scores for all 105 pairs of randomized vectors are computed for each property bin φpqr. This distribution of scores serves to elucidate the probability of having a
scores for all 105 pairs of randomized vectors are computed for each property bin φpqr. This distribution of scores serves to elucidate the probability of having a 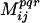 score greater than a given cutoff score (Mc) by chance. This probability, also known as the P value, is calculated as the ratio of the total number of pairs yielding scores above Mc divided by the total number of pairs in the distribution. The
score greater than a given cutoff score (Mc) by chance. This probability, also known as the P value, is calculated as the ratio of the total number of pairs yielding scores above Mc divided by the total number of pairs in the distribution. The  score corresponding to a P value of 5 × 10–3 is chosen as the cutoff.
score corresponding to a P value of 5 × 10–3 is chosen as the cutoff.
A clash is defined to occur between two statistically significant residue positions i, j in the hybrid (residue at position i retained parental sequence p1 and j from p2) if at least one of the following criteria is met:
 |
[3] |
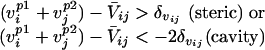 |
[4] |
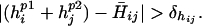 |
[5] |
Because cavity formation tends to be less problematic than steric hindrances (see ref. 14) a more relaxed cutoff for cavity formation is chosen. Here, C̄ij, V̄ij, and H̄ij are the mean charge, volume, and hydrophobicity, respectively, found to be conserved between positions i and j in the protein family members. Assessing the departure away from the mean property values for any pair of positions i, j, identified as conserved, requires the definition of cutoff ranges for volume (δvij) and hydrophobicity (δhij) as follows:
 |
[6] |
A lower bound on the cutoff ranges is set at 10% of the mean values to prevent denoting small deviations in the properties as clashes. Table 1 summarizes the steps of FamClash procedure.
Table 1. Summary of the FamClash procedure.
| Step 1. | Identify all pairs of positions i,j in the sequence alignment where at least 20% of the residue pairs have change, volume, and hydrophobicity that lie in the same 3D property bin φpqr. | |
| Step 2. | Evaluate mutual information 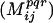 score for all pairs of positions i,j denoted as conserved for the corresponding bin φpqr. score for all pairs of positions i,j denoted as conserved for the corresponding bin φpqr. |
|
| Step 3. | Perform bootstrap replicate analysis and select positions i,j that meet the P value cutoff of 5 × 10-3. | |
| Step 4. | Investigate the selected residue positions in the hybrids for clashes based on the following criteria: | |
|
||
| Step 5. | Hybrids are given a score equal to the number of clashes identified in step 4. |

Results and Discussion
Library Construction and Hybrid Isolation. Two ITCHY libraries were constructed from the E. coli/B. subtilis (EB) or the B. subtilis/E. coli (BE) DHFR pairs sharing a 44% sequence identity at the protein level. The naive library sizes were 1.9 × 106 and 2.0 × 106 members, respectively, providing complete coverage of the minimum library size of 7.3 × 104 [(270 bp)2]. A genetic selection for functional hybrids was developed by using an E. coli strain containing a complete deletion of DHFR (43). The nature of the selection required the use of inactive DHFR fragments to make ITCHY libraries, which limited the crossover window to residues 31–120 (see Materials and Methods). After selection, hybrids were picked at random and sequenced, 55 from the EB library and 10 from the BE library. DNA sequencing showed that 30 of the EB library members had duplications of various sizes, and that all of the BE library members had duplications.
The number of DHFR hybrids with duplications was somewhat unexpected, especially considering how rarely they were identified in ITCHY libraries of GAR transformylases (40, 42). In the BE library, attempts were made to identify perfect crossovers (i.e., containing no duplications) by removing hybrids larger than WT DHFR by gel electrophoresis (data not shown). However, even after sorting, all BE hybrids contained at least one or two amino acid duplications, many with considerably larger ones. The stringency of the genetic selection was designed to be very low, accepting DHFR hybrids with kcat values 103-fold lower than WT (data not shown), which may have contributed to the high number of duplications observed. To simplify the analysis, 13 perfect crossovers from the EB library were selected for further studies. These DHFR hybrids were chosen to provide the best distribution across the 90-aa crossover window (see Fig. 2), and all hybrids containing duplications were not pursued further.
Fig. 2.

Predicted clashes in EB hybrids are shown for all single crossover EB hybrids. A clash between any two residue positions is shown as an arc. The specific activity (μmol/min per mg) and number of clashes in each hybrid are also shown. Note that the 0 and 159 crossover positions correspond to the parental B. subtilis and E. coli DHFR sequences, respectively.
FamClash Analysis of EB Library. Conserved pairs of positions for the two aligned DHFR sequences were identified by evaluating the  scores, as outlined in Materials and Methods. The DHFR protein family sequence alignment was obtained by using the pfam database (45), including a total of 265 DHFR sequences (as of Nov. 15, 2003). Statistically significant residue positions were identified by the bootstrap replicate analysis. Residue pairs in the EB and BE libraries corresponding to the statistically important residue positions (P <5 × 10–3) were identified, and their properties were investigated for consistency with the protein family sequence data. Specifically, we found that 14 residue pairs for the EB hybrids showed significant deviations in the property triplet from what is found to be conserved among the corresponding residue positions in the protein family sequences (see Fig. 2 and Table 2). Only six such pairs were identified for hybrids with a BE directionality (Table 2). We observed that most of these clashes are caused by large changes in the total volume of the residue pairs. In fact, many of the identified clashes in the hybrids are a direct consequence of reversed orientation of residue pairs in the two parental sequences. For example, the residue pair 36/135 in E. coli is a lysine and a serine, whereas in B. subtilis the same pair involves the same residues but in a reversed order (see Table 2). This consequently results in a steric hindrance in the EB hybrid and a cavity formation in the BE hybrid. Both hydrophobicity and charge were found to be fairly conserved, and thus very few clashes caused by deviation from charge and hydrophobicity values were identified. Table 2 lists all of the identified clashes between residue pairs in the hybrids (also see Fig. 2). Notably, we found that many of the predicted clashes are between distant residue pairs.
scores, as outlined in Materials and Methods. The DHFR protein family sequence alignment was obtained by using the pfam database (45), including a total of 265 DHFR sequences (as of Nov. 15, 2003). Statistically significant residue positions were identified by the bootstrap replicate analysis. Residue pairs in the EB and BE libraries corresponding to the statistically important residue positions (P <5 × 10–3) were identified, and their properties were investigated for consistency with the protein family sequence data. Specifically, we found that 14 residue pairs for the EB hybrids showed significant deviations in the property triplet from what is found to be conserved among the corresponding residue positions in the protein family sequences (see Fig. 2 and Table 2). Only six such pairs were identified for hybrids with a BE directionality (Table 2). We observed that most of these clashes are caused by large changes in the total volume of the residue pairs. In fact, many of the identified clashes in the hybrids are a direct consequence of reversed orientation of residue pairs in the two parental sequences. For example, the residue pair 36/135 in E. coli is a lysine and a serine, whereas in B. subtilis the same pair involves the same residues but in a reversed order (see Table 2). This consequently results in a steric hindrance in the EB hybrid and a cavity formation in the BE hybrid. Both hydrophobicity and charge were found to be fairly conserved, and thus very few clashes caused by deviation from charge and hydrophobicity values were identified. Table 2 lists all of the identified clashes between residue pairs in the hybrids (also see Fig. 2). Notably, we found that many of the predicted clashes are between distant residue pairs.
Table 2. Positions, residue pairs, and nature of clashes in the hybrids.
| Hybrid | Residue positions | Residue pair, parent 1 | Residue pair, parent 2 | Residue pair, hybrid | Nature of clash* |
|---|---|---|---|---|---|
| EB | 17/63 | ES | DT | ET | Steric |
| EB | 30/97 | WG | YA | WA | Steric/hyd |
| EB | 36/135 | LS | SL | LL | Steric/hyd |
| EB | 47/149 | WH | FY | WY | Steric/hyd |
| EB | 62/63 | LS | VT | LT | Steric |
| EB | 62/69 | LD | VE | LE | Steric |
| EB | 62/78 | LV | VL | LL | Steric |
| EB | 62/99 | LV | VL | LL | Steric |
| EB | 62/104 | LL | VF | LF | Steric |
| EB | 80/127 | ED | DE | EE | Steric |
| EB | 80/156 | EL | DY | EY | Steric |
| EB | 98/146 | RQ | QK | RK | Chg/steric/hyd |
| EB | 127/129 | DE | ED | DD | Cavity |
| EB | 129/156 | EL | DY | EY | Steric |
| BE | 17/63 | DT | ES | DS | Cavity |
| BE | 36/135 | SL | LS | SS | Cavity/hyd |
| BE | 80/127 | DE | ED | DD | Cavity |
| BE | 92/103 | FL | MF | FF | Hyd |
| BE | 98/146 | QK | RQ | Chg/steric/hyd | |
| BE | 127/129 | ED | DE | EL | Steric |
Clashes formed may be caused by departure from volume (steric hindrance-steric or cavity formation), charge (chg), or hydrophobicity (hyd) values observed in the protein family.
Fig. 3 shows the total number of identified clashes for the single crossover incremental truncation EB and BE libraries. Notably, the BE hybrids have about half as many clashes as the EB hybrids. Also, five of the six clashes identified in BE hybrids are also present in the EB hybrids (see Table 2). Interestingly, in four of five cases of volume clashes common to both libraries, EB hybrids retain residue pairs with larger side chains presumably leading to steric hindrances, whereas in the BE library a corresponding volume reduction was observed. This result suggests that BE hybrids, by avoiding steric clashes, are more likely to retain functionality in comparison with their EB mirror chimeras. This finding is consistent with the experimental results in which BE hybrids are found to be much more tolerant to insertions.
Fig. 3.

The number of clashes in each of the single crossover EB (solid line) and BE (dashed line) DHFR hybrids are plotted against crossover position.
DHFR Hybrid Characterization and Analysis. Specific activities of the EB hybrids were determined in lysates of the E. coli DHFR mutant MH829. The hybrids with the lowest activity, crossovers 55–96, all reside in the adenosine binding subdomain. This region of DHFR is directly involved in NADPH binding (50), and splicing together residues in this subdomain from divergent DHFRs could have dramatically affected cofactor binding, implying the thermodynamic dissociation constants, Kd values, are significantly affected. Molecular dynamics simulations have identified anticorrelations between the 55–96 region and both the Met-20 loop (residues 14–24) and βF-G loop (residues 116–125), suggesting that the protein dynamics of these hybrids also might have been perturbed (51). Further, functional connectivities between the cofactor and substrate binding sites have been observed for DHFR (52, 53), which could be affected by crossovers in the NADPH binding region.
The DHFR activity was plotted against crossover position and compared with the FamClash predictions (Fig. 4). Log–log plots are frequently used to correlate activity versus mutational data. This relationship implies that the change in free energy is proportional to the log of the total number of mutations alluding to a continuously diminishing effect of additional mutations. Also, schema results (12) have demonstrated that the logarithm of the fraction of functional recombinants is proportional to the negative of the logarithm of schema disruptions. In analogy with these results, we decided to use a log-log plot to contrast activities and total number of clashes. As shown in Fig. 4, the trend of DHFR activities correlated surprisingly well with the number of clashes in each hybrid and appears to exhibit a V shape, although the small sample size could have contributed to this observation. It is possible that many perfect crossovers in the gaps shown in Fig. 4 are active DHFR hybrids, and the activities of these potential hybrids may deviate from the observed trend. The stringency of the selection could be raised to enrich for only the most active hybrids. However, the results from previous ITCHY libraries suggested there would be valleys of low activity (40), and the goal of this work was to obtain the most complete crossover distribution possible for comparison with computational predictions.
Fig. 4.

Plot of specific activities (▪) of the 13 EB DHFR hybrids against crossover position. The total number of identified clashes (▵) [i.e., log(1 + number of clashes)] for each one of these hybrids is also shown. Note that the 0 and 159 crossover positions correspond to the parental B. subtilis and E. coli DHFR sequences, respectively. The specific activity and number of clashes for hybrid 62 are shown separately.
Notably, as shown in Fig. 4, EB hybrid 79 has fewer clashes than the neighboring hybrids. The FamClash method predicted that residue 62 from E. coli clashes with residue 78 from B. subtilis and residue 80 from E. coli clashes with residues 127 and 156 from B. subtilis. Both of these clashes are absent in EB hybrid 79, and consistent with these predictions, this hybrid showed considerably better activity than flanking hybrids 73 and 81. In addition, crossover position 62 was predicted to have the maximum number of clashes. This hybrid was subsequently constructed and assayed, and the activity of this hybrid was poor, consistent with the downward trend observed in the plot. However, the activity of hybrid 62 was noticeably higher than hybrid 73, which was predicted to have fewer clashes. This finding is consistent with a diminishing effect of increasing number of clashes in analogy with the observation that increased number of mutations do not additively effect activity (54). Also, increasing numbers of clashes may not have the predicted additive effect on enzyme activity, perhaps because of the inability at this time to rank the importance of each clash and to capture higher-order effects.
Summary
In the current implementation of FamClash, all clashes are considered equally deleterious. One would expect that some clashes may be more severe than others and, therefore, may have significant impact on activity, sometimes even greater than the combined effect of more than one clash. Moreover, more than two residues may be involved in retaining a particular property that cannot be identified when analyzing just pairs of residues, alluding to the limitations of the FamClash procedure. Nevertheless, the results presented here show that FamClash is quite successful at qualitatively predicting the pattern of the specific activity of the hybrids. Similar trends have been observed for other systems not presented here. More importantly, by identifying these clashes, this method provides valuable insights for protein engineering interventions to remedy these clashes. Specifically, by appropriately substituting residues at the clashing positions, significant improvement in the activity of the hybrids can be achieved.
Acknowledgments
We thank Gregory L. Moore and Piyush Agarwal for valuable help and suggestions. This work was supported by National Science Foundation Award BES0331047 (to C.D.M.) and National Institutes of Health Grant GM 24129 (to S.J.B.). A.R.H. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation Grant DRG-1729-02.
Abbreviations: DHFR, dihydrofolate reductase; ITCHY, incremental truncation for the creation of hybrid enzymes; EB, Escherichia coli/Bacillus subtilis; BE, B. subtilis/E. coli.
See Commentary on page 3997.
References
- 1.Ostermeier, M., Nixon, A. E. & Benkovic, S. J. (1999) Bioorg. Med. Chem. 7, 2139–2144. [DOI] [PubMed] [Google Scholar]
- 2.Sieber, V., Martinez, C. A. & Arnold, F. H. (2001) Nat. Biotechnol. 19, 456–460. [DOI] [PubMed] [Google Scholar]
- 3.Richardson, T. H., Tan, X., Frey, G., Callen, W., Cabell, M., Lam, D., Macomber, J., Short, J. M., Robertson, D. E. & Miller, C. (2002) J. Biol. Chem. 277, 26501–26507. [DOI] [PubMed] [Google Scholar]
- 4.Lutz, S., Ostermeier, M., Moore, G. L., Maranas, C. D. & Benkovic, S. J. (2001) Proc. Natl. Acad. Sci. USA 98, 11248–11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hiraga, K. & Arnold, F. H. (2003) J. Mol. Biol. 330, 287–296. [DOI] [PubMed] [Google Scholar]
- 6.Ostermeier, M. (2003) Trends Biotechnol. 21, 244–247. [DOI] [PubMed] [Google Scholar]
- 7.Ness, J. E., Kim, S., Gottman, A., Pak, R., Krebber, A., Borchert, T. V., Govindarajan, S., Mundorff, E. C. & Minshull, J. (2002) Nat. Biotechnol. 20, 1251–1255. [DOI] [PubMed] [Google Scholar]
- 8.Moore, G. L. & Maranas, C. D. (2004) AIChE J., in press.
- 9.Kuhlman, B., Dantas, G., Ireton, G. C., Varani, G., Stoddard, B. L. & Baker, D. (2003) Science 302, 1364–1368. [DOI] [PubMed] [Google Scholar]
- 10.Bogarad, L. D. & Deem, M. W. (1999) Proc. Natl. Acad. Sci. USA 96, 2591–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voigt, C. A., Martinez, C., Wang, Z. G., Mayo, S. L. & Arnold, F. H. (2002) Nat. Struct. Biol. 9, 553–558. [DOI] [PubMed] [Google Scholar]
- 12.Meyer, M. M., Silberg, J. J., Voigt, C. A., Endelman, J. B., Mayo, S. L., Wang, Z. G. & Arnold, F. H. (2003) Protein Sci. 12, 1686–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore, G. L. & Maranas, C. D. (2003) Proc. Natl. Acad. Sci. USA 100, 5091–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saraf, M. C. & Maranas, C. D. (2003) Protein Eng. 16, 1025–1034. [DOI] [PubMed] [Google Scholar]
- 15.Joern, J. M., Meinhold, P. & Arnold, F. H. (2002) J. Mol. Biol. 316, 643–656. [DOI] [PubMed] [Google Scholar]
- 16.Kikuchi, M., Ohnishi, K. & Harayama, S. (2000) Gene 243, 133–137. [DOI] [PubMed] [Google Scholar]
- 17.Hansson, L. O., Bolton-Grob, R., Widersten, M. & Mannervik, B. (1999) Protein Sci. 8, 2742–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saraf, M. C., Moore, G. L. & Maranas, C. D. (2003) Protein Eng. 16, 397–406. [DOI] [PubMed] [Google Scholar]
- 19.Lockless, S. W. & Ranganathan, R. (1999) Science 286, 295–299. [DOI] [PubMed] [Google Scholar]
- 20.Gaucher, E. A., Gu, X., Miyamoto, M. M. & Benner, S. A. (2002) Trends Biochem. Sci. 27, 315–321. [DOI] [PubMed] [Google Scholar]
- 21.del Sol Mesa, A., Pazos, F. & Valencia, A. (2003) J. Mol. Biol. 326, 1289–1302. [DOI] [PubMed] [Google Scholar]
- 22.Lichtarge, O. & Sowa, M. E. (2002) Curr. Opin. Struct. Biol. 12, 21–27. [DOI] [PubMed] [Google Scholar]
- 23.Landgraf, R., Xenarios, I. & Eisenberg, D. (2001) J. Mol. Biol. 307, 1487–1502. [DOI] [PubMed] [Google Scholar]
- 24.Liang, M. P., Brutlag, D. L. & Altman, R. B. (2003) Pac. Symp. Biocomput. 8, 204–215. [DOI] [PubMed] [Google Scholar]
- 25.Gobel, U., Sander, C., Schneider, R. & Valencia, A. (1994) Proteins 18, 309–317. [DOI] [PubMed] [Google Scholar]
- 26.Larson, S. M., Di Nardo, A. A. & Davidson, A. R. (2000) J. Mol. Biol. 303, 433–446. [DOI] [PubMed] [Google Scholar]
- 27.Gaucher, E. A., Thomson, J. M., Burgan, M. F. & Benner, S. A. (2003) Nature 425, 285–288. [DOI] [PubMed] [Google Scholar]
- 28.Fukami-Kobayashi, K., Schreiber, D. R. & Benner, S. A. (2002) J. Mol. Biol. 319, 729–743. [DOI] [PubMed] [Google Scholar]
- 29.Govindarajan, S., Ness, J. E., Kim, S., Mundorff, E. C., Minshull, J. & Gustafsson, C. (2003) J. Mol. Biol. 328, 1061–1069. [DOI] [PubMed] [Google Scholar]
- 30.Di Nardo, A. A., Larson, S. M. & Davidson, A. R. (2003) J. Mol. Biol. 333, 641–655. [DOI] [PubMed] [Google Scholar]
- 31.Taylor, W. R. & Hatrick, K. (1994) Protein Eng. 7, 341–348. [DOI] [PubMed] [Google Scholar]
- 32.Altschuh, D., Lesk, A. M., Bloomer, A. C. & Klug, A. (1987) J. Mol. Biol. 193, 693–707. [DOI] [PubMed] [Google Scholar]
- 33.Ptitsyn, O. B. & Finkel'shtein, A. V. (1970) Dokl. Akad. Nauk SSSR 195, 221–224. [PubMed] [Google Scholar]
- 34.Shindyalov, I. N., Kolchanov, N. A. & Sander, C. (1994) Protein Eng. 7, 349–358. [DOI] [PubMed] [Google Scholar]
- 35.Lesk, A. M. & Chothia, C. (1980) J. Mol. Biol. 136, 225–270. [DOI] [PubMed] [Google Scholar]
- 36.Rod, T. H., Radkiewicz, J. L. & Brooks, C. L., 3rd (2003) Proc. Natl. Acad. Sci. USA 100, 6980–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gong, X. S., Wen, J. Q., Fisher, N. E., Young, S., Howe, C. J., Bendall, D. S. & Gray, J. C. (2000) Eur. J. Biochem. 267, 3461–3468. [DOI] [PubMed] [Google Scholar]
- 38.Suel, G. M., Lockless, S. W., Wall, M. A. & Ranganathan, R. (2003) Nat. Struct. Biol 10, 59–69. [DOI] [PubMed] [Google Scholar]
- 39.Lutz, R. & Bujard, H. (1997) Nucleic Acids Res. 25, 1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ostermeier, M., Shim, J. H. & Benkovic, S. J. (1999) Nat. Biotechnol. 17, 1205–1209. [DOI] [PubMed] [Google Scholar]
- 41.Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A. & Struhl, K. (2002) Short Protocols in Molecular Biology (Wiley, New York).
- 42.Lutz, S., Ostermeier, M. & Benkovic, S. J. (2001) Nucleic Acids Res. 29, E16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herrington, M. B. & Chirwa, N. T. (1999) Can. J. Microbiol. 45, 191–200. [PubMed] [Google Scholar]
- 44.Rajagopalan, P. T., Lutz, S. & Benkovic, S. J. (2002) Biochemistry 41, 12618–12628. [DOI] [PubMed] [Google Scholar]
- 45.Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., Griffiths-Jones, S., Howe, K. L., Marshall, M. & Sonnhammer, E. L. (2002) Nucleic Acids Res. 30, 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klein, P., Kanehisa, M. & DeLisi, C. (1984) Biochim. Biophys. Acta 787, 221–226. [DOI] [PubMed] [Google Scholar]
- 47.Krigbaum, W. R. & Komoriya, A. (1979) Biochim. Biophys. Acta 576, 204–248. [DOI] [PubMed] [Google Scholar]
- 48.Cid, H., Bunster, M., Canales, M. & Gazitua, F. (1992) Protein Eng. 5, 373–375. [DOI] [PubMed] [Google Scholar]
- 49.Engelman, D. M., Steitz, T. A. & Goldman, A. (1986) Annu. Rev. Biophys. Biophys. Chem. 15, 321–353. [DOI] [PubMed] [Google Scholar]
- 50.Sawaya, M. R. & Kraut, J. (1997) Biochemistry 36, 586–603. [DOI] [PubMed] [Google Scholar]
- 51.Radkiewicz, J. L. & Brooks, C. L., 3rd (2000) J. Am. Chem. Soc. 122, 225–231. [Google Scholar]
- 52.Fierke, C. A., Johnson, K. A. & Benkovic, S. J. (1987) Biochemistry 26, 4085–4092. [DOI] [PubMed] [Google Scholar]
- 53.Pan, H., Lee, J. C. & Hilser, V. J. (2000) Proc. Natl. Acad. Sci. USA 97, 12020–12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang, Z., Wagner, C. R. & Benkovic, S. J. (1994) Biochemistry 33, 11576–11585. [DOI] [PubMed] [Google Scholar]